

Abstract
The underlying mechanisms linking antisense RNA, chromatin architecture and gene expression have not been fully elucidated. Here we show that long transcripts encoded from the Kcnq1ot1 antisense promoter silence the flanking genes more efficiently than short antisense transcripts. Interestingly, the antisense RNA-mediated deposition of inactive chromatin-specific histone modifications was higher with the longer antisense transcripts than with the shorter antisense transcripts. The kinetic studies of expression and chromatin remodeling of overlapping and nonoverlapping genes in response to antisense transcription revealed that the overlapping gene was rapidly silenced due to decrease in the occupancy of basal transcription machinery and simultaneous enrichment of its promoter with inactive chromatin modifications. The nonoverlapping gene, initially enriched with histone modifications specific to active chromatin, was subsequently silenced. Surprisingly, the flanking sequences were initially enriched with H3K9 monomethylation, as compared to di- and trimethylation, with a subsequent shift to trimethylated H3K9 enrichment. Our data provide a new perspective into antisense RNA-mediated gene silencing, and, more importantly, provide an explanation for why the antisense transcripts encoded from imprinting control regions are of significant length.
Keywords: antisense RNA, chromatin, epigenetics, gene silencing, imprinting control regions
Introduction
Several lines of evidence have been accumulated to suggest that antisense RNA-mediated gene silencing can be a key regulatory step in multi-step transcriptional regulatory mechanisms (Lavorgna et al, 2004). Examples of this include X-chromosome inactivation (Lee et al, 1999a), gene regulation in imprinted clusters (Sleutels et al, 2002; Thakur et al, 2004) and, more recently, evidence of individual gene regulation (Tufarelli et al, 2003). In silico studies based on mouse and human EST databases have predicted the presence of more than 2500 antisense transcripts (Yelin et al, 2003). Recent investigations by the FANTOM3 consortium have predicted that antisense transcripts are 1.5–2-fold greater than that estimated from previous studies in mouse and humans, suggesting that antisense transcription-mediated gene regulation is a widespread mechanism (Katayama et al, 2005).
Based on current literature, antisense transcripts can be grouped into two classes: small antisense transcripts and long antisense transcripts. The length of small antisense transcripts, also termed micro-RNAs, range between 21 and 23 nucleotides, while the length of long antisense transcripts varies significantly, that is, from 100 bp to several hundreds of kilobases. While the functional significance of some of small antisense transcripts during development has recently been unraveled (Pasquinelli and Ruvkun, 2002), the functional role of long antisense transcripts in development has not been thoroughly investigated.
Chromatin, the result of the intimate association of histone proteins and DNA into repeating nucleosomal units, is organized into euchromatin and heterochromatin. Emerging evidence suggests a functional role for noncoding RNA in influencing euchromatin and heterochromatin organization (Bernstein and Allis, 2005). However, the underlying mechanisms largely remain unknown. Euchromatin and heterochromatin are distinguished by characterizing the relative presence of various histone modifications. For example, euchromatin is enriched with histone modifications such as acetylated H3K9 (H3K9Ac) and dimethylated H3K4 (H3K9Me2), while heterochromatin is marked predominantly by trimethylated H3K9 (H3K9Me3) and trimethylated H3K27 (H3K27Me3). Interestingly, the three methylated forms of H3K9 (mono- (H3K9Me1), di- (H3K9Me2) and H3K9Me3) are differentially distributed in the silent domains located in euchromatic and pericentric heterochromatin regions. The silent domains in euchromatic regions are enriched with H3K9Me1 and -Me2, while the pericentric heterochromatin is enriched with H3K9Me3 (Peters et al, 2003; Rice et al, 2003).
Recent investigations have invoked antisense transcription in establishment of both active and inactive chromatin states. For example, in the α-globin locus of thalassemic patients, the globin promoter is epigenetically inactivated by antisense transcription from a widely expressed LUC7L promoter, which is juxtaposed in antisense orientaion to the α-globin promoter due to a translocation event (Tufarelli et al, 2003). Interestingly, in a recent investigation, it has been shown that in embryonic stem cells the Tsix antisense transcription induces H3K4Me2 over the entire Xist locus, which overlaps the Tsix antisense transcript, thereby making the Xist/Tsix locus transcriptionally competent. It has been proposed that the Tsix-induced transcriptionally competent chromatin may play a pivotal role in the transition from imprinted to random X-chromosome inactivation during early mammalian embryogenesis (Navarro et al, 2005). Interestingly, however, the antisense transcription from imprinting control regions (ICRs), which are associated with gene clusters exhibiting the parent of origin-specific expression patterns, leads to the silencing of not only overlapping genes but also nonoverlapping genes (Sleutels et al, 2002; Thakur et al, 2004). Although several imprinted antisense transcripts have been identified in imprinted clusters, only the antisense transcripts Air and Kcnq1ot1 have been functionally implicated in regulating imprinting thus far (Sleutels et al, 2002; Thakur et al, 2004). Another interesting feature associated with these antisense transcripts is that they are generally of more than 50 kb in length (Mitsuya et al, 1999; Lyle et al, 2000; Landers et al, 2004). The functional role of the antisense transcript length in the maintenance of gene expression in imprinted gene clusters remains, however, unclear.
The promoter of the Kcnq1ot1 antisense transcript is mapped to an ICR located in intron 10 of the Kcnq1 gene on mouse chromosome 7. The ICR is methylated on the maternal chromosome (Lee et al, 1999b; Smilinich et al, 1999), while it remains unmethylated on the paternal chromosome. The paternal inheritance of a deletion of the Kcnq1 ICR, encompassing the Kcnq1ot1 promoter, resulted in the activation of the normally silent genes Osbp15, Phld2a, Slc22a1l, Cdkn1c (telomeric) and Kcnq1, Tssc4, Cd81, Ascl2 (centromeric), indicating a crucial role for the ICR in maintaining the parent of origin-specific expression of neighboring genes as far as 200 kb distant from the antisense promoter (Fitzpatrick et al, 2002). In a previous report, we have documented that the Kcnq1 ICR executes a bidirectional silencing property in controlling the expression of neighboring genes. By selectively deleting sequences encompassing the antisense promoter, or by truncating antisense transcription by inserting polyadenylation sequences at different distances from the antisense transcription start site, we have shown that the bidirectional silencing property of the Kcnq1 ICR is controlled by antisense RNA (Pandey et al, 2004; Thakur et al, 2004). Although the 3.6 kb Kcnq1 ICR fragment (containing 1.7 kb sequence 3′ of the antisense promoter) independently silences the flanking reporter genes in an episomal-based system, we could not detect any significant silencing when we inserted a polyadenylation sequence 1.7 kb downstream of the antisense transcription start site, that is, at the end of the ICR sequence (Thakur et al, 2004). This observation indicates that antisense transcription beyond the ICR fragment is required for achieving efficient bidirectional silencing. Based on these observations, we have previously proposed that an increase in the length of the antisense transcript or in the duration of antisense transcription would perhaps increase the recruitment of repressor complexes by the antisense RNA. This in turn would create a nucleation site for heterochromatin formation over the sequences that overlap the antisense transcription, followed by the spreading of the heterochromatin to the other areas from the nucleation site (Pandey et al, 2004; Thakur et al, 2004).
We were therefore keen to address the aforementioned outstanding issues concerning the antisense transcription-mediated bidirectional silencing using an episomal-based system. First, by using RT–PCR we have shown that the antisense transcript spans more than 10 kb in length in PS4 episome. By incorporating the SV40 polyadenylation (PolyA) sequences at 4.9, 5.5, 6.2 and 9.2 kb from the antisense transcription start site, we have shown that the degree of silencing is directly proportional to the length of the transcript. In addition, we have shown that longer antisense transcripts could more efficiently propagate the epigenetic modifications specific to inactive chromatin than that shorter transcripts could do.
By analyzing the kinetics of silencing of overlapping and nonoverlapping genes in relation to antisense transcription, we have demonstrated that the antisense RNA initially silences the overlapping gene, and subsequently the nonoverlapping adjacent gene, by spreading an inactive chromatin conformation. The kinetic studies of histone modifications in relation to antisense transcription, analyzed using chromatin immunopurification (ChIP), revealed that all three methylated forms of H3K9 could be detected over the flanking sequences. Surprisingly, however, the flanking sequences were initially enriched with H3K9Me1 as compared to H3K9Me2 or H3K9Me3, with a subsequent shift to H3K9Me3 enrichment. Together, these observations provide significant insights into the Kcnq1ot1 antisense RNA-mediated bidirectional silencing, and, more importantly, the present study provides a strong mechanistic link between antisense RNA and heterochromatin.
Results
The length of the antisense RNA encoded from the Kcnq1ot1 promoter determines the degree of bidirectional silencing
To address the functional role of the antisense transcript length encoded from the Kcnq1ot1 promoter of the Kcnq1 ICR, we initially mapped the antisense transcript in PS4 episome (see PS4 in Figure 1B for details) using the RT–PCR approach. As shown in Figure 1A–C, by using primer pairs covering various regions of PS4 episome (see Figure 1B for the positions of RT–PCR primers, which are shown as dashed lanes), we have shown that the antisense transcript spans sequences of more than 10 kb. We truncated the antisense RNA by inserting the polyA at 4.9 kb (see PS4polyA4.9 in Figure 1B), 5.5 kb (see PS4polyA5.5 in Figure 1B), or 9.2 kb (see PS4polyA9.2 in Figure 1B) downstream of the antisense transcription start site. We have transiently transfected these episomal constructs along with a previously characterized PS4polyA1.7 episome construct (the polyA sequence was inserted 1.7 kb downstream of the antisense transcription start site; see Thakur et al, 2004 for details) into the JEG-3 cell line (derived from human placenta) for 8 days. Initially, we carried out RT–PCR assays on RNA extracted from these transfections using primers that map pre- and post-polyA sequence insertion to check whether the polyA sequence truncated the antisense transcript. We used OripA and OripB RT–PCR primers to check whether the antisense transcript was truncated due to insertion of the PolyA sequence at 9.2 or 5.5 kb downstream of the antisense transcription start site, respectively. Similarly, RT–PCR primers, H19 Pro, were used to check truncation of the antisense transcript due to the PolyA sequence inserted 4.9 kb downstream of the antisense transcription start site (Figure 1B and C). There was no detectable antisense transcription beyond the PolyA sequence insertion site (see the left panel for PS4polyA9.2, middle panel for PS4polyA5.5 and right panel for PS4polyA4.9 in Figure 1C).
Figure 1.
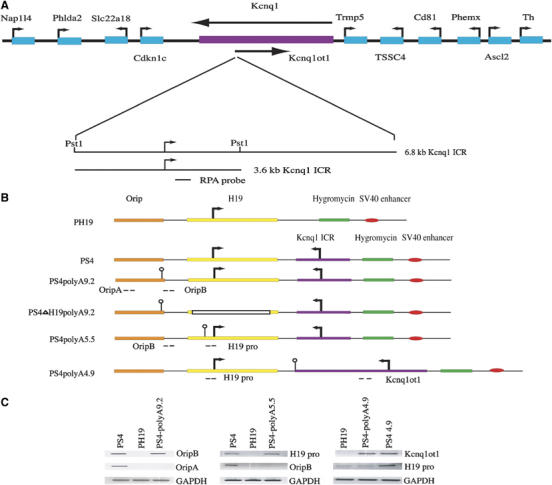
Mapping of the antisense transcript in PS4 episomal plasmid. (A) The physical map of an imprinted cluster at the distal end of the mouse chromosome 7. An extended map of the 6.8 kb region encompassing the Kcnq1 ICR is shown below. The curved arrows depict the transcription start sites. (B) Physical maps of the various episome constructs PH19, PS4, PS4polyA9.2, PS4polyA5.5, PS4ΔH19polyA9.2 and PS4 polyA4.9, depicting the position of the PolyA sequence (shown by the flag) relative to the antisense transcription start site. RT–PCR primers, OriPA, OripB, H19pro and Kcnq1ot1, shown by the dashed lines, were used for both mapping of the antisense transcript and to check truncation of the antisense transcript due to the PolyA sequence insertion. (C) Finemapping of the antisense transcript by RT–PCR showed that the length of the antisense transcript is more than 10 kb. The analyses also showed truncation of the antisense transcript at the PolyA sequence.
We next analyzed the activity of H19 reporter gene by RNase protection assay (RPA) and the hygromycin gene activity by counting the resistant colonies obtained after selection with hygromycin. Together, these analyses enabled us to assess the bidirectional silencing activity of the Kcnq1 ICR, as these reporter genes are located on both sides of the ICR in PS4polyA5.5 and PS4polyA9.2. We observed pronounced silencing of reporter genes in PS4polyA9.2 as compared to PS4polyA5.5 and PS4polyA1.7, indicating that an increase in the length of antisense RNA increases the degree of silencing (see the left panel in Figure 2A). Insertion of the PolyA sequence, however, did not interfere with the antisense transcription (see the right panel in Figure 2A). These results clearly suggest a critical role for the length of transcript encoded from the Kcnq1ot1 antisense promoter.
Figure 2.
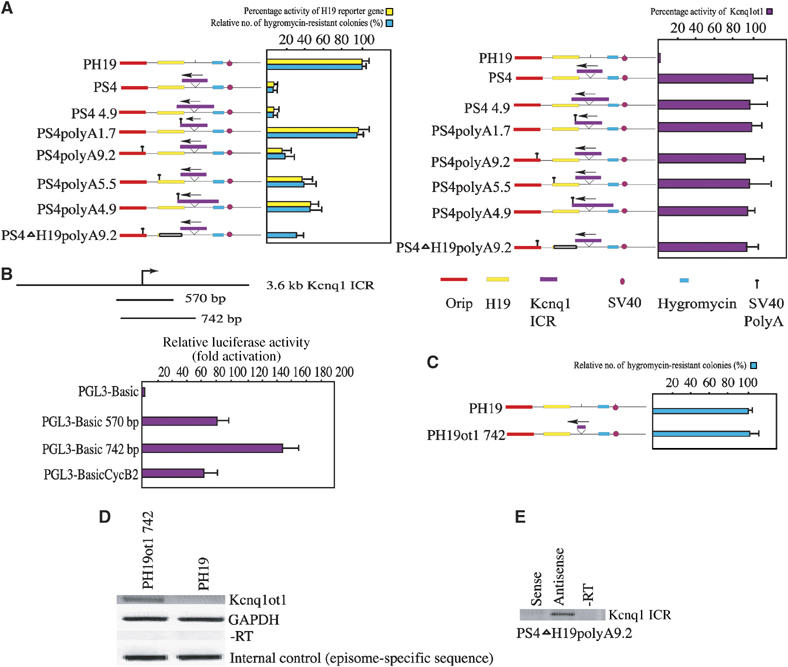
The length of the antisense transcript determines the degree of bidirectional silencing. (A) The left panel contains a bar graph showing the activity of the H19 and hygromycin genes in various episomal constructs, as depicted beside the bar graph. For each construct, the activity of H19 is represented by yellow bars and that of hygromycin by green bars. The activity of each reporter gene is represented as percentage expression levels. The activity of the control vectors (PH19 for H19 and hygromycin genes) were, for convenience, assigned a value of 100% and the values obtained with mutant episomal plasmids were related to this value. The activity of H19 reporter gene was quantified by RPA using RNA obtained from JEG-3 cells harvested 8 days after transient transfection with episomes. Percentages of expression levels were calculated after normalization against total input RNA, using GAPDH mRNA as an internal standard, and episomal copy numbers, quantified by Southern hybridization of DNA obtained from transfected cells probed with both an episome-specific probe and an internal control probe, PDGF gene. The number of hygromycin-resistant clones (green bars) was used to assess bidirectional silencing by the Kcnq1 ICR. The right panel depicts a bar graph showing the expression levels of the antisense transcript in various PolyA constructs, in relation to the control construct PS4. The percentage expression levels for the antisense transcript activity were calculated as described above. The data represent the means±s.d. of three independent experiments. (B) Functional mapping of the Kcnq1ot1 antisense promoter within the Kcnq1 ICR. Cartoon showing the fragments of the Kcnq1 ICR that were cloned into promoter-less luciferase vector (PGL3-Basic). The bar graph shows the relative luciferase activity. The strength of the promoter activity of PGL3-Basic570 and PGL3-Basic742 is presented relative to PGL3-BasicCycB2. All of the luciferase constructs were transfected into JEG-3 cell line. The data represent means±s.d. of three independent experiments performed in duplicate. (C) The 742 bp fragment encompassing the Kcnq1ot1 antisense promoter does not show any bidirectional silencing activity. The graph shows the hygromycin gene activity in PH19ot1 742 relative to the control PH19 plasmid. The data represent means±s.d. of three independent experiments. (D) RT–PCR assay shows that the 742 bp fragment encodes the antisense transcript in PH19ot1 742 episome construct. The RT–PCR was performed using the H19pro primers (see Figure 1B for primer details). (E) Strand-specific RT–PCR, performed using the Kcnq1ot1 primers (see Figure 1B for primer details), shows the presence of the antisense transcript in the Kcnq1 ICR region, but not the presence of any transcript encoded from the episomal sequences.
We next investigated whether the antisense transcript encoded from the sequence spanning the Kcnq1ot1 antisense promoter alone is sufficient to induce bidirectional silencing. To address this issue, we fine mapped the functional antisense promoter in the 3.6 kb Kcnq1 ICR by PCR cloning the several fragments spanning the various regions of the antisense promoter into promoter-less luciferase vector (PGL3-Basic) and assaying their promoter activity by analyzing the luciferase activity. We used CycB2 promoter (B2-Luci) as a positive control for this assay (Lange-zu Dohna et al, 2000). The 742 bp fragment showed maximum promoter activity in contrast to the 570 bp fragment and CycB2 promoter (Figure 2B). We then replaced the 3.6 kb Kcnq1 ICR with the 742 bp fragment in PS4 polyA9.2 (see PH19ot1 742 in Figure 2C) and transfected this plasmid along with PH19 into the JEG-3 cell line. RT–PCR analysis of the RNA obtained from these transfected cells revealed that the 742 bp fragment encodes the antisense transcript, suggesting that the 742 bp fragment contains a functional antisense promoter (Figure 2D). We next analyzed its bidirectional silencing properties by measuring the hygromycin gene activity, by counting the hygromycin-resistant clones. As can be seen in Figure 2C, insertion of the sequences spanning the antisense promoter alone cannot spread the bidirectional silencing.
In both PS4polyA9.2 and PS4polyA5.5 constructs, the antisense RNA overlaps the H19 reporter gene and therefore there is potential for the formation of double-stranded RNA (dsRNA) due to bidirectional transcription from the opposing promoters. This in turn could trigger the dsRNA-mediated RNA interference silencing pathway. In a previous study, however, we ruled out such a possibility by selectively deleting the H19 transcription unit in PS4 episome. Since the antisense transcript overlaps with more than 10 kb sequence in PS4 episome, there is a formal possibility for pairing up with an episome-specific transcript such as EBNA (Thakur et al, 2004). To investigate this possibility, we have deleted 3.0 kb of the H19 transcription unit, covering the H19 promoter and coding regions, from PS4polyA9.2 construct (see PS4ΔH19 polyA 9.2 in Figure 1B). Since we could not analyze the H19 reporter gene activity in PS4ΔH19 polyA 9.2 construct, we assayed the activity of hygromycin gene and found that significant silencing of hygromycin gene had occurred (see the left panel in Figure 2A). More importantly, in PS4ΔH19polyA9.2 construct, the antisense transcript does not overlap with any known episomal-specific transcript. In addition, strand-specific RT–PCR analysis within the Kcnq1 ICR in PS4ΔH19polyA9.2 construct did not reveal any bidirectional transcription, which could be due to cryptic promoter activity from the episomal sequences (Figure 2E). Consistent with our earlier results, the present data obtained with PS4ΔH19 polyA 9.2 rule out the potential involvement of dsRNA, formed as a result of bidirectional transcription from opposing promoters, in the Kcnq1 ICR-mediated bidirectional silencing. Interestingly, the degree of hygromycin gene silencing was relatively low in PS4ΔH19polyA9.2 as compared to PS4polyA9.2. This is consistent with the fact that the total length of the antisense transcript in PS4ΔH19polyA 9.2 is 6.2 kb.
The mouse H19 transcription unit has been shown to contain conserved secondary structures (Tilghman et al, 1995). Though studies thus far have not attributed any known function to the mouse H19 transcript, the antisense transcription through these repeats could generate secondary structures, which in turn could be responsible for silencing. In other words, the observed bidirectional silencing could be as a result of the antisense transcription through the H19 or episome-specific sequences. To address this issue, we added an extra 3.2 kb native sequence from the mouse Kcnq1 locus to the already existing 1.7 kb sequence downstream of the antisense promoter, making the total length of the native Kcnq1ot1 gene sequence downstream of the antisense promoter 4.9 kb. We then inserted the polyA sequence into the unique Not1 site at the end of the 4.9 kb sequence (see PS4polyA4.9 in Figure 1B). This antisense transcript does not overlap with any known episome-specific sequence or transcript from episome. Analysis of reporter gene expression in this construct showed a significant reduction in the activity of both H19 and hygromycin genes, suggesting that the observed bidirectional silencing is a result of 3.6 kb Kcnq1 ICR and that H19 or episome-specific sequences have no role in the silencing (Figure 2A). More importantly, this observation emphasizes the fact that the 1.7 kb downstream sequence harbors critical regions involved in spreading the bidirectional silencing and that these critical regions could effectively propagate silencing signals in longer antisense transcripts by interacting with RNP complexes (Figure 5). The degree of silencing of reporter genes by the antisense RNA was relatively low in PS4polyA4.9 when compared to the PS4 polyA9.2, consistent with the functional role of antisense transcript length in the bidirectional silencing.
Figure 5.
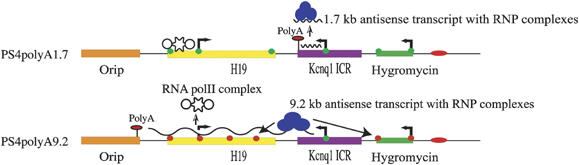
Model describing the functional role of the antisense transcript's length in bidirectional silencing mediated by the Kcnq1ot1 antisense RNA. As the antisense promoter requires shorter duration to synthesize 1.7 kb Kcnq1ot1 antisense transcript in the PS4polyA1.7, it detaches from the chromatin soon after its formation, even though it has the sequences required for the bidirectional silencing. In contrast, the antisense promoter takes a longer duration to synthesize 9.2 kb antisense transcript in the PS4polyA9.2, thus allowing the antisense RNA, complexed with heterochromatic machinery, to remain present for a relatively longer time at the site of antisense transcription. This in turn provides an opportunity for the antisense transcript to spread inactive chromatin structures in cis. The curved line denotes the antisense transcript. H3K9Me3 is depicted with red circles, whereas H3K9Ac is depicted with green circles.
Spreading of epigenetic modifications over the overlapping H19 reporter gene promoter was tightly linked to the antisense transcript length
Since the above observations clearly documented that the longer antisense transcripts could more efficiently silence the flanking genes than the shorter antisense transcripts, we wanted to investigate how the length of the antisense transcript contributes to the silencing of flanking genes. First, we wanted to know whether the Kcnq1ot1 antisense transcription is linked to the establishment of inactive chromatin over the flanking sequences. To this end, we performed ChIP using the wild-type PS4 (PS4polyA9.2) and the mutant PS4 episome carrying the Kcnq1 ICR with mutations at the NF-Y binding sites of the antisense promoter (PS4CAT1–3; for details, see Pandey et al, 2004), which supports only 30% of the antisense transcription compared to that of wild-type PS4. As shown in Figure 3A, B and E, the overlapping H19 promoter was enriched with H3K9Me3 in PS4polyA9.2 in comparison to PS4CAT1–3, suggesting that antisense RNA is involved in modification of the flanking active chromatin into inactive chromatin. We next addressed whether an increase in the length of the antisense transcript has any effect on the establishment of inactive chromatin. As can be seen from Figure 3C–E, the overlapping promoter was enriched with H3K9Me3 in PS4polyA9.2 compared to PS4polyA1.7, suggesting that the longer antisense transcripts encoded from the Kcnq1ot1 promoter could efficiently modify the flanking active chromatin into inactive chromatin.
Figure 3.
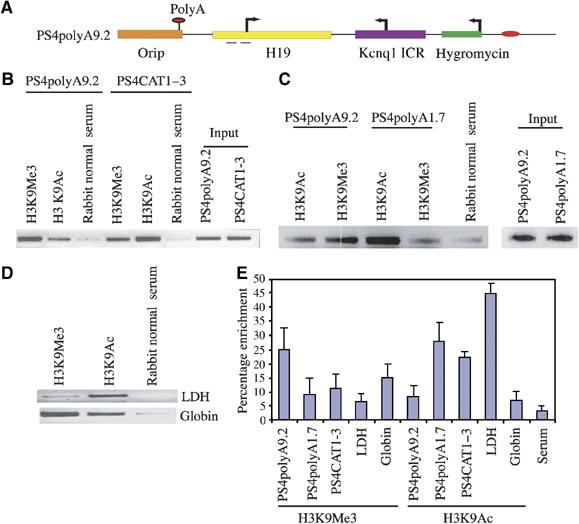
The longer antisense transcripts efficiently induce inactive chromatin over the overlapping promoter as compared to the shorter transcripts. (A) Schematic representation of the PS4polyA9.2 episomal construct. (B) Semiquantitative PCR analysis of ChIP material obtained from the JEG-3 cells, transfected with PS4polyA9.2 and PS4CAT1–3, revealed that the inactive chromatin is established over the overlapping promoter in the antisense transcription-dependent manner. (C) Semiquantitative PCR analysis of ChIP material obtained from the JEG-3 cells transfected with PS4polyA9.2 and PS4polyA1.7 episome constructs revealed that the longer, but not the shorter antisense transcript, modifies the active flanking chromatin over the H19 reporter gene promoter (enriched in H3K9Ac) into inactive chromatin (enriched in H3K9Me3). (D) β-Globin (used as a control to measure the histone modifications specific to inactive chromatin) and LDH (housekeeping gene, used as a control to measure the histone modifications specific to active chromatin) promoters were used as internal controls to assess the quality of the ChIPs. (E) Quantitative analysis of ChIP material used in (B–D). The graph shows percentage enrichment and the data were quantified as described in Materials and methods. The data represent means±s.d. of three independent ChIP experiments.
The antisense RNA silences overlapping genes prior to nonoverlapping genes
In mouse, the antisense transcript Kcnq1ot1 overlaps with only the Kcnq1 gene, but nevertheless affects the regulation of several nonoverlapping genes (see Figure 1A). Kinetic experiments, where the overlapping and nonoverlapping genes are silenced in vivo by means of antisense RNA, have not been performed. This could be partly due to difficulty in performing such kinetic experiments when the silencing is regulated in a spatio-temporal manner during early mouse development. In PS4polyA9.2, the antisense transcript overlaps with H19, but not hygromycin gene, which is on the other side of the Kcnq1 ICR (see PS4polyA9.2 in Figure 1B). An episome-based system (PS4polyA9.2), therefore, provides an opportunity to address the temporal order of the silencing of the overlapping and the nonoverlapping genes in relation to the antisense transcription. To investigate this issue in detail, we propagated PH19 and PS4 polyA 9.2 plasmids into the JEG-3 cell line for 2, 4 and 8 days after transfection. The RNA extracted from these transient transfections was subjected to RPA using the RNA probes specific to H19 and hygromycin reporter genes. As shown in Figure 4A, significant silencing of the overlapping gene could be seen within 2 days after transfection. Whereas the nonoverlapping gene was marginally silenced at around 2 days, marked silencing was detected only at 8 days. Interestingly, we obtained similar results when we performed the latter experiment on NS11 episome construct (where the hygromycin gene is the overlapping gene to the antisense promoter and the H19 gene is the nonoverlapping gene; for details, see Thakur et al, 2004), which rules out the possible involvement of promoter specificity (Figure 4B). Collectively, these results suggest that the antisense RNA silences the overlapping gene much earlier than the nonoverlapping gene.
Figure 4.

The antisense transcription silences the overlapping gene prior to the nonoverlapping gene. Bar graphs showing the activity of H19 and hygromycin genes in PS4polyA9.2 (A) and NS11 (B) plasmids, propagated for 2, 4 and 8 days post-transfection. The H19 and hygromycin gene activity was assessed using RPA. The percentage activity of the H19 and hygromycin genes was calculated after normalization against total input RNA and episomal copy number, as described in the legend of Figure 2. (C) Schematic representation of PS4polyA9.2 episomal construct. The dashed lines indicate the position of the primers used for quantitative PCR reactions. (D, E) Quantitative analyses of ChIP material (bar graphs showing percentage enrichment, the data represent means±s.d. of three independent ChIP experiments and quantification was performed as described in Figure 3E) using antibodies such as H3K9Me1, H3K9Me2 and H3K9Me3, H3K27Me3, H3K4Me2 and H3K9Ac on the crosslinked chromatin obtained from PH19- and PS4polyA9.2-transfected cells harvested at 2 and 8 days post-transfection. The ChIP analyses revealed that the H19 overlapping promoter (D) is more enriched with inactive chromatin-specific histone modifications in the 2-day sample as compared to the nonoverlapping hygromycin promoter (E). The bar graphs showing the real-time quantification of enrichment of H3K9Me3 and H3K9Ac in the EBNA (Ec, F) and H19 (H19c1 and H19c2, G) coding regions of PS4polyA9.2 and PH19 constructs. Real-time quantification was performed as described in Figure 3E. The data represent means±s.d. of three independent ChIP experiments. (H, I) Antisense transcription interferes with the occupancy of RNAPolII preinitiation complex. Quantitative PCR analysis of ChIP material obtained using RNAPolII and TFIIB antibodies on crosslinked chromatin from JEG-3 cells transfected with PS4polyA9.2 and PH19 for 2 and 8 days over the promoters of the overlapping H19 (H) and the nonoverlapping hygromycin gene (I). The graphs show percentage enrichment. Real-time quantification was performed as described in Figure 3E. The data represent means±s.d. of three independent ChIP experiments.
The silencing of the overlapping gene, but not the nonoverlapping gene, by the antisense RNA 2 days after transfection was an intriguing finding, which prompted us to investigate the kinetics of inactive chromatin formation over the overlapping and nonoverlapping genes in relation to antisense transcription. To this end, we assayed the temporal order of histone modifications specific to active and inactive chromatin over the overlapping and nonoverlapping promoters by ChIP analysis using various antibodies (H3K9Me1, H3K9Me2, H3K9Me3, H3K9Ac, H3K27Me3 and H3K4Me2) on crosslinked chromatin obtained from the JEG-3 cells transfected with PH19 and PS4PolyA9.2 for 2 and 8 days. Quantitative analyses of the immunopurified material show that, in the 2-day sample, the overlapping promoter was enriched with H3K9Me1, H3K4Me2 and H3K9Ac, but not with H3K9Me2, H3K9Me3 and H3K27Me3. The 8-day sample, however, was relatively enriched with H3K9Me3 in comparison to H3K4Me2, H3K9Ac, H3K9Me1 and H3K9Me2. H3K27Me3 at the overlapping promoter was significantly more enriched in the 8-day sample as compared to the 2-day sample (Figure 4C and D).
In contrast, the chromatin surrounding the nonoverlapping promoter was more enriched with H3K9Ac and H3K4Me2 than with H3K9Me1, H3K9Me2 and H3K9Me3 in the 2-day sample. In the 8-day sample, however, the chromatin was relatively enriched in H3K9Me3 compared with other histone modifications (Figure 4C and E). Since there is a significant silencing of the overlapping promoter compared with the nonoverlapping promoter, even though the overlapping promoter was enriched with H3K9Me1, H3K4Me2 and H3K9Ac, we presumed that the antisense transcription, in addition to epigenetically modifying the flanking chromatin, might also interfere with formation of RNAPolII preinitiation complex. To test this possibility, we performed the kinetic analysis of occupancy of RNAPolII and TFIIB, which are major constituents of RNAPolII preinitiation complex, on both the overlapping and nonoverlapping promoters in relation to the antisense transcription. Interestingly, RNAPolII and TFIIB occupancy is significantly disturbed in the overlapping promoter in 2- and 8-day samples as compared to the nonoverlapping promoter (Figure 4H and I).
We next analyzed whether the antisense RNA-mediated heterochromatinization is restricted to the promoter regions or whether it includes nonpromoter sequences as well. We have addressed this issue by analyzing the levels of H3K9Me3 and H3K9Ac in the EBNA (Ec) and H19 (H19c1 and H19c2) coding regions of PS4polyA9.2 and PH19 constructs (Figure 4C). The EBNA and H19 coding regions showed increased levels of H3K9Me3 and low levels of H3K9Ac in PS4polyA9.2 compared to PH19, suggesting that antisense RNA-mediated bidirectional silencing involves linear spreading of epigenetic modifications (Figure 4F and G). Collectively, these results indicate that H3K9Ac and H3K9Me profiles are primarily affected by the antisense transcription. More importantly, these findings indicate that antisense RNA-mediated heterochromatinization involves the processivity of H3K9Me1 and H3K9Me2 into H3K9Me3.
Discussion
Antisense transcription-mediated bidirectional silencing is a poorly explored area in biology. The antisense transcripts encoded from ICRs are generally longer than 50 kb in length and, more importantly, are implicated in the bidirectional silencing of flanking genes spread over several hundreds of kilobases. However, studies aimed at unraveling the functional significance of antisense transcript length have not been carried out. In this report, we show, for the first time, that antisense transcript length is intimately linked to the degree of bidirectional silencing and chromatin remodeling. By inserting the polyA sequence at different locations relative to the antisense transcription start site in PS4 episome, we have demonstrated that longer antisense transcripts silence flanking reporter genes more efficiently than do shorter antisense transcripts, suggesting that the length of the antisense transcript plays a critical role in antisense RNA-mediated bidirectional silencing. This observation raises an important question, namely, how does an increase in the length of antisense transcript determine the intensity of silencing? We envisage two different scenarios to explain this intriguing observation. (1) If the antisense RNA is longer, the antisense transcriptional process requires more time to synthesize RNAs, thus allowing the antisense RNA to remain present for a longer time at the site of transcription. In turn, this helps to increase the recruitment of heterochromatic complexes by the crucial cis-acting sequences in the antisense RNA to the regions that overlap antisense transcription, thus forming a nucleation site for the spreading of the heterochromatin in cis (see Figure 5). (2) Alternatively, the longer antisense transcripts would bind more efficiently to chromatin in cis than do the shorter antisense transcripts. The binding of the antisense RNA to the chromatin must be mediated through either the binding of RNP complexes to the crucial sequences in the RNA or through direct interaction of certain sequences in the RNA itself. We presume that the spreading of heterochromatin in this situation could be analogous to Xist-mediated X-inactivation in mammalian females.
Although recent investigations have shown the presence of allele-specific chromatin modifications in the entire mouse Kcnq1domain (Lewis et al, 2004; Umlauf et al, 2004), the relevance of allele-specific histone modifications to antisense transcription has not been established. By exploiting the wild-type Kcnq1 ICR (PS4polyA9.2) and the Kcnq1 ICR harboring mutant antisense promoter (PS4CAT1–3), the present data implicate the antisense RNA in the remodeling of active chromatin into inactive chromatin. More importantly, an enrichment of histone modifications specific to inactive chromatin by the longer antisense transcripts, but not by the shorter antisense transcripts, further underscores the functional role of the length of antisense RNA in recruitment of heterochromatic machinery. The presence of inactive chromatin not only at the promoter sequences but also at the nonpromoter sequences suggests that antisense RNA-mediated bidirectional silencing involves linear spreading of inactive chromatin.
Our parallel kinetic studies of expression and chromatin organization of overlapping and nonoverlapping genes in relation to antisense transcription suggest that the overlapping promoter enriched with H3K9Me1, H3K9Ac and H3K4Me2 is silenced initially, whereas the nonoverlapping promoter initially enriched with H3K4Me2 and H3K9Ac is silenced later. However, within 8 days, both the overlapping and nonoverlapping promoters are substantially silenced and are enriched with H3K9Me3, deacetylated H3K9 and H3K27Me3, suggesting that methylation of H3K9 and deacetylation of H3K9 are early events that take place in response to the antisense transcription and seem to coincide with the silencing process. Interestingly, significant silencing of the overlapping promoter in the presence of high levels of H3K9Me1, H3K4Me2 and H3K9Ac suggests that initial silencing of the overlapping promoter could be the result not only of antisense RNA-mediated heterochromatinization but also of a mechanism that is related to the antisense transcription. We presume that the basal transcription machinery at the antisense promoter could, by being stronger, occlude the transcription machinery at the overlapping promoter, thus contributing to the significant silencing of the overlapping promoter. The significant loss in the occupancy of RNApolII and TFIIB over the overlapping promoter, but not over the nonoverlapping promoter in the 2-day sample, renders support to the suggestion that antisense transcription through the overlapping promoter directly interferes with the occupancy of the RNAPolII preinitiation complex. Based on these observations, we presume that the silencing of the overlapping gene involves both the antisense transcription-mediated epigenetic modification of chromatin and interference in formation of the RNAPolII preinitiation complex. Silencing of the nonoverlapping gene, however, is primarily due to the antisense RNA-mediated bidirectional spreading of heterochromatin in cis. In a previous report, we have ruled out the potential involvement of dsRNA, formed as result of transcription between the overlapping H19 and the antisense promoters, in bidirectional silencing (Thakur et al, 2004). The presence of the bidirectional silencing in PS4ΔH19polyA9.2 further supports our previous data. However, we cannot rule out the possibility that stemloop structures within the Kcnq1ot1 antisense RNA could trigger repeat associated small-interfering RNA-mediated heterochromatinization that subsequently spreads to neighboring regions.
Previously, we have demonstrated that DNA methylation of the overlapping promoter occurs much later than the silencing process in response to antisense transcription (Thakur et al, 2003). Together, these findings suggests that changes in H3K9Me and H3K9Ac is the primary event in response to antisense transcription and that DNA methylation occurs as a consequence of histone modifications. Interestingly, changes in H3K9Me1 and H3K9Me2 and deacetylation of H3K9 were detected in relation to antisense transcription in the 2-day PS4polyA9.2 sample. The kinetic data presented here do not allow us to determine whether histone deacetylation occurs prior to histone methylation, as both occurred at about the same time. Previously, the kinetic data on integrated transgene silencing in in vitro cultured cells have suggested that deacetylation of H3K9 occurs first, followed by H3K9Me3 and DNA methylation (Mutskov and Felsenfeld, 2004). Taken together, these observations indicate that the temporal order of histone modifications in relation to gene silencing occurs in multiple pathways.
The enrichment of H3K9Me1 and H3K9Me2 after 2 days and H3K9Me3 after 8 days suggests the processivity of H3K9Me1 and H3K9Me2 into H3K9Me3. Interestingly, a recent report showed that the less stable silent domains in the euchromatin are marked by H3K9Me1 and H3K9Me2, while the more stable pericentric heterochromatin is predominantly marked by H3K9Me3 (Lehnertz et al, 2003; Peters et al, 2003; Rice et al, 2003). The gradual enrichment of H3K9Me3 from H3K9Me1 and H3K9Me2 in this investigation suggests that the silent chromatin structure that is formed initially as a result of the antisense transcription is of a less stable character and is gradually transformed into a more stable silent chromatin structure, resembling pericentric heterochromatin. More importantly, it is speculated that the trimethylated state, rather than mono- and dimethylated states of a selective lysine position in the histone N-terminus, represents a stable epigenetic mark (Lehnertz et al, 2003; Peters et al, 2003). Based on these observations, we speculate that the degree of silencing generated by the antisense RNA may be linked to the degree of methylation at the H3K9.
It has been shown that G9a histone methyltransferase catalyzes the formation of the majority of global H3K9Me1 and H3K9Me2, whereas H3K9Me3 is catalyzed specifically by Suv39h1 and Suv39h2 (Peters et al, 2003; Rice et al, 2003). The presence of all three methylated forms of H3K9 in response to the antisense transcription indicates that G9a and Suv39h histone methyltransferases are part of the heterochromatic machinery that is recruited through the antisense RNA. Interestingly, ES cells lacking Suv39h1 and Suv39h2 methyltransferases showed a marked decrease in the levels of H3K9Me3 with a simultaneous increase in the levels of H3K9Me1 in the pericentric heterochromatin, indicating that H3K9Me1 could act as an in vivo substrate for Suv39h1 and Suv39h2 (Lehnertz et al, 2003; Peters et al, 2003; Rice et al, 2003). The present observation, that there is a gradual shift from H3K9Me1 and H3K9Me2 into H3K9Me3 in antisense RNA-mediated silencing, provides a strong support for the suggestion that H3K9Me1 and H3K9Me2 can act as in vivo substrates for Suv39h1 and Suv39h2.
Materials and methods
Plasmid constructs
PS4polyA9.2 and PS4polyA5.5 episomes were constructed by inserting the SV40 polyadenylation sequence into Hpa1 and Pml1 sites of PS4 episome, respectively. The SV40 polyadenylation sequence was amplified using primers as described earlier (Thakur et al, 2004). PS4ΔH19polyA9.2 was made by selectively deleting the 3.0 kb H19 transcript unit by cutting the PS4polyA9.2 with Bstz17I and re-ligating the plasmid. PS4polyA4.9 epsiomal plasmid was made by amplifying 3.2 kb of native Kcnq1 sequence (see Supplementary data for primer details) and cloning the amplified fragment at the 3′end of the antisense transcription unit of the 3.6 kb Kcnq1 ICR in PS4 episome, making the total antisense transcript unit 4.9 kb long. The polyA sequence, amplified with primers flanked with Not1 site, was cloned into the unique Not1 site at the end of the 4.9 kb antisense transcription unit.
The episome silencer/insulator assay
The pREP4-based episomal vectors were transfected into JEG-3 cells, according to the methods described previously (Kanduri et al, 2000). In order to assess the role of length of antisense transcription in silencing, total RNA was extracted 8 days after transfection, whereas for analysis of overlapping and nonoverlapping gene silencing, episomal plasmids were transfected for 2, 4 and 8 days. RPA was performed as described previously (Kanduri et al, 2000) using 365 bp H19 antisense probe, 320 bp hygromycin antisense probe, 150 bp glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antisense probe as control and a 270 bp Kcnq1ot1 probe. Quantification of individual protected fragments was done using a Fuji FLA 3000 Phosphorimager. The H19 and Kcnq1ot1 expression were corrected with respect to both internal control (GAPDH) and episome copy number as determined by Southern blot analysis of BglII restricted DNA, hybridized with H19 and PDGFB probes (Kanduri et al, 2000).
To assess the effect of Kcnq1 ICR-mediated bidirectional silencing on hygromycin resistance gene, equimolar concentrations of episomal-based plasmids were transfected into JEG-3 cells. Following transfection, the cells were selected with 150 μg/ml of hygromycin until all the cells died in the control plate, which contained the cells incubated with transfection reagent without any episomal plasmid DNA. Following selection, the drug-resistant colonies were stained with hematoxylin and counted.
RT–PCR
Random primed cDNA (using random primer, Promega) and strand-specific cDNA templates (using specific primer) were synthesized using Omniscript reverse transcriptase (Quiagen), using 2 μg of DNase I (RQ1, Promega)-treated RNA as a template. The cDNAs were PCR amplified for 25 cycles (94°C for 45 s, 57°C for 30 s and 72°C for 1 min) using primers listed in Supplementary data. The conditions for stand-specific cDNA synthesis were followed as per the protocol published previously (Newall et al, 2001).
Luciferase assays
JEG-3 cells were transiently transfected with the PGL3-Basic, PGL3-Basic-containing fragments spanning the antisense promoter and PGL3-BasicCycB2 vectors. After 36 h, cells were lysed and luciferase activities in duplicate samples were determined as described by the manufacturer (Promega). The pCH110 vector carrying the β-galactosidase gene under the control of the SV40 promoter was used as an internal control for transfection efficiency. Luciferase values were calculated by dividing the luciferase activity by the β-galactosidase activity.
Chromatin immunoprecipitation
Chromatin immunoprecipitation assays were performed using the protocol described earlier (Kuo and Allis, 1999) and the following antibodies: H3K9Ac and H3K4Me2 (Upstate), H3K9Me1, H3K9Me2 and H3K9Me3 and H3K27Me3 (Abcam), RNApolII (Euromedex) and TFIIB (Santa Cruz). We performed quantitative real-time PCR measurements for input and immunopurified material using Quantitect SYBR green PCR mix in an iCycler real-time PCR machine (Biorad). Each PCR reaction was run in triplicate to control for PCR variation. We calculated the percentage of immunoprecipitation (IP) by dividing the average value of the IP by the average value of the corresponding input. The primers used in the semiquantitative and quantitative PCRs are listed in Supplementary data. The primers for LDHA and β-globin promoters used as controls have been described previously (Diaz-Meyer et al, 2005).
Supplementary Material
Supplementary Material
Acknowledgments
We thank Professor Rolf Ohlsson for suggestions, Gerard Malsher and Joanne Whitehead for critical review of the article. We thank Roberto Mantovani for providing PGL3-Basic CycB2 (B2-Luci) for our studies. This work was supported by the grants from Göran Gustafssons Stiftelse, Swedish Research Council (VR-NT) and the Swedish Cancer Research foundation (Cancerfonden), Stiftelsen för Zoologisk Forskning to CK. CK is a Senior Research Fellow (Epigenetics) supported by the Swedish Research Council (Medicine).
References
- Bernstein E, Allis CD (2005) RNA meets chromatin. Genes Dev 19: 1635–1655 [DOI] [PubMed] [Google Scholar]
- Diaz-Meyer N, Yang Y, Sait SN, Maher ER, Higgins MJ (2005) Alternative mechanisms associated with silencing of CDKN1C in Beckwith–Wiedemann syndrome. J Med Genet 42: 648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick GV, Soloway PD, Higgins MJ (2002) Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet 32: 426–431 [DOI] [PubMed] [Google Scholar]
- Kanduri C, Holmgren C, Franklin G, Pilartz M, Ullerås E, Kanduri M, Liu L, Ginjala V, Ulleras E, Mattsson R, Ohlsson R (2000) The 5′-flank of the murine H19 gene in an unusual chromatin conformation unidirectionally blocks enhancer–promoter communication. Curr Biol 10: 449–457 [DOI] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, Wells C, Frith M, Ravasi T, Pang KC, Hallinan J, Mattick J, Hume DA, Lipovich L, Batalov S, Engstrom PG, Mizuno Y, Faghihi MA, Sandelin A, Chalk AM, Mottagui-Tabar S, Liang Z, Lenhard B, Wahlestedt C (2005) Antisense transcription in the mammalian transcriptome. Science 309: 1564–1566 [DOI] [PubMed] [Google Scholar]
- Kuo M, Allis C (1999) In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19: 425–433 [DOI] [PubMed] [Google Scholar]
- Landers M, Bancescu DL, Le Meur E, Rougeulle C, Glatt-Deeley H, Brannan C, Muscatelli F, Lalande M (2004) Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic Acids Res 32: 3480–3492. (print 2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange-zu Dohna C, Brandeis M, Berr F, Mossner J, Engeland K (2000) A CDE/CHR tandem element regulates cell cycle-dependent repression of cyclin B2 transcription. FEBS Lett 484: 77–81 [DOI] [PubMed] [Google Scholar]
- Lavorgna G, Dahary D, Lehner B, Sorek R, Sanderson CM, Casari G (2004) In search of antisense. Trends Biochem Sci 29: 88–94 [DOI] [PubMed] [Google Scholar]
- Lee JT, Davidow LS, Warshawsky D (1999a) Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 21: 400–404 [DOI] [PubMed] [Google Scholar]
- Lee MP, DeBaun MR, Mitsuya K, Galonek HL, Brandenburg S, Oshimura M, Feinberg AP (1999b) Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith–Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci USA 96: 5203–5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH (2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13: 1192–1200 [DOI] [PubMed] [Google Scholar]
- Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, Higgins M, Feil R, Reik W (2004) Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 36: 1291–1295 (Epub 2004 Oct 31) [DOI] [PubMed] [Google Scholar]
- Lyle R, Watanabe D, te Vruchte D, Lerchner W, Smrzka OW, Wutz A, Schageman J, Hahner L, Davies C, Barlow DP (2000) The imprinted antisense RNA at the Igf2r locus overlaps but does not imprint Mas1. Nat Genet 25: 19–21 [DOI] [PubMed] [Google Scholar]
- Mitsuya K, Meguro M, Lee MP, Katoh M, Schulz TC, Kugoh H, Yoshida MA, Niikawa N, Feinberg AP, Oshimura M (1999) LIT1, an imprinted antisense RNA in the human KvLQT1 locus identified by screening for differentially expressed transcripts using monochromosomal hybrids. Hum Mol Genet 8: 1209–1217 [DOI] [PubMed] [Google Scholar]
- Mutskov V, Felsenfeld G (2004) Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J 23: 138–149 (Epub 2003 Dec 11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P, Pichard S, Ciaudo C, Avner P, Rougeulle C (2005) Tsix transcription across the Xist gene alters chromatin conformation without affecting Xist transcription: implications for X-chromosome inactivation. Genes Dev 19: 1474–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newall AE, Duthie S, Formstone E, Nesterova T, Alexiou M, Johnston C, Caparros ML, Brockdorff N (2001) Primary non-random X inactivation associated with disruption of Xist promoter regulation. Hum Mol Genet 10: 581–589 [DOI] [PubMed] [Google Scholar]
- Pandey RR, Ceribelli M, Singh PB, Ericsson J, Mantovani R, Kanduri C (2004) NF-Y regulates the antisense promoter, bidirectional silencing, and differential epigenetic marks of the Kcnq1 imprinting control region. J Biol Chem 279: 52685–52693 (Epub 2004 Sep 29) [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Ruvkun G (2002) Control of developmental timing by microRNAs and their targets. Annu Rev Cell Dev Biol 18: 495–513 [DOI] [PubMed] [Google Scholar]
- Peters AH, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577–1589 [DOI] [PubMed] [Google Scholar]
- Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD (2003) Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell 12: 1591–1598 [DOI] [PubMed] [Google Scholar]
- Sleutels F, Zwart R, Barlow DP (2002) The non-coding air RNA is required for silencing autosomal imprinted genes. Nature 415: 810–813 [DOI] [PubMed] [Google Scholar]
- Smilinich N, Day C, Fitzpatrick G, Caldwell G, Lossie A, Cooper P, Smallwood A, Joyce J, Schofield P, Reik W, Nicholls R, Weksberg R, Driscoll D, Maher E, Shows T, Higgins M (1999) A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith–Wiedemann syndrome. Proc Natl Acad Sci USA 96: 8064–8069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur N, Kanduri M, Holmgren C, Mukhopadhyay R, Kanduri C (2003) Bidirectional silencing and DNA methylation sensitive methylation spreading properties of the KcnqI ICR map to the same regions within the KcnqI imprinting control region. J Biol Chem 273: 9514–9519 [DOI] [PubMed] [Google Scholar]
- Thakur N, Tiwari VK, Thomassin H, Pandey RR, Kanduri M, Gondor A, Grange T, Ohlsson R, Kanduri C (2004) An antisense RNA regulates the bidirectional silencing property of the kcnq1 imprinting control region. Mol Cell Biol 24: 7855–7862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman SM, Bartolomei MS, Webber AL, Brunkow ME, Saam J, Leighton PA, Pfeifer K (1995) Genomic imprinting of the H19 and Igf2 genes in the mouse. In Genomic Imprinting: Causes and Consequences, Ohlsson R, Hall K, Ritzen M (eds) Cambridge: Cambridge University Press [Google Scholar]
- Tufarelli C, Stanley JA, Garrick D, Sharpe JA, Ayyub H, Wood WG, Higgs DR (2003) Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet 34: 157–165 [DOI] [PubMed] [Google Scholar]
- Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, Feil R (2004) Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 36: 1296–1300 (Epub 2004 Oct 31) [DOI] [PubMed] [Google Scholar]
- Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, Nemzer S, Pinner E, Walach S, Bernstein J, Savitsky K, Rotman G (2003) Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol 21: 379–386 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material