

Abstract
The microtubule binding domain (MTBD) of dynein is separated from the AAA core of the motor by an ~15 nm stalk that is predicted to consist of an anti-parallel coiled coil. However, the structure of this coiled-coil and the mechanism it uses to mediate communication between the MTBD and ATP-binding core are unknown. Here, we sought to identify the optimal alignment between the hydrophobic heptad repeats in the two strands of the stalk coiled-coil. To do this, we fused the MTBD of mouse cytoplasmic dynein, together with 12-36 residues of its stalk, onto a stable coiled-coil base, provided by Thermus thermophilus seryl tRNA-synthetase, and tested these chimeric constructs for microtubule binding in vitro. The results identified one alignment that yielded a protein displaying high-affinity for microtubules (2.2 μM). The effects of mutations applied to the MTBD of this construct paralleled those previously reported (Koonce, M. P. and Tikhonenko, I. (2000) Mol. Biol. Cell 11, 523–529) for an intact dynein motor unit in the absence of ATP, suggesting that it resembles the tight-binding state of native intact dynein. All other alignments showed at least 10-fold lower affinity for microtubules with the exception of one, which had an intermediate affinity. Based on these results and on amino-acid sequence analysis, we hypothesize that dynein utilizes small amounts of sliding displacement between the two strands of its coiled-coil stalk as a means of communication between the AAA core of the motor and the MTBD during the mechanochemical cycle.
Dyneins are a family of microtubule based motor proteins consisting of 1-3 heavy chains (>500 kDa) along with assorted light and intermediate chains (1, 2). The cytoplasmic isoform is responsible for many types of movement within the cell, including transport of small vesicles and large organelles, mRNA and protein complexes, as well as formation of the mitotic spindle and separation of chromosomes (2, 3). Numerous other isoforms are found in axonemes where they produce the microtubule sliding motion that underlies the beating movement of cilia and flagella (1, 4, 5). In comparison to other motor proteins, such as kinesin and myosin, much less is known about the mechanism of the dynein family.
The motor activity of dynein is located in a ~380kD fragment (6, 7) at the C-terminus of the heavy chain. Sequence analysis indicates that this motor domain contains six AAA1 domains (8, 9), and electron microscopy reconstructions (10, 11) suggest that these six domains are arranged in a ring (Fig. 1). As originally predicted on the basis of vanadate-mediated photocleavage (12), the first AAA domain (AAA1) is the principal site of ATP hydrolysis (13). In addition, nucleotide hydrolysis is also required at AAA3 (14) and nucleotide binding at sites AAA2 and AAA4 may also contribute to dynein function (13–15). The sequences of the AAA5 and AAA6 domains have highly degenerate ATP-binding motifs, indicating that these domains are unlikely to bind nucleotide and they presumably serve primarily structural roles.
FIG. 1. Molecular structure of intact cytoplasmic dynein.
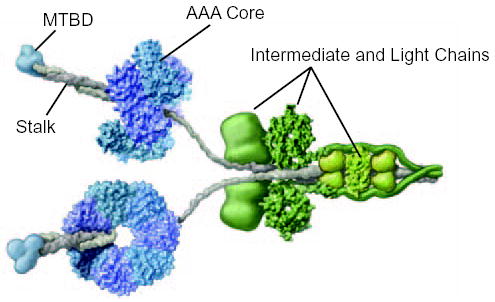
The cytoplasmic dynein motor is a dimer containing two identical heavy chain subunits of Mm ~520 kDa. The core of the motor, formed by the C-terminal two-thirds of the heavy chain, comprises a ring of six AAA ATPase domains, depicted here in blue and purple. The microtubule-binding domain (MTBD) (blue) protrudes from the AAA core on a coiled coil stalk (grey). The attachment of cargo to the dynein motor involves light and intermediate chain subunits (green) that are associated with the N-terminal third of the heavy chain. Adapted from (2) with permission.
Dynein is thought to bind microtubules by a small globular microtubule-binding domain (MTBD) located at the tip of a long (10–15 nm) and slender stalk (Fig. 1) (16). Thus, unlike myosins and kinesins that have their polymer binding sites on the surface of the catalytic core, the MTBD of dynein is well separated from the enzymatic sites. This raises the intriguing question of how transitions in the nucleotide hydrolysis cycle are communicated to the MTBD by the ATP-binding sites.
Sequence analysis of the dynein heavy chain reveals two stretches of predicted coiled coil (CC1 and CC2), that are located between AAA4 and AAA5, and separated by a small globular region of mass ~14 kDa (17–19). An expressed fragment of dynein containing this region of the heavy chain formed a rod-like structure similar to the stalk. The ~14 kDa globular region was identified as the MTBD by its ability to bind to microtubules (18) and confirmed by the demonstration that mutagenesis of conserved residues within the MTBD interferes with microtubule binding (20). More recently, a smaller expressed fragment, containing principally the MTBD, has been shown to decorate microtubules (21). However, the details of how CC1 and CC2 interact to form the stalk, as well as the mechanism by which the coiled-coil couples the dynein ATP hydrolysis cycle with changes in the affinity of the MTBD, remain unknown.
Here, we addressed these questions by making a series of chimeric constructs in which the different lengths of the dynein stalk are given a stable base by splicing them to a portion of the antiparallel coiled-coil side arm found in the native structure of seryl tRNA-synthetase (SRS). We hypothesised that the MTBD in these constructs would be able to bind microtubules if the dynein sequence emerging from the SRS was properly phased to align the two strands of the putative coiled-coil in their native conformation. Our microtubule-binding data, combined with sequence analysis, suggest that an anti-parallel coiled coil in the distal region of the stalk is required for tight microtubule binding and that sliding movements between the two strands of the coiled-coil structure constitute a possible mechanism for regulating the affinity of microtubule-binding during the ATPase cycle.
EXPERIMENTAL PROCEDURES
Construction of SRS-MTBD plasmids
DNA fragments encoding the MTBD of cytoplasmic dynein and desired lengths of its coiled-coil stalk were PCR-amplified from mouse testis cDNA (See-Gene, Inc.) using primers with in-frame SalI and HindIII restriction sites to facilitate cloning into the SalI and HindIII sites in a synthetic construct encoding a monomeric form of SRS (GenBank accession number AY881245). The resultant clones contained a continuous open reading frame encoding the SRS-MTBD chimeric protein with the MTBD and coiled-coil of the dynein fused between residues Leu33 and Lys93 of the native SRS coiled-coil side arm (Fig. 2).
FIG. 2. Design and construction of chimeric SRS-MTBD coiled coil.
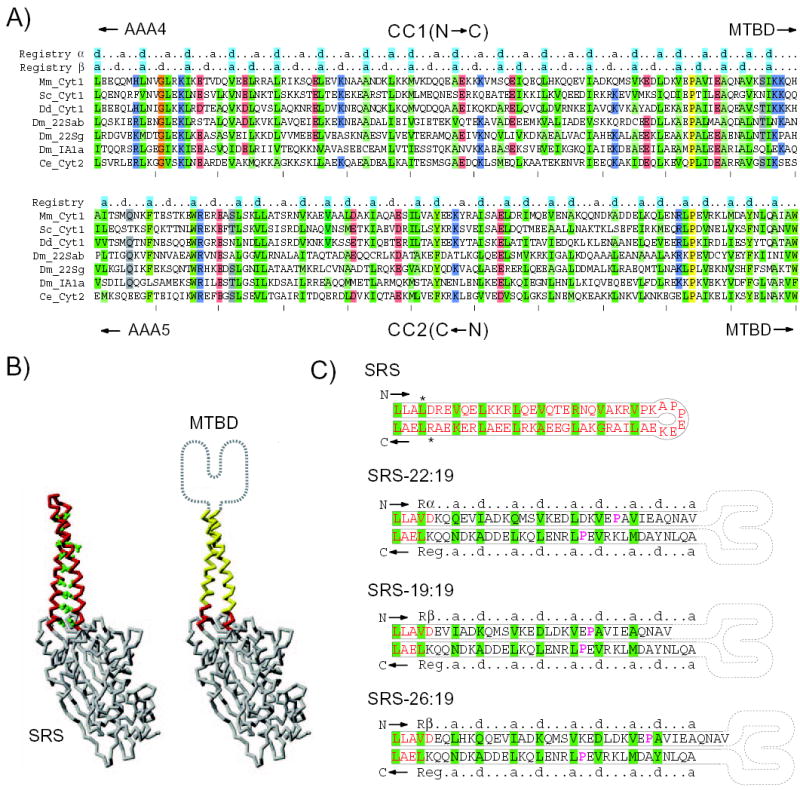
A, Sequence alignment of CC1 and CC2 region of dynein heavy chains. Species used are: mouse (Mm), Saccharomyces cerevisiae (Sc), Dictyostelium discoideum (Dd), Drosophila melanogaster (Dm), and Caenorhabditis elegans (Ce). Dynein isoforms used are: cytoplasmic (Cyt1), intra-flagellar transport (Cyt2), axonemal outer arm (22Sab and 22Sg), and axonemal inner arm (1A1). The sequences of CC2 are written in reverse order so that the ends of CC1 and CC2 adjoining the MTBD are both situated on the same (right) side of the alignment. In the mouse sequence (top line) the alignment shows CC1 residues from Leu3192 to His3301 and CC2 residues from Trp3393 to Ala3502. Positions in the alignment are shaded colours to indicate amino-acid type where the similarity is greater than 70%. The conserved residues Pro3285 and Pro3409 are highlighted yellow. Heptad repeats are indicated above the alignment: a, first position of heptad; d, fourth position of heptad. These heptad markers are shaded cyan at positions where there is >50% consensus of similar hydrophobic amino acids. The amino acids Ile, Leu, Val, Ala and Met are considered similar hydrophobic residues. B, Backbone trace of the SRS molecule used in SRS-MTBD constructs (PDB: 1SRY). Side chains of the amino acids forming the heptad repeat in the SRS coiled coil (left) are shown in green. In the SRS-MTBD cartoon (right), residues belonging to the SRS coiled-coil base are indicated in red and residues replaced by dynein are indicated in yellow. The MTBD located at the tip of the stalk is indicated schematically. C, Diagram showing the amino acid sequence of the coiled-coil stalk in native SRS and in 3 chimeric SRS-MTBD constructs. Asterisks adjacent to the SRS sequence indicate residues Leu33 and Arg93 that become mutated to Val and Lys, respectively, in order to generate SalI and HindIII restriction sites. The SRS-derived amino acids are indicated in red. Rα and Rβ α and β registries of CC1; Reg, registry of CC2. Amino acids comprising the “a” and “d” positions of the heptad repeats are highlighted in green. The conserved residues Pro3285 and Pro3409 are in magenta. The structure of the dynein MTBD is represented in cartoon form.
Bacterial expression of SRS-MTBD constructs
The SRS-MTBD chimeric protein was expressed in E. coli strain BL21(DE3) by induction with 0.5 mM IPTG for 4 hr at 16°C. After being washed into homogenisation solution (50 mM Na-phosphate, pH 8.0, 200 mM NaCl, 10% glycerol, 0.1% 2– mercaptoethanol, 0.5 mM PMSF), the bacteria were lysed using a French press, and the homogenate clarified by centrifugation. The expressed protein was purified by adsorption on Ni-beads and eluted with solution containing 50 mM Na-phosphate, pH 8, 200 mM NaCl, 10% glycerol, 0.1% 2– mercaptoethanol, 150–200 mM imidazole. The yield of purified protein was ~10 mg/(liter of culture) for most monomeric constructs. A sample of each expression culture was processed for nucleotide sequencing in order to confirm its identity. Gel electrophoresis of typical preparations showed that the SRS-MTBD was ~95% pure. In typical monomeric preparations, ~90% of the protein eluted as a single symmetrical peak upon size-exclusion chromatography (Supplemental Data). Some preparations were further purified by chromatography on a Superdex 200 column.
Microtubule binding assay
Polymerized bovine or porcine brain microtubules were pelleted by centrifugation and resuspended gently in assay buffer (50 mM Tris-Cl pH 8, 50 mM potassium acetate, 2 mM MgCl2, 1 mM EGTA, 1 mM DTT, 10 μM paclitaxel and 10% glycerol) at 2.0 mg/ml. Samples of SRS-MTBD were precipitated with 40% saturated (NH4)2SO4 and redissolved in assay buffer at 2.0 mg/ml. Proteins concentrations were determined from the A280 of aliquots diluted into 6M guanidine HCl, by assuming extinction coefficients of 1.04 and 1.15 (mg/ml)−1 cm−1 for SRS-MTBD and tubulin, respectively. Binding of SRS-MTBD was assayed by incubating appropriate concentrations of SRS-MTBD with a suspension of microtubules (5 μM dimeric tubulin) in assay buffer for 15 min at room temperature. After centrifugation for 10 min at 200,000 × g, the pellet was rinsed before resuspension in an equal volume of buffer. Samples of the uncentrifuged suspension and of the supernatant and pellet fractions were analysed by gel electrophoresis. The intensities of Coomassie-stained bands corresponding to SRS-MTBD and tubulin were quantified by digital scanning and integration with the IBM-PC version of NIH Image (Scion Corp). The fraction of total SRS-MTBD that co-sedimented with microtubules was computed for each sample and used to calculate values for the unbound SRS-MTBD concentration and the microtubule occupancy. Values of the binding affinity and of the saturation binding level were calculated by using the program Prism 4 (GraphPad Software Inc.) to fit a single-site binding hyperbola to a plot of microtubule occupancy as a function of unbound SRS-MTBD concentration. In the fitting of the plot, points were weighted by 1/(microtubule occupancy) to compensate for the magnified effect of small errors at high occupancy levels. Control experiments showed that <1% of a 20 μM solution of unmodified monomeric SRS, containing no MTBD, bound to microtubules under the conditions of the assay (data not shown).
Throughout this paper, numbers used to identify particular amino-acids refer to positions in the full-length sequence of cytoplasmic dynein heavy chain from mouse (GenBank accession number AAF91078).
RESULTS
Sequence Analysis of the Dynein MTBD Stalk
To gain insight into the structure of the dynein stalk, we first analysed the sequence of this region. While prediction programs clearly identify coiled-coil heptad repeats (hydrophobic residues in the first “a” and fourth “d” positions of a seven position repeat) in CC1 and CC2, the length of coiled-coil and the residues belonging to the heptad repeat are difficult to establish with certainty in individual dynein sequences (22). We therefore carried out an alignment of stalk regions from a wide variety of dyneins to determine whether a conserved heptad motif would emerge in CC1 and CC2. The alignment of a subset of dyneins is shown in Fig. 2A (for complete alignment see Supplemental Data). The alignment reveals that the sequence and length of CC1 and CC2 are strongly conserved, although the precise boundaries of an anti-parallel coiled-coil remain hard to predict. A pair of conserved proline residues is present close to the junction between the stalk and the MTBD (23); however, the sequence alignment shows that the heptad repeat continues past these residues. At the basal end of the stalk, adjacent to AAA4 and AAA5, the boundary of the coiled coil is again difficult to define from the sequence.
We also attempted to decipher the phasing of the heptad repeat in CC1 and CC2. In the case of CC2, the alignment shows a pattern of conserved “a” and “d” position hydrophobic residues that create a continuous registry of heptad repeats over the full length of the stalk (Fig. 2A). CC1 shows a different pattern in which conserved hydrophobic residues repeat only once per seven amino acids over most of stalk length. Thus, unlike CC2, CC1 appears to contain just one, rather than two, conserved hydrophobic residues every heptad. This pattern can be fit into two equally probable registries for the phasing of the heptad (Fig. 2A, registries α and β). As a result of the ill-defined boundaries of the putative coiled coil and the ambiguity in the phasing of CC1, it is difficult to predict the structure of the stalk (i.e. which amino acids in CC1 pack against those in CC2 to form the core of the coiled coil).
In Vitro Microtubule Binding of Dynein MTBD-SRS Fusion Proteins
In order to investigate the structure of the stalk, we designed a series of chimeric constructs in which the MTBD along with a portion of its predicted coiled-coil stalk is fused onto a stable anti-parallel coiled-coil base. We hypothesised that these constructs would bind microtubules tightly if the heptad repeats in the two strands of the dynein portion were in correct phase with those in the coiled-coil base. For the base coiled coil, we chose a monomeric form of the seryl tRNA-synthetase (SRS) from Thermus thermophilus, a thermostable protein with a known atomic level structure (Fig. 2B) (24).
In a first set of constructs, we maintained the heptad repeat of CC2 in synchronous phase with that of the SRS by keeping the number of dynein residues in CC2 constant at 19, while the corresponding number of residues in CC1 was varied from 15 to 29 (all lengths are counted from the conserved residues Pro3285 and Pro3409). This synchronisation of phasing is illustrated in Fig. 2C which shows the coiled-coil sequences of native SRS and of three chimeric constructs, SRS-22:19, SRS-19:19, and SRS-26:19. Assays comparing the relative abilities of these constructs to bind microtubules indicate that they separate into three groups: a high level of binding in SRS-22:19; an intermediate level of binding in SRS-19:19; and only low levels of binding in all the other constructs of the set (Figs. 3A, C). More detailed assays to quantify the affinity of microtubule binding in SRS-22:19, SRS-19:19 and three representative examples of the weak-binding constructs are shown in Figures 3A, B. In three independent preparations of the strong-binding construct SRS-22:19, the binding affinity averaged 2.2 ± 0.2 μM, with 57 ± 3 percent of the dimeric tubulin sites in the microtubule appearing occupied at saturation. In preparations of SRS-19:19, the binding affinity averaged 12.1 ± 1.5 μM with a saturation binding of 60 ± 4 percent of the microtubule sites. The apparent saturation of SRS-22:19 binding when approximately half of the binding sites become occupied may be due to steric interference from the SRS component of the construct. In the weak binding constructs, the available data were inadequate to determine saturation binding levels, but constraining this level to be equal to that of SRS-22:19 yielded average binding affinities of 26 - 62 μM for SRS-15:19, SRS-21:19 and SRS-23:19.
FIG. 3. Binding of chimeric SRS-MTBD constructs to microtubules.
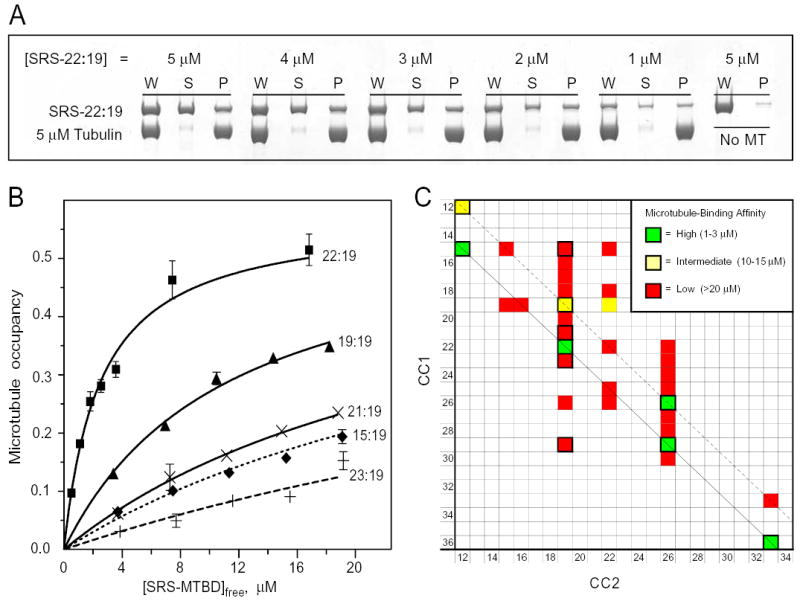
A, Polyacrylamide electrophoresis gels showing the binding of different concentrations of SRS-22:19 to microtubules. Binding was assayed by co-sedimentation after incubation with a suspension of 5 μM microtubules and the indicated concentrations of SRS-22:19 for 15 min at room temperature. See Methods for assay details. W, uncentrifuged sample, S, supernatant; P, pellet. A parallel sample of SRS-22:19 incubated and centrifuged with no microtubules was used as a blank (typically 1–2%). Recovery of SRS-MTBD and tubulin averaged (94 ± 10)%. B, Microtubule binding affinity of SRS-MTBD constructs with a fixed CC2 length of 19 amino acids and different lengths of CC1. Assays and gel electrophoresis were performed essentially as in Fig. 3A. Error bars indicate standard error of 2 replicate gels of the same microtubule-binding assay. Averages of affinity data from multiple independent preparations are given in text. C, Affinity of microtubule binding by SRS-MTBD constructs with different lengths of CC1 and CC2. The affinity of 33 constructs with CC1/CC2 lengths ranging from 12 to 36 amino acids was classified as high, medium or low by comparing the fraction of SRS-MTBD bound to microtubules in side-by-side assays with one or more of the constructs whose affinity had been assayed in detail as shown in Fig 3B. Assays were performed with 3 μM and 10 μM SRS-MTBD with 5 μM microtubules. The shortest constructs, SRS-12:12 and SRS-15:12, were less stable than the others and assays involving them have been corrected for presence of a non-binding aggregated form. Diagonal lines indicate constructs related to SRS-22:19 (solid black) and SRS-19:19 (dashed black). Cells with thick black border indicate constructs for which Rs has been determined (Supplemental data).
In order to study a broader range of alignments between CC1 and CC2, we prepared additional constructs in which the lengths of both CC1 and CC2 were varied. The results in Fig. 3C reveal the presence of a series of high-affinity constructs, SRS-15:12, SRS-22:19, SRS-29:26, and SRS-36:33, the members of which are all related to SRS-22:19 by a change of one or more full heptads in the lengths of both CC1 and CC2. The constructs in this series all have high levels of affinity for binding to microtubules, close or identical to that of SRS-22:19 itself. In a second series of constructs that are similarly related to SRS-19:19, the binding affinities appear substantially less uniform, with only that of SRS-12:12 being comparable to that of SRS-19:19, whereas the affinity of SRS-26:26 is as high as that of SRS-22:19 and that of SRS-33:33 is down within the range of the other low-affinity constructs. Apart from the constructs in these two series, none of the other constructs examined had more than a low affinity for binding to microtubules. A change of as little as a single residue added to or subtracted from the length of CC1 in SRS-22:19 or SRS-29:26 was sufficient to reduce the binding affinity by more than 10-fold. Moreover, constructs such as SRS-26:19, SRS-29:19 and SRS-15:19 (Figs. 2C, 3C) that were formed by adding or subtracting either one-half or one- full heptad to CC1 while keeping CC2 unchanged showed a similarly low affinity for microtubules. These results demonstrate that the affinity of the MTBD for binding to microtubules is very sensitive to the configuration of the coiled-coil residues in the adjacent region of stalk.
It is notable that the series of constructs related to SRS-22:19 all have their CC2 registry aligned with registry α of CC1, while the series of constructs related to SRS-19:19 all have their CC2 registry aligned with registry β of CC1 (Figs. 2A, C), so that the members of both series have the potential to form a well-structured coiled coil in the stalk. In summary, we have identified the alignment of CC1 and CC2 in constructs related to SRS-22:19 as stabilizing a strong binding conformation of the MTBD and the corresponding alignment of SRS-19:19 as stabilizing a second moderate–high affinity conformation. All other alignments tested had only a low affinity for microtubules, regardless of whether they were predicted to be favourable (SRS-15:19, SRS-26:19, SRS-29:19, SRS-22:26) or unfavourable (all other constructs outside the two series) for coiled-coil formation.
Structural characterization of high- and low-affinity MTBD constructs
In order to examine whether the observed affinity differences were associated with local or global structural changes in the MTBD, we used gel-filtration chromatography to determine the Stokes radius (Rs) of a selection of ten SRS-MTBD constructs. The values of Rs obtained increased gradually with stalk length, from 3.7 nm for the shortest construct (SR-12:12) up to 4.2 nm for the longest (SRS-36:33). No detectable variation in Rs was observed to correlate with the affinities of the different constructs (Supplemental data). These data suggest that the different affinities observed are the result of local conformational changes in the vicinity of the microtubule binding site rather than being due to a global disruption of the MTBD structure. This conclusion is further supported by the fact that both high- and low-affinity constructs appeared almost identical in their pattern and speed of fragmentation upon limited digestion with trypsin (data not shown). However, we cannot exclude the possibility that such large-scale effects as twisting or bending of the stalk may play a role by influencing the steric accessibility of the microtubule-binding site on an SRS-MTBD construct to a microtubule in some cases.
Effect of Point Mutants in the Globular MTBD on Microtubule Binding Affinity
To test the hypothesis that the MTBD in SRS-22:19 represents a tight-binding conformation similar to that occurring in native dynein, we examined the effects of mutations that have previously been shown (20) to modulate the affinity of an intact dynein motor domain in the absence of ATP (Fig. 4). The mutation E3289A (corresponding to E3370A in Dictyostelium dynein), which increases the affinity of intact dynein for microtubules in the absence of ATP, increases the amounts of SRS-22:19 bound to microtubules by 37%. Similarly, the mutations E3304A, and R3382A (E3385A, R3464A in Dictyostelium dynein), as well as the double mutation K3298A K3299A (K3379A, K3380A in Dictyostelium dynein), which all inhibit the binding of intact dynein to microtubules in the absence of ATP, inhibit the binding of SRS-22:19 to microtubules by 60–85%. We also examined the effects of the above mutations applied to SRS-19:19 and found that they qualitatively paralleled that of the same mutation applied to SRS-22:19 (Fig. 4). Collectively, these results show that the effect of each of the four mutations on microtubule binding of SRS-22:19 paralleled the effect of the same mutation on the binding of the intact dynein motor to microtubules in the absence of ATP (20). The data strongly suggest that the conformation of the MTBD in SRS-22:19 represents the native tight-binding conformation of the MTBD of intact dynein in the absence of ATP and that its high affinity is not an artefact of the chimeric construct. This conclusion is further supported by the finding that the values of Rs for the above mutated forms of SRS-22:19 are identical to those of the wild-type construct (data not shown), as well as by the fact that the binding affinity of wild-type SRS-22:19 (2.2 μM) is similar to that reported previously (21) for a small (~14 kDa) fragment of yeast dynein that decorates microtubules (1.6 μM).
FIG. 4. Effect of site-directed MTBD mutations on microtubule binding by SRS-MTBD constructs.
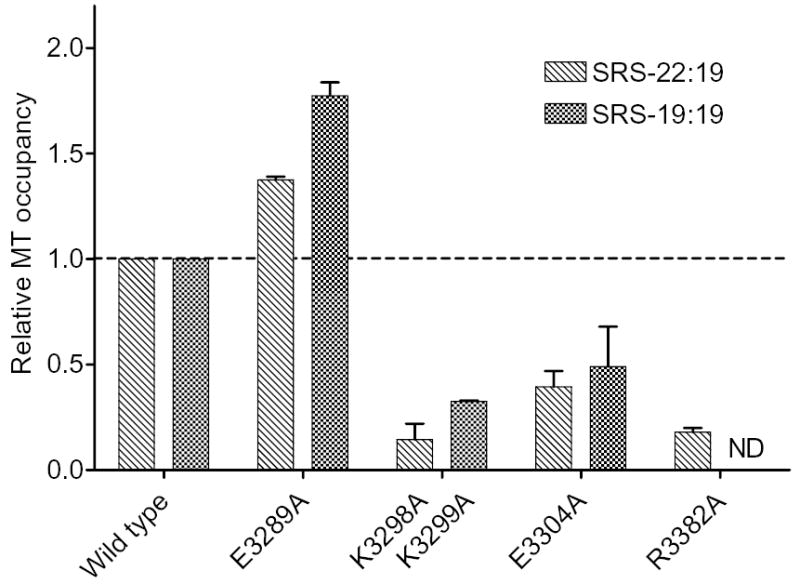
The microtubule occupancy of SRS-22:19 and SRS-19:19 constructs containing the indicated MTBD mutations is shown relative to that of corresponding constructs containing the wild-type MTBD. Microtubule occupancy was determined in side-by-side assays of mutant and wild-type constructs containing 3 μM SRS-22:19 or 10 μM SRS-19:19 with 5 μM microtubules. Bar heights show mean of 2 independent preparations for each mutated SRS-MTBD construct. Error bars show standard errors. ND, no data.
DISCUSSION
Our results demonstrate that the affinity of the MTBD for binding to microtubules is very sensitive to the configuration of the putative coiled-coil residues in the adjacent region of the stalk. Two factors in the structure of this region appear to be important for stabilizing a high-affinity conformation of the MTBD. One is a requirement for a well-structured coiled-coil in the region of the stalk adjacent to the MTBD. The second is a requirement for a correct alignment between the two strands of the coiled coil, CC1 and CC2.
The stringency of the requirement for a coiled-coil structure is indicated by the small number of sharply-defined stalk configurations that appear able to stabilize a high-affinity conformation of the MTBD. In the series of high-affinity constructs related to SRS-22:19, the phasing of the hydrophobic heptad register passes smoothly from its establishment in the SRS base over the length of CC1 and CC2 to the junction of the stalk with the MTBD. Changes in the length of CC1 that interrupt the continuity of the heptad phasing by as little as one residue cause a greater than 10-fold reduction in microtubule-binding affinity, whereas changes that maintain the heptad phasing by addition or subtraction of one or more full heptads to the lengths of both CC1 and CC2, as in SRS-29:26, SRS-36:33 and SRS-15:12, form a series of uniformly high affinity. The functional importance of an uninterrupted heptad repeat is supported by sequence alignments showing that the regularity of this repeat is highly conserved in the stalks of axonemal and cytoplasmic dyneins (Fig. 2A).
However, the presence of a continuous heptad repeat is not by itself sufficient to stabilize a high affinity conformation in the MTBD. It is also necessary that the alignment of CC1 and CC2 be correct. For example, in SRS-15:19 and SRS-29:19, the CC2 registry is aligned with the α-registry of CC1, the same as in SRS-22:19, yet these constructs have only a low binding affinity (Figs. 3B, C). The low affinity of these constructs suggests that a correct spatial relationship between coiled-coil residues at the top of the stalk and residues in the MTBD is an essential second requirement for high-affinity binding to microtubules.
A particularly intriguing outcome of our microtubule-binding data is that CC1 can potentially adopt a second position relative to CC2, corresponding to SRS-19:19 and the series of constructs related to it, in which the CC2 registry is aligned with the β-registry of CC1. Structural models of the dynein stalk coiled-coil in the 22:19 and 19:19 configurations (Supplemental data) illustrate the relationship between the two structures. In the 22:19 model, the hydrophobic residues repeating every seven amino acids in CC1 (Fig. 2A) pack against one side of CC2, whereas in the 19:19 structure they pack to the other side (Fig. 5A). In both cases, the side of the hydrophobic core opposite from these hydrophobic residues of CC1 is occupied by hydrophilic amino acids (Fig. 5B). The models also raise the possibility that the stalk could undergo a sliding transition, either between the 22:19 and 19:19 structures by a sliding of the hydrophobic residues in CC1 along the hydrophobic groove in CC2 (Fig. 5) or, possibly, by sliding in the opposite direction along the rather shallower grove between their positions in the structures of 22:19 and 26:19.
FIG. 5. Structural models of the dynein stalk.
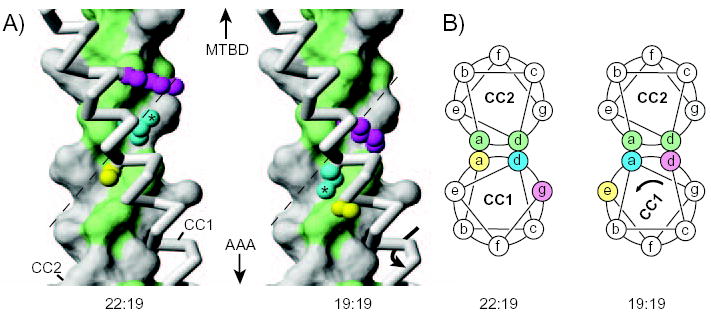
A, Models of a section of the dynein stalk in configurations expected to correspond to those occurring in SRS-22:19 or SRS19:19. CC2 is shown in surface representation with hydrophobic residues (Val, Ile, Leu, Ala, Met, Tyr) in the coiled-coil core (“a” and “d” positions) coloured green. CC1 is shown in backbone representation (grey), with side chains included for Ser3224 (yellow), Leu3227 (cyan; with asterisk) and Lys3230 (magenta). In passing from the configuration of SRS-22:19 to that of SRS-19:19, the side chain of Leu3227 shifts from packing against one side of CC2 (left) to packing against the other side (right), potentially by following the hydrophobic-lined groove, formed by Ile3459, Leu3463, Val 3466 and Val 3470, in the surface of CC2 (dashed line). Similar hydrophobic grooves in the core interface of CC2 occur in most other heptads along the length of the stalk coiled coil (Supplemental Data). B, Cartoons depicting transverse sections through the coiled-coil stalk in the region shown in Fig. 5A. Residues in CC1 are coloured as in Fig. 5A to illustrate that Leu3227 (cyan) shifts from an “a” heptad position to a “d” position in going between the 22:19 and 19:19 configurations (see also Fig. 2A). Part A of this figure was prepared with the program MOLMOL (28).
Our results strongly suggest that changes in the configuration of the dynein stalk are able to modulate the affinity of the MTBD. However, a remaining substantial question concerns the nature of the coiled-coil stalk transition that propagates the information for a state change over the 15 nm length of the stalk, between the AAA motor unit at one end and the MTBD at the other. One possible model is that the transition involves a small sliding movement, similar to the 1.5 Å displacement simulated by removal of one residue from CC1, which might generate a low-affinity binding state by distorting the coiled-coil structure in the stalk region adjacent to the MTBD in a manner similar to that occurring in our SRS-21:19 and SRS-23:19 constructs. However, this model appears to lack a mechanism for propagating the distortion of an ~1.5 Å displacement over the full length of the intact stalk. A more probable model, indirectly supported by our data, involves CC1 sliding by half a heptad (~4.5 Å axial displacement with ~58º rotation) relative to CC2 as shown in Fig. 5. In this model, the transition between the ADP/no nucleotide (tight-binding) and the ATP/ADP-Pi (weak-binding) states of dynein (25) would correspond to a shift from a 22:19-like to either a 19:19-like or possibly a 26:19-like configuration of the stalk. In either case, the change in registry could be mediated by the AAA4 domain of the catalytic core pulling and pushing on CC1, in a manner similar to that involved in the mechanical functioning of other AAA ATPases (26, 27).
Acknowledgments
IRG thanks Dr. Beth Burnside for so generously accommodating him within her research space at the University of California Berkeley. We thank Dr. Barbara Gibbons for her careful reading of the manuscript, Dr Nico Stuurman for initial help with the sequence analysis and Dr. Eva Nogales for the use of the SMART chromatography equipment in her laboratory.
Footnotes
This work was supported by the National Institutes of Health, Grants GM30401, P01-AR42895-10 and Postdoctoral Fellowship GM67403-01.
The on-line version of this article is available at http://www.jbc.org.
The nucleotide sequences reported in this paper have been submitted to the GenBank/EBI Data Bank with accession numbers AY881245, and AY870659.
The atomic coordinates and structure factors (codes 2BOR, and 2BOT) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The abbreviations used are: AAA, ATPase with any other activity; CC1 and CC2, coiled coil strands on the N-terminal and C-terminal sides, respectively, of the dynein microtubule-binding domain; DTT, dithiothreitol; MTBD, microtubule-binding domain; Rs, Stokes radius; SRS, seryl tRNA-synthetase; SRS-MTBD, chimeric construct of SRS with MTBD.
References
- 1.Gibbons IR. Cell Motil Cytoskeleton. 1995;32:136–144. doi: 10.1002/cm.970320214. [DOI] [PubMed] [Google Scholar]
- 2.Vale RD. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 3.Vallee RB, Williams JC, Varma D, Barnhart LE. J Neurobiol. 2004;58:189–200. doi: 10.1002/neu.10314. [DOI] [PubMed] [Google Scholar]
- 4.Porter ME, Sale WS. J Cell Biol. 2000;151:F37–F42. doi: 10.1083/jcb.151.5.f37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamiya R. Int Rev Cytol. 2002;219:115–155. doi: 10.1016/s0074-7696(02)19012-7. [DOI] [PubMed] [Google Scholar]
- 6.Koonce MP, Samso M. Mol Biol Cell. 1996;7:935–948. doi: 10.1091/mbc.7.6.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishiura M, Kon T, Shiroguchi K, Ohkura R, Shima T, Toyoshima YY, Sutoh K. J Biol Chem. 2004;279:22799–22802. doi: 10.1074/jbc.M313362200. [DOI] [PubMed] [Google Scholar]
- 8.Neuwald AF, Aravind L, Spouge JL, Koonin EV. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 9.Mocz G, Gibbons IR. Structure (Camb) 2001;9:93–103. doi: 10.1016/s0969-2126(00)00557-8. [DOI] [PubMed] [Google Scholar]
- 10.Burgess SA, Walker ML, Sakakibara H, Oiwa K, Knight PJ. J Struct Biol. 2004;146:205–216. doi: 10.1016/j.jsb.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Samso M, Koonce MP. J Mol Biol. 2004;340:1059–1072. doi: 10.1016/j.jmb.2004.05.063. [DOI] [PubMed] [Google Scholar]
- 12.Gibbons IR, Lee-Eiford A, Mocz G, Phillipson CA, Tang WJY, Gibbons BH. J Biol Chem. 1987;262:2780–2786. [PubMed] [Google Scholar]
- 13.Kon T, Nishiura M, Ohkura R, Toyoshima YY, Sutoh K. Biochemistry. 2004;43:11266–11274. doi: 10.1021/bi048985a. [DOI] [PubMed] [Google Scholar]
- 14.Reck-Peterson SL, Vale RD. Proc Natl Acad Sci U S A. 2004;101:1491–1495. doi: 10.1073/pnas.2637011100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Takahashi Y, Edamatsu M, Toyoshima YY. Proc Natl Acad Sci U S A. 2004;101:12865–12869. doi: 10.1073/pnas.0403429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodenough U, Heuser J. J Mol Biol. 1984;180:1083–1118. doi: 10.1016/0022-2836(84)90272-9. [DOI] [PubMed] [Google Scholar]
- 17.Gibbons IR, Gibbons BH, Mocz G, Asai DJ. Nature. 1991;352:640–643. doi: 10.1038/352640a0. [DOI] [PubMed] [Google Scholar]
- 18.Gee MA, Heuser JE, Vallee RB. Nature. 1997;390:636–639. doi: 10.1038/37663. [DOI] [PubMed] [Google Scholar]
- 19.Koonce MP. J Biol Chem. 1997;272:19714–19718. doi: 10.1074/jbc.272.32.19714. [DOI] [PubMed] [Google Scholar]
- 20.Koonce MP, Tikhonenko I. Mol Biol Cell. 2000;11:523–529. doi: 10.1091/mbc.11.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizuno N, Toba S, Edamatsu M, Watai-Nishii J, Hirokawa N, Toyoshima YY, Kikkawa M. EMBO J. 2004;23:2459–2467. doi: 10.1038/sj.emboj.7600240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gee MA, Heuser JE, Vallee RB. Nature. 1997;390:636–639. doi: 10.1038/37663. [DOI] [PubMed] [Google Scholar]
- 23.Gee M, Vallee R. Eur Biophys J. 1998;27:466–473. doi: 10.1007/s002490050157. [DOI] [PubMed] [Google Scholar]
- 24.Biou V, Yaremchuk A, Tukalo M, Cusack S. Science. 1994;263:1404–1410. doi: 10.1126/science.8128220. [DOI] [PubMed] [Google Scholar]
- 25.Porter ME, Johnson KA. Annu Rev Cell Biol. 1989;5:119–151. doi: 10.1146/annurev.cb.05.110189.001003. [DOI] [PubMed] [Google Scholar]
- 26.Vale RD. J Cell Biol. 2000;150:F13–F20. doi: 10.1083/jcb.150.1.f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogura T, Wilkinson AJ. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 28.Koradi R, Billeter M, Wuthrich K. J Mol Graph. 1996;51:25–33. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]