

Abstract
Angiogenesis is a coordinated sequence of cellular responses that result in the outgrowth of new blood vessels. The angiogenic program is regulated by extracellular factors, whose input is integrated at least in part at the level of signal transduction pathways driven by phosphoinositide 3 kinase (PI3K) and phospholipase Cγ (PLCγ). Using an in vitro angiogenesis model, we discovered that PI3K was essential for tube formation, whereas PLCγ promoted regression. The underlying mechanism by which PLCγ antagonized tube formation appeared to be by competing with PI3K for their common substrate, phosphatidylinositol-4,5-bisphosphate. These studies are the first to identify signaling enzymes involved with vessel regression, and reveal that the angiogenic program can be coordinated by the availability of a membrane lipid.
Keywords: angiogenesis; PI3K; PLCγ; PtdIns-4,5-P2; tube regression
Introduction
Angiogenesis results from a quantitative, qualitative and temporal balance of cell proliferation, migration and differentiation. Although the mechanism by which this balance is achieved has not been elucidated, some of the signaling enzymes that are required for the angiogenic program have been identified (Matsumoto and Claesson-Welsh, 2001). For instance, phosphoinositide 3 kinase (PI3K) and its downstream effector Akt are essential for many of the cellular responses within the angiogenic program including proliferation, migration and survival (Gerber et al, 1998; Gille et al, 2000; Adini et al, 2003). Similarly, phospholipase Cγ (PLCγ) is required for vascular endothelial cell growth factor (VEGF)-dependent migration and proliferation of cultured endothelial cells (Takahashi et al, 2001). Furthermore, genetic studies have demonstrated that PLCγ is essential for angiogenesis (Lawson et al, 2003). PLCγ-deficient mice die during embryogenesis and display a ‘vascular' defect (Liao et al, 2002). Moreover, knocking in a VEGF receptor 2 (VEGFR2) mutant that cannot recruit or activate PLCγ results in lethality between embryonic days 8.5 and 9.5 and defective hematopoietic development (Sakurai et al, 2005). Although these studies clearly show that PLCγ is essential for angiogenesis, its precise contribution to the angiogenic program is unknown. Thus, although some of the relevant signaling enzymes have been identified, their contribution to each step of the angiogenic program and their participation in the overall coordination of events remain an open question.
The transition of a stable vessel to one that is capable of entering the angiogenic program is of interest both scientifically and therapeutically. This destabilization step is an early event that is required for all subsequent phases of the angiogenic program and for the regression of vessels (Carmeliet, 2000). At the present time, little is known regarding the molecular nature of this decision or the intracellular signaling events that direct this choice. This information will be invaluable for the treatment of angiogenic diseases (such as solid tumors, diabetic complications and age-related macular degeneration) because current antiangiogenic therapies seem to target immature/unstable vessels (Rasmussen et al, 2001; Shimizu and Oku, 2004). Being able to induce this state will be an important complement to target and potentiate antiangiogenic intervention.
Results and discussion
Preventing activation of PLCγ enhanced the tube response
One strategy to elucidate the molecular nature of the vessel regression is to identify the relevant signaling enzymes and to determine how they are regulated. To this end, we used an in vitro angiogenesis assay to investigate the role of PI3K and PLCγ on tube formation and regression. When cultured between two layers of collagen in the presence of VEGF-A, primary bovine retinal endothelial cells (BRECs) formed tubes (Figure 1A and B) (Im et al, 2005). The tubes spontaneously regressed, despite the presence of freshly added VEGF-A (Figure 1A and B). As outlined below, we suspected that PLCγ promoted regression of tubes. Consequently, we compared vessel formation and regression in BRECs expressing either the wild-type (WT) or mutant VEGFR2 that did not activate PLCγ (Y1175F) (Takahashi et al, 2001). Somewhat more tubes were consistently observed in the cells expressing the mutant VEGFR2 (Figure 1C). The more striking observation was that the Y1175F tubes did not regress as did the WT tubes (Figure 1C). We repeated these studies with a second primary endothelial cell type (human umbilical vein endothelial cells (HUVECs)) and observed the same phenomenon (Supplementary Figure S1A–C). Furthermore, as an alternative approach to attenuate the contribution of PLCγ, we used siRNA to reduce the amount of PLCγ (Figure 1G). Tubes that formed from these cells were resistant to regression, whereas cells expressing a control siRNA (which did not reduce the level of PLCγ; Figure 1G) regressed normally (Figure 1E). These observations were not unique to the VEGF-driven setting; PLCγ-dependent regression was also a feature of tubes formed in response to basic fibroblast growth factor (bFGF) (Figure 1F). These findings indicate that the in vitro assay is a model for both tube formation and regression. Furthermore, proangiogenic factors (such as VEGF-A and bFGF) engage a PLCγ-dependent signaling pathway that promotes tube regression.
Figure 1.
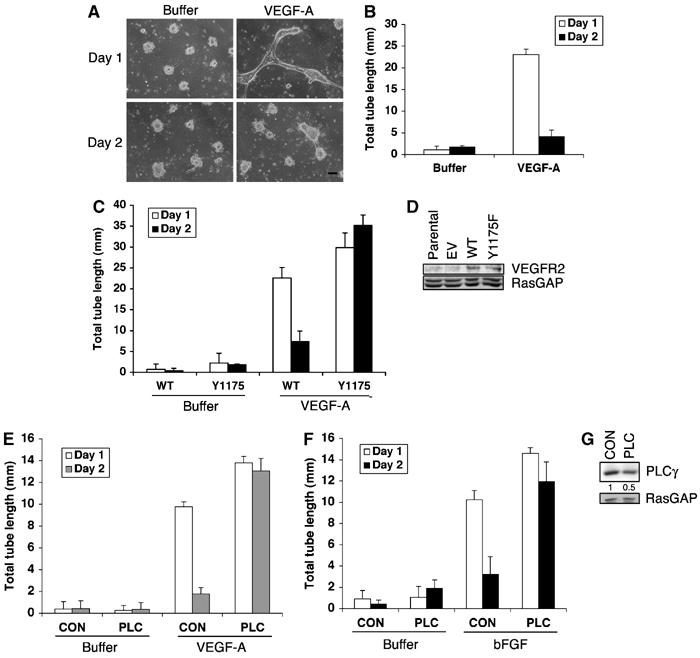
(A, B) Tubes formed in the presence of VEGF-A, and then spontaneously regressed. BRECs were plated in a collagen sandwich gel, and then medium containing 2.5 ng/ml VEGF-A or buffer was added. The medium was changed every day. At the indicated times, three randomly selected fields were photographed and tube lengths were measured (B). Representative photos are shown. Bar, 100 μm (A). The bar graph is the mean±s.d. of 3 wells/treatment. (C, D) Tubes failed to regress when the cells expressed a VEGFR2 mutant that was unable to activate PLCγ. BRECs expressing WT or mutant VEGFR2 (Y1175F) were subjected to a tube assay. A Western blot of total cell lysates shows that the expression level of the introduced WT and Y1175F VEGFR2 was similar (D). (E–G) Reducing the level of PLCγ-stabilized tubes. HUVECs were transfected with either siRNA for PLCγ (PLC) or non-targeting control siRNA (CON) and subjected to a tube assay in the presence of 2.5 ng/ml VEGF-A (E) or 25 ng/ml bFGF (F). Total cell lysates were subjected to Western blot analysis using an anti-PLCγ antiserum. The blot was reprobed with an anti-RasGAP antiserum to verify equivalent protein loading. PLCγ expression was decreased by an average of 50% in siRNA PLCγ-transfected cells (G).
PI3K was required for tube formation, whereas PLCγ promoted tube regression
To investigate the role of PI3K and its relationship with PLCγ in the context of tube formation and regression, we developed a model system that allowed the conditional activation of each of these signaling enzymes. More specifically, we used platelet-derived growth factor receptor β (PDGFR) WT or PDGFR phosphorylation site mutants that do or do not activate PI3K and/or PLCγ. A comparable set of VEGFR2 or FGFR mutants is not available. BRECs do not express endogenous PDGFRs (Supplementary Figure S5A), although they do express PDGF (Supplementary Figure S2A and B). Stable expression of the WT PDGFR resulted in the spontaneous formation of tubes that were of similar morphology and length to the tubes that formed in parental cells stimulated with VEGF-A (Figure 2A and B). Tube formation was dependent on the extracellular PDGF, as adding a neutralizing PDGF antibody blocked the tube response (Supplementary Figure S2C and D). Supplementing the PDGFR-expressing cells with VEGF-A further enhanced the tube response (Figure 2A and B). Thus, expressing the PDGFR established an autocrine loop that drove spontaneous tube formation.
Figure 2.
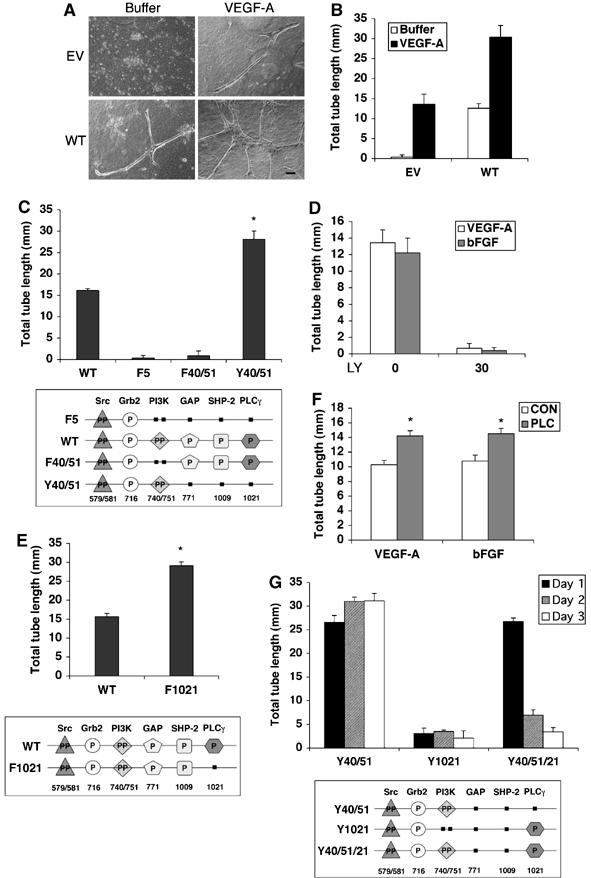
(A, B) Expressing the WT PDGFR induced tubes. BRECs stably expressing the WT PDGFR (WT) or an empty vector (EV) were subjected to a tube assay as described in the legend of Figure 1. The tubes that formed in the WT-expressing cells were of similar morphology and length as those that formed when EV-expressing cells were cultured in the presence of VEGF-A. Bar, 100 μm. (C) PI3K was required for tube formation. The top panel shows the results of tube formation driven by BRECs expressing several mutant PDGFRs. No PDGF or VEGF-A was added to the cultures; the response is driven by endogenously produced PDGF. The difference between WT and Y40/51 was statistically significant (*P<0.001). The bottom panel is a diagram of the signaling proteins that bind to the PDGFR and the tyrosine phosphorylation sites that are required for binding. The filled squares symbolize tyrosine to phenylalanine mutations, whereas intact phosphorylation sites are represented by ‘P'. WT associates with Src, Grb2, GAP, SHP-2, PI3K and PLCγ. The F40/51 mutant selectively fails to engage PI3K, but associates with all of the other four signaling molecules. The F5 receptor does not efficiently recruit GAP, SHP-2, PI3K or PLCγ. In this experiment, the F5 receptor is the control for the Y40/51 mutant. The Y40/51 mutant associates with Src, Grb2 and PI3K but not with GAP, SHP-2 or PLCγ. (D) Blocking PI3K activity prevented tube formation. HUVECs were subjected to the tube assay in the presence of LY294002 (0 and 30 μM) and supplemented with 2.5 ng VEGF-A or 25 ng/ml bFGF. (E) PLCγ negatively regulated tube formation. BRECs expressing the WT or F1021 PDGFR were subjected to the tube assay. The difference between WT and F1021 was statistically significant (*P<0.001). (F) Silencing PLCγ increased tube formation induced by VEGF-A or bFGF. HUVECs were transfected with either siRNA for PLCγ (PLC) or non-targeting control (CON) siRNA. The resulting cells were subjected to a tube assay in the presence of 2.5 ng/ml VEGF-A or 25 ng/ml bFGF. The difference between CON and PLC was statistically significant (*P<0.05). (G) PLCγ did not alter the formation of tubes; however, it promoted their regression. BRECS expressing the indicated PDGFRs were subjected to the tube assay and the results at the indicated days are shown in the bar graph. The Y1021 receptor was a control showing that restoring PLCγ to the F5 receptor did not rescue tube formation.
In certain tumor cell lines, PDGF can stimulate the production of VEGF (Wang et al, 1999). We tested if the tubes that formed in the PDGFR-expressing cells were driven by endogenously produced VEGF. Despite the use of several experimental approaches, we were unable to detect any evidence for such a scenario. Soluble extracellular VEGFR2 blocked VEGF-A-dependent tube formation in parental cells, but had no effect on the tubes that were formed by WT PDGFR-expressing cells (Supplementary Figure S2E). Furthermore, expression of the WT PDGFR did not alter the level of VEGF-A mRNA (Supplementary Figure S2F). We conclude that the PDGF/PDGFR-driven system induced a comparable tube response to that observed when parental cells were stimulated with VEGF-A.
The PDGF/PDGFR-driven system allowed us to evaluate the role of PI3K in tube formation without global inhibition of PI3K activity. A PDGFR mutant that was unable to recruit PI3K (F40/51) failed to induce tubes (Figure 2C). Similarly, the F5 mutant, which lacks bindings sites for PI3K, RasGAP, SHP-2 and PLCγ, was unable to drive tube formation (Figure 2C). The tube response was restored when we repaired the binding sites for PI3K in the F5 mutant to generate the Y40/51 receptor (Figure 2C). Thus, PI3K was required for tube formation in the PDGF/PDGFR-driven system. Importantly, the same requirement for PI3K was observed with HUVECs stimulated with VEGF-A or bFGF; a PI3K inhibitor blocked tube formation stimulated by either of the proangiogenic factors (Figure 2D). These findings indicated a critical role for PI3K in tube formation, which is consistent with previous reports (Gerber et al, 1998; Hamada et al, 2005).
We routinely noticed that tube formation with Y40/51 cells was better than WT cells (Figure 2C). This suggested that one or more of the signaling enzymes recruited by the WT receptor (RasGAP, SHP-2 or PLCγ) were suppressing the response. Given the known involvement of PLCγ in angiogenesis (Takahashi et al, 2001; Liao et al, 2002; Lawson et al, 2003) and our findings in the VEGF-A- and bFGF-driven systems (Figure 1C, E and F and Supplementary Figure S1B), we focused on PLCγ. Tube formation in cells expressing a receptor that did not activate PLCγ (F1021) was better than in cells expressing the WT receptor (Figure 2E). The same trend was observed in HUVECs stimulated with either VEGF or bFGF; reducing the level of PLCγ with siRNA boosted tube formation (Figure 2F). These studies revealed that PI3K promoted tube formation, and that PLCγ negatively impacted one or more steps in the overall tube response.
The negative impact of PLCγ on the overall tube response could be from blocking tube formation and/or promoting regression of tubes after they have formed. To investigate this issue, we examined both tube formation and regression using PDGFR mutants that activated either or both PI3K and PLCγ. Within the first day of the experiment, tubes formed comparably for both the Y40/51 and Y40/51/21 cells (Figure 2G), indicating that PLCγ did not prevent the formation of tubes. Instead, PLCγ appeared to promote tube regression because at the latter time points the Y40/51/21 tubes regressed, whereas the Y40/51 tubes persisted (Figure 2G). Similarly, cells expressing PDGFR mutants that recruited PI3K in combination with RasGAP or SHP-2 (instead of PLCγ) formed stable tubes (Supplementary Figure S2G). These experiments indicate that PLCγ promoted regression.
Taken together, the results in Figure 2 indicate that PI3K was essential for tube formation, whereas PLCγ reduced the overall tube response by inducing tube regression.
Activation of PLCγ reduced the output of PI3K
Our findings in Figure 2 suggested an antagonistic relationship between PI3K and PLCγ. These two enzymes require the same substrate (phosphatidylinositol-4,5-bisphosphate (PtdIns-4,5-P2)) raising the possibility that the antagonism resulted from a competition for substrate. More specifically, we speculated that activation of PLCγ reduced the level of PtdIns-4,5-P2 available for PI3K and thereby attenuated the output of the PI3K pathway.
A prediction of the substrate competition hypothesis is that phosphoAkt (a downstream target of PI3K) would be reduced in situations where PLCγ is activated. This is what we observed using several experimental settings. In PDGF- or VEGF-stimulated monolayers, phosphoAkt was lower in cells expressing receptors that activated PLCγ as compared with receptors that did not (Figure 3A and Supplementary Figure S3A). A similar phenomenon was observed when the cells were organized into tubes; the extent of phosphoAkt was greater when the receptor (PDGFR or VEGFR2) failed to activate PLCγ or when the level of PLCγ was reduced with siRNA (Figure 3B and Supplementary Figure S3D–F). For the latter time points (Figure 3B and Supplementary Figure S3B), the tubes were harvested before regression, and so the decline in phosphoAkt was not the result of regression. Furthermore, other signaling systems (PLCγ tyrosine phosphorylation) did not fall at the latter time points, arguing against a global decline in signaling (Figure 3C and Supplementary Figure S3C). Thus, all three experimental approaches indicate that PLCγ antagonized PI3K/Akt signaling.
Figure 3.
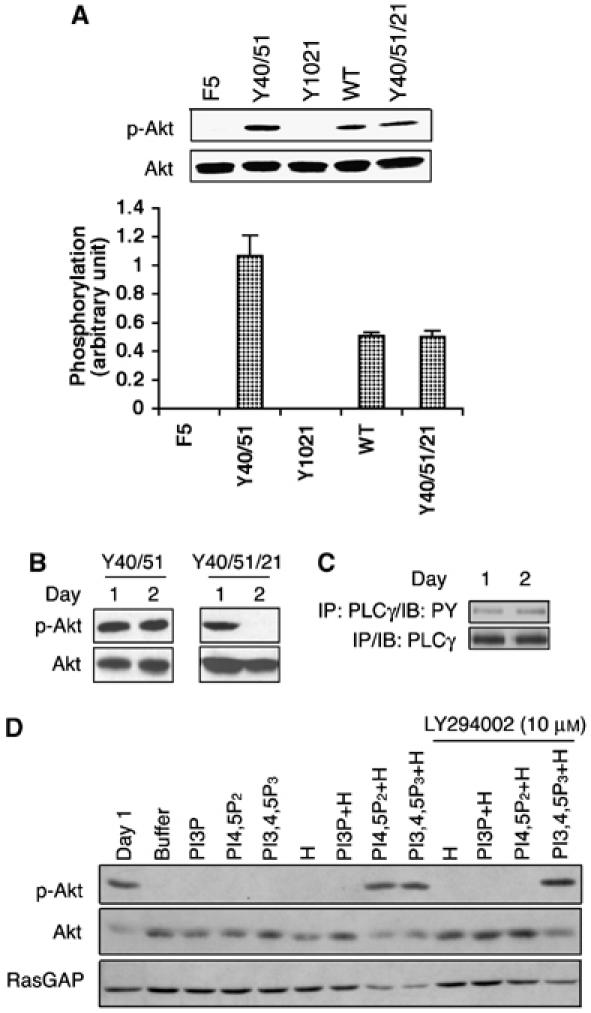
(A) Recruiting PLCγ attenuated activation of Akt. Monolayers of cells expressing the indicated receptors were stimulated with 100 ng/ml PDGF for 5 min. The cells were harvested and total cell lysates were subjected to a Western blot using an anti-phosphoAkt (Ser473) antibody. The blot was then stripped and reprobed with an anti-Akt antibody. The bar graph shows the ratio of the phosphoAkt/Akt signal; the error bars are standard deviation of three independent experiments. (B) Activation of PLCγ correlated with a decline in phosphoAkt. BRECs expressing the indicated receptor were organized into tubes as described in the legend of Figure 1. At the desired times, total cell lysates were made and subjected to Western blot analysis using anti-phosphoAkt (Ser473) and anti-Akt antibodies. (C) Phosphotyrosine of PLCγ did not change when tubes regressed in the Y40/51/21 cells. The Y40/51/21 cells were subjected to a tube assay. At the indicated times, total cell lysates were made, immunoprecipitated with a PLCγ antibody and the phosphotyrosine content of PLCγ was assessed by Western blot analysis using an anti-phosphotyrosine antibody. (D) Synthetic lipids restored the level of phosphoAkt. The Y40/51/21 cells were subjected to a tube assay. Synthetic lipids and/or histone (H) were added every 12 h beginning at 12 h and ending at 48 h. For the treatment of LY294002 (10 μM), a mixture of synthetic lipids and histone was added with LY294002 simultaneously. Total lysates were made and subjected to Western blot analysis using anti-phosphoAkt and anti-Akt antibodies. The blot was stripped and reprobed with RasGAP as a loading control.
PLCγ is linked to Erk activation in VEGF-stimulated endothelial cells (Takahashi et al, 2001). Yet in our hands, Erk activation was not significantly impaired when we interfered with PLCγ (Supplementary Figure S3G–I). This may be because neither of the experimental systems completely eliminated PLCγ activation, and thus the remaining PLCγ activity may have been sufficient to fully activate Erk.
A second prediction of the substrate competition hypothesis is that boosting the cellular level of PtdIns-4,5-P2 would restore the output of the PI3K pathway. Indeed, adding synthetic PtdIns-4,5-P2 to Y40/51/21 tubes increased phosphoAkt to the day 1 level (Figure 3D). The finding that this response was blocked by a PI3K inhibitor (LY294002) (Figure 3D) further supported the idea that PLCγ created a shortage of PtdIns-4,5-P2. Adding the PI3K lipid product (PtdIns-3,4,5-P3) also prevented the fall in phosphoAkt, but this response was not sensitive to PI3K inhibitors (Figure 3D), as it was downstream of PI3K. The results in Figures 3 and Supplementary Figure S3 support the hypothesis that PLCγ antagonized the PI3K pathway at the level of lipid substrate.
Synthetic PI3K lipid substrates prevented tube regression
We also tested if increasing the cellular level of PtdIns-4,5-P2 prevented regression of tubes. In both the PDGF/PDGFR and VEGF/VEGFR systems, PtdIns-4,5-P2 stabilized tubes and this event was sensitive to inhibition of PI3K activity (Figure 4). PtdIns-3,4,5-P3 also stabilized tubes, but in this case the PI3K inhibitor had no effect. Time-lapse photography of the tubes indicated that the synthetic lipids did not induce new tube formation, but rather prevented existing tubes from disappearing (unpublished observation). We concluded that, like the decline in phosphoAkt, the PLCγ-mediated regression of tubes resulted from an attenuation of the PI3K/Akt output.
Figure 4.
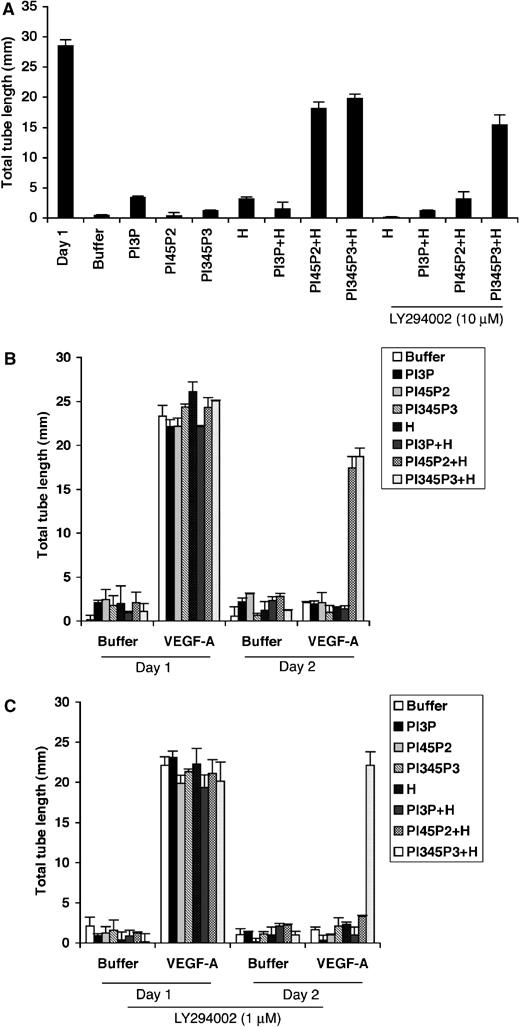
(A) Synthetic lipids prevented tube regression. The Y40/51/21 cells were treated as described in the legend of Figure 3D, except that instead of isolating the cells from the tubes, the total tube length was measured. (B) The regression of tubes in a VEGF-A-driven setting was also ablated by synthetic lipids. BRECs were subjected to a tube assay as described in the legend of Figure 1. The treatment with synthetic lipids was exactly as outlined in the legend of Figure 3D. (C) The synthetic PI3K lipid substrate did not prevent tube regression in the presence of a PI3K inhibitor. The treatment was same as panel B, except that a sub-saturating dose of LY294002 was added simultaneously with the lipids. As in panel A, the chosen dose of LY294002 was insufficient to inhibit tube formation.
PI3K/Akt promoted survival of vessels (Gerber et al, 1998; Adini et al, 2003), which prompted us to consider whether tube regression was the result of apoptosis. Three different measures of apoptosis (cell number, caspase 3 cleavage and TUNEL staining) indicated that apoptosis was occurring (Supplementary Figure S4). However, the tubes regressed before they underwent apoptosis. When we chemically induced apoptosis in stable tubes using a high concentration of a PI3K inhibitor, it resulted in apoptotic cells geographically arranged in the tube structures. Whereas, when the tubes regressed in our system, the cells within the tubes retracted into aggregates, and then underwent apoptosis (Supplementary Figure S4C). As the tubes collapsed into aggregates, there was an expected change in the actin (phalloidin staining; Supplementary Figure S4D), which may be casually related to apoptosis. Thus, although the cells in regressing tubes did eventually undergo apoptosis, it appeared that apoptosis was not the cause for regression. Others have also reported that tubes/vessels first regress, and then apoptose (Bayless and Davis, 2004; Saunders et al, 2005).
Partial inhibition of PI3K induced tube regression without affecting tube formation
The competition hypothesis provided an explanation for why tubes regressed, but left open the question of why they were capable of forming initially. One possibility was that the antagonism (i.e. PLCγ activation) was engaged only after tubes formed. However, Figure 3C and Supplementary Figure S3C show comparable tyrosine phosphorylation of PLCγ as tubes formed and regressed. Thus, the degree of PLCγ-dependent antagonism appeared to be constant. A second possibility was that more PI3K signaling was necessary for stabilization of tubes as opposed to their formation. Thus, PLCγ-dependent attenuation of PI3K output was insufficient to prevent the tubes from forming. This idea is consistent with previous findings that PI3K inhibitors promoted tube regression without affecting their formation using endothelial cells of the adrenal cortex (Qi et al, 1999). To test this idea, we compared the effect of sub-maximal doses of LY294002 on vessel formation and stability. Partially blocking PI3K (using 2 or 5 μM of LY294002) had no effect on tube formation, yet it was sufficient to induce tube regression (Figure 5). Higher doses of LY294002 inhibited formation of tubes in BRECs and HUVECs (Figures 5 and 2D), which was consistent with the PDGFR mutant data showing that PI3K was necessary for tube formation (Figure 2C). These observations suggested that tube stability was more dependent on PI3K signaling than the tube formation phase of the angiogenic program.
Figure 5.
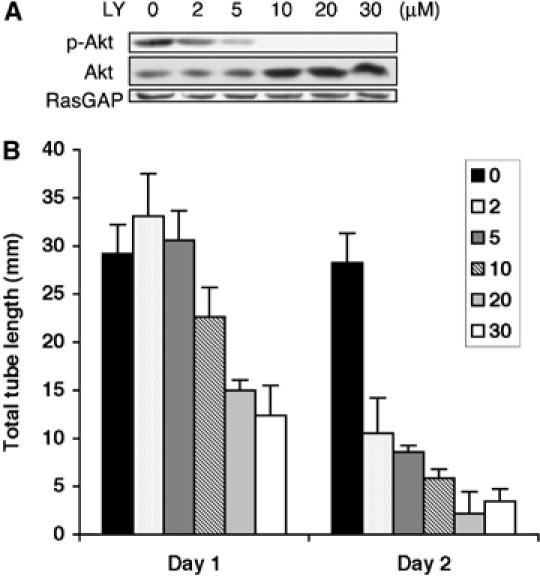
(A) LY294002 reduced the level of phosphoAkt in a concentration-dependent manner. The Y40/51 cells were subjected to a tube assay in the presence of LY294002 at the indicated concentrations. Total cell lysates were made at day 1 and subjected to Western blot analysis using anti-phosphoAkt and anti-Akt antibodies. The blot was stripped and reprobed with RasGAP. (B) Partial inhibition of PI3K promoted tube regression without altering tube formation. Same as panel A, except the total tube length was measured at days 1 and 2 instead of harvesting the cells for Western blot analysis.
The data presented support the idea that PLCγ promoted vessel regression by antagonizing PI3K. However, they did not rule out other possibilities. For instance, PLCγ may promote the synthesis/secretion of antiangiogenic factors, whose input is masked by flooding the system with synthetic lipids. Although our studies leave open this (and other) possibilities, they do strongly suggest that the amount of PtdIns-4,5-P2 is limiting, and support our overall conclusion that the angiogenic program can be regulated at the level of a membrane lipid.
Our finding that PLCγ controls tube formation by reducing the cellular level of PtdIns-4,5-P2 further increases our appreciation of how PtdIns-4,5-P2 contributes to cell signaling. It is not only a depot for the protein kinase C family activator (diacylglycerol) and intracellular calcium booster (inositol triphosphate) (Irvine, 2000), it can also inhibit tyrosine kinases (c-abl) (Plattner et al, 2003) and thereby modify a variety of cellular responses. Finally, the data presented herein indicate that PtdIns-4,5-P2 can act as an interface between PI3K and PLCγ to coordinate the various phases of the angiogenic program.
Materials and methods
Antibodies and reagents
Anti-mouse and anti-rabbit antibodies conjugated to horseradish peroxidase were obtained from Amersham Biosciences (Piscataway, NJ). Rabbit polyclonal anti-phospho-Akt antibody and anti-Akt antibody were obtained from Cell Signaling Technology (Beverly, MA). Mouse monoclonal anti-phosphotyrosine antibodies PY20 and 4G10 were purchased from Transduction Labs (Lexington, KY) and Upstate Biotechnology Inc. (Lake Placid, NY), respectively. The RasGAP, PDGFR, PLCγ and VEGFR2 antibodies were crude polyclonal rabbit antisera that were previously described (Valius et al, 1993; Rahimi and Kazlauskas, 1999). LY294002 was purchased from Calbiochem (San Diego, CA). The WT VEGFR2 and Y1175F VEGFR2 cDNAs were kindly provided by Dr Lena Claesson-Welsh (Uppsala University, Uppsala, Sweden). Recombinant VEGF-A was purchased from Upstate Biotechnology Inc. PDGF BB was purchased from R&D system (Minneapolis, MN). All other chemicals and reagents were obtained from Sigma (St Louis, MO) unless otherwise indicated.
Cell culture
BRECs were isolated from bovine eyes as described previously (Gitlin and D'Amore, 1983; Im et al, 2005). BRECs were maintained in EBM (Clonetics, Walkersville, MD) supplemented with 10% horse serum (Clonetics), 80 U/ml penicillin/streptomycin C (Irvine Scientific, Santa Ana, CA), and 12 μg/ml bovine brain extract (Clonetics). The cells were plated on plastic coated with 50 μg/ml bovine fibronectin and incubated at 37°C in 5% CO2. For all experiments, cells were used between passages 7 and 10. HUVECs were purchased from Clonetics and maintained in EGM-2 (Clonetics) with low serum growth factor supplement (Clonetics). For all experiments, HUVECs were used between passages 5 and 7.
Tube formation assay
Tube formation assay was performed as previously described (Im et al, 2005). The average tube length was routinely 15–30 mm in either BRECs or HUVECs exposed to VEGF-A, or bFGF, or PDGFR-expressing BRECs responding to endogenous PDGF.
Transfection of siRNA-PLCγ oligonucleotides
siRNA oligonucleotides that target PLCγ and non-targeting siRNA pool were purchased from Dharmacon (Lafayette, CO) and resuspended according to the manufacturer's instructions. For transfection, 1 × 105 HUVECs were plated into each well of a six-well dish and incubated for 16–18 h in culture medium. A 100 nM portion each of siRNA-PLCγ and siRNA-control oligonucleotides was mixed with TransPass™ R2 transfection reagent (New England BioLabs, Beverly, MA) 20 min before transfection. Cells were washed once with DMEM (GIBCO BRL, Gaithersburg, MD) and then the transfection reagent mixture was added. After a 4 h incubation, 2 ml of culture medium was added and the cultures were incubated overnight. Cells were then incubated for 48 h in freshly added culture medium.
Stable expression of PDGFR mutants
The WT, F40/51, F1021, Y40/51, Y1021, Y40/51/21 and F5 PDGFR constructs were previously constructed and characterized (Valius and Kazlauskas, 1993). Briefly, the F5 receptor has tyrosines 740, 751, 1009 and 1021 mutated to phenylalanine. The Y40/51 receptor is the same as F5, except that the mutations at 740 and 751 have been repaired. Similarly, the Y1021 receptor has tyrosine in place of phenylalanine at position 1021, whereas all the other mutations are as in F5. The F5 PDGFR does not efficiently associated with PLCγ, RasGAP, PI3K or SHP-2 and fails to activate either PI3K or PLCγ. The Y40/51 and Y1021 PDGFRs recruit PI3K and PLCγ, respectively. The PDGFR cDNAs were subcloned into the retroviral vector pLXSN. The PLSXN empty vector and PDGFR mutants/pLXSN constructs were transfected into 293GPG cells. The supernatant was collected for 5 days, concentrated (25 000 g, 90 min, 4°C) and used as described previously (Ory et al, 1996). Cells were infected and selected on the basis of proliferation in the presence of G418 (1 mg/ml).
Western blot analysis and immunoprecipitation
For Western blot analysis of VEGFR2, 2 × 106 BRECs or HUVECs were plated into a 10 cm tissue culture plate and incubated for at least 18 h in culture medium. Total cell lysates were prepared by adding lysis buffer containing 20 mM Tris (pH 8.0), 150 mM NaCl, 1 mM dithiothreitol, 1% deoxycholic acid, 0.5% sodium dodecyl sulfate (SDS), 1% Nonidet P-40 and protease inhibitors (2 μg/ml aprotinin, 5 μg/ml leupeptin, 10 μg/ml phenyl methyl sulfonyl fluoride and 10 mM sodium fluoride) and incubating for 1 h on ice. After centrifugation, the supernatants were collected and the protein concentration was determined. Proteins (10–30 μg) were separated on 10% SDS–polyacrylamide gels, and Western blot analysis was performed as described previously (Im et al, 2005).
To reprobe a blot, the blot was first stripped by incubating for 30 min at 60°C in a buffer containing 6.25 mM Tris–HCl, pH 6.8, 2% SDS and 100 mM β-mercaptoethanol and then reprobed with the desired primary antibody.
For Western blot analysis of phospho-Akt and Akt, the PDGFR-expressing cells or VEGFR2 WT/Y1175F-expressing cells or siRNA PLC/control siRNA-transfected cells were plated in a collagen sandwich gel and incubated for the desired time period. The cells were recovered following a collagenase treatment (collagenase type I-S from Sigma, 281 U/well for 20 min at 37°C), which dissolved the collagen gel. The cells were rinsed three times with ice-cold PBS, total cell lysates were made and then subjected to Western blot analysis as described above.
To analyze tyrosine phosphorylation of PLCγ in the tubes, cells were recovered from tubes and lysed as described above. The lysate was precleared with non-immune antibodies coupled to protein A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA), and then PLCγ was immunoprecipitated. Immunocomplexes were collected on protein A/G plus agarose and washed three times with the lysis buffer described above. The immunoprecipitated proteins were subjected to an anti-phosphotyrosine Western blot as described above.
Synthetic lipids rescue assay
The delivery of synthetic lipids to the cells was performed as previously described (Weiner et al, 2002). Briefly, parental or PDGFR-expressing BRECs were plated in a collagen sandwich gel and incubated for 12 h before adding lipids. Di-C16 synthetic phospholipids PtdIns-4,5-P2 and PtdIns-3,4,5-P3 (Echelon, Salt Lake City, UT) were freshly prepared at 25 μM in 150 mM sodium chloride, 4 mM potassium chloride and 20 mM HEPES at pH 7.2, and resuspended by vigorous vortexing. For histone–phospholipid complexes, 25 μM phospholipids was mixed with 100 μM freshly prepared histone (Echelon), vortexed vigorously and incubated for 5 min at room temperature. Histone–phospholipid complexes were diluted 1:10 with modified Hanks buffered saline solution immediately before addition to the media on top of the collagen gel. These media were replaced every 12 h until the end of the experiment.
Statistics
The Student's t-test was used to assess statistical significance.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Acknowledgments
We thank Dr Lena Claesson-Welsh for kindly providing the WT and Y1175F VEGFR2 cDNAs. We also thank Drs Laura E Benjamin, Chris Carpenter, Lena Claesson-Welsh, Giulio Romeo, Rita N Barcia, Hetian Lei and Sang Hoon Rhee for critically reading the manuscript. This study was supported by a Pediatric Ophthalmology Research Grant of the Knights Templar Eye Foundation Inc. (to EI), and by an NIH grant EY016385 (to AK).
References
- Adini I, Rabinovitz I, Sun JF, Prendergast GC, Benjamin LE (2003) RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev 17: 2721–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayless KJ, Davis GE (2004) Microtubule depolymerization rapidly collapses capillary tube networks in vitro and angiogenic vessels in vivo through the small GTPase Rho. J Biol Chem 279: 11686–11695 [DOI] [PubMed] [Google Scholar]
- Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6: 389–395 [DOI] [PubMed] [Google Scholar]
- Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N (1998) Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 273: 30336–30343 [DOI] [PubMed] [Google Scholar]
- Gille H, Kowalski J, Yu L, Chen H, Pisabarro MT, Davis-Smyth T, Ferrara N (2000) A repressor sequence in the juxtamembrane domain of Flt-1 (VEGFR-1) constitutively inhibits vascular endothelial growth factor-dependent phosphatidylinositol 3′-kinase activation and endothelial cell migration. EMBO J 19: 4064–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin JD, D'Amore PA (1983) Culture of retinal capillary cells using selective growth media. Microvasc Res 26: 74–80 [DOI] [PubMed] [Google Scholar]
- Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, Yajima N, Horie Y, Hasegawa G, Naito M, Miyazaki J, Suda T, Itoh H, Nakao K, Mak TW, Nakano T, Suzuki A (2005) The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev 19: 2054–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im E, Venkatakrishnan A, Kazlauskas A (2005) Cathepsin B regulates the intrinsic angiogenic threshold of endothelial cells. Mol Biol Cell 16: 3488–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine R (2000) Nuclear lipid signaling. Sci STKE 2000: RE1. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Mugford JW, Diamond BA, Weinstein BM (2003) Phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev 17: 1346–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao HJ, Kume T, McKay C, Xu MJ, Ihle JN, Carpenter G (2002) Absence of erythrogenesis and vasculogenesis in Plcg1-deficient mice. J Biol Chem 277: 9335–9341 [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Claesson-Welsh L (2001) VEGF receptor signal transduction. Sci STKE 2001: RE21. [DOI] [PubMed] [Google Scholar]
- Ory DS, Neugeboren BA, Mulligan RC (1996) A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA 93: 11400–11406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R, Irvin BJ, Guo S, Blackburn K, Kazlauskas A, Abraham RT, York JD, Pendergast AM (2003) A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-gamma1. Nat Cell Biol 5: 309–319 [DOI] [PubMed] [Google Scholar]
- Qi JH, Matsumoto T, Huang K, Olausson K, Christofferson R, Claesson-Welsh L (1999) Phosphoinositide 3 kinase is critical for survival, mitogenesis and migration but not for differentiation of endothelial cells. Angiogenesis 3: 371–380 [DOI] [PubMed] [Google Scholar]
- Rahimi N, Kazlauskas A (1999) A role for cadherin-5 in regulation of vascular endothelial growth factor receptor 2 activity in endothelial cells. Mol Biol Cell 10: 3401–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HS, Rasmussen CS, Durham RG, King CR, Wei L (2001) Looking into anti-angiogenic gene therapies for disorders of the eye. Drug Discov Today 6: 1171–1175 [DOI] [PubMed] [Google Scholar]
- Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M (2005) Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci USA 102: 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders WB, Bayless KJ, Davis GE (2005) MMP-1 activation by serine proteases and MMP-10 induces human capillary tubular network collapse and regression in 3D collagen matrices. J Cell Sci 118: 2325–2340 [DOI] [PubMed] [Google Scholar]
- Shimizu K, Oku N (2004) Cancer anti-angiogenic therapy. Biol Pharm Bull 27: 599–605 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Yamaguchi S, Chida K, Shibuya M (2001) A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 20: 2768–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valius M, Bazenet C, Kazlauskas A (1993) Tyrosines 1021 and 1009 are phosphorylation sites in the carboxy terminus of the platelet-derived growth factor receptor beta subunit and are required for binding of phospholipase C gamma and a 64-kilodalton protein, respectively. Mol Cell Biol 13: 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valius M, Kazlauskas A (1993) Phospholipase C-gamma 1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor's mitogenic signal. Cell 73: 321–334 [DOI] [PubMed] [Google Scholar]
- Wang D, Huang HJ, Kazlauskas A, Cavenee WK (1999) Induction of vascular endothelial growth factor expression in endothelial cells by platelet-derived growth factor through the activation of phosphatidylinositol 3-kinase. Cancer Res 59: 1464–1472 [PubMed] [Google Scholar]
- Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR (2002) A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol 4: 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5