

Abstract
T7 DNA primase is composed of a catalytic RNA polymerase domain (RPD) and a zinc-binding domain (ZBD) connected by an unstructured linker. The two domains are required to initiate the synthesis of the diribonucleotide pppAC and its extension into a functional primer pppACCC (de novo synthesis), as well as for the extension of exogenous AC diribonucleotides into an ACCC primer (extension synthesis). To explore the mechanism underlying the RPD and ZBD interactions, we have changed the length of the linker between them. Wild-type T7 DNA primase is 10-fold superior in de novo synthesis compared to T7 DNA primase having a shorter linker. However, the primase having the shorter linker exhibits a two-fold enhancement in its extension synthesis. T7 DNA primase does not catalyze extension synthesis by a ZBD of one subunit acting on a RPD of an adjacent subunit (trans mode), whereas de novo synthesis is feasible in this mode. We propose a mechanism for primer initiation and extension based on these findings.
Keywords: DNA replication, primer synthesis mechanism, RNA polymerase domain, zinc-binding domain
Introduction
DNA replication depends on a class of RNA-synthesizing enzymes designated DNA primases that are distinct from the classical RNA polymerases. The protein to which primase activity was first ascribed is the product of gene 4 of bacteriophage T7 (Scherzinger et al, 1977b). This protein has been used as a model for studying DNA primase structure and the DNA priming mechanism. The multifunctional T7 gene 4 helicase–primase along with only three other proteins, T7 gene 5 DNA polymerase, its processivity factor Escherichia coli thioredoxin, and the T7 gene 2.5 single-stranded DNA (ssDNA)-binding protein mediate the basic reactions at the T7 replication fork (Richardson, 1983).
The gene 4 protein is composed of a helicase domain located in the C-terminus half of the protein and a primase domain located in the N-terminus half. The crystal structures of the gene 4 protein (Toth et al, 2003), as well as that of the helicase (Sawaya et al, 1999; Singleton et al, 2000) and primase (Kato et al, 2003) domains have been solved. The primase domain is modular and contains two major domains, a Cys4 zinc-binding domain (ZBD) (residues 1–54) and an RNA polymerase domain (RPD) (residues 71–245). An unstructured polypeptide of 16 residues (55–70), referred to as the linker, connects the ZBD and the RPD (Kato et al, 2003) (Figure 1). The small ZBD is located at the extreme N-terminus of the protein. The larger RPD is located adjacent to the C-terminal helicase domain. The T7 helicase domain (residues 272–566) has a globular fold with a catalytic core of mononucleotide-binding fold (Sawaya et al, 1999). A region of 26 residues (246–271) between the helicase and primase domains connects the two domains and is essential for the formation of functional gene 4 protein hexamers (Guo et al, 1999; Lee and Richardson, 2004).
Figure 1.
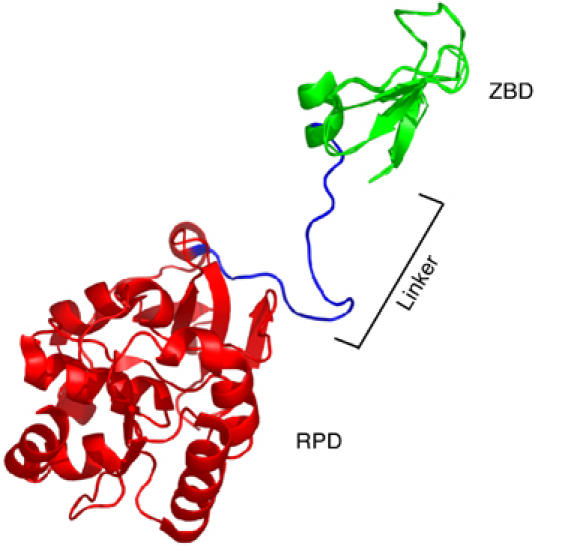
Crystal structure of the T7 primase fragment, PDB code 1NUI (Kato et al, 2003), showing one of the subunits of the crystallized dimer. The ZBD is shown in green. The RPD is shown in red and the linker between them is shown in blue.
The coexistence of the helicase and primase activities on the same polypeptide provides several advantages. The helicase activity exposes single-stranded lagging strand template for primase to initiate Okazaki fragment synthesis. In addition, the physical linkage between the two domains provides a mechanism for the primase to translocate on the DNA in search for recognition sites. Moreover, the hexameric ring formed by the helicase allows the individual subunits to interact with each other; the ZBD of one subunit can interact with the RPD catalytic site on another subunit of the hexamer (Lee and Richardson, 2002). The covalent linkage between primase and helicase does not occur in E. coli and T4 bacteriophage. The primase and helicase proteins of these organisms are encoded by separate genes, but have evolved to form stable complexes with each other (Schaeffer et al, 2005).
An additional form of gene 4 protein is also found in T7 phage-infected cells. The 56-kDa gene 4 protein (56-kDa-gp4) is a truncated form lacking the N-terminal 63 residues found in the 63-kDa protein, as its translation is initiated at residue 64. The 56-kDa-gp4 cannot catalyze template-directed RNA synthesis although it does form hexamers and exhibits helicase activity. Consequently, the 56-kDa-gp4 alone cannot support growth of T7 phage whereas the 63-kDa protein can.
The T7 primase catalyzes the synthesis of tetraribonucleotides in a template-directed reaction (Tabor and Richardson, 1981). Tetraribonucleotide synthesis is initiated from a trinucleotide recognition site, 5′-GTC-3′. The 3′-cryptic C is essential for recognition but is not copied into the product (Mendelman and Richardson, 1991). The resulting diribonucleotide is then extended, in a template directed manner, to yield the functional tetraribonucleotide primers, pppACCC, pppACAC, and pppACCA, provided that appropriate complementary nucleotides are present in the recognition sequence (Frick and Richardson, 1999).
The ZBD plays a critical role in the recognition of the trinucleotide site (Kusakabe et al, 1999) and in the transfer of this site to the RPD (Kato et al, 2003). The inability of the 56-kDa-gp4 to synthesize primers in a template-directed manner is due to the lack of its ZBD. The RPD, as shown by biochemical data, contains the nucleotide-binding site and catalytic core for oligoribonucleotide synthesis (Lee and Richardson, 2001). The flexible linker between the ZBD and the RPD domains could play a significant role in primer synthesis by forming the optimal distance for interactions between the two domains and for interaction with the lagging strand DNA polymerase. We previously demonstrated that the linker between the ZBD and RPD is sufficiently flexible to allow the ZBD in one subunit to functionally interact with an RPD on an adjacent subunit within a hexamer (Lee and Richardson, 2002). Modeling of the structures of a full-length gene 4 protein lacking the ZBD show that such a trans reaction can readily occur (Toth et al, 2003).
Although there are several crystal structures of either the catalytic domain of E. coli DNA primase (Keck et al, 2000; Podobnik et al, 2000) or the ZBD of Bacillus stearothermophilus DNA primase (Pan and Wigley, 2000), the only two crystal structures of prokaryotic DNA primase encompassing both the ZBD and the RPD are those of T7 DNA primase fragment (Kato et al, 2003) and of Aquifex aeolicus (Corn et al, 2005). Therefore, only little is known about the linker length between the ZBD and RPD in other prokaryotic DNA primases. Nevertheless, such linkers that enable the ZBD and the RPD to functionally interact in other prokaryotic DNA primases must exist.
As mentioned above, T7 DNA primase catalyzes the synthesis of the diribonucleotide pppAC at the core recognition sequence 5′-GTC-3′. These diribonucleotides cannot serve as primer for T7 DNA polymerase; nonetheless, they are synthesized in large amounts relative to the functional tetraribonucleotide (Mendelman and Richardson, 1991; Kusakabe and Richardson, 1997). Although small amounts of diribonucleotide have been observed with other primases, their abundance is far less than that seen with gene 4 protein (Hinton and Nossal, 1987; Swart and Griep, 1995). An intriguing possibility is the association of diribonucleotides with the primase until extended to a functional primer at the recognition site for synthesis of the functional tetraribonucleotide.
In this report, we examine the interactions between the ZBD and RPD for the synthesis of oligoribonucleotides by changing the linker of T7 DNA primase. Large changes in the length of the linker, not surprisingly, have drastic affects on primer synthesis. Smaller changes in the length, however, allow one to distinguish between diribonucleotides synthesis and their subsequent extension. The different mechanisms underlying diribonucleotides synthesis and their subsequent extension are further shown by examining these two activities of gene 4 proteins in a trans mode.
Results
The crystal structure of the primase fragment of the T7 gene 4 protein revealed its bipartite structure (Kato et al, 2003). The small N-terminal ZBD and the larger RPD are connected by an extended polypeptide linker (residues 55–70) (Figure 1). The RPD catalyzes the formation of phosphodiester bonds, whereas the ZBD is involved in sequence-specific recognition of template DNA (Kusakabe et al, 1999). In addition, the ZBD is necessary for both de novo oligonucleotide synthesis and for di- and triribonucleotides extension in a template-dependent manner (Kusakabe and Richardson, 1997). Therefore, for proper priming, the RPD and ZBD must come together to engage nucleotides and the DNA template. In order to understand the importance of the ZBD engagement with the RPD during primer synthesis by the T7 primase, we have characterized T7 DNA primases containing linkers of various lengths.
Complementation of phage growth by altered T7 gene 4 proteins
Some residues of the linker between the ZBD and the RPD might be permutable and some segments might be deleted without a drastic affect on gene 4 protein activity. On the other hand, alteration of other segments of the linker might result in a nonfunctional protein. To map these residues, we designed several deletions and an insertion in the linker (Table I). Deletions of residues 55–70, 55–60, 55–64, 58–63, 64–69, and 61–70 were introduced into the cloned T7 gene 4 on a plasmid. Five residues, SGGKP, were inserted after position 58. On the assumption that the two proline residues in the linker could be essential for forming a hinge-like motif structure, we made the more conservative substitution of valine for proline at either position P58 or P63. In addition, we constructed a gene 4 protein in which both P58 and P63 were replaced with valine. All plasmids were transformed into an E. coli C600 strain. The transformed strains were infected with phage T7Δ4, lacking the gene 4 protein-coding region in order to determine the ability of each of the genetically altered gene 4 proteins to support T7 growth. The region in the linker between residues 55 and 64 is not essential for primase function (Table I). Deletion, insertion and point mutations in the linker had no effect on the plating efficiency of T7Δ4 phage (Table I).
Table 1.
Ability of altered gene 4 proteins to complement growth of T7 phage
 |
| Gene 4 proteins containing the indicated changes in the linker were expressed in E. coli C600. Gray sequences represent deletions, boxed sequence represents insertion, and black boxes represent substitutions. After infection with T7Δ4 phage, the number of plaques were counted and normalized to the value obtained with wild-type gene 4 protein (EOP=efficiency of plating). |
Three deletions (Δ55–70, Δ64–69, and Δ61–70) render the protein inactive. The common feature of these deletions is that they all lack residues 64–69. We have purified the gene 4 protein with a deletion of residues 64–69. This protein cannot catalyze the synthesis of oligoribonucleotides (data not shown). Surprisingly, the protein is devoid of helicase activity as well. In order to further characterize this altered protein, we purified a 56-kDa-gp4, in which residues 2–6 are deleted. The 56-kDa-gp4 lacks the first 63 residues found in the wild-type gene 4 protein (wt-gp4), and therefore residues 2–6 are similar to residues 65–69 found in the full-length protein. Indeed, the altered 56-kDa-gp4 lacking residues 2–6 is also defective in helicase activity. This issue is addressed in the Supplementary data (Supplementary Figure S2A). These results suggest that some or all residues within residues 65–69 are crucial for the overall functioning of the protein, whereas residues 55–64 play a role in fine-tuning the gene 4 protein for optimal function.
None of the altered proteins shown in Table I had a dominant negative effect on the function of the wild-type protein expressed by the phage. Upon wild-type T7 infection of E. coli strain expressing each of the altered genes, the plating efficiency was unchanged compared to infection of E. coli cells expressing wt-gp4.
Although all of the gene 4 proteins having alterations outside of the essential region between residues 64 and 69 supported the growth of T7Δ4 phage, we nonetheless elected to characterize them biochemically. Previous studies have shown that rather drastic alterations in primase activity do not alter the ability of the protein to complement for T7 phage growth (Rosenberg et al, 1996). Since the purpose of this study was to address the importance of the length of the linker between the ZBD and the RPD, we chose to purify altered gene 4 proteins having deletions or insertions in the linker. An increase in the length of the linker was provided by purification of gp4-link(ins5) containing a five-residue insertion after residue 58. In order to examine the effect of shortening the linker, we purified gp4-link(del6) and gp4-link(del10), lacking 6 (residues 58–63) and 10 (residues 55–64) residues, respectively (Table I). In addition, as control, we purified the 56-kDa-gp4 lacking the ZBD as well as the full-length 63-kDa gene 4 protein.
De novo primer synthesis
Gene 4 protein synthesizes oligoribonucleotides when provided with ATP and CTP, divalent cations, and an appropriate ssDNA template (Scherzinger et al, 1977a). Primers are most efficiently synthesized at specific DNA sequences designated ‘primase recognition sites' which contain the sequence 5′-(G/T)GGTC-3′ or 5′-GTGTC-3′ (Tabor and Richardson, 1981). The essential 3′-cytosine found in the recognition site is not copied into the primer and is designated as ‘cryptic' (Tabor and Richardson, 1981). Oligoribonucleotide synthesis begins opposite the dT in the recognition site, and the resulting products are 5′-pppACCA-3′, 5′-pppACCC-3′, or 5′-pppACAC-3′.
Using a synthetic 15-nt oligonucleotide template containing the primase recognition sequence 5′-GGGTC-3′, we have measured the ability of each of the altered gene 4 proteins to catalyze the synthesis of oligoribonucleotides in the presence of ATP and [α-33P]CTP over a range of gene 4 protein concentration (22–200 nM). In this assay, the radioactive products of the reaction, pppAC, pppACC, and pppACCC, can be separated and analyzed on a polyacrylamide gel (Figure 2A). The amount of tetraribonucleotide synthesized by each protein is plotted as a function of protein concentration in Figure 2B. Wt-gp4 catalyzes the synthesis of predominantly the tetraribonucleotide, although at higher concentrations both the di- and triribonucleotides are present. As shown in Figure 2, gp4-link(del6), lacking six residues, has 10% of the activity of the wt-gp4 whereas the larger deletion in gp4-link(del10) lacks activity. The insertion mutant of gene 4 primase, gp4-link(ins5), displays approximately 50% of the activity observed with wt-gp4. The 56-kDa-gp4, as expected (Kusakabe and Richardson, 1997), does not catalyze the synthesis of tetraribonucleotides.
Figure 2.
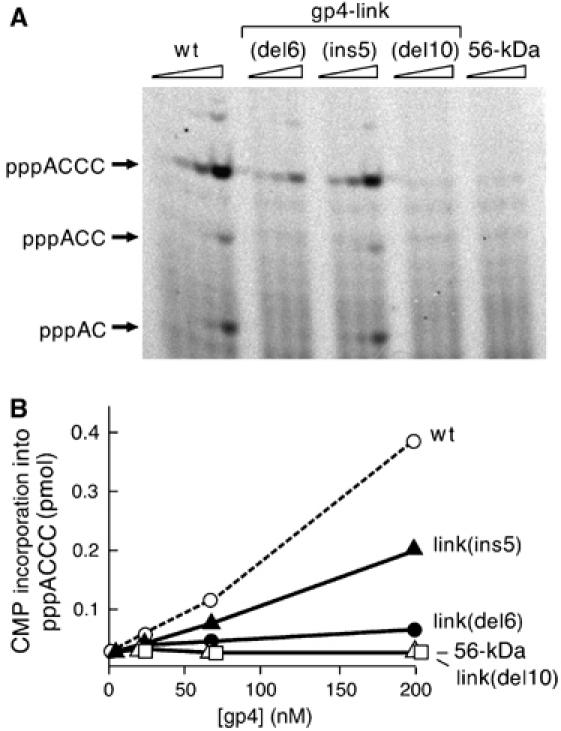
Template-directed de novo oligoribonucleotide synthesis. (A) Increasing amounts (0, 22, 66, and 200 nM) of the indicated T7 primases were incubated with ATP and [α-33P]CTP in the presence of template DNA 5′-GGGTCAAAAAAAAAA-3′ for 20 min at 37°C. The reaction products were analyzed on 25% denaturing polyacrylamide gel. Reaction products are indicated to the left of the gel. (B) Quantitative analysis of the products of primase synthesis was carried out by measuring the amount of radioactivity in each of the lanes of the gel above using a Fuji BAS 2500 Bioimaging analyzer. Total [α-33P]CMP incorporation into the tetraribonucleotide product is plotted against the gene 4 protein concentration.
Similar results are obtained over a large range of ATP concentrations (70–2000 μM) (Supplementary Figure S3A). In addition, we have carried out experiments comparing the de novo synthesis of the different gene 4 proteins on a template 5′-GTCN12-3′ that includes only the core recognition sequence, thereby enabling only the formation of AC diribonucleotide and not the ACCC tetramer. These assays show similar pattern to that observed with a template supporting ACCC formation (Supplementary Figure S4).
Diribonucleotide extension
In the presence of a primase recognition sequence, T7 primase can extend a preformed diribonucleotide, AC (Kusakabe and Richardson, 1997; Frick and Richardson, 1999). Since oligoribonucleotide synthesis is initiated with ATP, substitution of ATP in the reaction with a preformed oligoribonucleotide AC, ACC, or ACA allows one to monitor the ability of the primase to extend the di- or triribonucleotide to a longer tetraribonucleotide. For example, in the presence of the template 5′-GGGTC-3′ and the addition of AC or ACC and CTP, the products of the primase will be ACC and ACCC. It has been shown that T7 DNA primase has the same affinity for ATP and AC even though the latter is not phosphorylated (Kusakabe and Richardson, 1997). Moreover, the 56-kDa-gp4 lacking the ZBD extends di- and triribonucleotides far less efficiently than does the wt-gp4, suggesting an essential role for the ZBD in di- and triribonucleotide extension. In these reactions, there is an absolute requirement for the primer recognition site including the cryptic cytidine (Kusakabe and Richardson, 1997).
The ability of each altered protein to extend either AC or ACC was carried out as determined above for the de novo synthesis, except that ATP was omitted from the reaction. The template again was the 15-nt oligonucleotide containing the recognition sequence 5′-GGGTC-3′. As shown in Figure 3, gp4-link(del6) catalyzes both AC extension (Figure 3A) and ACC extension (Figure 3B) reactions two-fold more efficiently than does the wt-gp4. This result is surprising in that de novo synthesis of tetraribonucleotides by this protein is 10-fold lower than that observed with the wt-gp4. Consistent with its two-fold decrease in de novo primer synthesis, gp4-link(ins5) shows a two-fold decrease in the extension assay compared to the wt-gp4. The 56-kDa-gp4 protein is inactive in extending the oligoribonucleotides under these conditions, as previously reported (Kusakabe and Richardson, 1997). Gp4-link(del10) likewise is inactive. Similar results are obtained over a large range of AC concentrations (100–3000 μM) (Supplementary Figure S3B).
Figure 3.
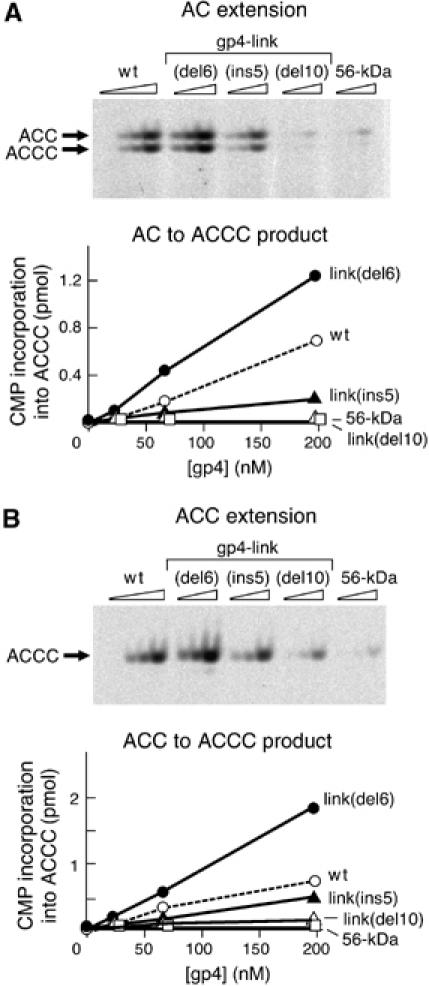
Template-directed oligoribonucleotide extension. Increasing amounts (0, 22, 66, and 200 nM) of the indicated T7 primases were incubated with AC (A) or ACC (B) ribonucleotides and [α-33P]CTP in the presence of template DNA 5′-GGGTCAAAAAAAAAA-3′ for 20 min at 37°C. The reaction products were analyzed on 25% denaturing polyacrylamide gel. The products are indicated to the left of the gel. Quantitative analysis of the products of primase synthesis, shown under each gel, was carried out as described in Figure 2B.
Taken together, the results show that linker of 16 residues, found in the wt-gp4, is optimal for the overall production of the functional tetraribonucleotide. Remarkably, a shorter linker of 10 residues, gp4-link(del6), allows for a better extension of the di- or tri-ribonucleotides, but is deficient in initiating de novo synthesis and consequently in overall primer synthesis. However, if the linker is reduced to six residues, neither de novo synthesis nor the extension of diribonucleotides occurs. A longer linker of 21 residues results in reduced extension capability of the protein as well as an overall reduction in de novo synthesis of primers.
Diribonucleotide initiation and extension in cis
Since the T7 primase domain is covalently linked to the helicase domain, it normally functions within the hexameric assembly (Patel and Hingorani, 1993; Toth et al, 2003). We have shown previously that the ZBD of one subunit of the hexamer can interact with the RPD of an adjacent subunit in trans to catalyze the synthesis of oligoribonucleotides (Lee and Richardson, 2002). Such an interaction is clearly feasible from modeling studies of the crystal structure of the gene 4 protein (Toth et al, 2003), but, in part, must be dependent on the flexible linker.
In order to study diribonucleotide synthesis and its extension in a cis-only mode, we constructed primase fragments derived from the wt-gp4, gp4-link(del6), and gp4-link(ins5). These protein were annotated wt-PF, PF-link(del6), and PF-link(ins5), respectively. The primase fragment is unable to form hexamers since it lacks the helicase domain required for oligomerization. Indeed, at concentrations lower than 20 μM, the primase fragment does not function in trans; a primase fragment with a catalytically deficient RPD cannot complement the activity of the 56-kDa-gp4 (Lee and Richardson, 2002). Moreover, mixing a primase fragment containing a catalytically defective RPD with a primase fragment containing an intact RPD but lacking the ZBD does not restore primer synthesis activities unless a concentration of over 20 μM of both primase fragments is used (S-J Lee and CC Richardson, unpublished results). The experiments in Figure 4 show a de novo synthesis assay (Figure 4A) and a diribonucleotide extension assay (Figure 4B) carried out with the indicated primase fragments as described above for the gene 4 proteins over a range of primase fragment concentration (25–200 nM). The amount of tetraribonucleotide synthesized by each primase fragment is plotted as a function of primase fragment concentration below each image. The results in Figure 4A show that in cis, the de novo synthesis of the wt-PF is enhanced as compared to PF-link(del6) and to PF-link(ins5). PF-link(del6) catalyzes tetraribonucleotide synthesis at 10% the rate of the wt-PF. The activity of PF-link(ins5) is approximately 33% that of the wt-PF. These results are comparable to the results obtained with the full-length gene 4 proteins. Assays for diribonucleotide extension (Figure 4B) with the primase fragments provide results similar to those found with the full-length proteins, as well. PF-link(del6) extends AC two-fold better than the wt-PF, whereas PF-link(ins5) extends AC three-fold less efficiently than does the wt-PF. These results indicate that the differences between de novo synthesis of AC and the extension of AC are observed even for a single subunit (in cis).
Figure 4.
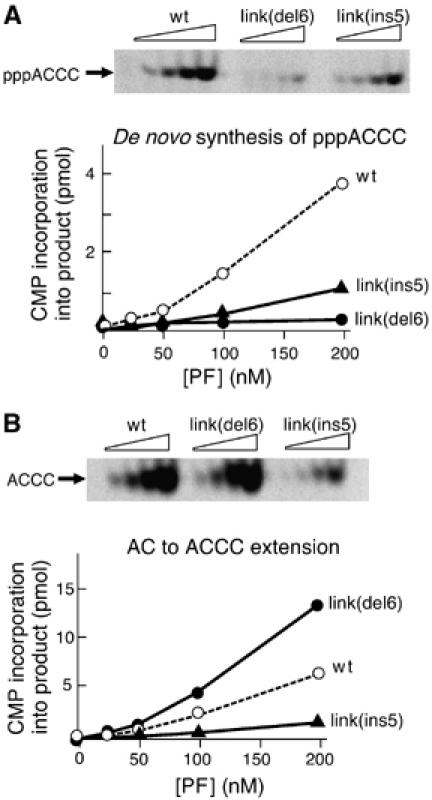
Oligoribonucleotide synthesis by primase fragments. (A) Template-directed de novo synthesis. Increasing amounts (0, 25, 50, 100, and 200 nM) of the indicated primase fragments were incubated with ATP and [α-33P]CTP in the presence of template DNA 5′-GGGTCAAAAAAAAAA-3′ for 30 min at 37°C. The reaction products were analyzed on 25% denaturing polyacrylamide gel. The major product, 5′-pppACCC-3′, is indicated to the left of the gel. (B) Template-directed AC extension. Increasing amounts (0, 25, 50, 100, and 200 nM) of the indicated primase fragments were incubated with AC ribonucleotide and [α-33P]CTP in the presence of template DNA 5′-GGGTCAAAAAAAAAA-3′ for 30 min at 37°C. The reaction products were analyzed on 25% denaturing polyacrylamide gel. The major product, 5′-ACCC-3′, is indicated to the left of the gel.
Diribonucleotide initiation and extension in trans
The dichotomy between diribonucleotide formation and diribonucleotide extension observed above for the full-length gp4-link(del6) is observed even in a cis-only mode as seen with PF-link(del6). It is likely that the distance between a ZBD and the RPD of the same subunit differs from the distance between a ZBD and the RPD of an adjacent subunit. We have shown above that changing the distance between the ZBD and the RPD on the same subunit can have a negative affect on the synthesis of diribonucleotides and a positive affect on their extension. The distances required for optimal diribonucleotides synthesis and their extension in trans may well differ from those required in cis.
In order to study the trans-only mode, we have made use of a genetically altered gene 4 protein, gp4-D237A, that is unable to catalyze the synthesis of phosphodiester bonds due to the single amino-acid replacement in the active site of the RPD (Lee and Richardson, 2005). This mutation does not affect the ZBD and was used in earlier studies to demonstrate the trans interaction of the ZBD and the RPD (Lee and Richardson, 2005). We introduced the D237A mutation into the genes encoding the wt-gp4, gp4-link(del6), and gp4-link(ins5) in order to obtain the respective proteins, gp4-D237A, gp4-link(del6)/D237A, and gp4-link(ins5)/D237A. By introducing the inactive RPD into each of the DNA primases, it is then possible to determine if their ZBD can function in trans when heterohexamers containing subunits of the 56-kDa-gp4 lacking a ZBD are constructed. Thus, de novo synthesis of oligoribonucleotides or extension of preformed di- or triribonucleotide can only occur via a trans mode.
We have measured each of the altered gene 4 proteins containing the D237A substitution for their ability to restore in trans the de novo synthesis and diribonucleotide extension activities to the 56-kDa-gp4. Gene 4 proteins were each mixed with an equimolar amount of 56-kDa-gp4 and incubated for 10 min to allow oligomerization. The protein mixture was then incubated with either a reaction mixture containing ATP and CTP for measuring de novo synthesis or to a reaction mixture containing AC and CTP for measuring diribonucleotide extension. Figure 5 shows the de novo synthesis products of the indicated gene 4 protein mixed with the 56-kDa-gp4 (top) and the diribonucleotide extension products of the indicated proteins mixed with the 56-kDa-gp4 (bottom). Remarkably, extension of exogenous diribonucleotide in trans is barely detected for gp4-link(del6)/D237A, as is the case for all of the other gene 4 proteins. The diribonucleotide extension activity of gp4-link(del6)/D237A+56-kDa-gp4 is merely 1% the activity of gp4-link(del6)+56-kDa-gp4 (Figure 5, bottom). De novo synthesis assay with the same protein mixtures shows that the diribonucleotide synthesis activity of the same gp4-link(del6)/D237A+56-kDa-gp4 is about 20% of the activity gp4-link(del6)+56-kDa-gp4 (Figure 5, top). The observation that de novo synthesis can occur in trans while extension of exogenous diribonucleotide cannot occur in trans is demonstrated by comparing gp4-link(del6)/D237A+56-kDa-gp4 to gp4-link(del6)+56-kDa-gp4. The same results are obtained with wt-gp4 and gp4-link(ins5). Both gp4-D237A and gp4-link(ins5)/D237A, like gp4-link(del6)/D237A, show very weak (∼1%) restoration of trans diribonucleotide extension activity as compared to the corresponding proteins without the D237A substitution (Figure 5, bottom), whereas ∼20% of de novo synthesis reaction is restored in trans (Figure 5, top). Controls for the experiment are shown in Supplementary Figure S5. These results are surprising since the primase can extend diribonucleotides synthesized de novo in trans. However, as discussed in the Discussion, the diribonucleotides arising from de novo synthesis are already properly positioned in the active site for subsequent extension. The inability of any of the gene 4 proteins to extend an exogenous diribonucleotide in trans suggests that in this mode, diribonucleotide extension is deficient as compared to de novo synthesis. The fact that de novo synthesis occurs in trans by all the gene 4 proteins whereas exogenous diribonucleotide extension does not suggests again that the mechanism underlying de novo synthesis and the mechanism of diribonucleotide extension are distinct steps that require specific distances between the ZBD and the RPD.
Figure 5.
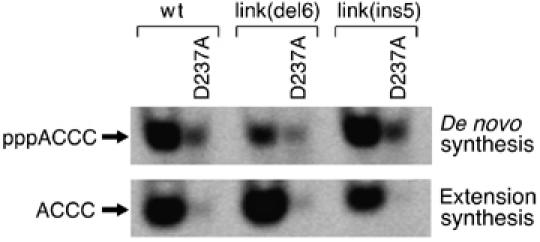
Oligoribonucleotide synthesis catalyzed by an RPD and a ZBD of different subunits. A fixed amount (200 nM) of the indicated gene 4 protein was mixed with 200 nM 56-kDa-gp4. The mixture was incubated at room temperature for 10 min and then with DNA template 5′-GGGTCAAAAAAAAAA-3′ in the standard de novo synthesis reaction with ATP and [α-33P]CTP (top panel) or in the standard AC extension assay with AC and [α-33P]CTP (bottom panel) for 30 min at 37°C. Reaction products were analyzed on a 25% denaturing polyacrylamide gel. The major products, 5′-pppACCC-3′ and 5′-ACCC-3′, are indicated to the left of the gel.
Other activities of the altered gene 4 proteins
The ability of both gp4-link(del6) and gp4-link(ins5) to provide functional primer to T7 DNA polymerase is reduced as compared to the wt-gp4 (Supplementary Figure S1). The alterations of gp4-link(del6) and gp4-link(ins5) in primer synthesis are confined to the primase domain as no significant changes were observed when the helicase unwinding activity, dTTPase hydrolysis, and ssDNA binding of these proteins were compared to that of the wt-gp4 (Supplementary Figure S2B).
Discussion
DNA primases play an essential role in DNA replication. These enzymes catalyze the synthesis of RNA primers at specific recognition sites for the initiation of synthesis of all Okazaki fragments replicated by the lagging strand DNA polymerase (Frick and Richardson, 2001). The DNA primase encoded by phage T7 synthesizes predominantly RNA primers of the sequences 5′-pppACCC-3′, 5′-pppACAC-3′, and 5′-pppACCA-3′ when provided with ribonucleotides and an appropriate ssDNA template. These tetraribonucleotides are in fact found at the 5′ termini of Okazaki fragments synthesized in phage-infected cells (Shinozaki and Okazaki, 1977). Convincing information is available on the structure of the prokaryotic primases and the mechanism of synthesis of the oligonucleotide products (Frick and Richardson, 2001). However, the mechanism by which they recognize specific sequences in DNA and their interactions with the other components of the replisome remain unresolved.
An optimal length of linker is required for optimal activity
In an effort to further elucidate the mechanism of tetraribonucleotide synthesis, we have examined the role of the linker of 16 residues that connects the ZBD with the RPD (Figure 1). Since both the RPD and the ZBD are required for tetraribonucleotide synthesis (Lee and Richardson, 2005), we postulated that their interaction must be affected by the length of the linker connecting them. This linker, at various lengths, is found in several prokaryotic primases such as E. coli, T3, and T4 DNA primases. In earlier studies, we have shown that the linker is sufficiently flexible to allow a ZBD of one subunit to functionally interact with an adjacent RPD within a hexamer (Lee and Richardson, 2002), a finding supported by structural studies (Toth et al, 2003).
Genetic examination of the linker, using phage complementation analyses, shows that at least 10 of these residues, residues 55–64 are not essential for phage growth (Table I). However, shortening the linker to six residues eliminates the ability of the protein to synthesize oligonucleotides. We consider it likely that such a short linker does not allow for functional association of the ZBD with the RPD. Lengthening the linker to 21 residues reduces primase activity as well, although to a lesser extent than shortening the linker to six residues. Most likely, the effective concentration of the ZBD near the RPD is reduced once the linker lengthened to 21 residues or shortened to six residues. These findings suggest that an optimal interaction of the ZBD with the RPD is achieved by a linker longer than six residues but shorter than 21 residues.
Different mechanisms exist for the synthesis of diribonucleotides and their subsequent extension
The most surprising observation arising from the present study is the effect of shortening the linker to 10 residues. Whereas the altered protein, gp4-link(del6), is defective in de novo synthesis of tetraribonucleotides, it is enhanced in its ability to extend a partially formed di- or triribonucleotide to a functional tetraribonucleotide primer. It has been shown that extension of di- and triribonucleotides by wt-gp4 to a tetraribonucleotide is as efficient as de novo synthesis of the tetraribonucleotide. Moreover, the ZBD of the T7 primase is required for di- and triribonucleotide extension as is the recognition site (Kusakabe and Richardson, 1997); substitution of the cryptic cytidine in the trinucleotide recognition sequence with thymidine eliminates both de novo and extension synthesis. The enhanced extension of di- and triribonucleotide by gp4-link(del6) together with its decreased de novo synthesis of oligoribonucleotides provides clear biochemical evidence for two modes of synthesis.
Proposed mechanism for primer synthesis
We envision a model (Figure 6A) in which the ZBD recognizes the core recognition sequence 5′-GTC-3′, and secures it (Kato et al, 2003) for the initiation of diribonucleotide synthesis. Only at this position of the ZBD relative to the RPD and with the correct template secured, is the formation of pppAC catalyzed. Following diribonucleotide synthesis, the ZBD changes its position relative to the RPD while still binding the same core nucleotide sequence. This conformational change moves the template such that the catalytic site can add the next nucleotide. Consequently, gp4-link(del6) with the shorter linker extends diribonucleotides with better efficiency than primases having longer linkers because its ZBD is constantly facing the RPD active site at the ‘extension' position and thus the template is readily delivered for the extension synthesis.
Figure 6.
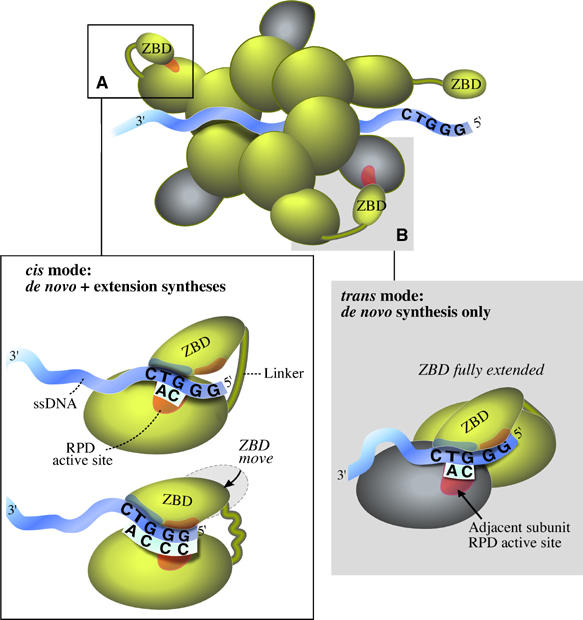
Model for the synthesis of primer by the hexamer gene 4 protein. Based on the studies presented in this communication, we can now explain in full, as elaborated in the Discussion, all of the steps in primer synthesis. The cis mode is shown in panel (A) and the trans mode is shown in panel (B).
The model explains the stringency observed for diribonucleotide synthesis at the core recognition site 5′-GTC-3′. We propose that the ZBD dictates that only AC formation is allowed when it is in the position for diribonucleotide synthesis. In the extension step, in contrast, the ZBD is not directly involved in the catalysis of the phosphodiester bond, but rather in the movement of the template for subsequent extension. Therefore, the extension sequence is dictated by the DNA and RPD alone and other, less-restricted, products are allowed.
The model also explains a yet unresolved question regarding the accumulation of relative large amounts of the diribonucleotide, pppAC, during primer synthesis. Since the extension of these diribonucleotides is accomplished through separate interactions of the ZBD with the RPD, any dissociation of these two steps would result in accumulation of abortive diribonucleotides.
We find that the length of the linker between the RPD and the ZBD found in wt-gp4 is optimal for the synthesis of functional tetraribonucleotides. This length best allows for the ZBD to transition between the ‘initiation' position to its subsequent ‘extension' position. Shorter lengths, but not too short, are better for AC extension, but do not allow for efficient synthesis of pppAC. A longer length is suitable for both de novo synthesis and diribonucleotide extension, but renders the T7 DNA primase less efficient in both activities probably due to improper alignment of the ZBD to its catalytic site.
Proposed interactions of the hexamer subunits
The above discussion has focused on the ZBD of gene 4 protein interacting with the RPD on the same polypeptide. However, the functional form of the gene 4 protein is a hexamer in which the helicase domain is responsible for the assembly of the subunits. As a result, the primase domain is present at the amino half of each of the six subunits. As mentioned earlier and shown in this report, the ZBD of one subunit can interact with the RPD of an adjacent subunit to catalyze the synthesis of oligoribonucleotides. Therefore, in one scenario, the primase catalyzes the de novo synthesis of the initial diribonucleotide via interactions between two adjacent subunits (trans mode). Once the diribonucleotide has been synthesized, it can be extended to the functional tetraribonucleotide while the subunits are in a trans mode. However, when preformed diribonucleotides are used in order to bypass the initial diribonucleotide synthesis step, their extension can only occur in a cis mode. Why can the primase extend diribonucleotides in trans that arise from de novo synthesis and not those provided exogenously? Clearly, the diribonucleotide arising from de novo synthesis is initially positioned within the active site and is available for immediate extension whereas the preformed diribonucleotide must be first positioned in the active site prior to the extension step. The extension synthesis in trans is thus less favorable than diribonucleotide initiation synthesis in this mode. This result fits the model in which the ZBD contacts the RPD via a motif for diribonucleotide initiation. The template is then repositioned such that another motif of the ZBD faces the RPD active site for diribonucleotide extension (Figure 6A). In trans, the ‘initiation' motif of the ZBD can contact the RPD of an adjacent subunit, but cannot move for extension synthesis. The inability of the ZBD to move with the template arises from the linker being fully extended in the trans mode (Figure 6B). In addition to the limitation imposed by the length of the linker, the movement of the ZBD from the ‘initiation' position to the ‘extension' position requires different conformations in cis versus trans. The ‘extension' conformation in trans is less active than the ‘extension' conformation in cis, as it approaches the adjacent subunit in a mode less favorable for extension. The deficiency observed for trans extension synthesis even when using gp4-link(ins5) having a five residues longer linker results from the less-favored ‘extension' conformation in trans.
Several intriguing observations lead us to postulate that in vivo the synthesis of the tetraribonucleotides occurs, at least in part via a trans mode. The gene 4 protein is actually found in phage-infected cells in two molecular weight forms, the 56-kDa gene 4 protein and the 63-kDa gene 4 protein. The former arises as the result of an internal start codon and a strong ribosomal-binding site located within the full-length gene 4. Thus, as pointed out earlier, the 56-kDa gene 4 protein lacks ZBD. In vivo the two forms are found in equal amounts and we speculate that the functional hexamers consist of alternating 56- and 63-kDa subunits. Hence, if the RPD of the 56-kDa protein is utilized, it would be dependent on the ZBD of an adjacent subunit. Recently, single molecule experiments have shown that leading strand synthesis pauses for several seconds whenever primer synthesis occurs even in the absence of a lagging strand DNA polymerase (Lee et al, 2006). Cessation of helicase translocation and hence unwinding of the duplex provides the only reasonable explanation for such a pause. What leads to the inhibition of helicase movement? We propose that the interaction of the ZBD of one subunit with the RPD of an adjacent subunit triggers a conformation change that halts the sequential hydrolysis of dTTP and movement of the DNA around the hexameric ring (Crampton et al, 2006). A trans interaction of the ZBD that serves as a ‘molecular brake' of the RPD in primer synthesis was most recently reported for the E. coli primase DnaG (Corn et al, 2005). The model proposed by Corn et al (2005) depicts a trans interaction between a ZBD of one subunit on a RPD of another subunit. Their model, however, does not address the subsequent extension step of the primer. Thus, the trans mode of oligonucleotide synthesis might serve as a switch to temporarily halt the movement of the replication fork and thus maintain coordination of leading and lagging strand synthesis to await the initiation of a new Okazaki fragment.
Materials and methods
Materials
Oligonucleotides were purchased from Integrated DNA Technologies. QuikChange II Site-Directed Mutagenesis Kit was purchased from Stratagene. T4 polynucleotide kinase was purchased from Amersham Bioscience. Radiolabeled nucleotides were purchased from Perkin Elmer. Agarose and β,γ-methylene dTTP were purchased from USB Corp. Polyethyleneimine cellulose thin layer chromatography (TLC) plates were purchased from JT Baker. ATP-agarose resins were purchased from Sigma-Aldrich. T7 DNA polymerase (T7 gene 5 protein–E. coli thioredoxin complex) and M13mp18 ssDNA were kindly provided by Donald Johnson (Harvard Medical School).
Site-directed mutagenesis, protein overproduction and purification
Plasmids encoding T7 gene 4 protein with alterations in the linker between the ZBD and the RPD were constructed using Stratagene QuikChange II Site-Directed Mutagenesis Kit according to the manufacturer's instructions. The plasmid used for gene 4 cloning contained a methionine substituted for glycine at residue 64 which was introduced to prevent secondary initiation at this site, and has been shown not to affect the activity of the gene 4 protein (Mendelman and Richardson, 1991). The entire gene 4 protein-coding region was confirmed after each manipulation by DNA sequence analysis. After transformation of the plasmid into E. coli HMS174(DE3) or BL21(DE3), gene 4 proteins were overproduced by induction of the gene with 1 mM IPTG for 3 h at 37°C or for 5 h at 30°C. Gene 4 proteins were purified as described (Lee and Richardson, 2001).
Phage complementation assay
E. coli C600 containing a plasmid that expresses gene 4 protein under a T7 promoter (pET24-gp4 or its derivatives constructed in this study) was grown to an OD600 of 1. Serially diluted T7 phage stocks were mixed with an aliquot of the E. coli culture in 0.7% soft agar and poured onto LB plates. After an overnight incubation at room temperature or after 2–4 h at 37°C, the number of plaques appearing on the plate was determined.
Biochemical assays of T7 primase
Most of the assays used in this study have been described (Lee and Richardson, 2001, 2004). All assays (template-directed oligoribonucleotide synthesis, template-directed oligoribonucleotide extension, and RNA-primed DNA synthesis by DNA polymerase) were carried out in a reaction buffer containing 40 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 10 mM DTT, and 50 mM potassium glutamate. Additional components were added as described for each assay. All reactions were carried out at 37°C for the indicated time period.
Template-directed oligoribonucleotide synthesis
The ability of gene 4 protein to catalyze de novo synthesis of oligoribonucleotides was determined by measuring the incorporation of [α-33P]CMP into oligoribonucleotides using a synthetic DNA template containing a primase recognition site. The reaction (10 μl) included 10 μM template DNA 5′-GGGTCAAAAAAAAAA-3′, 0.1 mM each of ATP and [α-33P]CTP (0.1 μCi), and the indicated amount of gene 4 protein. After incubation at 37°C for 20 min, the reaction was terminated by the addition of 5 μl sequencing dye and loaded onto a 25% denaturing polyacrylamide sequencing gel containing 3 M urea. Electrophoresis was carried out at 1800 V for 3 h and the gel was dried for autoradiography. Radioactive oligoribonucleotide products were analyzed using a Fuji BAS 2500 Bioimaging analyzer. In some cases, where indicated, proteins were mixed with other proteins in the reaction buffer prior to addition of the reaction components and were incubated for 10 min at room temperature to allow oligomerization.
Template-directed oligoribonucleotide extension
T7 gene 4 protein can also catalyze the extension of the diribonucleotide AC to the tri- and tetraribonucleotide when provided with a primase recognition site 5′-GGGTC-3′. The reaction was as described above for a template-directed oligoribonucleotide synthesis assay except the diribonucleotide AC or ACC were substituted for ATP at 1 mM final concentration. In some cases, where indicated, proteins were mixed with other proteins in the reaction buffer prior to addition of the reaction components and were incubated for 10 min at room temperature to allow oligomerization.
Supplementary Material
Supplementary Material
Supplementary Figures
Acknowledgments
We thank Ben Beauchamp for help with purifying the proteins. This work was supported by United States Public Health Services Grant GM 54397 and by US Department of Energy Grant DE-FG02-96ER62251.
References
- Corn JE, Pease PJ, Hura GL, Berger JM (2005) Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol Cell 20: 391–401 [DOI] [PubMed] [Google Scholar]
- Crampton DJ, Mukherjee S, Richardson CC (2006) DNA-induced switch from independent to sequential dTTP hydrolysis in bacteriophage T7 DNA helicase. Mol Cell 21: 165–174 [DOI] [PubMed] [Google Scholar]
- Frick DN, Richardson CC (1999) Interaction of bacteriophage T7 gene 4 primase with its template recognition site. J Biol Chem 274: 35889–35898 [DOI] [PubMed] [Google Scholar]
- Frick DN, Richardson CC (2001) DNA primases. Annu Rev Biochem 70: 39–80 [DOI] [PubMed] [Google Scholar]
- Guo S, Tabor S, Richardson CC (1999) The linker region between the helicase and primase domains of the bacteriophage T7 gene 4 protein is critical for hexamer formation. J Biol Chem 274: 30303–30309 [DOI] [PubMed] [Google Scholar]
- Hinton DM, Nossal NG (1987) Bacteriophage T4 DNA primase-helicase. Characterization of oligomer synthesis by T4 61 protein alone and in conjunction with T4 41 protein. J Biol Chem 262: 10873–10878 [PubMed] [Google Scholar]
- Kato M, Ito T, Wagner G, Richardson CC, Ellenberger T (2003) Modular architecture of the bacteriophage T7 primase couples RNA primer synthesis to DNA synthesis. Mol Cell 11: 1349–1360 [DOI] [PubMed] [Google Scholar]
- Keck JL, Roche DD, Lynch AS, Berger JM (2000) Structure of the RNA polymerase domain of E. coli primase. Science 287: 2482–2486 [DOI] [PubMed] [Google Scholar]
- Kusakabe T, Hine AV, Hyberts SG, Richardson CC (1999) The Cys4 zinc finger of bacteriophage T7 primase in sequence-specific single-stranded DNA recognition. Proc Natl Acad Sci USA 96: 4295–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe T, Richardson CC (1997) Gene 4 DNA primase of bacteriophage T7 mediates the annealing and extension of ribo-oligonucleotides at primase recognition sites. J Biol Chem 272: 12446–12453 [DOI] [PubMed] [Google Scholar]
- Lee JB, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM (2006) DNA primase acts as a molecular brake in DNA replication. Nature 439: 621–624 [DOI] [PubMed] [Google Scholar]
- Lee SJ, Richardson CC (2001) Essential lysine residues in the RNA polymerase domain of the gene 4 primase–helicase of bacteriophage T7. J Biol Chem 276: 49419–49426 [DOI] [PubMed] [Google Scholar]
- Lee SJ, Richardson CC (2002) Interaction of adjacent primase domains within the hexameric gene 4 helicase–primase of bacteriophage T7. Proc Natl Acad Sci USA 99: 12703–12708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Richardson CC (2004) The linker region between the helicase and primase domains of the gene 4 protein of bacteriophage T7. Role in helicase conformation and activity. J Biol Chem 279: 23384–23393 [DOI] [PubMed] [Google Scholar]
- Lee SJ, Richardson CC (2005) Acidic residues in the nucleotide-binding site of the bacteriophage T7 DNA primase. J Biol Chem 280: 26984–26991 [DOI] [PubMed] [Google Scholar]
- Mendelman LV, Richardson CC (1991) Requirements for primer synthesis by bacteriophage T7 63-kDa gene 4 protein. Roles of template sequence and T7 56-kDa gene 4 protein. J Biol Chem 266: 23240–23250 [PubMed] [Google Scholar]
- Pan H, Wigley DB (2000) Structure of the zinc-binding domain of Bacillus stearothermophilus DNA primase. Struct Fold Des 8: 231–239 [DOI] [PubMed] [Google Scholar]
- Patel SS, Hingorani MM (1993) Oligomeric structure of bacteriophage T7 DNA primase/helicase proteins. J Biol Chem 268: 10668–10675 [PubMed] [Google Scholar]
- Podobnik M, McInerney P, O'Donnell M, Kuriyan J (2000) A TOPRIM domain in the crystal structure of the catalytic core of Escherichia coli primase confirms a structural link to DNA topoisomerases. J Mol Biol 300: 353–362 [DOI] [PubMed] [Google Scholar]
- Richardson CC (1983) Bacteriophage T7: minimal requirements for the replication of a duplex DNA molecule. Cell 33: 315–317 [DOI] [PubMed] [Google Scholar]
- Rosenberg AH, Griffin K, Washington MT, Patel SS, Studier FW (1996) Selection, identification, and genetic analysis of random mutants in the cloned primase/helicase gene of bacteriophage T7. J Biol Chem 271: 26819–26824 [PubMed] [Google Scholar]
- Sawaya MR, Guo S, Tabor S, Richardson CC, Ellenberger T (1999) Crystal structure of the helicase domain from the replicative helicase–primase of bacteriophage T7. Cell 99: 167–177 [DOI] [PubMed] [Google Scholar]
- Schaeffer PM, Headlam MJ, Dixon NE (2005) Protein–protein interactions in the eubacterial replisome. IUBMB Life 57: 5–12 [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lanka E, Hillenbrand G (1977a) Role of bacteriophage T7 DNA primase in the initiation of DNA strand synthesis. Nucleic Acids Res 4: 4151–4163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzinger E, Lanka E, Morelli G, Seiffert D, Yuki A (1977b) Bacteriophage-T7-induced DNA-priming protein. A novel enzyme involved in DNA replication. Eur J Biochem 72: 543–558 [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Okazaki T (1977) RNA-linked nascent DNA pieces in T7 phage-infected Escherichia coli cells. I. Role of gene 6 exonuclease in removal of the linked RNA. Mol Gen Genet 154: 263–267 [DOI] [PubMed] [Google Scholar]
- Singleton MR, Sawaya MR, Ellenberger T, Wigley DB (2000) Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 101: 589–600 [DOI] [PubMed] [Google Scholar]
- Swart JR, Griep MA (1995) Primer synthesis kinetics by Escherichia coli primase on single-stranded DNA templates. Biochemistry 34: 16097–16106 [DOI] [PubMed] [Google Scholar]
- Tabor S, Richardson CC (1981) Template recognition sequence for RNA primer synthesis by gene 4 protein of bacteriophage T7. Proc Natl Acad Sci USA 78: 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth EA, Li Y, Sawaya MR, Cheng Y, Ellenberger T (2003) The crystal structure of the bifunctional primase–helicase of bacteriophage T7. Mol Cell 12: 1113–1123 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Figures