

Abstract
We have investigated the communication between subunits in replication factor C (RFC) from Archaeoglobus fulgidus. Mutation of the proposed arginine finger in the small subunits results in a complex that can still bind ATP but has impaired clamp-loading activity, a process that normally only requires binding of nucleotide. The small subunit alone forms a hexameric ring that is six-fold symmetric in the absence of ATP. However, this symmetry is broken when the nucleotide is bound to the complex. A conformational change associated with nucleotide binding may relate to the opening of PCNA rings by RFC during the loading reaction. The structures also reveal the importance of the N-terminal helix of each subunit at the ATP-binding site. Analysis of mutant protein complexes containing subunits lacking this N-terminal helix reveals key distinct regulatory roles during clamp loading that are different for the large and small subunits in the RFC complex.
Keywords: clamp-loader, DNA replication, PCNA, RFC
Introduction
Replicative polymerases from eukaryotes to prokaryotes obtain processivity using ring-shaped DNA sliding clamps that are loaded onto DNA by clamp-loader proteins in ATP-dependent reactions. Although the amino-acid sequences and the protein complex compositions differ between the various systems, both the overall structure of the clamp/clamp-loader proteins and the molecular mechanisms of the clamp-loading process appear to be conserved (Jeruzalmi et al, 2002). The Escherichia coli γ complex, which has an in vivo composition of γ3δδ′χψ (Pritchard et al, 2000), is the most thoroughly analysed clamp loader. A heteropentamer consisting only of γ3δδ′ has been shown to be able to load the dimeric β sliding clamp onto DNA (Onrust and O'Donnell, 1993). Crystal structures of the γ3δδ′ complex and of the δ subunit in a complex with a mutant monomeric form of β have been published (Jeruzalmi et al, 2001a, 2001b). Together with earlier biochemical data (Naktinis et al, 1995), these crystal structures suggested that the δ subunit contacts the β clamp and traps β in a conformation where one of the two clamp interfaces is open. ATP binding alone, but not hydrolysis, is essential for the E. coli clamp loader to open the sliding clamp and to load it onto a primer/template junction of DNA (Turner et al, 1999). Biochemical studies have shown that this is also true for replication factor C (RFC), the eukaryotic and archaeal clamp loader (Gomes and Burgers, 2001; Seybert and Wigley, 2004). Unlike the E. coli clamp loader, eukaryotic RFC consists of five different subunits (RFC1–RFC5) (Cullmann et al, 1995). However, like the minimal E. coli clamp loader (γ3δδ′), the composition of RFC is heteropentameric (Lee et al, 1991). The crystal structure of a mutant yeast RFC complexed with PCNA revealed details of the interactions within the complex (Bowman et al, 2004). Despite being crystallised with a very poorly hydrolysable ATP analogue (ATPγS), the structure determined was of a catalytically inactive mutant protein in which the four ‘arginine fingers' of the small subunits were replaced by glutamine. Furthermore, even though the structure contains bound nucleotides and it has been established that clamp loading by RFC requires ATP binding but not hydrolysis (Gomes et al, 2001; Seybert and Wigley, 2004), the complex is clearly not in a conformation that could load PCNA onto DNA because the PCNA ring is topologically closed.
With sequence identities of 30–40%, the clamp loaders of Archaea seem to be a closely related, yet simplified, version of their eukaryotic counterparts. Thus, in contrast to eukaryotic clamp loaders in which each of the five different RFC subunits have been found to be individually essential (Cullmann et al, 1995), only two RFC-like subunits are found in each of the completely sequenced archaeal genomes (Cann and Ishino, 1999). The conserved RFC boxes II–VIII can be found in both the large and the small subunits that constitute archaeal clamp loaders (Cann and Ishino, 1999). Archaeal clamp loaders from Sulfolobus solfataricus, Methanobacterium thermoautotrophicum, Pyrococcus furiosus and Archaeoglobus fulgidus have been isolated and shown to stimulate the processivity of their replicative polymerases in PCNA-dependent reactions (Kelman and Hurwitz, 2000; Pisani et al 2000; Cann et al, 2001; Seybert and Wigley, 2004). The two subunits of archaeal RFC form a complex with 1:4 (large to small) stoichiometry (Pisani et al, 2000; Cann et al, 2001; Seybert et al, 2002).
Electron microscopy (EM) studies of the wild-type archaeal RFC/PCNA complex from P. furiosus (Shiomi et al, 2000; Miyata et al, 2004, 2005) have revealed important insights into the assembly of the complex, with results that differed from the crystal structure of the mutant yeast RFC/PCNA (Bowman et al, 2004). Although the earlier low-resolution (23 Å) images were misinterpreted, once higher resolution (12 Å) images were available it became clear exactly how the complex was assembled. Unlike the yeast structure, the PCNA ring is opened in the EM images. This appears to be a consequence of the PCNA interacting with multiple subunits of RFC that are arranged in a spiral, as seen in all clamp-loader structures. This results in a conformation of the PCNA that is reminiscent of a ‘lock washer'. However, the crack in the PCNA ring is not wide enough (only 5 Å) to allow passage of duplex DNA, but could allow single-stranded DNA to enter the ring, although the EM analysis also showed that duplex DNA was bound at the centre of the RFC/PCNA complex. As a result, the authors suggest that either PCNA is loaded onto the single-strand DNA ahead of the template and the RFC/PCNA complex, then slides back onto the primer template junction or, alternatively, that there is a conformational change that might open up the ring further to allow entry of duplex DNA directly, the nature of which is presently unknown. However, just such a conformational change was observed upon binding of ATP using EM, atomic force microscopy and biochemical methods (Shiomi et al, 2000), although the limited resolution of these studies precluded details of the nature of this conformational change.
Using mutant archaeal clamp loaders deficient in either ATP binding or hydrolysis in different subunits, it was revealed that the different subunits use ATP binding and hydrolysis in distinct ways at different steps in the loading process (Seybert and Wigley, 2004). Binding of nucleotide by the large subunit and three of the four small subunits is sufficient for clamp loading. However, ATP hydrolysis by the small subunits is required for release of PCNA to allow the subsequent formation of the complex between PCNA and the polymerase. By contrast, ATP hydrolysis by the large subunit is required for catalytic clamp loading.
In the present study, we have investigated communication between subunits in the RFC holoenzyme from A. fulgidus. Mutation of the arginine finger in the small subunits results in a complex that is able to bind ATP but has impaired ATPase activity. However, quite unexpectedly, the mutant complex is also deficient in clamp loading, an activity that does not require ATP hydrolysis. Crystal structures of the small subunit alone reveal that it forms a hexameric ring that, in the absence of bound nucleotide, is six-fold symmetric. However, this symmetry is broken when nucleotide is bound to the complex. The large conformational change observed may relate to the opening of PCNA rings that is required for them to be loaded onto DNA substrates. The structures also suggest a key role for the N-terminal helix of each subunit, a feature that is conserved across clamp-loading proteins and is known as RFC box II (Cullmann et al, 1995). Analysis of mutant protein complexes containing subunits lacking this N-terminal helix reveal different regulatory roles in each subunit, both of which are key to coupling ATP hydrolysis with clamp loading.
Results
Arginine finger mutant
Many oligomeric NTPases have the NTP-binding site located between protein subunits, with residues from both subunits contributing to the NTPase activity. Commonly, there is an arginine residue extending from a neighbouring subunit into the NTP-binding site that interacts with the γ-phosphate, thereby promoting catalysis, often referred to as an ‘arginine finger' (Scheffzek et al, 1997). RFC small subunits contain a conserved sequence motif (SRC) in which the central arginine residue is implicated as an arginine finger (Bowman et al, 2004) By contrast, the RFC large subunit lacks this motif. In order to test the effect of replacing this residue in the small subunits, we mutated Arg-152 of the small subunits to alanine and reconstituted RFC complexes containing a wild-type large subunit and mutant small subunits (denoted LaWT/SmSAC).
Our first experiments were to determine the stoichiometry of nucleotide binding by the LaWT/SmSAC complex using a spin-column assay described previously (Seybert and Wigley, 2004) (Table I). These data revealed that the mutant complex, in the presence of PCNA, was able to bind four nucleotide molecules in common with the wild type, albeit with slightly reduced nucleotide affinity. We have shown previously that these four nucleotides are bound to the large subunit and three of the four small subunits (Seybert and Wigley, 2004). Although competent to bind nucleotide, the LaWT/SmSAC complex had an ATPase activity that was severely compromised (Table I), consistent with the yeast arginine finger mutant RFC complex (Bowman et al, 2004). However, we went on to investigate whether the archaeal mutant complex was able to catalyse clamp loading and primer extension, neither of which was analysed for the mutant yeast enzyme. Interestingly, the archaeal mutant complex was completely deficient in loading of PCNA onto circular DNA substrates (Figure 1) and, hence, was also unable to support primer extension (data not shown). The inability to load clamps is particularly interesting since we, and others, have shown previously that ATP binding without hydrolysis is sufficient for clamp loading (Turner et al, 1999; Gomes and Burgers, 2001; Seybert and Wigley, 2004). In particular, an RFC complex with wild-type large subunits and mutant small subunits that are able to bind, but not hydrolyse ATP, and which retain the arginine finger (LaWT/SmBmut), are fully competent in stoichiometric loading of PCNA (Figure 1A). Although deficient in PCNA release after loading, the LaWT/SmBmut complex is able to support primer extension under conditions where the polymerase level is not rate limiting (Seybert and Wigley, 2004). By contrast, the defect in the primer extension assay for the Lawt/SmSAC cannot be overcome even when the polymerase concentration is raised 10-fold (Figure 1B). Since both the the LaWT/SmSAC and LaWT/SmBmut mutant complexes are able to bind nucleotide, yet the LaWT/SmSAC alone is unable to load PCNA, this suggests that there is a step after ATP binding, but preceding hydrolysis, that is required for clamp loading and that this step requires an intact arginine finger. We sought information on the nature of this step from structural studies of the RFC small subunits, since these appear to be the key to understanding the link between the arginine finger at the ATP-binding site and loading of PCNA.
Table 1.
Nucleotide-binding and ATPase assays
| RFC/PCNA complex | No. of ATPγS bound per RFC complex | KD ATPγS (μM) | kcat (s−1) | Km ATP (μM) |
|---|---|---|---|---|
| LaWTSmWT | 4.1±0.3 | 0.5 | 1.7 | 25 |
| LaWTSmSAC | 3.6±0.2 | 4.0 | <0.1 | nd |
| Molecules of ATPγS bound to wild-type RFC (LaWTSmWT) or RFC-containing small subunits with a mutation in the arginine finger (LaWTSmSAC) were determined using a spin-column assay described previously (Seybert and Wigley, 2004). The KD ATPγS represents an average for the whole complex and does not distinguish between different sites. | ||||
Figure 1.
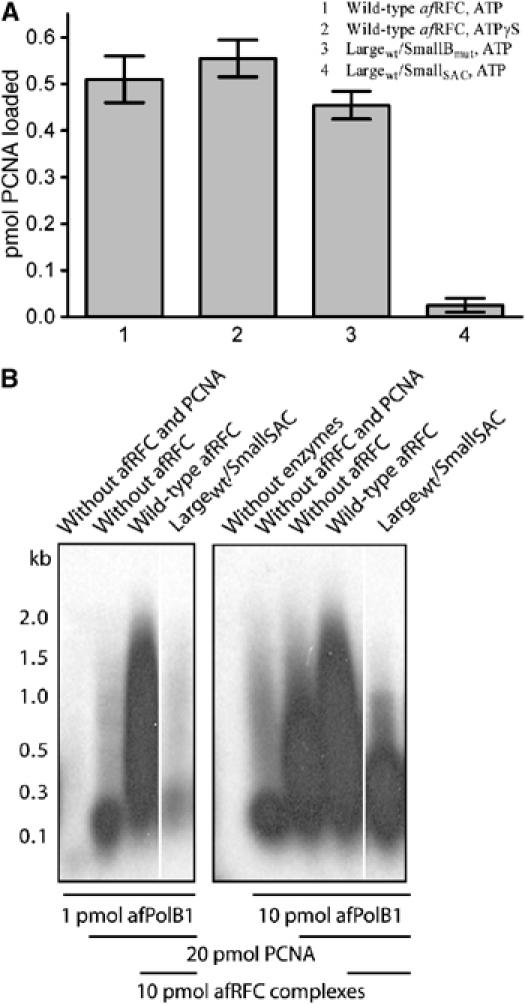
(A) PCNA-loading assays were performed using a spin-column assay. Reactions contained 8 pmol 32P-PCNA, 4 pmol RFC complexes and 4 pmol nicked pUC19 DNA. Samples 1, 3 and 4 contained 5 mM ATP, sample 2 contained 5 mM ATPγS. The spin columns were pre-equilibrated in 200 mM NaCl. The eluates of the reactions were separated by SDS–PAGE in a 15% gel and quantified on a phosphorimager. Samples 1 and 2 contained wild-type RFC, sample 3 contained LaWT/SmBmut RFC complexes with small subunits that contained a mutation in the Walker B motif, which we have shown previously to be able to bind ATP, but has severely impaired ATPase activity (Seybert and Wigley, 2004), and sample 4 contained complexes in which the small subunits had the arginine finger mutation (LaWT/SmSAC). (B) DNA primer extension assay showing a defect of the Lawt/SmSAC complex in stimulating DNA synthesis catalysed by PolB1. Although this defect appears to be partially compensated by increasing the level of PolB1 polymerase in the assay, the presence of the mutant complex actually supports less primer extension than in the absence of RFC in the control lane. By contrast, the Lawt/SmBmut mutant complex that is able to bind but not hydrolyse ATP at the small subunits shows a stimulatory effect under similar conditions (Seybert and Wigley, 2004). The bars below the gels indicate how much PolB1, PCNA or RFC was present in each lane.
The structure of RFCSm
As expected, the overall fold of the A. fulgidus RFC small subunit monomer is the same as that of the P. furiosus enzyme (Oyama et al, 2001). In brief, the protein is crescent shaped with three subdomains (Figure 2). Domains 1 (residues 1–161) and 2 (residues 162–226) comprise the canonical AAA+ fold (Ogura and Wilkinson, 2001), while domain 3 (residues 227–318) forms a five-helix bundle. The high-resolution structure of the domain 3 deletion protein (RFCSm1−226) is consistent with that of domains 1 and 2 of the full-length crystal forms and has clear density for a bound ADPNP molecule, with associated magnesium ion, located in the nucleotide-binding pocket. Given the relatively low resolution of the structures of apo RFCSm and the ADPNP complex, this higher resolution structure of RFCSm1−226 confirms the overall fold of the protein around the important ATP-binding site and provides a good initial model for refinement of the ADPNP complex structure.
Figure 2.
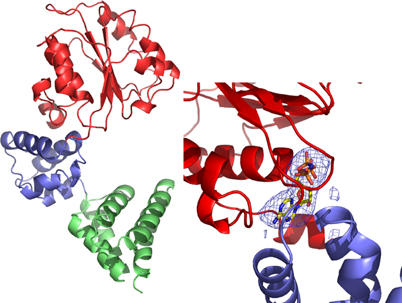
Overall fold of a monomer of the A. fulgidus RFC small subunit. The N-terminal domain 1 is coloured in red, domain 2 is blue and domain 3 is green. The inset shows a zoom in the ATP-binding site, with difference (Fo−Fc) electron density for one of the ADPNP molecules bound to the complex.
Comparison of the A. fulgidus RFC small subunit structure with that from P. furiosus (Oyama et al, 2001) reveals a number of differences. First, the structures of both the apo and ADPNP complexes form hexameric rings, whereas the P. furiosus protein crystallises as a dimer of trimers. However, EM images of the P. furiosus RFC small subunit reveals that at physiological pH the protein forms hexameric rings (Mayanagi et al, 2001). Second, we observe nucleotide bound at all six sites in the hexamer. By contrast, for one of the P. furiosus RFC small subunit trimers, there are ADP molecules bound at all three sites in one trimer, but at only one site in the other trimer. Finally, two of the subunits in the P. furiosus complex undergo domain swapping of their C-terminal helices at the subunit interface. Similar interactions are not observed in any other clamp loader subunit structures (Jeruzalmi et al, 2001a; Bowman et al, 2004). Although we do not observe any such swapping in either of our structures, the limited resolution of our data does not allow us to preclude such an interaction.
The nucleotide-free complex
The structure of nucleotide-free RFCSm (apo-RFCSm) was determined by molecular replacement using various fragments of the ADPNP–RFCSm complex (see below) as search models. Given the limited resolution of the data (4 Å), it was not appropriate to attempt a full atomic crystallographic refinement. However, rigid-body refinement of the structure was undertaken, treating each domain of every subunit as a separate rigid body. These results allow an analysis of global conformation changes in the complex when compared to the nucleotide-bound complex, but preclude detailed analysis of molecular interactions.
The apo-RFCSm crystal structure has six monomers in the asymmetric unit, forming a hexameric ring with six-fold rotational symmetry (Figure 3). The six subunits adopt almost identical conformations, with a maximum r.m.s.d. of 1.9 Å over all Cα positions between any pair of subunits. The overall structure is consistent with the model of the P. furiosus apo-RFCSm hexamer derived by cryo-EM (Mayanagi et al, 2001), but differs from that obtained by crystallography (Oyama et al, 2001). The ring is held together around a collar structure at the base by interactions between the C-terminal domains (domain 3), with domains 1 and 2 projecting upwards and away from this collar (Figure 3). This structural feature is very similar to that seen in the Saccharomyces cerevisiae RFC/PCNA–ATPγS complex (Bowman et al, 2004), despite the differing subunit composition (six-fold homohexamer of RFCSm compared to five-fold heteropentamer of RFC). Consequently, to some extent, the assembly of the RFCSm hexamer can be considered to resemble that of the RFC holoenzyme.
Figure 3.
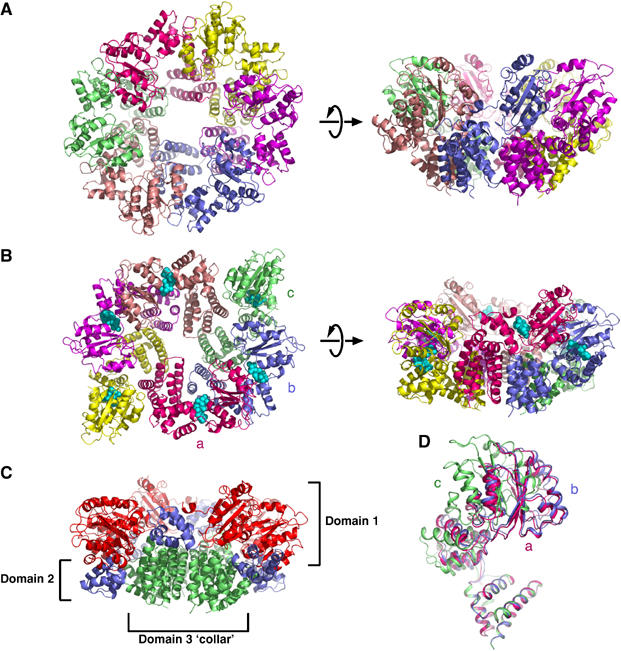
(A) RFC small-subunit apo-protein hexamer viewed from above the plane of the ring and from the side, with the different subunits in separate colours. (B) Similar views of the RFC small-subunit hexamer complexed with ADPNP. The subunits are denoted as a, b and c in accordance with the text. (C) The arrangement of domains in the hexamer coloured with the same colour scheme as in Figure 2. (D) Superposition of the three different conformations of independent subunits in the ADPNP complex. The subunits are labelled a, b and c as in the text.
The ADPNP complex
The crystal structure of the ADPNP complex has three subunits in the asymmetric unit, forming one half of a hexamer that is completed by a crystallographic two-fold symmetry axis. The complex as a whole adopts an elliptical conformation (Figure 3).
There is a large conformational change between the nucleotide-bound and nucleotide-free structures. The motions occurring can be described as (a) an ‘iris' type opening and closing of the structure, and (b) a set of coordinated domain movements in the C subunit(s) that pivot domains 1 and 2 of these subunits in a rigid body radial movement away from the centre of the hexamer (Figure 4). This motion results in expansion of the gap between the PCNA-interaction helices of subunits C and A′ by approximately 40 Å.
Figure 4.
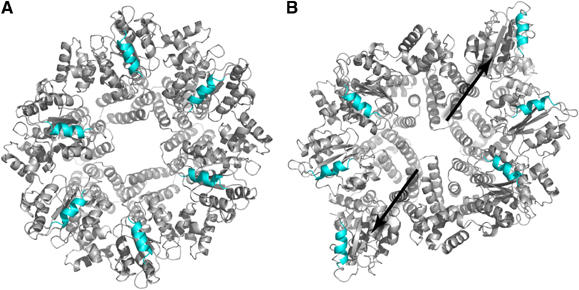
Diagram highlighting the conformational changes between the apo-protein (A) and that complexed with ADPNP (B). The arrows show the direction of movement of the N-terminal domains of the pair of subunits that show the largest conformational change. The helices that interact with PCNA are highlighted in blue.
As expected, the ADPNP is bound in the cleft between domains 1 and 2, and is visible in all subunits. In all bound nucleotides, the adenine ring is in the anti conformation with respect to the ribose, as seen in other clamp loaders (Podobnik et al, 2003; Bowman et al, 2004). However, the asymmetric nature of the ADPNP complex results in the formation of three different types of site (designated A, B, C), between the subunit pairs A:B, B:C and C:A′, respectively.
In all of the ATP-binding sites, the arginine finger (Arg-152) is located some distance away from the terminal phosphate, approximately 13 Å in the A and B sites and 40 Å in the C site. Clearly, the disposition of the subunits in the ADPNP complex is some way from catalytic competence, consistent with the lack of ATPase activity of the small subunits alone (Seybert et al, 2002). The structure suggests that one factor contributing to this separation of the two parts of the ATP-binding sites is the position of RFC motif II helix, located at the N-terminus of the protein. This helix extends back from domain 1 to sit over the cleft between domains 1 and 2 restricting the closing of the inter-subunit interface over the nucleotide (Figure 5). Consequently, the helix appears to function as a ‘door-stop', preventing the catalytically active alignment of subunits (see below).
Figure 5.
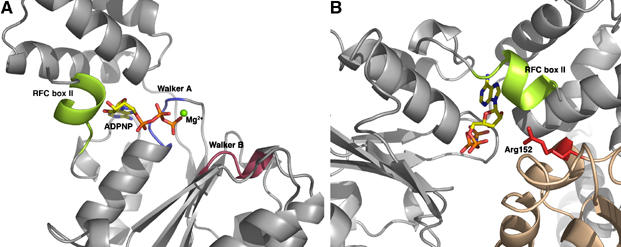
(A) The location of the N-terminal helix (RFC box II) within the ATP-binding site. (B) Diagram showing how the N-terminal helix blocks access of the arginine finger (Arg152) to the ATP-binding site.
N-terminal helix deletions
Although the small subunits alone are able to bind nucleotides and to form rings, they are devoid of ATPase activity. Despite this, we can observe that the ATP-binding sites are located between the subunits, are similar to those seen in the yeast RFC holoenzyme complex, and are competent to bind ATP. However, we showed above that the arginine finger of the small subunits is essential for ATPase activity in RFC holoenzyme and the crystal structures of the small subunit hexamer reveal that this residue is displaced from the active site, explaining why the small subunits are able to bind ATP, but lack ATPase activity. At each ATPase site, entry of the arginine finger from the neighbouring subunit is blocked by a short helix located at the N-terminus of the subunit. A similar interface is observed at each of the ATP-binding sites in the yeast RFC/PCNA/ATPγS structure (Bowman et al, 2004). Importantly, the amino-acid sequence of this helix is conserved across both large and small subunits of RFC and in fact between all clamp loaders (Figure 6), to the extent that it is one of the characteristic RFC boxes (RFC box II) identified in clamp loaders (Cullmann et al, 1995). Given the key role that this N-terminal helix appears to play in controlling access of the arginine finger to the active site, we decided to delete this helix in both the large and small subunits and investigate the contribution that each makes to the biochemical activities in reconstituted RFC complexes. The truncated large and small subunits will be referred to as LaΔN and SmΔN, respectively.
Figure 6.

Alignment of the RFC box II (Cullmann et al, 1995) amino-acid sequences of A. fulgidus (af), human (h) and S. cerevisiae (sc), as well as the functionally homologous sequences from bacteriophage T4 (T4gp44) and from E. coli γ/τ (ecTAU) and δ′ (ecDELTA′). The colours indicate the level of homology between the aligned sequences: (1) yellow, 33–50%, (2) cyan, 50–75%, (3) magenta, between 75 and 99% and (4) blue, 100% homology.
The ATPase activity of the various mutant complexes is presented in Table II. Surprisingly, all three mutant complexes have enhanced maximal ATP turnover rates compared to the wild type. Although the complex containing a truncated large subunit and wild-type small subunits has a similar Km for ATP as wild type, it is higher for the mutant complexes containing truncated small subunits, suggesting that a step after ATP hydrolysis has become limiting. Previous studies have shown that the small subunits account for most of the ATPase activity of the complex (Seybert and Wigley, 2004).
Table 2.
ATP hydrolysis by wild-type and mutant RFC complexes in the presence of DNA and PCNA
| Complex | kcat (s−1) | KM (μM ATP) | KD (μM ATPγS) |
|---|---|---|---|
| Wild-type afRFC | 1.7 | 25 | 0.5 |
| LargeΔN/SmallΔN | 6.8a | >1000 | >50 |
| LargeΔN/Smallwt | 3.4 | 26 | 1.0 |
| Largewt/SmallΔN | 6.8a | >>200 | 69 |
| aSince the assays were carried out at 2 mM ATP, which is saturating for the wild type and complexes, given the high KM for these other mutant complexes, the actual rate will be higher; so these rates should be taken as an apparent kcat at 2 mM ATP, rather than a true kcat. | |||
Given the enhanced ATP turnover of the mutant complexes, we decided to investigate how this had affected clamp loading. The PCNA-loading assay that we employed utilises a circular plasmid DNA containing a single nick site that allows single-turnover loading of PCNA onto the substrate (Seybert and Wigley, 2004). Once loaded, the PCNA is topologically linked to the DNA. However, if the plasmid is linearised, any PCNA that has been loaded onto the DNA, but released from the RFC complex, is able to diffuse along the DNA and then dissociate from the free ends. In light of the enhanced turnover of ATP, we anticipated that the N-terminal deletion mutants would be proficient in loading PCNA. However, the results were surprising. The two mutant complexes containing truncated small subunits were in fact severely compromised in loading PCNA (Figure 7A). We therefore deduce that ATP hydrolysis has become uncoupled from clamp loading in these mutants, indicating a role for the N-terminal helix of the small subunits in linking these processes. By contrast, one mutant complex (LaΔNSmWT) showed almost three-fold enhancement of loading of PCNA in this assay. It has been shown previously (Gomes and Burgers, 2001; Seybert and Wigley, 2004) that while loading of PCNA onto DNA merely requires ATP binding, the release of the clamp requires hydrolysis by the small subunits. Consequently, if the circular DNA substrate is linearised, we observe release of PCNA from the DNA. However, if ATPγS is used in place of ATP to prevent hydrolysis, the PCNA remains bound to the RFC and hence complexed with the DNA. Interestingly, there is a complete loss of PCNA from the LaΔNSmwt complex once the DNA is linearised, irrespective of whether ATP or ATPγS is present, indicating that the link between ATP hydrolysis by the small subunits and PCNA release is now broken in this mutant.
Figure 7.
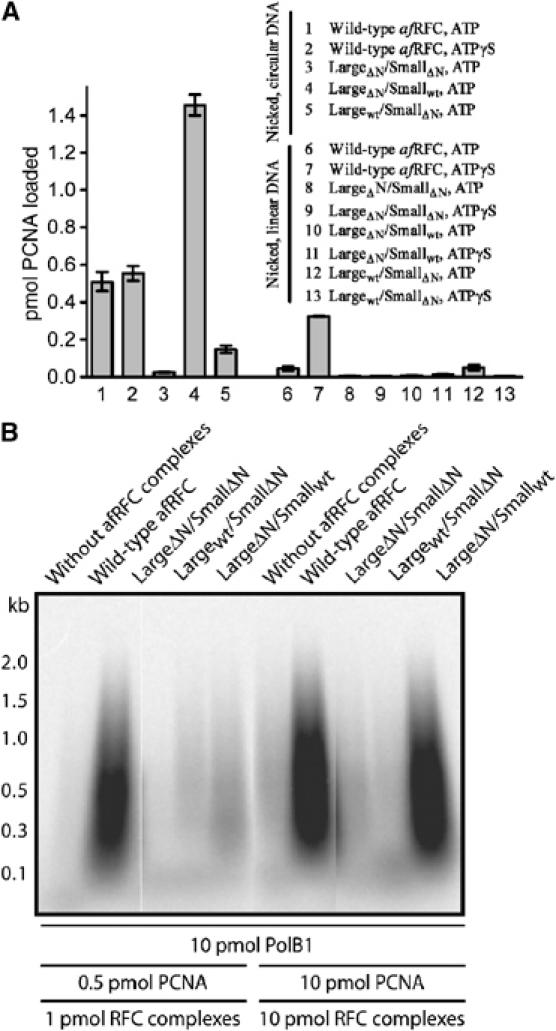
(A) Deletion of the N-terminal helix in the RFC small subunits inhibits the formation of stable PCNA–DNA complexes, whereas deletion of the N-terminal helix in the RFC large subunit stabilizes PCNA–DNA complexes. Loading assays were performed using a spin-column assay. Reactions contained 8 pmol 32P-PCNA, 4 pmol nicked pUC19 DNA (reactions 1–5), or 4 pmol nicked, linearised pUC19 DNA (reactions 6–13). Reactions 1, 3–5, 6, 8, 10 and 12 contained 5 mM ATP. Reactions 2, 7, 9, 11 and 13 contained 5 mM ATPγS. The mixtures also contained 4 pmol wild-type (samples 1, 2, 6 and 7) or mutant (samples 3–5 and 8–13) RFC complexes. The spin columns were pre-equilibrated in 200 mM NaCl. The eluates of the reactions were separated by SDS–PAGE in a 15% gel and quantified on a phosphorimager. (B) Comparison of the ability of the N-terminal helix truncated proteins with wild-type and Walker B mutant RFC complexes to stimulate DNA synthesis catalysed by PolB1. Reaction mixtures (20 μl) were supplemented with 200 mM NaCl and singly primed, closed circular M13mp18 DNA (25 fmol). After incubation for 30 min at 65°C, the DNA was precipitated and analysed by alkaline gel electrophoresis. Autoradiograms of dried gels are shown.
Finally, we looked to see how these mutants had been affected in the primer extension assay. This assay looks at the complete process of clamp loading; so defects at any step in the reaction cycle result in reduced efficiency of product formation. As expected, the two mutant complexes that were deficient in clamp loading (LaWTSmΔN and LaΔNSmΔN) were also completely deficient in primer extension (Figure 7B). Under conditions where catalytic loading by RFC is rate limiting (Seybert and Wigley, 2004), primer extension was also significantly reduced with the LaΔNSmWT complex. However, by raising the concentration of RFC/PCNA so that this is no longer rate limiting, the amount of extended primer becomes the same as that of the wild type. These properties are shared by a mutant RFC that was shown to be deficient in catalytic clamp loading, in which the large subunits are able to bind, but not hydrolyse, ATP (Seybert and Wigley, 2004).
Discussion
Clamp loaders are multi-subunit complexes that utilise ATP to open and close DNA polymerase processivity factors onto DNA. The recent crystal structure determination of a complex between a mutant yeast RFC, PCNA and a nonhydrolysable ATP analogue gave important insights into the interactions between the different protein components (Bowman et al, 2004). However, certain aspects of the crystal structure were not consistent with biochemical observations, nor was it clear how ATP was utilised during the clamp-loading process. It is evident from biochemical studies that clamp loading is a highly regulated process involving communication between the different ATPase sites in the complex. We have undertaken a combination of structural and biochemical studies to understand better how these protein subunits communicate with each other.
The ATP-binding sites in multi-component ATPases are often located at the interface of subunits. Although the major component of the binding site is located on one subunit, residues essential for catalysis are located on the adjacent subunit. Most commonly, this takes the form of an arginine finger (Scheffzek et al, 1997) and such a mechanism has been observed in many GTPases and ATPases (Ahmadian et al, 1997; Singleton et al, 2000). The ATP-binding sites of clamp loaders also contain an arginine finger (Bowman et al, 2004). In order to assess the contribution of the arginine finger to catalysis, we prepared a mutant RFC in which the arginine of the small subunits was replaced by an alanine residue. Our biochemical data are consistent with those obtained for the yeast RFC mutant and confirm that this arginine finger mutant RFC remains competent to bind ATP, but is severely ATPase deficient. However, quite unexpectedly, we discovered that the arginine finger mutant of the archaeal RFC is unable to load clamps. Biochemical data have shown that clamp loading by RFC requires ATP binding at all four sites in the complex, but does not require hydrolysis (Gomes and Burgers, 2001; Seybert and Wigley, 2004). These observations are significant because the crystal structure of the yeast RFC–PCNA complex was that of the arginine finger mutant complexed with an ATP analogue, ATPγS (Bowman et al, 2004). The PCNA ring has to be opened in order to load onto DNA, and it was not evident from the crystal structure how this process might take place, since this complex should have been active for loading, yet the PCNA ring was closed. Our data now explain why the yeast RFC/PCNA/ATPγS complex is in an inactive conformation for loading because mutation of the arginine finger prevents a step after ATP binding that is required before the complex can become competent to load PCNA. A similar conclusion was drawn from recent work looking at conformational changes in yeast RFC/PCNA complex (Zhuang et al, 2006). Using FRET analysis, they were able to monitor conformational changes in the PCNA ring within the complex. They deduced that binding of ATP to the complex induces an in-plane opening of the PCNA ring of 34 Å. This is followed by an out-of-plane partial closing of the ring once the complex interacts with DNA (thought to be represented by the EM images of Miyata et al, 2005), and then finally a complete closure of the ring with subsequent release from RFC. The initial (34 Å) opening of the ring on binding of ATP precedes hydrolysis and it is this conformation that should have been observed in the mutant yeast RFC/PCNA/ATPγS crystal structure (Bowman et al, 2004). However, as discussed above, the PCNA ring is unexpectedly closed in the structure. Together with the data of Zhuang et al (2006), the data we present here for the role of the arginine fingers now explain that observation.
The role of arginine fingers has been investigated in the E. coli clamp loader (Johnson and O'Donnell, 2003; Snyder et al, 2004). There are four arginine fingers in this system, but one of these contributes to a site that does not bind ATP. Of the three ATP sites in the γ subunits the arginine finger is contributed by another γ subunit in two cases and by the δ′ subunit in the other. Mutation of these residues revealed that the order of ATP hydrolysis is controlled between the sites (Johnson and O'Donnell, 2003), but also that there was an effect of the muations upon the affinity of the complex for DNA (Snyder et al, 2004). Although it might be tempting to consider that RFC and the E. coli clamp loader work by similar mechanisms, there are a number of differences between the clamp loader and RFC. These differences include, but are not limited to, (i) the use of three rather than four ATP molecules to load the clamp, (ii) a dimeric rather than trimeric clamp, (iii) differences in the interactions between the clamp loaders and their clamps, and (iv) differences between the number and order of ATP hydrolysis events.
In the absence of a crystal structure of wild-type A. fulgidus RFC complexed with PCNA, we turned to looking at the small subunits because it is these at which ATP hydrolysis is required to release clamps after loading. We reasoned, therefore, that nucleotide-dependent structural changes in these subunits might be the key to understanding the loading mechanism. Our structures of the small subunit apo and ADPNP complex reveal a substantial conformational change associated with nucleotide binding. The effect of these changes is to open up the structure in a way that could be coupled to opening of a PCNA ring. While we need to emphasise that the structures we present here are of the small subunit hexamer alone rather than of the RFC holoenzyme, similarities between the assembly of the hexamer and the RFC complex suggest that the nature of the conformational changes we observe might reflect a similar process in intact RFC, albeit in a qualitative rather than quantitative manner. It may be that conformational changes that are tightly regulated in the intact RFC or RFC/PCNA complexes could become uncoupled in the small subunit hexamer.
The crystal structure of yeast RFC complexed with PCNA (Bowman et al, 2004) is not competent to load PCNA because the ring is topologically closed. In order to pass duplex DNA through a break in the ring, RFC must open up PCNA by at least 20 Å. The EM images of P. furiosus RFC/PCNA complex (Miyata et al, 2005) revealed that in this structure the PCNA ring was opened enough (5 Å) to allow passage of a single strand of DNA but not a duplex, even though duplex DNA was observed at the centre of the complex. The authors suggested that if RFC loads PCNA onto primer template junctions directly, then the complex would have to open up more (to at least 20 Å) to allow the passage of duplex DNA through the ring. They also conclude that the present structural data cannot fully explain the clamp-loading process. Since PCNA loading is associated with ATP binding rather than hydrolysis, it may be that the conformational changes we observe in our structure of the small subunits relate to this loading process. Conformational changes associated with the binding of ATP have been observed directly using low-resolution microscopy techniques (Shiomi et al, 2000).
The RFCSm ADPNP complex structure revealed that the N-terminal helix of the subunits is positioned close to the arginine finger and might be controlling access of this residue to the site and hence regulating ATP hydrolysis (Figure 5). The conservation of the amino-acid sequence of this helix across all clamp loaders and clamp-loader subunits (RFC box II; Cullmann et al, 1995) suggests an important role for this region of the proteins. We produced mutant RFC holoenzymes in which the N-terminus of either the small or large subunits (or both) was truncated to remove this helix. Biochemical analysis of these mutants revealed that all three had enhanced maximal ATP turnover compared to the wild type. However, RFC complexes containing truncated small subunits were unable to promote clamp loading, even at a stoichiometric level. Hence, ATPase activity in these mutants has become uncoupled from clamp loading.
Truncation of the large subunit produces a more subtle effect that is only evident when combined with wild-type small subunits. The mutant RFC actually has enhanced stoichiometric clamp loading, but the controlled release of PCNA that is promoted by ATP hydrolysis at the small subunits in the wild-type complex (Seybert and Wigley, 2004) has now become uncoupled with release taking place even without ATP hydrolysis (ATPγS). Interestingly, when it comes to primer extension, the ΔN mutation of the large subunit has very poor activity when compared to wild type under conditions where clamp loading is required to be catalytic, but shows wild-type activity when used under stoichiometric conditions. This behaviour mimics the phenotype of a mutant RFC in which the ATPase of the large subunit is inactivated, while leaving ATP binding intact (Seybert and Wigley, 2004).
Consequently, we deduce that, although the N-terminal helix plays a regulatory role in both subunits, the effects of its removal differ according to the different functions of ATP hydrolysis at each site. Thus, controlled ATPase activity at the small subunits is coupled to clamp loading, or more precisely to clamp release after loading, and when this becomes uncoupled in the mutant, PCNA is released prematurely due to unrestrained ATP hydrolysis. By contrast, ATP hydrolysis at the large subunit acts to recycle RFC in an active form after clamp loading and release. Many aspects of the biochemistry reveal that the large subunit in fact acts as the master controller in the clamp loader. For example, ATP hydrolysis is stimulated by DNA, but we know that even though DNA binding is mediated by the large subunit, the majority of ATPase activity arises from the small subunits (Seybert and Wigley, 2004). The role of the N-terminal helix of the large subunit in coupling PCNA release to controlled ATP hydrolysis in the small subunits is also consistent with this master role.
Materials and methods
Materials
Unless stated otherwise, all chemicals were supplied by Sigma. The radioactive stock solution used in the nucleotide binding assays was prepared as follows: 2.5 μl [35S]ATPγS (10 μCi/μl, >1000 Ci/mmol, Amersham Biosciences) and 25 μl cold 10 mM ATPγS were mixed in a final volume of 1 ml in 2 mM DTT and 10 mM Tris–HCl, pH 7.5. This stock solution was stored in small aliquots at −80°C and only thawed once.
Site-directed mutagenesis
A recombination-polymerase chain reaction (PCR) method (Yao et al, 1992) was used to introduce point mutations into the afRFC large and small subunits. The plasmid pET22-afRFCsmall (Seybert et al, 2002) was used to introduce the substitution Arg-152 to alanine into the coding sequence of the afRFC small subunit. The primers for the substitution Arg-152 to alanine, with the exchanged codon underlined, were as follows: 5′-CATTCAGAGCGCGTGTGCGGTTTTCAGATTCA AG-3′ and 5′-GAAAACCGCACACGCGCTCTGAATGGGCTC-3 ′.
afRFC large-subunit N-terminal deletion was amplified by PCR from the wild-type afRFC large subunit clone. The primers for amplification were RFC LSUΔN F 5′-GGAAATGCATATGGAAGAAGTCGTTGCAGAC AAGAGC-3′ and T7 R 5′GCTAGTTATTGCTCAGCGG-3′. This was cloned into pET28a (Novagen), which incorporates a N-terminal his tag, using NdeI and NotI restriction sites. This clone removes the first 12 amino-acid residues of the large-subunit sequence.
afRFC small-subunit N-terminal deletion was amplified by PCR using the wild type small subunit clone as a template. The primers for amplification were RFC SSUΔN2 F 5′-GGAAATGCATATGGATGAAGTCGTGGGGCAG GATG-3′ and the T7 R primer as shown above. This was cloned into pET22b (Novagen) using NdeI and NotI restriction sites. This removes the first 16 amino-acid residues of the RFC small subunit.
Nucleotide-binding assays
These were conducted using a spin-column method as described previously (Seybert and Wigley, 2004).
ATPase assays
Generally, the ATPase activity of afRFC was determined using a continuous spectrophotometric assay or the ammonium molybdate-malachite green assay as described previously (Seybert and Wigley, 2004).
PCNA-loading assays
These were carried out as described previously (Seybert and Wigley, 2004).
Primer extension assays
These were carried out as described previously (Seybert et al, 2002).
Protein expression, purification and crystallisation
A. fulgidus RFC small subunit (RFCSm) was expressed and purified as described previously (Seybert et al, 2002). Crystals of the RFCSm–ADPNP complex were prepared as follows. Protein was concentrated to 10 mg/ml in 10 mM Tris–HCl, pH 7.5, 200 mM NaCl, 1 mM DTT, 5 mM MgCl2 and 1 mM ADPNP. Crystals were grown by the hanging drop vapour diffusion method against a reservoir solution containing 100 mM NaOAc, pH 5.0, 0.9–1.6 M 1,6-hexanediol. Crystals reached a full size of approximately 300 × 200 × 200 μm within 1 week. For cryocooling purposes, the crystals were transferred in the same reservoir solution with 0.2 M increments in the 1,6-hexanediol concentration until a final concentration of 2.6 M, then plunged into liquid propane. The crystals were of the spacegroup P3121, with cell dimensions a=b=109.2, c=257.1 Å.
For the apo crystal form, protein was concentrated to 10 mg/ml in 10 mM Tris–HCl, pH 7.5, 200 mM NaCl and 1 mM DTT. Crystals were grown in hanging drops by vapour diffusion against a reservoir solution of 100 mM Tris–HCl, pH 8.0, 100 mM MgCl2, 10% (w/v) PEG 8K. Crystals reached a full size of approximately 600 × 400 × 200 μm within 1 week. Crystals were transferred to a cryoprotectant of the reservoir solution with the addition of 30% (v/v) glycerol prior to plunging into liquid propane. The crystals were of the space group P212121, with cell dimensions a=123.9, b=134.2 and c=146.4 Å.
The gene encoding RFCSm1−226 was cloned into pET22b (Novagen) and expressed in E. coli B834. Cells were lysed in buffer A (50 mM Tris–HCl, pH 7.5, 1 mM DTT, 1 mM EDTA, 100 mM NaCl) and heat-treated at 65°C for 10 min. The supernatant was ammonium sulphate fractionated (0–65% saturation) and precipitated protein re-dissolved in buffer A, then applied to a Superdex 75 gel filtration column (Amersham). RFC-containing fractions were diluted to a conductivity of <5 mS/cm, and applied to HiTrap heparin-Sepharose and eluted with buffer B (50 mM Tris–HCl, pH 7.5, 1 mM DTT, 1 mM EDTA and 1 M NaCl). For crystallisation, the protein was concentrated to 12 mg/ml in 10 mM Tris–HCl, pH 7.5, 1 mM DTT, 100 mM NaCl, 2 mM ADPNP and 5 mM MgCl2. Crystals were grown by hanging-drop vapour diffusion against a reservoir of 100 mM HEPES, pH 7.5, 10% (w/v) PEG 6K and 6% (v/v) 2-methyl-2,4-pentanediol. The crystals grew within 3 days to a size of 80 × 100 × 180 μm and were of space group P212121, with cell dimensions a=77.5, b=91.5, c=140.6 Å. Cryocooling was carried out by fast transfer to a cryoprotectant of 100 mM HEPES, pH 7.5, 15% (w/v) PEG 6K, 15% (v/v) 2-methyl-2,4-pentanediol, followed by freezing in the gas stream.
Data collection and structure solution
Data were integrated and reduced using programs from the HKL suite (Otwinowski and Minor, 1997). Model building was carried out using TURBO-FRODO (Roussel and Cambillau, 1989). Unless otherwise stated, all other operations were performed using the CCP4 suite of programs (CCP4, 1994). All crystallographic statistics are shown in Table III.
Table 3.
Crystallographic statistics
| ADPNP complex |
|||||
|---|---|---|---|---|---|
| Native | Hg1 | Hg2 | Se | Apo | |
| Resolution (Å) | 30–3.5 | 30–3.9 | 30–5.5 | 30–8 | 20–4.0 |
| Completeness (%) | 99.5 | 92.1 | 98.5 | 99.7 | 95.8 |
| Rsymm (%) | 6.4 | 6.6 | 6.2 | 6.0 | 8.8 |
| Rderiv (%) | — | 23 | 28 | 35 | — |
| No. of sites | — | 6 | 8 | 13 | — |
| Phasing power | — | 1.1 | 1.4 | 0.90 | — |
| Mean figure of merit | 0.36 | ||||
| R-factor (%) | 29.9 | ||||
| Rfree (%) | 32.4 | ||||
| R.m.s.d bond length (Å) | 0.017 | ||||
| R.m.s.d. bond angles (deg) | 1.85 | ||||
| Domain 3 deletion | Native | Pb1 | Pb2 | Se | |
|---|---|---|---|---|---|
| Resolution (Å) | 30–2.1 | 20–2.2 | 20–2.4 | 20–2.2 | |
| Completeness (%) | 92.6 | 94.5 | 96.0 | 95.1 | |
| Rsymm (%) | 3.3 | 4.0 | 6.5 | 3.3 | |
| Rderiv (%) | — | 11.3 | 15.9 | 9.1 | |
| No. of sites | — | 2 | 4 | 15 | |
| Phasing power | — | 0.33 | 0.51 | 1.28 | |
| Mean figure of merit | 0.34 | ||||
| R-factor (%) | 21.4 | ||||
| Rfree (%) | 25.6 | ||||
| R.m.s.d bond length (Å) | 0.012 | ||||
| R.m.s.d. bond angles (deg) | 1.54 |
RFCSm:ADPNP complex
Diffraction data were collected on beamline 14.4 of the ESRF, Grenoble. Attempts at solving the structure by molecular replacement, using various search models derived from the P. furiosus RFCSm structure (PDB ID code=1IQP), were unsuccessful. However, a low-occupancy mercury derivative was identified, and the locations of the heavy atoms allowed identification of NCS elements and approximate location of the three monomers in the asymmetric unit by matching the mercury positions to cysteines conserved between the A. fulgidus and P. furiosus proteins. The poor initial phases generated from the mercury derivative allowed identification of potential selenium positions in a selenomethionine-substituted protein, which were crosschecked against the previously positioned protomers. Phases from the mercury and selenium derivatives were improved by density modification and multi-domain averaging as implemented in the program DM (Cowtan, 1994), and an initial model was built. Subsequent refinement using TLS parameterisation in REFMAC (Murshudov et al, 1997) and rebuilding allowed construction of a complete model.
RFCSm apo structure
Again, initial attempts at molecular replacement with 1IQP or the RFCSm:ADPNP monomers were unsuccessful. However, a correct solution was found using a dimer of molecules A and B from the ADPNP complex, which had been truncated to remove domain 3. Searches using the partial structure information allowed location of domains 1 and 2 of molecule C and subsequent searches using domain 3 alone as a search model allowed completion of the structure. As data were only available to 4 Å resolution, the structure was simply refined as 18 rigid bodies (three domains per chain, six chains) in CNS (Brunger et al, 1998).
RFCSm1−226 structure
The structure of the truncated RFC small subunit was solved with two lead and one selenium isomorphous derivatives using the programs from the SOLVE suite (Terwilliger and Berendzen, 1999). There were four molecules in the asymmetric unit, with ADPNP bound to each monomer. Initial maps were improved by solvent flattening and four-fold averaging. Rounds of building were alternated with refinement in REFMAC to give a model with a final R-factor of 21.4% (Rfree=25.6%). Coordinates have been deposited in the PDB under accession codes 2CHG (RFCSm1−226), 2CHQ (RFCSm:ADPNP) and 2CHV (RFCSm apo).
Acknowledgments
This work was supported by Cancer Research UK and a postdoctoral fellowship from the European Molecular Biology Organization (to AS). We thank N Raven for providing A. fulgidus and cells and S Sandler for providing plasmid pSJS1240.
References
- Ahmadian MR, Stege P, Scheffzek K, Wittinghofer A (1997) Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat Struct Biol 4: 686–689 [DOI] [PubMed] [Google Scholar]
- Bowman GD, O'Donnell M, Kuriyan J (2004) Structural analysis of a eukaryotic sliding DNA clamp–clamp loader complex. Nature 429: 724–730 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Cann IK, Ishino S, Yuasa M, Daiyasu H, Toh H, Ishino Y (2001) Biochemical analysis of replication factor C from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol 183: 2614–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann IK, Ishino Y (1999) Archaeal DNA replication: identifying the pieces to solve a puzzle. Genetics 152: 1249–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Cowtan K (1994) Joint CCP4 and ESF-EACBM newsletter on protein. Crystallography 31: 34–38 [Google Scholar]
- Cullmann G, Fien K, Kobayashi R, Stillman B (1995) Characterization of the five replication factor C genes of Saccharomyces cerevisiae. Mol Cell Biol 15: 4661–4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes XV, Burgers PM (2001) ATP utilization by yeast replication factor C. I. ATP-mediated interaction with DNA and with proliferating cell nuclear antigen. J Biol Chem 276: 34768–34775 [DOI] [PubMed] [Google Scholar]
- Gomes XV, Schmidt SL, Burgers PM (2001) ATP utilization by yeast replication factor C. II. Multiple stepwise ATP binding events are required to load proliferating cell nuclear antigen onto primed DNA. J Biol Chem 276: 34776–34783 [DOI] [PubMed] [Google Scholar]
- Jeruzalmi D, O'Donnell M, Kuriyan J (2001a) Crystal structure of the processivity clamp loader gamma (γ) complex of E. coli DNA polymerase III. Cell 106: 429–441 [DOI] [PubMed] [Google Scholar]
- Jeruzalmi D, O'Donnell M, Kuriyan J (2002) Clamp loaders and sliding clamps. Curr Opin Struct Biol 12: 217–224 [DOI] [PubMed] [Google Scholar]
- Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O'Donnell M, Kuriyan J (2001b) Mechanism of processivity clamp opening by the δ subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell 106: 417–428 [PubMed] [Google Scholar]
- Johnson A, O'Donnell M (2003) Ordered ATP hydrolysis in the γ complex clamp loader AAA+ machine. J Biol Chem 278: 14406–14413 [DOI] [PubMed] [Google Scholar]
- Kelman Z, Hurwitz J (2000) A unique organization of the protein subunits of the DNA polymerase clamp loader in the archaeon Methanobacterium thermoautotrophicum H. J Biol Chem 275: 7327–7336 [DOI] [PubMed] [Google Scholar]
- Lee SH, Kwong AD, Pan ZQ, Hurwitz J (1991) Studies on the activator 1 protein complex, an accessory factor for proliferating cell nuclear antigen-dependent DNA polymerase delta. J Biol Chem 266: 594–602 [PubMed] [Google Scholar]
- Mayanagi K, Miyata T, Oyama T, Ishino Y, Morikawa K (2001) Three-dimensional electron microscopy of the clamp loader small subunit from Pyrococcus furiosus. J Struct Biol 134: 35–45 [DOI] [PubMed] [Google Scholar]
- Miyata T, Oyama T, Mayanagi K, Ishino S, Ishino Y, Morikawa K (2004) The clamp-loading complex for processive DNA replication. Nat Struct Mol Biol 11: 632–636 [DOI] [PubMed] [Google Scholar]
- Miyata T, Suzuki H, Oyama T, Mayanagi K, Ishino Y, Morikawa K (2005) Open clamp structure in the clamp-loading complex visualized by electron microscopic image analysis. Proc Natl Acad Sci USA 102: 13795–13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Naktinis V, Onrust R, Fang L, O'Donnell M (1995) Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. II. Intermediate complex between the clamp loader and its clamp. J Biol Chem 270: 13358–13365 [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure-diverse function. Genes Cells 6: 575–597 [DOI] [PubMed] [Google Scholar]
- Onrust R, O'Donnell M (1993) DNA polymerase III accessory proteins. II. Characterization of δ and δ′. J Biol Chem 268: 11766–11772 [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Oyama T, Ishino Y, Cann IK, Ishino S, Morikawa K (2001) Atomic structure of the clamp loader small subunit from Pyrococcus furiosus. Mol Cell 8: 455–463 [DOI] [PubMed] [Google Scholar]
- Pisani FM, De Felice M, Carpentieri F, Rossi M (2000) Biochemical characterization of a clamp-loader complex homologous to eukaryotic replication factor C from the hyperthermophilic archaeon Sulfolobus solfataricus. J Mol Biol 301: 61–73 [DOI] [PubMed] [Google Scholar]
- Podobnik M, Weitze TF, O'Donnell M, Kuriyan J (2003) Nucleotide-induced conformational changes in an isolated Escherichia coli DNA polymerase III clamp loader subunit. Structure 11: 253–263 [DOI] [PubMed] [Google Scholar]
- Pritchard AE, Dallmann HG, Glover BP, McHenry CS (2000) A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex: association of δδ′ with DnaX4 forms DnaX3δδ′. EMBO J 19: 6536–6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel A, Cambillau C (1989) TURBO-FRODO. In Silicon Graphics Geometry Partner Directory, Silicon Graphics (ed), pp 77–78. Mountain View, CA: Silicon Graphics [Google Scholar]
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A (1997) The Ras–RasGAP complex: structural basis for GTPase activation and its loss in oncogenic mutants. Science 277: 333–338 [DOI] [PubMed] [Google Scholar]
- Seybert A, Scott DJ, Scaife S, Singleton MR, Wigley DB (2002) Biochemical characterisation of the clamp/clamp loader proteins from the euryarchaeon Archaeoglobus fulgidus. Nucleic Acids Res 30: 4329–4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybert A, Wigley DB (2004) Distinct roles for ATP binding and hydrolysis at individual subunits of an archaeal clamp loader. EMBO J 23: 1360–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi Y, Usukura J, Masamura Y, Takeyasu K, Nakayama Y, Obuse C, Yoshikawa H, Tsurimoto T (2000) ATP-dependent structural change of the eukaryotic clamp-loader protein, replication factor C. Proc Natl Acad Sci USA 97: 14127–14132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Sawaya MR, Ellenberger T, Wigley DB (2000) Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 101: 589–600 [DOI] [PubMed] [Google Scholar]
- Snyder AK, Williams CR, Johnson A, O'Donnell M, Bloom LB (2004) Mechanism of loading the Escherichia coli DNA polymerase III sliding clamp. J Biol Chem 279: 4386–4393 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J, Hingorani MM, Kelman Z, O'Donnell M (1999) The internal workings of a DNA polymerase clamp-loading machine. EMBO J 18: 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Jones DH, Grose C (1992) Site-directed mutagenesis of herpesvirus glycoprotein phosphorylation sites by recombination polymerase chain reaction. PCR Methods Appl 1: 205–207 [DOI] [PubMed] [Google Scholar]
- Zhuang Z, Yoder BL, Burgers PMJ, Benkovic SJ (2006) The structure of a ring-opened proliferating cell nuclear antigen-replication factor C complex revealed by fluorescence energy transfer. Proc Natl Acad Sci USA 103: 2546–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]