

Abstract
The matrix protein (MA) of the Moloney murine leukemia virus (M-MuLV) was found to interact with IQGAP1, a prominent regulator of the cytoskeleton. Mutational studies defined residues of MA critical for the interaction, and tests of viruses carrying MA mutations revealed a near-perfect correlation between binding and virus replication. The replication-defective mutants showed defects in both early and late stages of the life cycle. Four viable second-site revertant viruses were isolated from three different replication-defective parental mutants, and in all cases the interaction with IQGAP1 was restored by the suppressor mutations. The interaction of MA and IQGAP1 was readily detected in vitro and in vivo. Virus replication was potently inhibited by a C-terminal fragment of IQGAP1, and impaired by RNAi knockdown of IQGAP1 and 2. We suggest that the IQGAPs link the virus to the cytoskeleton for trafficking both into and out of the cell.
Keywords: cytoskeleton, IQGAP, murine leukemia virus, retrovirus replication, trafficking
Introduction
Viruses must make long and complex journeys both on their way into the interior of the cell early in infection, and on their way to the periphery of the cell and outside in the late stages of infection (Sodeik, 2000). The Gag proteins of retroviruses, and the matrix protein (MA) domains of Gag in particular, have been long recognized as important for the intracellular trafficking of the viral genome (Yuan et al, 1993). Gag proteins are synthesized in the infected cell as a large precursor, and the N-terminal MA domain of that precursor directs Gag to the plasma membrane (Spearman et al, 1994; Zhou et al, 1994; Ono and Freed, 1999) using a multipartite membrane-binding signal: a myristic acid moiety covalently attached to the N-terminal glycine of MA (Rein et al, 1986), a stretch of hydrophobic residues, and basic residues thought to interact with the acidic phospholipids on the inner leaflet of the plasma membrane (Soneoka et al, 1997). The multimerization of Gag during assembly is thought to promote the higher affinity association with the membrane (Ono et al, 2000). After virion assembly and release, the Gag precursor is processed to release the mature virion proteins, and upon cleavage MA undergoes a conformational switch that reduces its affinity for membrane (Resh, 2004). Upon entry into a newly infected cell, MA is involved in distinct events such as virion uncoating and the initiation of reverse transcription in the cytoplasm (Crawford and Goff, 1984; Kiernan et al, 1998). The details of how MA mediates these various distinct steps in the life cycle, and what cellular machinery it may recruit in these processes, are not known.
To explore the potentially diverse roles of the MA protein in virus trafficking both in and out of the cell, we conducted a search for MA-binding proteins expressed from mouse cDNA libraries interacting with Moloney murine leukemia virus (M-MuLV) MA. The most frequently identified cDNA in the screen encoded IQGAP1 (Weissbach et al, 1994; Kuroda et al, 1996), a prominent regulator of the cytoskeleton (for a review, see Noritake et al, 2005). IQGAP1 (along with its close homolog IQGAP2) binds directly to actin and crosslinks actin filaments (Fukata et al, 1997). IQGAP1 also binds the G proteins Cdc42 and Rac, and is likely a mediator of their effects in reorganizing the cytoskeleton (Fukata et al, 2002; Watanabe et al, 2004). Furthermore, IQGAPs bind to myosin essential light chain, and so may act through that interaction to control the cytoskeleton, cell shape, and intracellular transport (Weissbach et al, 1998). IQGAP1 has been shown to interact with many other proteins—β-catenin, E-cadherin, calmodulin, CLIP-170, and the adenomatous polyposis coli gene product (APC) (Fukata et al, 1999, 2002; Li et al, 1999; Watanabe et al, 2004)—and so has been implicated in many aspects of cell physiology, including cell adhesion (Kuroda et al, 1998), polarization, and migration. The specific interaction of IQGAP1 with the M-MuLV MA protein is highly suggestive of its involvement in the intracellular trafficking of the virus. We have obtained strong genetic and biochemical evidence that this interaction is in fact essential for virus replication, and, remarkably, is involved in both early and late stages of the virus life cycle.
Results
Identification of murine IQGAP1 as an MA interacting protein
To identify cellular proteins that interact with the M-MuLV MA, the yeast two-hybrid method was used to screen Gal4AD fusion proteins from WEHI-3B, a murine macrophage/monocytic leukemia cell line, for interaction with a LexADB-MA ‘bait' construct. Out of more than 106 yeast transformants, a total of 43 hits were recovered, with four genes isolated repeatedly (Supplementary Table S1). IQGAP1, a protein implicated in regulation of the cytoskeleton (Kuroda et al, 1996), was recovered seven times. These clones were independent isolates, and encoded Gal4AD fusions to different C-terminal portions of the IQGAP1 protein.
To identify the residues of MA required for the interaction with IQGAP1 or the other binding proteins, a series of alanine substitution mutations were introduced into the central portion of the MA coding region in the LexADB-MA construct (Figure 1A). Western blots were used to confirm that all these mutant proteins were well expressed in yeast. The mutants were tested for their interaction with various Gal4AD fusions identified in the screen (Table I). Binding to IQGAP1 was sensitive to a distinctive set of mutations localized in two blocks: mutants T1–T6, and mutants T11, SMA6, and SMA8. These blocks were separated by mutations in the intervening sequences (T7–T10) that had no effect on IQGAP1 binding. Interactions with the other candidate genes showed different and distinctive patterns of disruption by the mutants (Table I).
Figure 1.
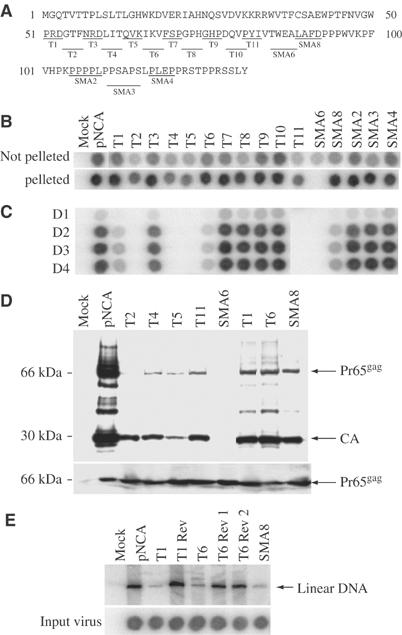
Construction and analysis of substitution mutants of M-MuLV MA. (A) Alanine-substitution mutations of MA. The complete sequence of MA is shown. Underlined resides were mutated to alanines in blocks of three, four, or five in the indicated mutants. (B) Assembly and release of mutant virions measured by RT assay. Spots indicate the amount of labeled nucleotide incorporated in an exogenous RT assay on a homopolymer template by virion-associated RT. Top row: 293T cells were transiently transfected by MA mutant proviral DNAs, and crude culture supernatants were harvested 48 h after transfection and analyzed by RT assay. Second row: virions were isolated from the medium by pelleting through a sucrose cushion, resuspended, and analyzed by RT assay. Mock, no viral DNA. pNCA, wild-type viral DNA. (C) Equal amounts of virus (normalized by RT assay of the crude harvests) were used to infect naïve Rat2 cells. Culture supernatants were harvested on successive days as indicated and virus yield was monitored by RT assay. Mock, no virus was applied. (D) Western blot analysis of CA-containing gag gene products in virions and lysates of transfected cells. The virus particles were prepared as described in Materials and methods. Virions and cells were lysed, and proteins were subjected to Western blot analysis, probing with anti-CA antisera. The cell lysates were normalized for transfection efficiency using β-galactosidase assay. The positions of Pr65gag and CA are indicated. (E) Southern blot detection of linear viral DNA synthesized in infected Rat2 cells. Supernatants from transfected 293T cells were used to acutely infect Rat2 cells. The virus preparations were normalized for equal RT activity. Low molecular weight DNAs were extracted by the Hirt method and the viral DNAs were detected by hybridization with a radiolabeled virus-specific DNA probe. The position of linear DNAs is marked. The levels of RT applied are shown in the lower panel.
Table 1.
MA mutations effect interaction with host proteinsa and virus spreadb
| Gal4AD-IQGAP1 | Gal4AD-VAV | Gal4AD-DNAJ/HSP | Gal4AD-transketolase | Gal4AD-DOK3 | Virus spread | |
|---|---|---|---|---|---|---|
| LexADBD | − | − | − | − | − | N/A |
| LexA-MA | +++ | +++ | +++ | ++++ | ++++ | +++ |
| LexA-MA T1 | + | +++ | ++++ | ++++ | +++ | + |
| LexA-MA T2 | − | − | ++++ | ++++ | − | − |
| LexA-MA T3 | + | ++ | ++++ | ++++ | +++ | +++ |
| LexA-MA T4 | − | +/− | ++++ | ++++ | ++ | − |
| LexA-MA T5 | − | +/− | ++++ | ++++ | ++++ | − |
| LexA-MA T6 | + | +++ | +++ | ++++ | ++++ | + |
| LexA-MA T7 | ++ | + | +/− | ++ | − | +++ |
| LexA-MA T8 | +++ | +++ | +++ | +++ | − | +++ |
| LexA-MA T9 | ++ | +++ | +++ | +++ | +++ | +++ |
| LexA-MA T10 | +++ | +++ | +++ | +++ | ++ | +++ |
| LexA-MA T11 | − | +/− | ++++ | +/− | +/− | − |
| LexA-MA SMA6 | − | − | ++++ | ++++ | +/− | − |
| LexA-MA SMA8 | − | −/+ | +++ | ++++ | − | + |
| LexA-MA SMA2 | +++ | +/− | +++ | +++ | +++ | +++ |
| LexA-MA SMA3 | +++ | +/− | +++ | +++ | ++++ | +++ |
| LexA-MA SMA4 | ++ | +/− | +/− | ++++ | ++++ | +++ |
| aPluses indicate level of β-galactosidase activity in yeast two-hybrid interaction assay. ++++ colonies turned blue (indicating β-galactosidase activity) in 0.5 h; +++ colonies turned blue in 1 h; ++ colonies turned blue in two hours; + colonies turned blue in 3–4 h; +/− colonies with trace of blue color in 6–8 h;−, colonies with no detectable blue color after more than 8 h. | ||||||
| bPluses indicate replication kinetics as shown in Figure 1C. +++, mutants were fully viable and exhibited wild-type kinetics; +, mutants were replication defective and exhibited a delay in virus replication; −, mutants were completely replication defective. | ||||||
| MA, matrix protein. | ||||||
Interaction of MA with IQGAP1 correlates with virus replication
If the interaction of any of the cellular proteins with MA is required for virus replication, those MA mutations that disrupt the binding should also block the virus replication in mammalian cells. To test the alanine substitution mutations for their effects on viral functions, the mutations were moved into the context of a complete infectious copy of the M-MuLV proviral DNA. The DNAs were then introduced into 293T cells, the culture medium was collected after 48 h, virions were pelleted through sucrose cushions, and the yield of virus was assessed by measurement of the DNA polymerase activity of the virion-associated reverse transcriptase (RT), both in the crude harvest and after pelleting (Figure 1B). All but one of the 16 mutants were able to induce the formation of pelletable virus. Several of the mutants produced significantly lower levels of virus (notably T2, T4, T5, and T11); we estimated the yields for these mutants to be 3–5 × less than the wild type. These mutations thus caused a moderate defect in some aspect of the late stage of the life cycle. Mutant SMA6 made essentially no pelletable virus.
To test the mutant virions for their ability to initiate infection and spread in an infected culture, the crude virus harvests were normalized to equal levels of RT activity and then used to infect the virus-sensitive Rat2-2 cell line. Culture medium was then collected from the infected cultures on successive days and monitored for virus by RT assays as before (Figure 1C). The yields of virus for the various mutants were assessed by quantitation of the RT levels on day 4 (Supplementary Figure S3A). MA mutants T2, T4, T5, T11, and SMA6 were completely replication defective and were unable to carry out a productive infection. Mutants T1, T6 and SMA8 were partially defective, initiating the production of low levels of virus. Mutants T3, T7, T8, T9, T10, SMA2, SMA3, and SMA4 were fully replication competent and spread through the cultures with kinetics and yield similar to the wild type. Comparison of the pattern of mutant virus replication with the pattern of mutant binding to IQGAP1 revealed a very strong correlation: the defective mutants showed no binding, the partially defective mutants showed weak binding, and the fully viable mutants showed wild-type binding (Table I). Only mutants T3 and SMA8 showed imperfect, but still positive, correlation of their ability to replicate with IQGAP1 binding. In contrast, the pattern of binding of the other host proteins was highly distinct from the pattern for replication. Most of the host proteins showed no correlation at all with replication, and only Vav showed even partial correlation. Vav binding with several mutants, however—notably T6, SMA2, SMA3, and SMA4—did not track with replication. The results suggest that the target of MA binding that is essential for virus replication is likely IQGAP1, and, surprisingly, that no other critical functions or binding targets are affected by these mutations in this portion of MA.
MA mutants that do not interact with IQGAP1 are defective for both late and early events
To determine the steps in the viral life cycle affected by the MA mutations, selected defective mutants were characterized further. The mutant DNAs, along with a reporter plasmid encoding β-galactosidase (pCMV-β-gal), were introduced into 293T cells as before, and the culture medium and cell lysates were collected after 72 h. The virus harvests were normalized to equal levels of β-galactosidase in the cell lysates, the virions were purified by ultracentrifugation through a sucrose cushion, and the virion proteins were analyzed by SDS–PAGE and Western blots with anti-capsid (CA) antibody to measure the yield of virus (Figure 1D upper panel). The yields of virus were further assessed by quantitation of the band corresponding to CA (Supplementary Figure S3B). The replication-defective mutants (T2, T4, T5, T11, and SMA6) showed a major reduction in virion yield compared to the wild type, in agreement with the RT assays; one of these (SMA6) showed no virion proteins. The partially defective mutants (T1, T6, and SMA8) showed a less dramatic reduction in yield. The Gag protein detected in all cases, however, showed relatively normal processing of the Pr65gag precursor to the mature p30 (CA) product; the variation in extent of processing was well within the range of variation seen in different clones of wild-type virus. Examination of the intracellular levels of Pr65gag in lysates showed that all mutants synthesized levels of the Gag precursor comparable to that of the wild-type virus (Figure 1D lower panel). These results suggest the region of MA interacting with IQGAP1 plays a significant, although not absolutely essential, role in late steps of the life cycle.
To determine if the MA mutants were affected in early events, virions were harvested from 293T cells transfected with the partially defective mutant DNAs and normalized to equal amounts of virus based on the RT levels. These virus preparations were used for an acute infection of Rat2-2 cells, and the synthesis of linear viral DNA was monitored by Southern blot of the low-molecular weight DNA isolated 24 h postinfection (Yuan et al, 1999). The mutants (T1, T6, and SMA8) showed a significant defect in formation of the viral DNA, producing roughly 5–10-fold less DNA than the wild-type virus (Figure 1E). The result indicates that these mutants had a substantial block during the very early phase of infection. The mutations could have acted either at the time of virion assembly in the producer cells—generating poorly infectious virions—or at the time of entry into the infected cells, but clearly demonstrate an important role for MA that results in low levels of viral DNA synthesis.
Revertant viruses carrying second-site suppressor mutations show restored interaction of MA with IQGAP1
The correlation of IQGAP binding with virus replication suggests that the IQGAP interaction might indeed be important for MA function. If this notion were correct, the MA of replication-competent revertants of the defective mutants should always reacquire their ability to interact with IQGAP. To isolate such revertants, 293T cells were transfected with the mutant DNAs, virus was harvested and applied to Rat2-2 cells, and the infected cultures were passaged repeatedly. The emergence of replication-competent virus was monitored by RT assay of the culture medium. After periods ranging from 12 to 20 weeks, high levels of RT activity appeared in four cultures, each initiated by infection with one of three different mutants (T1, T4, and T6). Infection of fresh Rat2-2 cells with these revertant virus preparations resulted in the release of high levels of RT with wild-type kinetics (data not shown).
To identify the mutations responsible for the reversion events, low-molecular weight viral DNAs were prepared from cultures after acute infection with the putative revertant virus stocks. The MA-p12-CA portion of the viral DNAs was amplified by PCR and cloned, and the DNA sequences of the inserts were determined (Figure 2A). Revertant T1Rev retained the three original T1 alanine substitutions (PRD51-53AAA), but contained a new substitution, F38L, further upstream. Revertants T4Rev and T6Rev1 retained two of their original three alanine substitutions, but in each revertant one of the three alanines was changed to valine. In one case (T4Rev), the original wild-type residue was not valine but rather isoleucine; in the other case (T6Rev1), the reversion represented a change back to the wild-type residue, valine. Finally, revertant T6Rev2 retained its original alanine mutations (IKV66-68AAA) but, remarkably, had acquired substitution F38L, the same mutation seen in T1Rev. The F38L mutation was thus a supersuppressor of more than one disruptive mutation.
Figure 2.
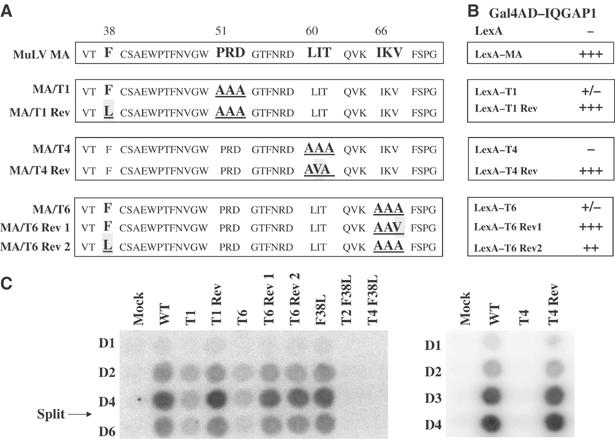
DNA sequences, interaction phenotypes, and replication of MA revertants. (A) The amino-acid sequences of the MA proteins encoded by the wild-type virus, the parental mutants, and the revertant viral genomes. (B) Interaction of MA mutants and revertants with IQGAP1 in the yeast two-hybrid system. Signal strengths are as indicated in Table I. (C) The replication of wild-type, mutant, and revertant viruses in infected Rat2-2 cells as judged by RT assay of culture supernatants collected on the indicated days postinfection.
To confirm that the putative suppressor mutations identified in these cloned DNAs of the revertant viruses were indeed responsible for the restored replication, the mutations were re-engineered by site-directed mutagenesis into the parental mutant DNAs, and these reconstructed DNAs were introduced into 293T cells to generate virus. These virus preparations were used to infect naive Rat2-2 cells and the rate of spread of the virus in the infected cultures was monitored by RT assays of the culture medium on successive days (Figure 2C). The yields of virus were determined by quantitation of the RT levels on day 4 (Supplementary Figure S3C). As before, the replication of the original mutants was either strongly attenuated (T1 and T6) or completely blocked (T4). In contrast, the replication kinetics of all the reconstructed revertants was indistinguishable from the wild-type virus (Figure 2C). Furthermore, the revertants showed restored ability to synthesize full levels of viral DNA after acute infection (Figure 1E). Thus, the reversion events identified by sequence were indeed responsible for the suppression of the mutations. Additional tests of constructs containing the F38L mutation showed that although it fully suppressed mutations T1 and T6, it was unable to rescue mutants T2 or T4 (Figure 2C). The F38L mutation in a wild-type background replicated like the wild-type virus (Figure 2C).
The key step in the analysis was then to test whether these suppressor mutations achieved their effect on replication by restoring the interaction of MA with IQGAP1. The DNA fragments containing the alanine and suppressor mutations were introduced into the LexA-MA constructs, and the resulting plasmids were introduced into yeast along with the Gal4AD-IQGAP1 expression plasmid. While the original alanine mutant MA proteins interacted poorly or not at all with IQGAP1, all four of the MA revertants interacted as strongly as the wild-type MA (Figure 2B). The F38L mutation in the context of mutants T2 and T4, which were not rescued by F38L, did not restore the interaction with IQGAP1 (data not shown). The F38L mutant in a wild-type background interacted well with IQGAP1 and also with Vav and Dok3 (not shown). Quantitative β-galactosidase assays of yeast cultures expressing several of the suppressor mutants fully confirmed the filter lift assays. Thus, selection for virus replication consistently resulted in suppressor mutations that restored the interaction with IQGAP1. These results strongly suggest that the interaction is required for proper MA function.
MA interacts with the C-terminus of IQGAP1
The IQGAPs are large multidomain proteins with binding sites for a number of distinct interacting partners. The region present on the smallest of the cDNAs from the two-hybrid clones contained a C-terminal fragment (amino acids 1440–1657), including the RasGAP-like domain. To further define the portion of IQGAP1 required for interaction with MA, a series of N- and C-terminal truncation mutants of IQGAP1 were generated by PCR, expressed as Gal4AD fusions in yeast, and tested with LexA-MA for reporter activation. Western blots confirmed that all the IQGAP1 fragments were well expressed. The minimal region required for strong interaction with MA consisted of residues 1500–1657, including the C-terminus of the protein (Figure 3A). Removal of as few as 17 amino acids from the C-terminus eliminated binding to MA. To further refine the edges of the minimal region, alanine-scanning mutations were introduced into regions spanning residues 1498–1529 and also 1642–1657. Mutants mt5, 6, and 7 near the N-terminal border showed strongly reduced binding (though mutant mt8 did not); and mutants mt9, 10, 11, and 12 at the very C-terminus were all completely defective for binding, suggesting that the C-terminus of IQGAP1 is essential for the interaction (Figure 3B). To confirm that the mutations did not cause a global disruption of the protein, we tested all the alanine mutants for interaction with a distinct partner and confirmed that the mutants all interacted (data not shown).
Figure 3.
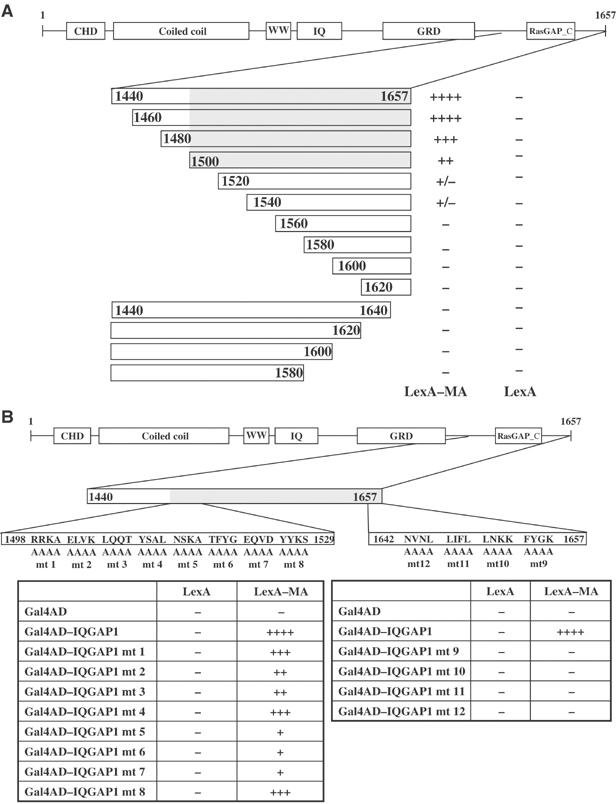
MA binding domain of IQGAP1 maps to the amino acids 1500–1657 (A) Deletion analysis. A schematic of the full-length IQGAP1 protein is shown at the top. The C-terminal region is expanded, and bars represent the portions expressed as Gal4AD fusions in yeast. Numbers refer to amino-acid residues of IQGAP1. Entries indicate level of β-galactosidase activity in yeast two-hybrid interaction assay, as given in Table I. (B) Alanine-scanning mutagenesis of selected regions of IQGAP1. Expanded views of amino acids 1440–1529 and 1642–1657 are shown. The position of the block of changes to alanine in each mutant is indicated. Yeast colonies expressing the indicated mutant fusion proteins were lifted and stained for β-galactosidase activity and scored as before.
MA interacts with IQGAP1 in vitro and in vivo
To confirm the binding of MA and IQGAP1 in settings distinct from that of the yeast two-hybrid system, we tested for interaction of the proteins in vitro and in mammalian cells. The wild-type and various MA mutants were expressed in bacteria as fusions with the maltose-binding protein (MBP-MA), bound to maltose–sepharose beads, and used as an affinity matrix for the binding of IQGAP1 expressed in bacteria as a fusion to glutathione S-transferase (GST-IQGAP1). The beads were washed and the bound proteins were analyzed by SDS–PAGE and Western blot (Figure 4A). Wild-type MBP-MA efficiently bound GST-IQGAP1, but not GST, from solution. MA mutants T4, T5, SMA6, and T11 bound GST-IQGAP1 very poorly or not at all. Furthermore, the four MA revertants, T1Rev, T4Rev, T6Rev1, and T6Rev2, all bound GST-IQGAP1 as well as the wild-type MA. Thus, the binding and the response to MA mutations and MA reversion mutations in vitro correlated well with the behavior of the proteins in yeast. However, an IQGAP1 mutation (Y1627H) that restored binding in yeast (see Supplementary Results) did not restore binding to MA T4 in vitro.
Figure 4.
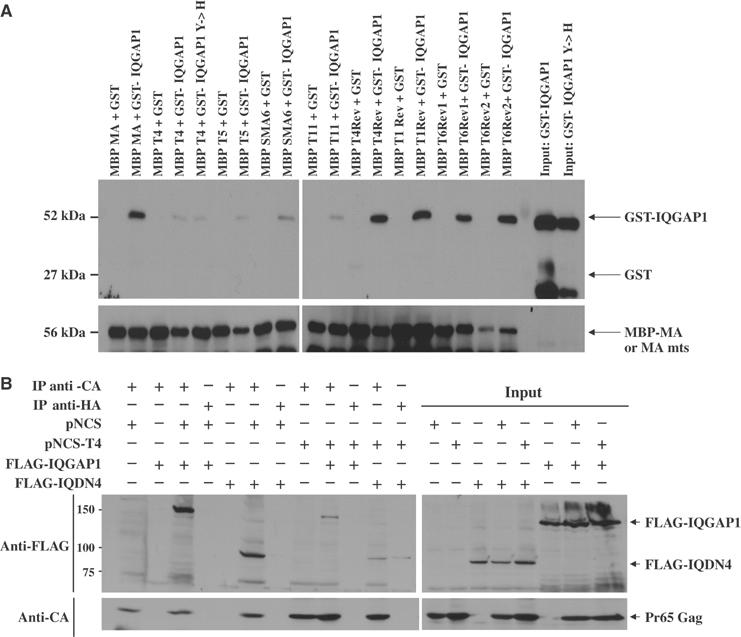
(A) Interaction of MA with IQGAP1 in vitro. Wild-type MA, MA mutants (T4, T5, SMA6, and T11), and MA revertants (T4Rev, T1 Rev, T6Rev1, and T6Rev2) were expressed in bacteria as fusions with the MBP, bound to amylose–sepharose beads, and used as an affinity matrix for the binding of IQGAP1 expressed in bacteria as a fusion to (GST-IQGAP1; aa 1440–1657) or (GST-IQGAP1 Y1627H; aa 1440–1657). The beads were washed and the bound proteins were analyzed by SDS–PAGE and Western blot with anti-MA antisera and GST antibodies. (B) Pr65gag and IQGAP1 associate in vivo. 293T cells were cotransfected with either wild-type proviral DNA (pNCS) or the T4 mutant (pNCS-T4), and with either Flag-tagged full-length IQGAP1 or the C-terminal fragment Flag-IQDN4. Lysates were prepared, the Gag proteins were immunoprecipitated with anti-CA antiserum, and the associated proteins were analyzed by SDS–PAGE and Western blot with anti-FLAG antibodies and anti-CA antisera. As a negative control, samples were also immunoprecipiated with anti-HA antibodies. A portion of the input samples were analyzed by Western blot without immunoprecipitation.
To test for association of MA and IQGAP1 in mammalian cells, the viral DNA genome and either Flag-tagged full-length IQGAP1 (FLAG-IQGAP1) or a C-terminal fragment (Flag-IQDN4; see below) were coexpressed in 293T cells. Lysates were prepared, the Gag proteins were immunoprecipitated with anti-CA antiserum, and the associated proteins were analyzed by SDS–PAGE and Western blot with anti-FLAG antibodies (Figure 4B). Wild-type Gag bound the full-length IQGAP1 and the C-terminal fragment efficiently, while the T4 mutant Gag bound them much less well. Immunoprecipitations carried out in the absence of virus, or with irrelevant antisera, did not bring down the IQGAP1 proteins. Thus, Pr65gag and IQGAP1 do associate in virus-producing cells.
IQGAP1 is required for M-MuLV replication
Expression of mutant or inactive versions of critical host components can result in dominant-acting inhibition of virus replication (Garrus et al, 2001; Strack et al, 2003). To test if IQGAP could induce such inhibition, we generated a series of DNA constructs designed to express FLAG-tagged C-terminal fragments of IQGAP1, all retaining the binding site for MA but lacking binding sites for other partners (Figure 5A). 293T cells were transfected with the viral DNA along with increasing amounts of these IQGAP constructs, virus was harvested after 48 h, and the yield was determined by SDS–PAGE and Western blot with anti-CA antiserum. Overexpression of IQDN4, containing approximately the C-terminal half of the complete protein, caused a strong and dose-dependent inhibition of virion production, with more than a 10-fold reduction at the highest DNA levels (Figure 5B; CA levels quantitated in Supplementary Figure S3D). There was no effect on the intracellular levels of the Gag precursor protein except for a very small reduction at the highest level of IQDN4. The levels of the IQDN fragments often seemed to plateau at higher transfected DNA levels; the basis for this is not clear but may reflect protein turnover. Overexpression of the three other constructs had no significant effect on virus production even at the highest levels (IQDN3, 5, and 6; data for IQDN3 shown in Figure 5C; CA levels quantitated in Supplementary Figure S3E). Overexpression of IQDN4 had no effect on production of HIV-1 virus-like particles, ruling out general toxicity as a mechanism (Figure 5D). These results indicate that the IQDN4 is a potent dominant negative inhibitor of M-MuLV virion production.
Figure 5.
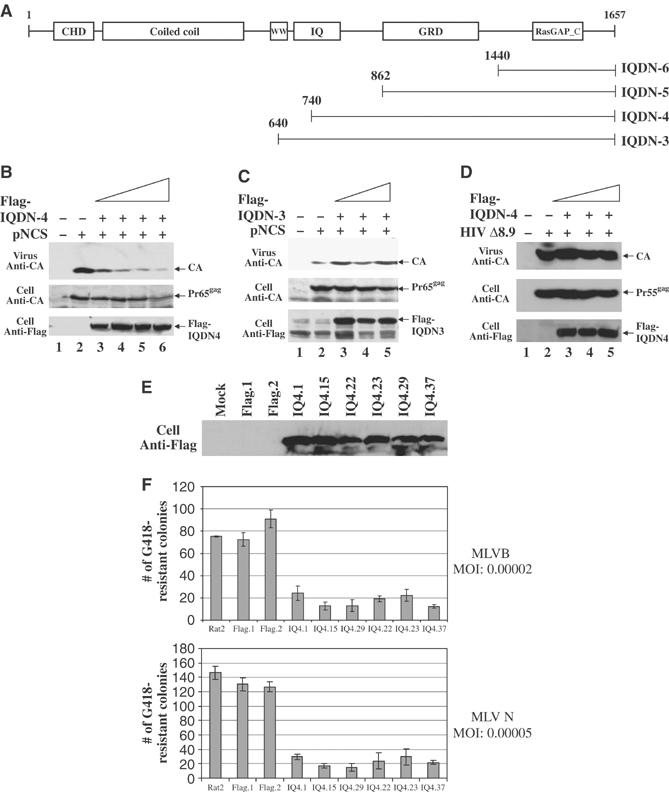
Overexpression of a C-terminal fragment of IQGAP1 (IQDN4) inhibits M-MuLV release. (A) Schematic diagram of N-terminal truncations of full-length IQGAP1. Numbers refer to the amino-acid residue of the protein. (B, C) 293T cells were cotransfected with 3 μg of the wild-type proviral DNA (pNCS) and either 6, 9, 12, or 15 μg of the Flag-tagged IQDN4 or IQDN3 expression construct as indicated. Virion proteins from culture media and cell lysates were subjected to Western blot analysis with anti-CA antisera and anti-FLAG antibodies. (D) 239T cells were cotransfected with 3 μg of an HIV-1 proviral DNA (Δ8.9) and increasing levels of IQDN4 as in (B). Virion proteins from culture media and cell lysates were subjected to Western blot analysis with anti-CA antisera and anti-FLAG antibodies. (E) Expression of IQDN4 increases retroviral resistance in Rat2 cells. Equal amounts of protein extracts from the indicated cell lines were subjected to SDS–PAGE, and immunoblotted with anti-FLAG antibodies to evaluate expression of IQDN4. (F) These same lines were then challenged by infection with MuLV vectors expressing the neor gene, and the number of G418-resistant colonies was determined after 14 days.
To determine if IQGAP1 mutants can block the early events of virus replication, we generated rodent cell lines that stably overexpress the FLAG-tagged C-terminal fragments of IQGAP1. The level of expression of the fragment in each clone was determined by Western blot with anti-FLAG antibodies (Figure 5E). The cultures were then challenged by infection with preparations of a retrovirus vector containing the neor gene. The infected cells were plated in medium containing G418, and the number of transduced colonies was determined after 10–12 days. While the wild-type Rat2 cells, cells expressing an empty vector control, and cells overexpressing full-length IQGAP1 were sensitive to virus, all six lines expressing IQDN4 were significantly resistant to MuLV infection, yielding 4–10-fold fewer colonies than the controls (Figure 5F). These results suggest that the IQDN4 fragment is a strong inhibitor of the early stages of infection as well as the late stages.
To test whether the expression of endogenous wild-type IQGAP1 is required for retroviral replication, we used RNAi methods to knock down the levels of mRNA and protein. We noted that there exist two highly related genes, IQGAP1 and IQGAP2, in both mouse and human, and tests in the yeast two-hybrid system showed that MA interacts equally well with both of the human gene products (data not shown). Thus, it might be necessary to eliminate products of both genes to detect an effect on the virus. We prepared three DNA constructs that would express hairpin RNAs targeted to downregulate each of the two human IQGAP cDNAs, and generated 293T lines that stably expressed six of the nine possible pairwise combinations of these siRNA constructs. Examination of lysates of these lines by SDS–PAGE and Western blot with antisera specific to either IQGAP1 or IQGAP2 revealed the specific downregulation of either one or both of the proteins in several of these lines (Figure 6A). To assess the lines for their virus susceptibility, we made use of an amphotropic MuLV, similar to Moloney MuLV but able to spread in human cells. Virus was applied at low multiplicity to amplify any effects on replication, and the rate of spread of the virus was assessed by measuring the appearance of virion-associated RT in the culture medium on successive days (Figure 6B). While replicating virus was detected in all the lines, those with the most profound decrease in both IQGAP1 and 2 showed a significant delay and a reduced maximal yield of virus (Figure 6B, lanes 3,4, and notably 8; viral yields across time are plotted in Supplementary Figure S3F). These results suggest that the IQGAPs at their normal levels of expression do play a positive role in virus replication.
Figure 6.
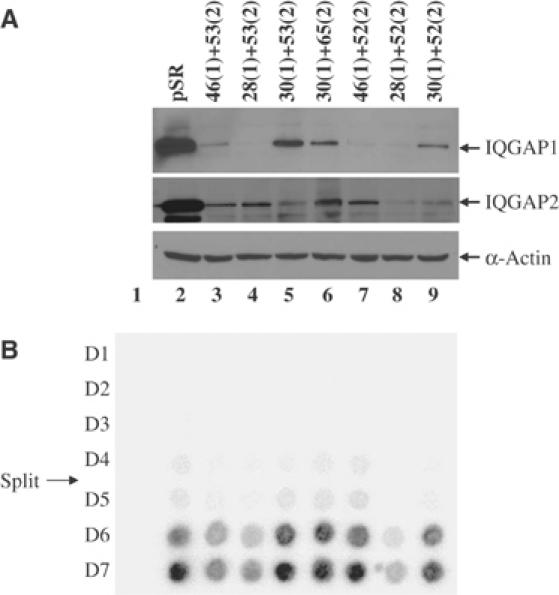
M-MuLV replication in cell lines with reduced IQGAP1 and IQGAP2 expression. Clonal cell lines expressing short hairpin RNAs specific for IQGAP1 and IQGAP2 were generated and tested for both knock down and ability to support virus replication. Individual hairpins are numbered, and the number in parentheses indicates the target gene as either IQGAP1 or 2. (A) Expression levels of IQGAP1, IQGAP2, or β-actin in knockdown cell lines as judged by Western blot analysis. (B) Replication kinetics in cell lines from (A), assessed by RT assay on the indicated days. Mock, infection without virus. The day at which cells were split is indicated.
To identify the stage of the viral life cycle most strongly affected by RNAi knockdown of IQGAP1 and 2, both late and early stages of replication were examined separately. The stable knockdown lines were transfected with proviral DNA encoding wild-type M-MuLV, and the yield of virus released at 48 h and 72 post-transfection was assessed by Western blots for CA protein. No significant inhibition of virus production as compared with wild-type control cells was detected (data not shown). To examine early events of the life cycle, the lines were infected with retroviral vectors expressing luciferase, and the efficiency of transduction was assessed by luciferase assays of cell lysates 48 h postinfection. The knockdown lines showed slightly lower levels than the control lines, but the differences were not statistically significant. To allow tests at low multiplicity, similar experiments were performed with retroviral vectors expressing hygromycin resistance, and the levels of transduction were assessed by counting hygromycin-resistant colonies after 10 days (Supplementary Figure S4). The knockdown lines consistently showed a 2–3-fold decrease in virus susceptibility. Thus, knockdown of IQGAP1 and 2 seems to have the largest effect on the early stage of the life cycle.
Discussion
The genetic and biochemical experiments described above provide strong evidence that the MuLV MA protein interacts with the IQGAP proteins, and that this interaction is required for virus replication. The binding was observed through the two-hybrid system in yeast, with recombinant proteins in vitro, and in mammalian cells. The murine viral MA interacts well with IQGAP1 from both mouse and human cells, and equally well with human IQGAP2. Nonbinding mutant viruses were uniformly replication-defective, with strong correlation between binding and virus viability. Most compellingly, revertants selected for restored virus replication all demonstrated a restoration of binding to IQGAP1. It is particularly significant that many different changes were found that led to this outcome; this argues strongly that the binding is not incidental. One suppressing mutation, F38L, acted as a supersuppressor, rescuing several MA mutants (T1 and T6) and restoring their interaction with IQGAP. In some cases, however (T2 and T4), this mutation did not rescue mutant virus replication or restore the interaction. Across all these cases, the ability to bind IQGAP correlated with virus viability. Thus, the interaction is apparently essential for virus replication.
It is likely that the wild-type IQGAP is performing a positive role in promoting virus replication, one that requires the intact and functional molecule; a C-terminal MA-binding fragment of IQGAP did not support virus replication but instead acted as a potent inhibitor, perhaps through competition with the endogenous protein. A further indication that the IQGAPs are important was the observation that RNAi-mediated knock down of both IQGAP1 and 2 simultaneously had a significant effect on virus replication. Knockdown of only IQGAP1 alone, or IQGAP2 alone, had no effect on the virus; thus, both family members are probably capable of supporting virus replication. This conclusion is also supported by the observation that mouse embryonic fibroblast cells from homozygous mutant mice lacking IQGAP1 but retaining IQGAP2 (Li et al, 2000) could support normal virus replication (data not shown). Analysis of the nonbinding mutant viruses, and of wild-type virus in cells expressing dominant negative fragments of IQGAP1, indicated that the IQGAPs are utilized both early and late in the infectious cycle. The precise role of IQGAP in virus replication is not clear, but the known functions of the IQGAPs suggests that they are involved in mediating interactions of the virus with the cytoskeleton, perhaps with trafficking into and out of the cell through that interaction.
The region of IQGAP1 that interacts with MA is the very C-terminal portion, roughly the last 150 amino acids (Figure 3A). Critical residues are found near residue 1500 and at the very C-terminus. The function of this region is not very well understood, but this domain has sequence similarity to RasGAP and interacts with CLIP-170, β-catenin, and the APC protein. The binding of MA could thus displace or block the interaction with these proteins, but would not be expected to prevent the interaction of IQGAP with actin, calmodulin, or Rho family members. In turn, the residues of MA that interact with IQGAP1 lie in several stretches separated by intervening residues. The status of the MA domain at the time of the interaction—whether in the context of the Gag precursor, or after processing to the mature protein—and the oligomeric state of MA—whether as a monomer or trimer—are not known. However, during the early phase of the life cycle MA is likely to be present as a processed trimer, and there is detailed information on the trimeric structure of the related HIV-1 matrix protein (Hill et al, 1996). Examining the positions of the homologous residues of the HIV-1 MA trimer suggests that the critical contacts with IQGAP1 lie in a cluster on a side surface, away from the face thought to lie in contact with the lipid membrane in the virus (Supplementary Figure S1). Based on the residues required for the interaction, MA-IQGAP binding would thus not be expected to disrupt MA multimerization. It is intriguing that a single substitution in MA (F38L) was able to restore the binding of two very different MA mutants to IQGAP and also fully restore virus replication. The altered residue, F38, is not on the contact surface but lies on a helix running above this same region, and might stabilize the MA structure or might subtly alter the positioning of the helix (Supplementary Figure S1). We note that F38L did not restore IQGAP binding to all mutants (e.g. T2 and T4) nor rescue the replication of the corresponding viruses, indicating that it is not a global bypass mutation.
IQGAP1 and 2 are thought to mediate the effects of the Rho-family GTPases, including Rho, Rac, and Cdc42, on various aspects of cell physiology: cell shape, cell adhesion, polarization, and migration (see Noritake et al, 2005 for a recent review). There are indications that the IQGAPs mediate these processes by direct contact with specific components of the cytoskeleton. These include actin itself, the extracellular signal-related kinases (ERKs), calmodulin, β-catenin, E-cadherin, CLIP-170, and the myosin essential light chain. In most cases, it is not clear what the precise role of the IQGAPs might be in mediating Rho-family signals. In the control of cell–cell adhesion, the IQGAPs are known to be localized to the regions of contact and are apparently required for the proper accumulation of actin, E-cadherin, and β-catenin at these sites of contact (Noritake et al, 2004). Knock down and expression of dominant negative fragments of IQGAP can either block or promote the normal dissociation of cell–cell adhesion (Kuroda et al, 1998), perhaps mediated by IQGAPs' ability to dissociate α-catenin from an ordered complex of β-catenin and E-cadherin. Why would the viral MA protein need to interact with IQGAP in this context? It has been speculated that the actin cytoskeleton is a physical barrier to virus entry (Campbell et al, 2004; Lehmann et al, 2005), and that incoming viruses need to locally disrupt the cytoskeleton for successful infection. Perhaps the interaction of MA with IQGAP is triggering such essential changes in the cytoskeleton. The failure of the nonbinding MA mutant viruses to successfully initiate very early events of infection is consistent with this notion.
IQGAP1 is also thought to control cell polarity and directional migration. Besides the actin cytoskeleton itself, the major players in these processes are thought to be proteins at the plus-ends of microtubules such as the CLIP-170 family, APC, and the dynein–dynactin complex. Importantly, IQGAP1 interacts with both CLIP-170 and APC, and colocalizes with them at the polarized leading edge. These proteins are likely involved in cytokinesis and vesicle trafficking (Pierre et al, 1992). Why might the MA protein need to interact with IQGAP in this setting? The simplest notion is that the interaction results in key movement of the viral genome during infection. This could in turn involve interactions with various motors, including myosin light chain, which interacts with IQGAP1 (Weissbach et al, 1998), or Kif4, which interacts directly with MA (Kim et al, 1998; Tang et al, 1999), and may also be involved in vesicular transport (Basyuk et al, 2003; Dong et al, 2005). Such movement could be needed by the virus either on the way in or out of the cell.
The interaction of MuLV MA with IQGAP detected here is not universally conserved for all retroviruses. Preliminary tests suggest that HIV-1 MA did not interact with IQGAP1 in yeast; other proteins, however, may be utilized by such retroviruses. A recent report suggests that HIV-1 MA interacts with another host protein, AP-3, during virus assembly (Dong et al, 2005). Viral Gag proteins depend on a large collection of host proteins that help virus budding and release, and though most of these players are conserved and important, different viruses make contacts with different subsets of the components of the machinery. We would suspect that IQGAPs, here seen as critical for the MuLVs, may be utilized by many other retroviruses and even other virus families.
Materials and methods
Bacterial and yeast strains
Saccharomyces cerevisiae strain CTY10-5d (MATa ade2 trp1-901 leu2-3,112 his3-200 gal4 gal80 URA3∷lexA-lacZ) contains an integrated GAL1-lacZ gene with the lexA operator. KC8 (Clontech, Mountain View, CA), an auxotrophic leuB, trpC, and hisB Escherichia coli strain, was used to isolate plasmids from yeast. E. coli strain DH5-α was used to express GST and MBP fusion constructs.
Yeast Two-hybrid library screening
The yeast two-hybrid library of mouse cDNAs from WEHI-3B was as described (Bacharach et al, 2000). Transformation of yeasts and scoring for LacZ expression by 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal) assay on nitrocellulose lifts were as described (Breedon and Nasmyth, 1985). Blue transformants were restreaked and retested, and cDNA was recovered and analyzed by sequencing.
Plasmid constructs
Plasmid pNCA contains an infectious copy of M-MuLV proviral DNA (Colicelli and Goff, 1988); pNCS is a version of pNCA that carries a simian virus 40 (SV40) replication of origin to allow high level of expression in 293T cells. The construction of MA mutants in pNCA and various fragments of murine IQGAP1 for expression in mammalian cells and in the yeast two-hybrid system is described in Supplementary data.
Cell culture
293T cells are 293 cells that stably express SV40 large T-Ag. Rat2-2 cells are rat embryonic fibroblast cells. 293T and Rat2-2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% calf serum.
Mammalian cell transfection, viral infections, and virion protein analysis
To examine virus viability, 293T cells were transiently transfected with wild-type (pNCS) or mutant proviral DNAs. Virus was harvested and used to infect naive Rat2-2 cells. Culture supernatants were collected on successive days and monitored for virus production by RT assay (Goff et al, 1981). To examine virion proteins, viral particles were harvested from transfected 293T cells and purified by ultracentrifugation, and the proteins were analyzed by Western blot as described previously (Yuan et al, 1999).
Isolation of revertant viruses
Virus supernatants were harvested at the peak of RT activity from 293T cultures transfected with T1, T4, and T6 mutants. The virus stocks were used to infect fresh Rat2-2 cells. The infected cells were repassaged every 4 days until RT activity reached levels similar to the wild type. The emerging viruses were used to infect fresh Rat2-2 cells, and low MW DNA was harvested (Hirt, 1967). A 945 bp fragment spanning the MA-p12-CA coding region was PCR amplified from the DNA.
MA/5′ Eco: 5′-ATCCGAATTCATGGGCCAGACTGTTACCACT-3′
XhoI/R2: 5′-ATCCCAGTCTGGGCGCTCGAGGGGAAAAGCG-3′
In total, 30 cycles of PCR were performed with approximately 10 ng of input DNA under the following conditions: 94°C for 45 s, 50°C for 30 s, and 72°C for 1 min. Amplified DNA was digested with EcoRI and XhoI. The EcoRI–XhoI fragments were cloned into the EcoRI and XhoI sites of a modified pBluescript vector and sequenced in their entirety.
Generation and characterization of lines stably expressing IQGAP1 fragments
To generate cell lines stably expressing Flag-tagged IQDN4, 3 × 106 Rat2 fibroblast cells were transfected with 3 μg of the wild-type IQDN4 (pcDNA4-Flag-IQDN4), or the empty vector (pcDNA4) using FuGENE 6 transfection reagent (Roche, Indianapolis, IN) and multiple clones selected and screened for IQDN4 expression by Western blot. MuLV viral vectors carrying selectable markers (Towers et al, 2000) were prepared by transfection of 293T cells using a combination of three expression vectors as described previously (Besnier et al, 2002). VSV-G expression vector (Naldini et al, 1996) was used to supply envelope for MuLV preparation. Throughout the text, viruses are named according to the origin of the Gag-Pol (N-MuLV or B-MuLV) and the neomycin or hygromycin reporter gene encoded within the packageable genome. Thus, an N-tropic or B-tropic MuLV particle carrying a neor gene is referred to as MuLV-N-neo or MuLV-B-neo, respectively. Target cells (1 × 105) were seeded in either six-well plates or 10-cm dishes 1 day before infection. All infections were performed in the presence of 8 μg/ml polybrene for 6 h. MuLV-neo or -hygro titer was measured by infection of cells and colony counting after selection in G418 (500 μg/ml) or hygromycin (200 μg/ml).
Generation of IQGAP1 and IQGAP2 knockdown lines by RNAi expression constructs
RNAi knockdown was performed using retroviral vectors encoding shRNAs. The viruses were generated by the cotransfection by Fugene 6 (Roche) of either pSR-hIQGAP1-Puro, pSR-hIQGAP2-Zeo, pSR-Puro, or pSR-Zeo cDNAs, and the vector plasmids, pCL-Eco and pMD.G, using a 5:5:1 ratio of the three plasmids in each transfection. All viruses were harvested 48 h post-transfection, filtered (0.45-μm filter, Pall Acrodisc), aliquoted, and stored at −80°C. Stocks were normalized by reverse transcriptase assay. Medium containing retrovirus vector pSR-hIQGAP1-Puro (encoding hairpins 46(1), 28(1), or 30(1)) and pSR-hIQGAP2-Zeo (encoding hairpins 53(2), 65(2), or 52(2)) was pelleted onto 293T cells (1200 g for 72 min) in 12-well plates. pSR-Puro and pSRZeo vectors were used to generate control cell lines. Infection was repeated 24 h later. Infected cells were selected with 1 μg/ml Puromycin or 50 μg/ml Zeocin.
Antibodies used to detect expression in the knockdown cell lines include: human IQGAP1 (Zymed/Invitrogen, Carlsbad, CA), IQGAP2 (Upstate Biotechnology, Charlottesville, VA), and β-actin (Sigma, St Louis, MO).
Amphotropic MuLV virus from a producer cell line (SL1439A), a kind gift of Alan Rein, NCI, was used to infect the knockdown cell lines. The culture supernatants were collected daily for 7 days and scored for RT activity.
Binding to MBP fusions in vitro
Amylose resin (NEB) was incubated with E. coli extract containing either MBP or MBP fusion proteins. Bound complexes were washed with Buffer A (200 mM NaCl, 50 mM Tris/HCl, 5 mM EDTA, and 5 mM DTT), and then the beads were subsequently incubated with E. coli extracts containing GST or GST fusion proteins. Bound complexes were washed with a modified Buffer A (500 mM NaCl, 50 mM Tris/HCl, 5 mM EDTA, 5 mM DTT, and 0.2% NP40). Beads were boiled in the presence of sample buffer and subjected to SDS–PAGE followed by immunoblot analysis with anti-MBP (Zymed/Invitrogen, Carlsbad, CA) and anti-GST (Covance Research Products, Berkeley, CA) monoclonal antibodies.
Coimmunoprecipitation
For Flag-IQGAP1 immunoprecipitation, 293T cells were transfected, harvested 72 h post-transfection, washed twice with cold phosphate-buffered saline (PBS), and then lysed with NP40 Buffer (150 mM NaCl, 0.5% NP40, 50 mM Tris, pH 8, 0.5 mM EDTA) and a protease inhibitor cocktail (Roche) at 4°C for 1 h. Clarified extracts were then incubated with Protein A and Protein G beads (Amersham Biosciences, Piscataway, NJ) plus either anti-HA (Roche, Indianapolis, IN) or anti-CA for 3 h, washed with NP40 buffer, boiled in sample buffer and subjected to SDS–PAGE and immunoblot analysis with anti-Flag (Sigma, St Louis, MO) and anti-CA antibodies.
Supplementary Material
Supplementary Information
Acknowledgments
We are grateful to Valmik K Vyas for helpful discussions and critical reading of early drafts. We also thank Barbara Studamire, Daniel Wolf, Mojgan Naghavi, Brian Houck-Loomis, and Lucia Nguyen for technical support and advice, as well as Jeremy Luban and Sarah Sebastian for RNAi reagents and helpful discussions. This work was supported by PHS grant R37 CA 30488 from the National Cancer Institute. AY is an Associate, and SPG is an Investigator of the Howard Hughes Medical Institute.
References
- Abramoff MD, Magelhaes PJ, Ram SJ (2004) Image processing with Image J. Biophoton Int 11: 36–42 [Google Scholar]
- Bacharach E, Gonsky J, Alin K, Orlova M, Goff SP (2000) The carboxy-terminal fragment of nucleolin interacts with the nucleocapsid domain of retroviral gag proteins and inhibits virion assembly. J Virol 74: 11027–11039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basyuk E, Galli T, Mougel M, Blanchard JM, Sitbon M, Bertrand E (2003) Retroviral genomic RNAs are transported to the plasma membrane by endosomal vesicles. Dev Cell 5: 161–174 [DOI] [PubMed] [Google Scholar]
- Besnier C, Takeuchi Y, Towers G (2002) Restriction of lentivirus in monkeys. Proc Natl Acad Sci USA 99: 11920–11925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breedon L, Nasmyth K (1985) Regulation of the HO gene. Cold Spring Harbor Symp Quant Biol 50: 643–650 [DOI] [PubMed] [Google Scholar]
- Campbell EM, Nunez R, Hope TJ (2004) Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J Virol 78: 5745–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicelli J, Goff SP (1988) Sequence and spacing requirements of a retrovirus integration site. J Mol Biol 199: 47–59 [DOI] [PubMed] [Google Scholar]
- Crawford S, Goff SP (1984) Mutations in gag proteins P12 and P15 of moloney murine leukemia virus block early stages of infection. J Virol 49: 909–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, Peters TR, Dermody TS, Woodruff E, Wang JJ, Spearman P (2005) AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell 120: 663–674 [DOI] [PubMed] [Google Scholar]
- Fukata M, Kuroda S, Fujii K, Nakamura T, Shoji I, Matsuura Y, Okawa K, Iwamatsu A, Kikuchi A, Kaibuchi K (1997) Regulation of cross-linking of actin filament by IQGAP1, a target for Cdc42. J Biol Chem 272: 29579–29583 [DOI] [PubMed] [Google Scholar]
- Fukata M, Kuroda S, Nakagawa M, Kawajiri A, Itoh N, Shoji I, Matsuura Y, Yonehara S, Fujisawa H, Kikuchi A, Kaibuchi K (1999) Cdc42 and Rac1 regulate the interaction of IQGAP1 with beta-catenin. J Biol Chem 274: 26044–26050 [DOI] [PubMed] [Google Scholar]
- Fukata M, Watanabe T, Noritake J, Nakagawa M, Yamaga M, Kuroda S, Matsuura Y, Iwamatsu A, Perez F, Kaibuchi K (2002) Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 109: 873–885 [DOI] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI (2001) Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107: 55–65 [DOI] [PubMed] [Google Scholar]
- Goff S, Traktman P, Baltimore D (1981) Isolation and properties of Moloney murine leukemia virus mutants: use of a rapid assay for release of virion reverse transcriptase. J Virol 38: 239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI (1996) Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci USA 93: 3099–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt B (1967) Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26: 365–369 [DOI] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51–59 [DOI] [PubMed] [Google Scholar]
- Kiernan RE, Ono A, Englund G, Freed EO (1998) Role of matrix in an early postentry step in the human immunodeficiency virus type 1 life cycle. J Virol 72: 4116–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Tang Y, Okada Y, Torrey TA, Chattopadhyay SK, Pfleiderer M, Falkner FG, Dorner F, Choi W, Hirokawa N, Morse HC III (1998) Binding of murine leukemia virus Gag polyproteins to KIF4, a microtubule-based motor protein. J Virol 72: 6898–6901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda S, Fukata M, Kobayashi K, Nakafuku M, Nomura N, Iwamatsu A, Kaibuchi K (1996) Identification of IQGAP as a putative target for the small GTPases, Cdc42 and Rac1. J Biol Chem 271: 23363–23367 [DOI] [PubMed] [Google Scholar]
- Kuroda S, Fukata M, Nakagawa M, Fujii K, Nakamura T, Ookubo T, Izawa I, Nagase T, Nomura N, Tani H, Shoji I, Matsuura Y, Yonehara S, Kaibuchi K (1998) Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin-mediated cell–cell adhesion. Science 281: 832–835 [DOI] [PubMed] [Google Scholar]
- Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W (2005) Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol 170: 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wang Q, Chakladar A, Bronson RT, Bernards A (2000) Gastric hyperplasia in mice lacking the putative Cdc42 effector IQGAP1. Mol Cell Biol 20: 697–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Kim SH, Higgins JM, Brenner MB, Sacks DB (1999) IQGAP1 and calmodulin modulate E-cadherin function. J Biol Chem 274: 37885–37892 [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Noritake J, Fukata M, Sato K, Nakagawa M, Watanabe T, Izumi N, Wang S, Fukata Y, Kaibuchi K (2004) Positive role of IQGAP1, an effector of Rac1, in actin-meshwork formation at sites of cell–cell contact. Mol Biol Cell 15: 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K (2005) IQGAP1: a key regulator of adhesion and migration. J Cell Sci 118: 2085–2092 [DOI] [PubMed] [Google Scholar]
- Ono A, Demirov D, Freed EO (2000) Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J Virol 74: 5142–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO (1999) Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol 73: 4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre P, Scheel J, Rickard JE, Kreis TE (1992) CLIP-170 links endocytic vesicles to microtubules. Cell 70: 887–900 [DOI] [PubMed] [Google Scholar]
- Rein A, McClure MR, Rice NR, Luftig RB, Schultz AM (1986) Myristylation site in Pr65gag is essential for virus particle formation by Moloney murine leukemia virus. Proc Natl Acad Sci USA 83: 7246–7250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh MD (2004) A myristoyl switch regulates membrane binding of HIV-1 Gag. Proc Natl Acad Sci USA 101: 417–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffel N, Harlos K, Iourin O, Rao Z, Kingsman A, Stuart D, Fry E (2002) Atomic resolution structure of Moloney murine leukemia virus matrix protein and its relationship to other retroviral matrix proteins. Structure (Camb) 10: 1627–1636 [DOI] [PubMed] [Google Scholar]
- Sodeik B (2000) Mechanisms of viral transport in the cytoplasm. Trends Microbiol 8: 465–472 [DOI] [PubMed] [Google Scholar]
- Soneoka Y, Kingsman SM, Kingsman AJ (1997) Mutagenesis analysis of the murine leukemia virus matrix protein: identification of regions important for membrane localization and intracellular transport. J Virol 71: 5549–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman P, Wang JJ, Vander Heyden N, Ratner L (1994) Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J Virol 68: 3232–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG (2003) AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114: 689–699 [DOI] [PubMed] [Google Scholar]
- Tang Y, Winkler U, Freed EO, Torrey TA, Kim W, Li H, Goff SP, Morse HC III (1999) Cellular motor protein KIF-4 associates with retroviral Gag. J Virol 73: 10508–10513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers G, Bock M, Martin S, Takeuchi Y, Stoye JP, Danos O (2000) A conserved mechanism of retrovirus restriction in mammals. Proc Natl Acad Sci USA 97: 12295–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, Nakagawa M, Izumi N, Akiyama T, Kaibuchi K (2004) Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell 7: 871–883 [DOI] [PubMed] [Google Scholar]
- Weissbach L, Bernards A, Herion DW (1998) Binding of myosin essential light chain to the cytoskeleton-associated protein IQGAP1. Biochem Biophys Res Commun 251: 269–276 [DOI] [PubMed] [Google Scholar]
- Weissbach L, Settleman J, Kalady MF, Snijders AJ, Murthy AE, Yan YX, Bernards A (1994) Identification of a human rasGAP-related protein containing calmodulin-binding motifs. J Biol Chem 269: 20517–20521 [PubMed] [Google Scholar]
- Yuan B, Li X, Goff SP (1999) Mutations altering the moloney murine leukemia virus p12 Gag protein affect virion production and early events of the virus life cycle. EMBO J 18: 4700–4710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Yu X, Lee TH, Essex M (1993) Mutations in the N-terminal region of human immunodeficiency virus type 1 matrix protein block intracellular transport of the Gag precursor. J Virol 67: 6387–6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Parent LJ, Wills JW, Resh MD (1994) Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J Virol 68: 2556–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information