

Abstract
The spatiotemporal regulation of cAMP can generate microdomains just beneath the plasma membrane where cAMP increases are larger and more dynamic than those seen globally. Real-time measurements of cAMP using mutant cyclic nucleotide-gated ion channel biosensors, pharmacological tools and RNA interference (RNAi) were employed to demonstrate a subplasmalemmal cAMP signaling module in living cells. Transient cAMP increases were observed upon stimulation of HEK293 cells with prostaglandin E1. However, pretreatment with selective inhibitors of type 4 phosphodiesterases (PDE4), protein kinase A (PKA) or PKA/A-kinase anchoring protein (AKAP) interaction blocked an immediate return of subplasmalemmal cAMP to basal levels. Knockdown of specific membrane-associated AKAPs using RNAi identified gravin (AKAP250) as the central organizer of the PDE4 complex. Co-immunoprecipitation confirmed that gravin maintains a signaling complex that includes PKA and PDE4D. We propose that gravin-associated PDE4D isoforms provide a means to rapidly terminate subplasmalemmal cAMP signals with concomitant effects on localized ion channels or enzyme activities.
Keywords: AKAP, cAMP, microdomain, PDE4, PKA
Introduction
It is now well accepted that cAMP signals can be compartmentalized to subcellular regions creating distinct pools or ‘microdomains' of the second messenger within the cell (Buxton and Brunton, 1983; Jurevicius and Fischmeister, 1996; Rich et al, 2001a; Zaccolo and Pozzan, 2002; Barnes et al, 2005). The compartmentalization of this freely diffusible messenger is fundamental to its ability to spatially segregate a bewildering array of downstream cAMP signaling events. Recent methodological advances have enabled the direct measurement of spatially and temporally segregated cAMP signals in the single cell (Rich et al, 2000; Goaillard et al, 2001; Zaccolo et al, 2002; Rochais et al, 2004; Dodge-Kafka et al, 2005). Mutant cyclic nucleotide-gated ion channels (CNGCs) can be used as cellular cAMP biosensors to measure changes in subplasmalemmal cAMP with high temporal resolution. Expression studies in HEK293 cells and cardiac myocytes have used these channels to show that agonists produce larger and faster changes in cAMP just beneath the plasma membrane than in the bulk cytosol. These findings have been used as evidence for the existence of cAMP ‘microdomains' where second messenger diffusion may be restricted by physical or enzymatic barriers (Rich et al, 2000, 2001b; Rochais et al, 2004).
Biochemical studies have also highlighted the role of multiprotein signaling complexes that localize the activity of the cAMP-dependent protein kinase (Tasken and Aandahl, 2004; Wong and Scott, 2004). These signaling complexes are organized by A-kinase anchoring proteins (AKAPs) that tether inactive protein kinase A (PKA) holoenzymes and other signaling molecules to defined subcellular locations (Coghlan et al, 1995; Klauck et al, 1996; Tasken et al, 2001). At these sites, AKAP signaling complexes function as cellular focal points for the propagation of specific cAMP-dependent phosphorylation events, as the anchored kinase is localized in close proximity to only a few of its many substrates (reviewed in Wong and Scott, 2004). Membrane targeting of PKA by AKAPs has been implicated in the cAMP-dependent regulation of several ion channels and the surface expression of AMPA-type synaptic glutamate receptors (Johnson et al, 1994; Rosenmund et al, 1994; Gao et al, 1997; Westphal et al, 1999; Dart and Leyland, 2001; Marx et al, 2002; Oliveria et al, 2003; Snyder et al, 2005). The local degradation of cAMP at such sites by cAMP-specific phosphodiesterases would create an efficient, tightly controlled signaling system.
Phosphodiesterases provide the sole means for degrading cAMP in cells and play a vital role in shaping intracellular gradients of the second messenger (Beavo and Brunton, 2002; Conti et al, 2003). The type 4 phosphodiesterase (PDE4) family provides a highly efficient and dynamic means of regulating cAMP as they exclusively hydrolyze cAMP and the activity of the ‘long' PDE4 isoforms (16 isoforms from four genes) is enhanced by PKA phosphorylation (Houslay and Adams, 2003). Previous co-immunoprecipitation studies provided the first evidence that specific PDE4 long isoforms and PKA can be complexed with AKAPs localized in the perinuclear and centrosomal regions of cells to limit the activation of downstream cAMP targets (Dodge et al, 2001; Tasken et al, 2001; recently reviewed by Baillie et al, 2005). In this report, we have taken a functional approach to investigate whether a similar type of molecular interaction shapes the spatiotemporal characteristics of subplasmalemmal cAMP changes in live HEK293 cells. Using CNGCs to monitor cAMP levels, we provide the first direct evidence that an AKAP/PKA/PDE4 complex governs the dynamic changes in cAMP levels at the plasma membrane under physiological conditions. Using RNA interference (RNAi) methods to selectively knockdown endogenous candidate AKAPs, we identify gravin as the AKAP responsible for the localized regulation of cAMP levels by PKA and PDE4 activity. Immunoprecipitation studies confirm that isoforms of PDE4D associate with gravin. These interactions are likely to play an essential role in terminating the actions of cAMP and the concomitant termination of PKA-mediated phosphorylation events at the plasma membrane.
Results
Subplasmalemmal cAMP dynamics
A mutant of the α-subunit of rat olfactory CNGCs (C460W/E583M) was expressed in HEK293 cells using an adenoviral vector to monitor subplasmalemmal cAMP changes in response to agonist stimulation. Heterologous expression of this mutant CNGC subunit produces a functional channel with 10-fold greater sensitivity to cAMP over cGMP (Rich et al, 2001b). CNGC activity was assessed by visualizing Ca2+ influx into fura-2-loaded cells, or by measuring activation of an inward current (ICNG) under voltage-clamp conditions. Figure 1A illustrates the typical Ca2+ influx in response to 1 μM PGE1 in a number of HEK293 cells expressing the CNGC. The same data are presented as pseudocolor images of fura-2 ratio intensity at selected time points after the addition of PGE1 (Figure 1B), and as the mean change in fura-2 signal over time (Figure 1C, n=23 cells). Superfusion of PGE1 for 2 min produced a transient rise in cytosolic Ca2+ levels (Figure 1A–C) representing a combination of cAMP synthesis and rapid hydrolysis/diffusion of cAMP away from its site of production. The dynamics of the cAMP signal generated in close proximity to the plasma membrane differ dramatically from the bulk cytosolic cAMP changes generated by the same stimulus (Figure 1D). In order to contrast subplasmalemmal and global cAMP changes, two different sensors were used to monitor cAMP changes in the different cellular compartments. The CNGCs localize to the plasma membrane where they detect rapid, transient cAMP changes (Figure 1D, black trace). In contrast, a cAMP sensor based on the cAMP-binding domain of an exchange protein directly activated by cAMP (Epac1) (Nikolaev et al, 2004) monitors cAMP changes in the cell cytosol. The FRET-based Epac-1 probe revealed a slower, more sustained rise in cAMP levels in the bulk cytosol (Figure 1D, red trace). Similar global cAMP changes were seen in all 26 cells tested, confirming compartmentalization of cAMP signals at the cell periphery to generate dynamic cAMP events.
Figure 1.
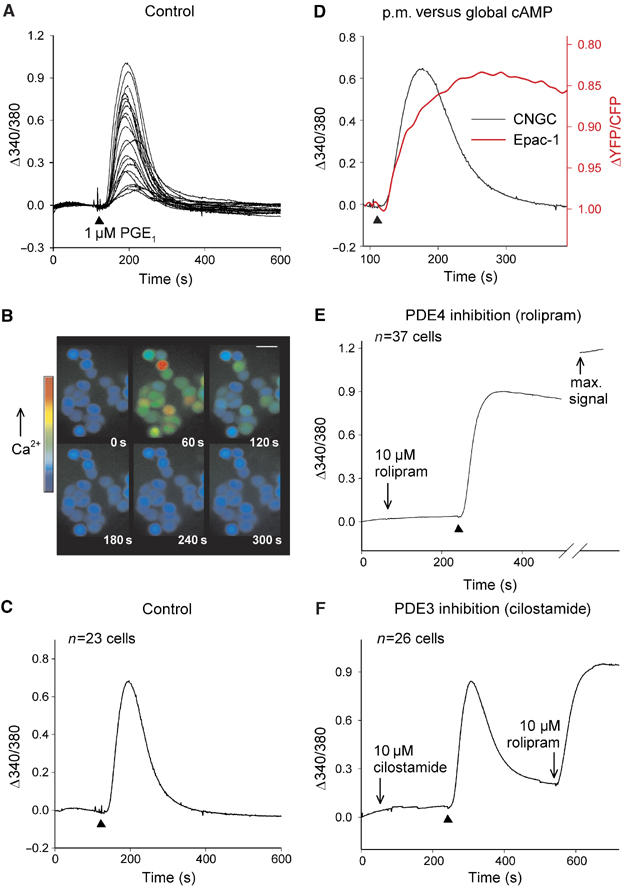
Subplasmalemmal cAMP dynamics. (A) A 1 μM portion of PGE1 produced a transient rise in Ca2+ levels as a consequence of AC activity in HEK293 cells expressing CNGCs. Agonist application is indicated by ▴. (B) Single-cell pseudocolor changes in fura-2 ratio intensity at 1 min intervals after PGE1 addition, or (C) average fura-2 ratio change over time. (D) Comparison of CNGC signal with FRET-based cAMP sensor signal from a single cell in response to 1 μM PGE1. (E) Pretreatment with 10 μM rolipram inhibited the return of cAMP levels to baseline. Maximum signal from the CNGCs obtained by addition of 10 μM PGE1+forskolin+rolipram. (F) Pretreatment with 10 μM cilostamide did not prevent cAMP recovery.
It has been previously suggested that the transient cAMP signals seen at the plasma membrane depend on phosphodiesterase activity (Rich et al, 2001a, 2001b). Therefore, cells were treated with the selective inhibitor rolipram (10 μM) for 3 min before PGE1 addition to examine the role of a cAMP-specific PDE (PDE4) in regulating the cAMP signal (Figure 1E, n=37 cells). Although rolipram did not increase basal cAMP levels, it profoundly inhibited the cAMP recovery profile (Figure 1E), suggesting that PDE4 catalyzes the metabolism of subplasmalemmal cAMP. Subsequent application of a cocktail to generate an additional cAMP rise (10 μM PGE1+10 μM forskolin+100 μM IBMX) amplified the signal further, confirming that the mutant CNGCs were not saturated under our experimental conditions. In cells treated with the PDE3 selective inhibitor (cilostamide, 10 μM), transient subplasmalemmal cAMP changes were still seen during stimulation with 1 μM PGE1 (Figure 1F) consistent with previous findings (Rich et al, 2001b). The subsequent addition of rolipram produced a dramatic and sustained rise in the cAMP signal detected by the CNGCs, again confirming that PDE4 activity shapes subplasmalemmal cAMP changes. In parallel experiments using uninfected HEK293 cells, PGE1 (or any other agent used in our study) did not produce any changes in Ca2+ independently of CNGC activation.
PDE4, PKA and AKAP interaction shapes cAMP changes
The pronounced effects of selective inhibition of PDE4 on subplasmalemmal cAMP signals (Figure 1E and F) suggest that local hydrolysis of cAMP levels by this enzyme is central to the compartmentalization of subplasmalemmal cAMP signals. It is well established that the activation of long isoforms of PDE4 is mediated via phosphorylation by a cAMP-dependent PKA (Sette and Conti, 1996). Furthermore, the PDE4D3 isoform is recruited to AKAP signaling complexes where it becomes preferentially activated by the anchored pool of PKA (Dodge et al, 2001; Tasken et al, 2001). To investigate the potential negative feedback regulation of cAMP levels by anchored PKA and PDE4 complexes, fura-2-loaded HEK293 cells were pretreated with inhibitors of PKA and PKA/AKAP interaction. As before, PDE4 inhibition generated a sustained cAMP signal compared to controls (Figure 2A and B). Pretreatment with the PKA inhibitor, H89 (10 μM), for 30 min produced a similar inhibitory effect on cAMP recovery during PGE1 stimulation (Figure 2C, n=28 cells). Cells were also pretreated with a stearoylated Ht31 peptide (St-Ht31; 20 μM) for 45 min before the addition of 1 μM PGE1 to assess whether PKA action at the plasma membrane depended on its interaction with an AKAP (Figure 2D, n=27 cells). This cell-permeant peptide competitively inhibits PKA–AKAP interactions, with a consequent displacement of PKA from its site of action (Carr et al, 1992; Vijayaraghavan et al, 1997). Under these conditions, PGE1 induced a clear rise in cAMP levels with very limited recovery, suggesting that PKA must interact with an AKAP in order to localize to, and activate, PDE4 at the membrane. The introduction of proline residues to the RII-binding region of Ht31 prevents its disruption of PKA binding to AKAPs (Carr et al, 1992). Pretreatment with this control peptide, St-Ht31-P (20 μM), did not affect recovery of the PGE1-induced cAMP rise (Figure 2E, n=30 cells). Analyses of cAMP recoveries from all Ca2+ influx measurements in CNGC-expressing HEK293 cells are presented as the percentage of cAMP recovery 5 min after PGE1 addition, compared to the peak agonist-evoked cAMP increase seen within the first minute (Figure 2F). The PDE4 inhibitor rolipram, the PKA inhibitor H89 and the anchoring disruptor peptide St-Ht31 each inhibited the recovery of cAMP levels when compared to controls (Figure 2F, P<0.001). In contrast, the control peptide St-Ht31-P did not decrease cAMP recovery. These findings support our hypothesis that an anchored PKA–PDE complex is responsible for the feedback regulation of subplasmalemmal cAMP levels.
Figure 2.
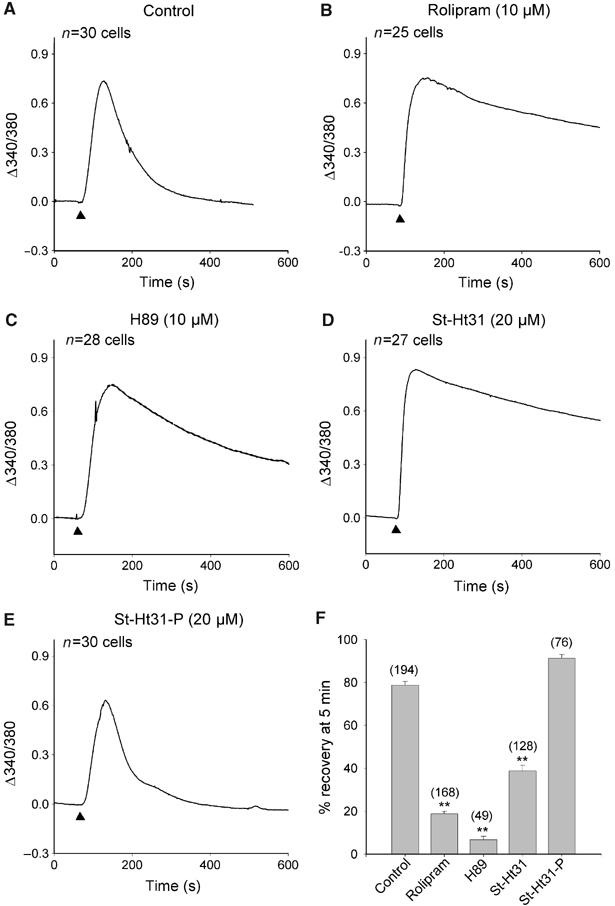
Effects of PDE4, PKA and PKA–AKAP interaction on cAMP dynamics. (A) A 1 μM portion of PGE1 (▴) produced a transient rise in Ca2+ levels as a consequence of AC activity. (B) Pretreatment with 10 μM rolipram, (C) 10 μM H89 or (D) 20 μM stearoylated Ht31 peptide (St-Ht31) inhibited the return of cAMP levels to baseline. (E) Proline-substituted Ht31 peptide (St-Ht31-P) had no effect. Applications of PGE1 were for 2 min. (F) Quantification of the effects of rolipram, H89 and St-Ht31 on cAMP recovery during the 5 min period after 1 μM PGE1 application. Data are plotted as mean±s.e.m., n-values are shown in brackets and * and ** represent values of P<0.01 and P<0.001, respectively, compared to controls.
Patch-clamp assessment of local cAMP dynamics
As monitoring Ca2+ influx through CNGCs to detect cAMP changes could theoretically be subject to artifactual effects of Ca2+ on CNGC activity (Kaupp and Seifert, 2002; Trudeau and Zagotta, 2003), we performed parallel experiments to assess CNGC activation by direct measurement of ionic currents using perforated patch-clamp electrophysiology. All the experiments were performed in nominally Ca2+-free solutions to remove any potential Ca2+ inhibition of the CNGCs, at a holding potential of −30 mV. Application of PGE1 for 2 min induced a transient inward current, signifying a transient rise in local cAMP levels (Figure 3A). When the experiment was repeated in the presence of 10 μM rolipram, a clear inward current was evoked, a minimal recovery was observed and ICNG reached a sustained plateau (Figure 3B). In agreement with the fura-2 experiments, inhibition of PKA activity with H89 or disruption of PKA anchoring with St-Ht31 also prevented ICNG recovery (Figure 3C and D). As before, the control peptide St-Ht31-P did not delay cAMP recovery (Figure 3E). The rates of ICNG recovery from several experiments in Ca2+-free conditions are compared and confirm inhibition of cAMP recovery by rolipram, H89 or Ht31 (Figure 3F, P<0.01). In contrast, the amplitude of ICNG activation was not significantly affected by any of the inhibitors tested (data not shown). In two additional experiments, we confirmed that addition of 1 μM PGE1 and 10 μM rolipram (Figure 3B) did not saturate our cAMP sensor. The amplitude of ICNG activation following inhibition of PDE4 (20.5 pA/pF on average) was further enhanced upon addition of a cocktail to maximally activate cAMP signals in the same cells (10 μM PGE1+10 μM forskolin+100 μM IBMX; 23.6 pA/pF on average). Collectively, these data suggest that anchored PKA and PDE4 function to remove cAMP during the recovery phase of the second messenger response.
Figure 3.
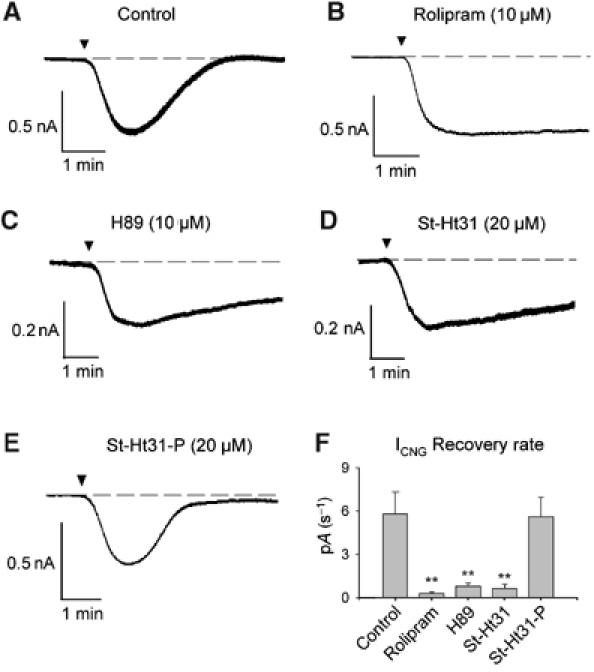
The effects of PDE4, PKA and PK–AKAP inhibition on PGE1-evoked ICNG activation. (A) Control application of 1 μM PGE1 (▾) produced a transient inward current in a CNGC-expressing HEK293 cell. (B) Pretreatment with 10 μM rolipram, (C) 10 μM H89 or (D) 20 μM St-Ht31 peptide inhibited recovery of the ICNG evoked by PGE1. (E) Negative control peptide 20 μM St-Ht31-P had no effect. Cells were held at −30 mV throughout. (F) Comparison of rates of current recovery. Data are plotted as mean±s.e.m. and ** represents values of P<0.01 compared to controls. Rolipram (n=4), H89 (n=4) and St-Ht31 (n=4) significantly reduced the rate of ICNG recovery compared to controls (n=5). St-Ht31-P (n=4) had no significant effect.
Gravin knockdown reduces subplasmalemmal cAMP hydrolysis
Several AKAPs target PKA and other enzymes to the plasma membrane (Wong and Scott, 2004). RII overlays performed on HEK293 subcellular fractions identified three major membrane-associated AKAP species (Figure 4A). Immunoblotting with AKAP-specific antibodies identified the proteins as AKAP79, AKAP149 and gravin (AKAP250). A significant proportion of the total gravin (AKAP250) pool was also present in the cytosolic fraction in HEK293 cells (Figure 4A). AKAP-Lbc and MAP2 were also present in our cells, but these AKAPs partition exclusively into the cytoplasmic fraction (data not shown). A short hairpin RNA (shRNA) plasmid that effectively knocks down endogenous AKAP79 in HEK293 cells has been extensively characterized by us (Hoshi et al, 2005). In addition, we developed shRNA plasmids to knock down endogenous AKAP149 and gravin. HEK293 cells were cotransfected with shRNA plasmid targeting AKAP79, AKAP149 or gravin and a CD4 expression plasmid. Three days post-transfection, CD4+ cells were identified and selected as described previously (Hoshi et al, 2005). Western blot analysis of whole-cell extracts from CD4+ cells showed that endogenous AKAP79, AKAP149 and gravin levels were reduced by ∼80% in cells transfected with the appropriate shRNA plasmid compared to pSilencer controls (Figure 4B).
Figure 4.
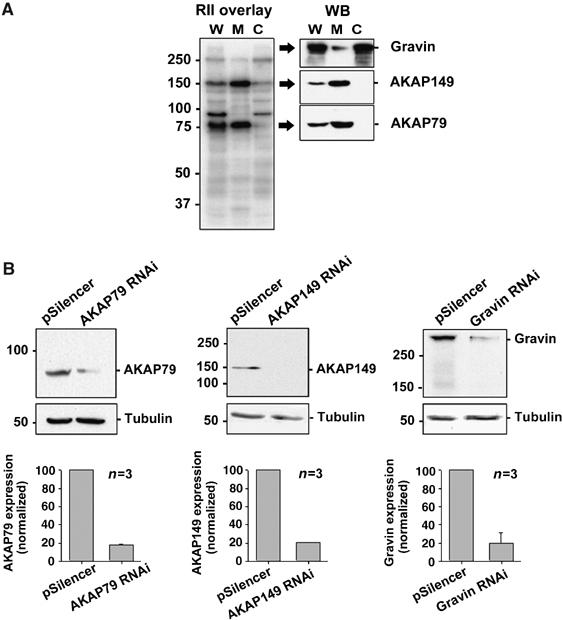
Identification and RNAi knockdown of candidate membrane-associated AKAPs. (A) RII overlays and immunoblotting identify AKAP79, AKAP149 and gravin (AKAP250) as the major membrane-associated AKAPs in HEK293 cells. W: whole-cell extract; M: membrane; C: cytosol. (B) Quantification of AKAP79, AKAP149 and gravin silencing in CD4+ cells at 3 days post-transfection. Lysates were immunoblotted for AKAP79, AKAP149 or gravin (upper blot) and tubulin (loading control, lower blot). AKAP expression levels were normalized to tubulin levels and expressed as a percentage of the amount of the AKAP in control cells. Amalgamated data from three independent experiments are presented as mean±s.e.m.
To identify the AKAP that was responsible for the termination of subplasmalemmal cAMP signals, CNGC activation was examined in HEK293 cells transfected with an shRNA plasmid targeting AKAP79, AKAP149 or gravin. Two days post-transfection, cells were infected with CNGC adenovirus, and cAMP changes in response to 1 μM PGE1 application were measured the following day. Significant changes in the dynamics of the cAMP signal were observed in cells where expression of gravin had been silenced (Figure 5A and D). Knockdown of this anchoring protein produced a marked reduction of cAMP recovery following agonist application (P<0.001). Selective AKAP149 or AKAP79 knockdown did not significantly alter subplasmalemmal cAMP recoveries (Figure 5A and D). An ensemble of real-time measurements of CNGC activity in individual HEK293 cells in response to PGE1 superfusion illustrates the range of effectiveness of gravin RNAi to inhibit the cAMP recovery phase in a single experiment when compared to the pSilencer controls (Figure 5B). The variability of response may reflect differences in the degree of gravin knockdown in each cell. These data demonstrate that at the single-cell level, suppression of endogenous gravin levels effectively slows the rate of recovery of subplasmalemmal cAMP. Thus, gravin may localize PKA to sites at the cell periphery to selectively enhance PDE4 activity and shape the cAMP signal in this region of the cell. Moreover, the fast recovery of cAMP levels under control conditions would suggest that the gravin signaling complex is functionally or physically coupled to PDE4 activity.
Figure 5.
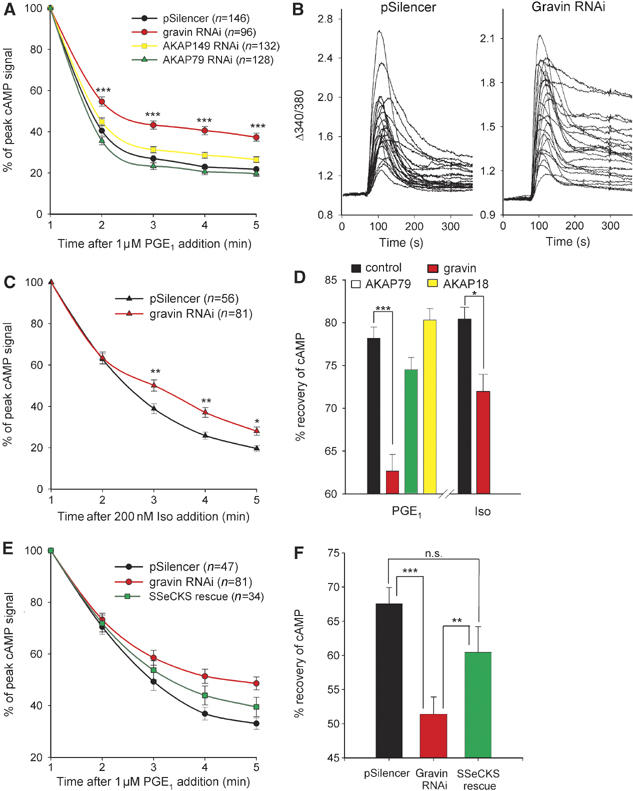
Gravin RNAi attenuates cAMP recovery following GPCR stimulation. (A) Comparison of cAMP recoveries at 1 min intervals following 1 μM PGE1 stimulation of CNGC-expressing HEK293 cells 3 days post-transfection of shRNA plasmids specific for gravin, AKAP149, AKAP79 or pSilencer (control). All data are normalized to the peak cAMP increase seen ∼1 min following agonist addition. (B) Examples of CNGC activation profiles in individual fura-2-loaded HEK293 cells under control conditions (pSilencer) and following gravin knockdown (gravin RNAi). (C) Inhibition of cAMP recovery in gravin-suppressed cells following addition of 200 nM isoprenaline. (D) Comparison of cAMP recoveries 5 min after stimulation with PGE1 (1 μM) or isoprenaline (200 nM) in cells where membrane-associated AKAPs had been selectively silenced. (E) Rescue of the effects of gravin knockdown (red trace) by expression of the murine orthologue, SSeCKS (green trace). (F) Quantification of the effects of gravin suppression and SSeCKS expression on cAMP recovery seen at 5 min after PGE1 addition. *P<0.05, **P<0.01, ***P<0.001 using ANOVA and Newman–Keuls.
Effects of gravin knockdown on β2 adrenoceptor-mediated signals
Previous studies have revealed that gravin is dynamically recruited to β2-adrenoceptors (β2-ARs) as a consequence of agonist stimulation (Lin et al, 2000; Fan et al, 2001) and PKA phosphorylation (Tao et al, 2003). This interaction plays an essential role in the desensitization, sequestration and resensitization of the G-protein-coupled receptor (Shih et al, 1999; Lin et al, 2000; Fan et al, 2001). Thus, we also examined the effects of gravin suppression on cAMP hydrolysis at the plasma membrane during application of isoprenaline (200 nM; a non-selective β-AR agonist). Knockdown of endogenous gravin levels significantly reduced the cAMP recovery rate following stimulation with isoprenaline (Figure 5C and D). This is consistent with decreased PDE activity in the vicinity of the β-AR. However, in contrast to the PGE1 data (Figure 5A), cAMP recovery following isoprenaline stimulation was generally slower under control conditions, and the effects of gravin suppression only become evident from 3 min onwards (Figure 5C, red trace). At 5 min after isoprenaline stimulation, the effects of gravin suppression were still not as robust as that seen during PGE1 stimulation (Figure 5D), signifying possible differences in the actions of the PDE-associated gravin complex at different GPCRs.
Expression of mouse gravin (SSeCKS) rescues cAMP recovery in gravin-silenced HEK293 cells
A standard means of confirming functional effects mediated by RNAi is to rescue the phenomenon by expression of a protein orthologue that is refractory to the gene silencing. Therefore, HEK293 cells selected for expression of the gravin shRNA plasmid were re-transfected with a murine gravin orthologue, SSeCKS (Src-suppressed C kinase substrate). The introduction of SSeCKS into HEK293 cells significantly reversed (∼60%) the effects of gravin knockdown with respect to the cAMP recovery profile (Figure 5E and F, P<0.01). The incomplete rescue by SSeCKS can be at least partly explained by limited transfection of the plasmid, estimated to be around 75–80%, in our cells. Another factor could be a reduced efficiency of PKA targeting by SSeCKS compared to endogenous gravin. In parallel experiments, AKAP18α, a membrane-targeted AKAP (Fraser et al, 1998) that is present at very low levels in HEK293 cells (FD Smith and JD Scott, personal communication), was re-transfected instead of SSeCKS as a negative control to assess the specificity of gravin/SSeCKS effects on cAMP recovery rates. Overexpression of AKAP18α did not reverse the effects of gravin knockdown (data not shown). Together, these results confirm an essential role for gravin in the regulation of subplasmalemmal cAMP dynamics.
Gravin interacts with PDE4D
Our data so far have implicated gravin and PDE4 activity in the suppression of subplasmalemmal cAMP signals. As PDE4 isoforms can associate with a growing number of targeting proteins (Baillie et al, 2005), we asked whether gravin was a PDE interacting protein. PDE4D immune complexes isolated from HEK293 cell lysates were screened for copurification of gravin by Western blot (Figure 6, lanes 1 and 2). Gravin copurified with PDE4D from HEK293 cell extract (Figure 6, upper blot, lane 2), but was not detected in the IgG control (Figure 6, upper blot, lane 1). Bands corresponding in size to PDE4D3 and PDE4D5 isoforms were present in the immune complexes (Figure 6, lower blot, lane 2), which is consistent with previous studies showing that PDE4D3 and PDE4D5 are the major PDE4D isoforms in HEK293 cells (Hoffmann et al, 1999; Lynch et al, 2005).
Figure 6.
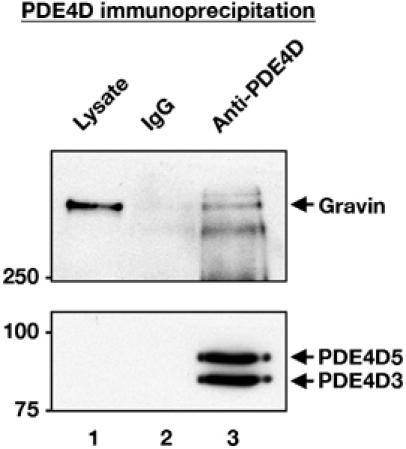
PDE4D co-precipitates with gravin. Endogenous PDE4D was immunoprecipitated from non-transfected HEK293 cell lysates (lane 3, bottom blot). Bands with molecular weights corresponding to PDE4D3 and PDE4D5 were observed. Immunoblotting demonstrates that gravin is present in immune complexes isolated with anti-PDE4D (lane 3; top blot; representative of three independent experiments).
Discussion
The production of cAMP by adenylyl cyclases is critical to the control of numerous cellular functions. It is now assumed that the diversity of action of this second messenger is dependent upon its localization and subsequent metabolism in discrete subcellular compartments. Previous studies have shown an important role for the cAMP-dependent phosphodiesterase, PDE4, in the compartmentalization of subplasmalemmal cAMP signals (Jurevicius and Fischmeister, 1996; Rich et al, 2001b; Rochais et al, 2004; Barnes et al, 2005). However, little is known about the mechanism of PDE4 localization to discrete cellular regions, where it plays an essential role in shaping the spatiotemporal dynamics of cAMP signals. Using recombinant CNGCs as biosensors to assess local changes in cAMP, we demonstrated the presence of a functional signaling complex involving PDE4, PKA and an AKAP. This complex localized cAMP-degrading enzyme activity to plasma membrane sites to generate transient cAMP changes in response to adenylyl cyclase activity. In contrast, global cAMP signals, detected with the cytosolic Epac1 FRET-based cAMP sensor, were slower and more sustained in response to the same stimulus. The ability of the PDE4 inhibitor rolipram, the PKA antagonist H89 and the AKAP-disrupting peptide St-Ht31 to inhibit recovery of PGE1-induced subplasmalemmal cAMP increases (Figure 2) suggests that each individual component of a PKA/PDE/AKAP signaling unit is essential to control the dynamics of cAMP changes at the plasma membrane. These data are consistent with a negative feedback regulation of cAMP by the activation of PDE4 via an anchored PKA (Sette and Conti, 1996; Dodge et al, 2001; Tasken et al, 2001; Dodge-Kafka et al, 2005). Pretreatment of cells with rolipram and H89 together did not augment inhibition of the recovery phase further (data not shown), suggesting that PDE4 and PKA actions are coupled. Thus, the modest decrease in cAMP levels that remained in the presence of selective inhibitors of the proposed signaling complex was most likely due to diffusion of cAMP away from its site of synthesis and into the bulk cytosol. Previous studies have estimated high diffusion rates of cAMP of ∼0.8 × 10−5 cm2/s (Bacskai et al, 1993) although cAMP diffusion between subplasmalemmal microdomains and the bulk cytosol is thought to be restricted to ∼5 × 10−9 cm2/s owing ‘permeability barriers' that hinder cAMP diffusion between cellular compartments (Rich et al, 2000).
The tight control of cAMP at its site of synthesis is likely to play an important role in regulating the effects of cAMP, or PKA, at numerous downstream targets. Indeed, these findings are supported by recent evidence that the cardiac selective anchoring protein, mAKAP, assembles an intricate multicomponent cAMP signaling module at the perinuclear membrane that includes three cAMP-dependent enzymes: PKA, PDE4D3 and Epac-1 (Dodge et al, 2001, Dodge-Kafka et al, 2005). The key regulatory enzyme in the complex is PDE4D3, as it counterbalances local accumulation of cAMP and the subsequent activation of PKA or Epac-1. Conversely, PKA phosphorylation of PDE4D3 increases its affinity for mAKAP and the Vmax for cAMP, to decrease localized cAMP levels that ultimately terminate PKA activity and Epac-1 signaling (Conti et al, 2003; Dodge-Kafka et al, 2005). In a separate study by Tasken et al (2001), co-immunoprecipitation revealed that a PKA-RIIα-associated AKAP450 localized PDE4D3 activity to the centrosomal area of rat testis Sertoli cells, where it was thought to regulate microtubule stability. Until now, the presence of a similar PKA/PDE4/AKAP complex at the plasma membrane was only a speculation (Conti et al, 2003; Malbon et al, 2004a). The RNAi approach that we employed to selectively silence membrane-associated AKAPs in HEK293 cells clearly demonstrated that gravin is responsible for the targeting of PKA and PDE4 activity to the plasma membrane. Gravin (also known as AKAP250, AKAP12 and SSeCKS) was first discovered to be an AKAP in 1997 and is thought to play a key role in the regulation of GPCRs through its direct interaction with PKA and protein kinase C (PKC) (Nauert et al, 1997; for recent review, see Malbon et al, 2004b). Co-immunoprecipitation studies using endogenous proteins in HEK293 cells revealed an interaction between gravin and PDE4D.
The slowing of the cAMP recovery phase reported here following PKA inhibition with H89 is consistent with PKA-dependent activation of long isoforms of PDE4 (Sette and Conti, 1996). This is supported by the presence of PDE4D3 and PDE4D5 as the major PDE4 isoforms in HEK293 cells (Hoffmann et al, 1999; Lynch et al, 2005; Figure 6A, lower blot) and the interaction of the PDE4D isoforms with gravin (Figure 6A, upper blot). The inability of H89 to enhance the effects of rolipram inhibition provides further evidence that PKA is acting through an association with PDE4. However, we cannot rule out possible effects of PKA inhibition on other PKA-dependent phosphorylation events such as gravin–receptor association (Tao et al, 2003), desensitization of prostanoid receptor isoforms (Nishigaki et al, 1996; Bastepe and Ashby, 1999) and inhibition of adenylyl cyclase isoforms (Iwami et al, 1995; Chen et al, 1997). It is not clear how additional actions of PKA might affect real-time subplasmalemmal cAMP dynamics, but we propose that a primary action of PKA in the gravin–PDE4D complex is its regulation of localized PDE4 activity to enhance cAMP hydrolysis.
The intimate relationship between gravin-anchored PKA and PDE4D activity allows the rapid hydrolysis of cAMP during stimulation with PGE1 and suggests that the gravin complex is in close proximity to (or directly interacts with) the Gs-coupled prostanoid receptor. Although previous studies have shown recruitment of gravin to β2-ARs as a consequence of agonist stimulation (Lin et al, 2000; Fan et al, 2001) and PKA phosphorylation (Tao et al, 2003), a direct association of gravin with prostanoid receptors mediating PGE1 effects has not been reported to date. The specific nature of the prostanoid receptor acted on by PGE1 is unknown, but candidates include EP2 and EP4 receptors, as these respond to PGE1 via GS (reviewed by Narumiya et al, 1999). Interestingly, the effects of gravin knockdown were apparent from the beginning of the cAMP recovery phase during stimulation with PGE1 (Figure 3A) but were delayed by about 3 min during stimulation with β2-AR agonist (Figure 3C). This is consistent with the hypothesis that gravin rapidly localizes to, or is pre-associated with, plasma membrane subdomains in the vicinity of Gs-coupled prostanoid receptors that may be distinct from regions where it is recruited to β2-ARs. It is conceivable that PDE4D could also associate with the gravin complex via β-arrestin and be recruited to GPCRs as a consequence of agonist stimulation (Lin et al, 2000; Fan et al, 2001). Previous work on β2-ARs revealed that recruitment of the scaffold protein, β-arrestin, serves to target PDE4D3/5 activity to the receptor following agonist occupancy (Perry et al, 2002; Baillie and Houslay, 2005) and that recruitment of β-arrestin to β2-ARs is abolished in gravin-deficient cells (Lin et al, 2000). This might explain some of the actions of rolipram and H89 reported in the present study, given that PKA phosphorylation is also central to GPCR desensitization by arrestin (Baillie et al, 2003) and gravin binding to the β2-AR (Tao et al, 2003). The findings presented here may illustrate both scenarios: one where β2-AR activation recruits a β-arrestin-associated gravin complex that can attenuate local cAMP signals after a delay of about 3 min, and the other where cAMP produced during activation of prostanoid receptors (potentially residing in different cellular microdomains) is rapidly hydrolyzed by a gravin-associated PDE4D that already resides at, or close to, the plasma membrane (see Figure 5). This is consistent with the recent work by Houslay and co-workers (Lynch et al, 2005), who suggested that gravin can be recruited more rapidly to membrane fractions (within 3 min) than to β2-ARs (5 min) following isoprenaline exposure, with some constitutive association of gravin with HEK293 cell membranes. Although AKAP79 is a principal AKAP associating with the plasma membrane of HEK293 cells (Li et al, 1996) that interacts with numerous signaling proteins (Wong and Scott, 2004) including the β2-AR (Fraser et al, 1998), RNAi data presented here do not support a role for this AKAP in the termination of cAMP signals. This is consistent with data suggesting that AKAP79 does not directly interact with PDEs (K Dodge and JD Scott, personal communication). Thus, although AKAP79 and gravin are both critical components of GPCR signaling, they appear to have quite different roles within this context. AKAP79 is involved in PKA-mediated receptor phosphorylation and switching of downstream signaling pathways (Lynch et al, 2005), whereas gravin appears central to receptor resensitization (Malbon et al, 2004b) and local cAMP hydrolysis as described here (Figure 7).
Figure 7.
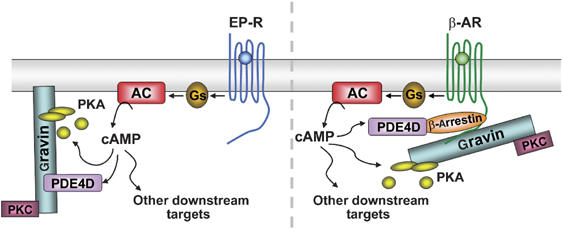
Two possible organizations for PDE4-associated gravin complexes regulating subplasmalemmal cAMP dynamics. Left-hand side, a gravin complex targets cAMP-specific phosphodiesterase (PDE4D3/5) activity to the plasma membrane for the rapid hydrolysis of cAMP produced during prostanoid receptor (EP-R) occupancy. The G-protein subunit, Gαs, stimulates adenylyl cyclase (AC) activity, increasing local cAMP levels that are rapidly hydrolyzed by gravin-associated PDE4 following PKA activation. Alternatively, as illustrated on the right hand side, a gravin complex, indirectly associated with PDE4D via β-arrestin, is recruited to GPCRs (e.g. β2-AR) following agonist stimulation. Gravin also binds to PKC.
In conclusion, the present study provides a particularly powerful means of assessing the elements of a cAMP signaling complex in fully functional cells; cAMP dynamics are seen at the plasma membrane in real time; the effects of pharmacological manipulation or molecular perturbation are also assessed in real time and then the elements of the complex are evaluated by traditional biochemical methods. Our results demonstrate the presence of an endogenous, functional complex of PDE4D, PKA and gravin localized at the plasma membrane where it generates discrete, dynamic cAMP signals, providing an elegant mechanism for localized actions of this signaling pathway.
Materials and methods
Cell culture and CNGC expression
HEK293 cells were grown in minimum essential medium supplemented with 10% (v/v) fetal bovine serum and maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were plated onto 25 mm poly-L-lysine-coated coverslips 24 h before infection with adenovirus encoding the α subunit of rat olfactory CNGC with mutations C460W and E583M. The adenovirus was constructed using the QuikChange site-directed mutagenesis kit (Stratagene) and purified as described previously (Rich et al, 2001b). A multiplicity of infection of 10 PFU/well was used and 2 mM hydroxyurea was added to each coverslip to inhibit adenovirus vector replication after 2 h. All cAMP measurements in CNGC-expressing cells were made 1 day post-infection.
cAMP measurements monitored by Ca2+ influx
CNGC-infected HEK293 cells were loaded with 4 μM fura-2/AM and 0.02% pluronic F-127 (Molecular Probes) for 45 min in extracellular buffer containing (mM) 140 NaCl, 4 KCl, 1 CaCl2, 0.1 MgCl2, 11 D-glucose and 10 Hepes, pH 7.4. After loading, cells were washed and then imaged using a CoolSnap CCD-camera (Photometrics) and monochromator system (Cairn Research) attached to a Nikon TMD microscope (× 40 objective). Emission images (D510/80M) at 340 and 380 nm excitation were collected at 1 Hz using MetaFluor software (Universal Imaging). Data were plotted as 340/380 nm ratio changes relative to the prestimulus fluorescence ratio (Δ340/380). Solutions were applied using a gravity-fed perfusion system at a flow rate of ∼4 ml/min. Percentage recoveries were calculated as the % decrease in Δ340/380 signal compared to the peak Δ340/380 change in each cell. The rates of cAMP change are expressed as the average change in Δ340/380 ratio/s over a 10 s period. The stearoylated Ht31 and Ht31-P peptides were purchased from Promega. All other agents were purchased from Sigma unless stated otherwise.
Whole-cell ICNG recording
Perforated patch-clamp technique was used to gain electrical access to the cell while retaining divalent cations and larger molecules within the cell. Recordings were made using an Axopatch 200-B amplifier (Axon Instruments). Pipettes were pulled from borosilicate glass and heat polished to give a final resistance of 2.5–3.5 MΩ when filled with solution containing (mM) 70 KCl, 70 potassium gluconate, 4 NaCl, 0.5 MgCl2, 10 Hepes, pH 7.4 and 200 μg/ml of nystatin. The solution was bath sonicated before use and pipettes were front filled with a small volume of nystatin-free pipette solution to aid gigaseal formation. Capacitative transients were elicited by applying 5 mV steps to determine access resistance. In the perforated patch-clamp configuration, a steady access resistance was obtained 10–15 min after gigaseal formation and was monitored at the end of each experiment to check that electrical access had been maintained. Cells were held at −30 mV throughout. The extracellular bath solution contained (mM) 140 NaCl, 4 KCl, 0.1 MgCl2, 11 D-glucose and 10 Hepes, pH 7.4. All agents were applied to cells using the same perfusion system as described above.
FRET measurements of Epac1 cAMP sensor
Fluorescent imaging of Epac1 sensor expressing HEK293 cells was performed using a CoolSNAP-HQ CCD camera and Optosplit (505DC) (Cairn Research, Kent) as described previously (Willoughby and Cooper, 2006) and analyzed using Metamorph software (Universal imaging).
RNAi against individual AKAPs
The AKAP79 shRNA has been previously described (Hoshi et al, 2005). Oligonucleotides containing (1) sequences from the human versions of gravin or AKAP149, (2) a hairpin loop and (3) an RNA Pol III termination signal were ligated into the ApaI and EcoRI sites of pSilencer 1.0-U6 (Ambion). The sequences were as follows: AKAP149, GAGTATGTAGCAGAGAAGT and gravin, ATAAAGAGATGGCTACTAA.
To induce RNAi, HEK293 cells were plated onto coverslips and cotransfected with the appropriate construct in the pSilencer vector and CD4 expression vector (pJPACD4) the next day using FuGene6 (Roche Diagnostics). To assess the efficacy of RNAi knockdown by immunoblotting, shRNA-positive cells were selected with anti-CD4 magnetic beads (Dynal) for 30 min at 37°C and lysed in 1% (v/v) Triton X-100, 10 mM Na2PO4, pH 7.4, 150 mM NaCl, 5 mM EDTA and 5 mM EGTA supplemented with protease inhibitors (1 mM benzamidine, 1 mM AEBSF, 0.2 μg/ml leupeptin/pepstatin). Equal amounts of protein were electrophoresed, transferred to nitrocellulose and immunoblotted (see below).
For fura-2 measurements, cells were infected with adenovirus encoding CNGC 2 days after transfection as described above and tested ∼24 h later. Before loading with fura-2, the CNGC-expressing cells were incubated with anti-CD4 magnetic beads and then washed thoroughly to identify cells positive for the shRNA (Hoshi et al, 2005). CD4+ cells were identified as cells within the camera field of view with non-fluorescent beads attached. Where indicated, cells were re-transfected with the murine form of gravin, SSeCKS, in pcDNA3.1 (Invitrogen) or murine AKAP18 in pcDNA3. For these rescue experiments, CD4+ cells were selected 48 h post-transfection using the anti-CD4 magnetic beads and replated onto coverslips 24 h before re-transfection with either SSeCKS or murine AKAP18. Cells were infected with CNGC 48 h later and tested the following day.
To create the SSeCKS/pcDNA3.1 expression plasmid, a fragment of SSeCKS corresponding to nucleotides 420–5052 of the coding region was PCR amplified from the NIH Mammalian Gene Collection cDNA clone IMAGE:4947266 and inserted into pcDNA3.1D-TOPO. The fragment of SSeCKS corresponding to nucleotides 1–447 of the coding region was PCR amplified from a mouse brain cDNA library and ligated in the KpnI and SwaI sites to create a full-length SSeCKS clone. To produce the AKAP18/pcDNA3 plasmid, the full-length coding region for the α isoform was ligated into the EcoRI and BamHI sites of pcDNA3 (Invitrogen).
RII overlays, immunoblotting and immunoprecipitation
RII overlays were performed as previously described (Carr et al, 1992). Immunoblotting was carried out with antibodies against AKAP79 (Hoshi et al, 2005), AKAP149 (BD Biosciences), AKAP-Lbc (Diviani et al, 2001), MAP2 (Sigma), gravin (Nauert et al, 1997), PDE4D (Abcam) and α-tubulin (Sigma). For immunoprecipitations of endogenous proteins, confluent 100 mm dishes of HEK293 cells were lysed with 1% CHAPS, 20 mM Tris pH 7.4, 150 mM NaCl, 10 mM EDTA, supplemented with protease inhibitors and equal amounts of protein were incubated with 5 μg anti-PDE4D antibody, 30 μg anti-gravin (Nauert et al, 1997) or an appropriate amount of rabbit IgG overnight at 4°C. Immunoprecipitates were washed once with lysis buffer and three times with 20 mM Tris pH 7.4 and 150 mM NaCl, then electrophoresed and transferred onto nitrocellulose. Bound proteins were detected by blotting with anti-PDE4D (ICOS) and anti-gravin.
Statistical analysis
Unless stated otherwise data were analyzed by one-way ANOVA followed by Newman−Keuls multiple comparisons tests (GraphPad Prism, GraphPad Software Inc.). Data are presented as means±s.e.m., with significance set at P<0.05.
Acknowledgments
cDNA for the fluorescently tagged Epac-1 cAMP sensor was a kind gift from Martin J Lohse. This work was supported by grants from The Wellcome Trust, the NIH (NS28389; DMFC and DK54441, JDS) and a fellowship from the Heart and Stroke Foundation of Canada (WW).
References
- Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER, Tsien RY (1993) Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science 260: 222–226 [DOI] [PubMed] [Google Scholar]
- Baillie GS, Houslay MD (2005) Arrestin times for compartmentalised cAMP signalling and phosphodiesterase-4 enzymes. Curr Opin Cell Biol 17: 129–134 [DOI] [PubMed] [Google Scholar]
- Baillie GS, Scott JD, Houslay MD (2005) Compartmentalisation of phosphodiesterases and protein kinase A: opposites attract. FEBS Lett 579: 3264–3270 [DOI] [PubMed] [Google Scholar]
- Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD (2003) beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA 100: 940–945 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Barnes AP, Livera G, Hunag P, Sun C, O'Neal WK, Conti M, Stutts MJ, Milgram SL (2005) Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J Biol Chem 280: 7997–8003 [DOI] [PubMed] [Google Scholar]
- Bastepe M, Ashby B (1999) Identification of a region of the C-terminal domain involved in short-term desensitization of the prostaglandin EP4 receptor. Br J Pharmacol 126: 365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavo JA, Brunton LL (2002) Cyclic nucleotide research—still expanding after half a century. Nat Rev Mol Cell Biol 3: 710–718 [DOI] [PubMed] [Google Scholar]
- Buxton IL, Brunton LL (1983) Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J Biol Chem 258: 10233–10239 [PubMed] [Google Scholar]
- Carr DW, Hausken ZE, Fraser ID, Stofko-Hahn RE, Scott JD (1992) Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem 267: 13376–13382 [PubMed] [Google Scholar]
- Chen Y, Harry A, Li J, Smit MJ, Bai X, Magnusson R, Pieroni JP, Weng G, Iyengar R (1997) Adenylyl cyclase 6 is selectively regulated by protein kinase A phosphorylation in a region involved in Gαs stimulation. Proc Natl Acad Sci USA 94: 14100–14104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD (1995) Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 267: 108–112 [DOI] [PubMed] [Google Scholar]
- Conti M, Richter W, Mehats C, Livera G, Park J-Y, Jin C (2003) Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem 278: 5493–5496 [DOI] [PubMed] [Google Scholar]
- Dart C, Leyland ML (2001) Targeting of an A kinase-anchoring protein, AKAP79, to an inwardly rectifying potassium channel, Kir2.1. J Biol Chem 276: 20499–20505 [DOI] [PubMed] [Google Scholar]
- Diviani D, Soderling J, Scott JD (2001) AKAP-Lbc anchors protein kinase A and nucleates Gα12-selective Rho-mediated stress fiber formation. J Biol Chem 276: 44247–44257 [DOI] [PubMed] [Google Scholar]
- Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD (2001) mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J 20: 1921–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD (2005) The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437: 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Shumay E, Wang H, Malbon CC (2001) The scaffold protein gravin (cAMP-dependent protein kinase-anchoring protein 250) binds the beta 2-adrenergic receptor via the receptor cytoplasmic Arg-329 to Leu-413 domain and provides a mobile scaffold during desensitization. J Biol Chem 276: 24005–24014 [DOI] [PubMed] [Google Scholar]
- Fraser IDC, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD (1998) A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J 17: 2261–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM (1997) cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19: 185–196 [DOI] [PubMed] [Google Scholar]
- Goaillard JM, Vincent PV, Fischmeister R (2001) Simultaneous measurements of intracellular cAMP and L-type Ca2+ current in single frog ventricular myocytes. J Physiol 530: 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD (1999) The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J 18: 893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD (2005) Distinct enzyme combinations in AKAP signaling complexes permit functional diversity. Nat Cell Biol 7: 1066–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD, Adams DR (2003) PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J 370: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwami G, Kawabe J, Ebina T, Cannon PJ, Homcy CJ, Ishikawa Y (1995) Regulation of adenylyl cyclase by protein kinase A. J Biol Chem 270: 12481–12484 [DOI] [PubMed] [Google Scholar]
- Johnson BD, Scheuer T, Catterall WA (1994) Voltage-dependent potentiation of L-type Ca2+ channels in skeletal muscle cells requires anchored cAMP-dependent protein kinase. Proc Natl Acad Sci USA 91: 11492–11496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius J, Fischmeister R (1996) cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc Natl Acad Sci USA 93: 295–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R (2002) Cyclic nucleotide-gated ion channels. Physiol Rev 82: 769–824 [DOI] [PubMed] [Google Scholar]
- Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD (1996) Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science 271: 1589–1592 [DOI] [PubMed] [Google Scholar]
- Li Y, Ndubuka C, Rubin CS (1996) A kinase anchor protein 75 targets regulatory (RII) subunits of cAMP-dependent protein kinase II to the cortical actin cytoskeleton in non-neuronal cells. J Biol Chem 271: 16862–16869 [DOI] [PubMed] [Google Scholar]
- Lin F, Wang H, Malbon CC (2000) Gravin-mediated formation of signaling complexes in beta 2-adrenergic receptor desensitization and resensitization. J Biol Chem 275: 19025–19034 [DOI] [PubMed] [Google Scholar]
- Lynch MJ, Baillie GS, Mohamed A, Li X, Maisonneuve C, Klussmann E, van Heeke G, Houslay MD (2005) RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with β-arrestin to control the PKA/AKAP79-mediated switching of the β2-adrenergic receptor to activation of ERK in HEK293 cells. J Biol Chem 280: 33178–33189 (M414316200) [DOI] [PubMed] [Google Scholar]
- Malbon CC, Tao J, Shumay E, Wang HY (2004a) AKAP (A-kinase anchoring protein) domains: beads of structure–function on the necklace of G-protein signalling. Biochem Soc Trans 32: 861–864 [DOI] [PubMed] [Google Scholar]
- Malbon CC, Tao J, Wang HY (2004b) AKAPs (A-kinase anchoring proteins) and molecules that compose their G-protein-coupled receptor signalling complexes. Biochem J 379: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS (2002) Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1–KCNE1 potassium channel. Science 295: 496–499 [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F (1999) Prostanoid receptors: structures, properties, and functions. Physiol Rev 79: 1194–1226 [DOI] [PubMed] [Google Scholar]
- Nauert JB, Klauck TM, Langeberg LK, Scott JD (1997) Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr Biol 7: 52–62 [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse ML (2004) Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem 279: 37215–37218 [DOI] [PubMed] [Google Scholar]
- Nishigaki N, Negishi M, Ichikawa A (1996) Two Gs-coupled prostaglandin E receptor subtypes, EP2 and EP4, differ in the desensitization and sensitivity of the metabolic inactivation of the agonist. Mol Pharmacol 50: 1031–1037 [PubMed] [Google Scholar]
- Oliveria SF, Gomez LL, Dell'Acqua ML (2003) Imaging kinase–AKAP79–phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol 160: 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ (2002) Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 298: 834–836 [DOI] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DMF, Karpen JW (2000) Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J Gen Physiol 116: 147–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DMF, Karpen JW (2001a) A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci USA 98: 13049–13054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Tse TE, Rohan JG, Schaack J, Karpen JW (2001b) In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J Gen Physiol 118: 63–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet J-L, Cooper DMF, Fischmeister R (2004) Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes. J Biol Chem 279: 52095–52105 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL (1994) Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature 368: 853–856 [DOI] [PubMed] [Google Scholar]
- Sette C, Conti M (1996) Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. J Biol Chem 271: 16526–16534 [DOI] [PubMed] [Google Scholar]
- Shih M, Lin F, Scott JD, Wang HY, Malbon CC (1999) Dynamic complexes of beta2-adrenergic receptors with protein kinases and phosphatases and the role of gravin. J Biol Chem 274: 1588–1595 [DOI] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF (2005) Role for A kinase-anchoring proteins (AKAPs) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem 280: 16962–16968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Wang HY, Malbon CC (2003) Protein kinase A regulates AKAP250 (gravin) scaffold binding to the beta2-adrenergic receptor. EMBO J 22: 6419–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasken K, Aandahl EM (2004) Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev 84: 137–167 [DOI] [PubMed] [Google Scholar]
- Tasken KA, Collas P, Kemmner WA, Witczak O, Conti M, Tasken K (2001) Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J Biol Chem 276: 21999–22002 [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Zagotta WN (2003) Calcium/calmodulin modulation of olfactory and rod cyclic nucleotide-gated ion channels. J Biol Chem 278: 18705–18708 [DOI] [PubMed] [Google Scholar]
- Vijayaraghavan S, Goueli SA, Davey MP, Carr DW (1997) Protein kinase A-anchoring inhibitor peptides arrest mammalian sperm motility. J Biol Chem 272: 4747–4752 [DOI] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD (1999) Regulation of NMDA receptors by an associated phosphatase–kinase signaling complex. Science 285: 93–96 [DOI] [PubMed] [Google Scholar]
- Willoughby D, Cooper DMF (2006) Ca2+ stimulation of adenylyl cyclase generates dynamic oscillations in cyclic AMP. J Cell Sci 119: 828–836 [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD (2004) AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol 5: 959–970 [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Magalhaes P, Pozzan T (2002) Compartmentalisation of cAMP and Ca2+ signals. Curr Opin Cell Biol 14: 160–166 [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Pozzan T (2002) Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295: 1711–1715 [DOI] [PubMed] [Google Scholar]