

Abstract
Although it is presynaptic, short-term plasticity has been shown at some synapses to depend upon the postsynaptic cell type. Previous studies have reported conflicting results as to whether Schaffer collateral axons have target-cell specific short-term plasticity. Here we investigate in detail the short-term dynamics of Schaffer collateral excitatory synapses onto CA1 stratum radiatum interneurones versus pyramidal cells in acute hippocampal slices from juvenile rats. In response to three stimulus protocols that invoke different forms of short-term plasticity, we find differences in some but not all forms of presynaptic short-term plasticity, and heterogeneity in the short term plasticity of synapses onto interneurones. Excitatory synapses onto the majority of interneurones had less paired-pulse facilitation than synapses onto pyramidal cells across a range of interpulse intervals (20–200 ms). Unlike synapses onto pyramidal cells, synapses onto most interneurones had very little facilitation in response to short high-frequency trains of five pulses at 5, 10 and 20 Hz, and depressed during trains at 50 Hz. However, the amount of high-frequency depression was not different between synapses onto pyramidal cells versus the majority of interneurones at steady state during 2–10 Hz trains. In addition, a small subset of interneurones (approximately 15%) had paired-pulse depression rather than paired-pulse facilitation, showed only depression in response to the high-frequency five pulse trains, and had more steady-state high-frequency depression than synapses onto pyramidal cells or the majority of interneurones. To investigate possible mechanisms for these differences in short-term plasticity, we developed a mechanistic mathematical model of neurotransmitter release that explicitly explores the contributions to different forms of short-term plasticity of the readily releasable vesicle pool size, release probability per vesicle, calcium-dependent facilitation, synapse inactivation following release, and calcium-dependent recovery from inactivation. Our model fits the responses of each of the three cell groups to the three different stimulus protocols with only two parameters that differ with cell group. The model predicts that the differences in short-term plasticity between synapses onto CA1 pyramidal cells and stratum radiatum interneurones are due to a higher initial release probability per vesicle and larger readily releasable vesicle pool size at synapses onto interneurones, resulting in a higher initial release probability. By measuring the rate of block of NMDA receptors by the open channel blocker MK-801, we confirmed that the initial release probability is greater at synapses onto interneurones versus pyramidal cells. This provides a mechanism by which both the initial strength and the short-term dynamics of Schaffer collateral excitatory synapses are regulated by their postsynaptic target cell.
The strength of an excitatory synapse in the brain is dynamically modulated by the pattern of activation it receives. Input patterns are transformed by presynaptic mechanisms of short-term plasticity, enabling information processing to occur (Zador & Dobrunz, 1997). As a result, synapses can act as frequency filters, novelty detectors and/or pattern detectors (Thomson, 2000). Variations in presynaptic properties enable synapses to be differentially ‘tuned’ depending upon their particular roles in the overall circuit. Synapses that are dynamically tuned by their patterns of prior activation have considerable power to process information (Liaw & Berger, 1996; Maass & Zador, 1999).
Although it is mediated by presynaptic mechanisms, short-term plasticity at synapses made by axons from the same cell type onto target neurones of different types can exhibit markedly different properties of short-term plasticity. This has been observed at the neuromuscular junction, synapses in invertebrates, synapses onto motor neurones, and some synapses in the mammalian neocortex (reviewed in Toth & McBain, 2000). This indicates that there must be a retrograde signal to the presynaptic terminal, although the nature of the signal has not yet been determined (Fitzsimonds & Poo, 1998). Because the properties of excitatory synapses can be both target specific and input specific, understanding the behaviour of a neural circuit such as the hippocampus requires a detailed description of the properties of each type of synapse in the circuit (Toth & McBain, 2000; Craig & Boudin, 2001).
At excitatory synapses onto hippocampal CA1 pyramidal cells, short-term plasticity causes synaptic strength to be modulated over a wide range in response to irregular stimulus patterns such as they receive in vivo (Dobrunz & Stevens, 1999). Much less is known, however, about short-term plasticity and frequency dependence of synaptic transmission at excitatory synapses onto CA1 interneurones. Like CA1 pyramidal cells, stratum (s.) radiatum interneurones in CA1 receive inputs from CA3 pyramidal cells via Schaffer collateral axons. However, the role of these interneurones in the hippocampal circuit is quite different, in that they provide critical feed-forward inhibition that can synchronize the firing of pyramidal cells (Cobb et al. 1995). Because each s. radiatum interneurone forms inhibitory synapses onto a very large number of CA1 pyramidal cells (Freund & Buzsaki, 1996), the short-term dynamics of their excitatory inputs will be fundamental in determining the overall balance of excitation and inhibition in the hippocampal circuit.
Previous studies have reported conflicting results as to whether Schaffer collateral axons show target-cell specificity of short-term plasticity onto CA1 pyramidal cells versus interneurones. Differences in paired-pulse plasticity were observed between Schaffer collateral inputs onto pyramidal cells versus s. oriens interneurones in CA1 (Scanziani et al. 1998). In contrast, another study reported no difference between Schaffer collateral inputs to CA1 pyramidal cells versus interneurones in either the amount of paired-pulse facilitation or in the plateau levels reached during short trains of stimuli (Wierenga & Wadman, 2003).
We investigated in detail the short-term dynamics of Schaffer collateral excitatory synapses onto CA1 s. radiatum interneurones versus pyramidal cells using acute hippocampal slices from juvenile rats. Using three different stimulus protocols, we show that there are differences in some but not all forms of presynaptic short-term plasticity, and that significant heterogeneity exists in the short-term plasticity of synapses onto interneurones. To investigate possible mechanisms for these differences in short-term plasticity, we developed a mathematical analysis of neurotransmitter release that fits the results of all three stimulus protocols. Our model incorporates features of several previous models (Tsodyks et al. 1998; Dittman et al. 2000), but extends these models to explicitly explore the contributions of changes in synaptic vesicle number and in calcium dynamics to different forms of short-term plasticity (Dobrunz & Stevens, 1997; Dobrunz, 2002). Results of our model suggest that the differences in short-term plasticity between synapses onto CA1 pyramidal cells and s. radiatum interneurones can be accounted for by a higher initial release probability per vesicle and larger readily releasable vesicle pool size at synapses onto interneurones. This results in a higher initial release probability at synapses onto interneurones versus pyramidal cells, which we confirmed using the MK-801 method (Huang & Stevens, 1997). By modulating the readily releasable vesicle pool and release probability per vesicle, both the initial strength and the short-term dynamics of Schaffer collateral excitatory synapses can be regulated by the postsynaptic target cell.
Methods
Slice preparation
Coronal slices 400 μm thick were cut using an oscillating tissue slicer (EMS-4000, Electron Microscopy Sciences, Fort Washington, PA, USA) from dorsal hippocampus of 11- to 15-day-old Long Evans rats (Dobrunz & Stevens 1997, 1999). Animals were deeply anaesthetized by inhalation of the volatile anaesthetic halothane (2-bromo-2-chloro-1,1,1-trifluoroethane, 0.2–0.4 ml in a 2 l container) and then decapitated using a guillotine. Slicing and dissection of the hippocampi were done in ice-cold dissecting solution containing (mm): NaCl, 120; KCl, 3.5; CaCl 2, 0.7; MgCl 2, 4.0; NaH2PO4, 1.25; NaHCO3, 26; and glucose, 10, bubbled with 95% O2–5% CO2, with pH 7.35–7.45. Slices were stored at room temperature in a holding chamber containing the dissecting solution and bubbled with 95% O2–5% CO2 for > 0.5 h prior to recording. During the experiment, slices were held in a submersion recording chamber perfused with external recording solution composed of (mm): NaCl, 120; KCl, 3.5; CaCl 2, 2.5; MgCl 2, 1.3; NaH2PO4, 1.25; NaHCO3, 26; and glucose, 10. The solution was bubbled with 95% O2–5% CO2, with pH 7.35–7.45. Picrotoxin (100 μm) was added to the external solution to block inhibitory synaptic responses mediated by GABAA receptors; the CA3 region of the hippocampus was removed to prevent recurrent excitation. The solution also contained 100 μm APV ([+]-2-amino-5-phosphonopentanoic acid) to block NMDA receptor-mediated currents and prevent postsynaptic short-term plasticity, as well as to prevent long-term potentiation and long-term depression (LTP and LTD). Experiments were performed at room temperature (approximately 24°C). APV was obtained from Tocris Cookson, and all other chemicals were obtained from Fisher Scientific or Sigma. All experiments were performed in accordance with the regulations of the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Electrophysiology
Pyramidal cells in CA1 s. pyramidale and interneurones in CA1 s. radiatum were identified visually using infrared differential inference contrast (IR-DIC) optics on a Nikon E600FN upright microscope (Nikon Inc.). Targeted neurones were patched in the voltage-clamp configuration and recorded at a holding potential of −60 mV using an Axopatch 200B amplifier (Axon Instruments). Patch electrodes (3–4.5 MΩ) were filled with internal solution composed of (mm): caesium gluconate, 100; EGTA, 0.6; MgCl2, 5.0; Hepes, 10. pH was adjusted to 7.2 with CsOH. The internal solution also contained 10 mm BAPTA to block interneurone LTP and LTD (Laezza et al. 1999), and to inhibit Ca2+-mediated G-proteins on the postsynaptic membrane and prevent postsynaptic short-term plasticity; QX-314 (5 mm) to improve space clamp and reduce non-linear effects caused by voltage-gated channels in dendrites while recording from the soma; 10 mm ATP to chelate intracellular polyamines and prevent possible postsynaptic short-term plasticity at calcium-permeable AMPA receptors (Bahring et al. 1997; McBain, 1998; Rozov & Burnashev, 1999; Toth et al. 2000); and 0.5% biocytin to enable post hoc morphological analysis of neurones recorded. The access resistance and holding current (< 200 pA) were monitored continuously. Recordings were rejected if either access resistance or holding current increased more than 20% during the experiment.
Excitatory postsynaptic currents (EPSCs) were recorded in response to extracellular stimulation of Schaffer collateral axons by a bipolar tungsten microelectrode (FHC, Bowdoinham, ME, USA) placed in s. radiatum. A low intensity of stimulation was used (10–50 μA) so that EPSCs recorded had only one peak. Stimulation was generated by a Master-8-cp stimulator (A.P.I, Jerusalem, Israel) and applied with a BSI-2 biphasic stimulus isolator (BAK Electronics, Mount Airy, MD, USA). There were three stimulation patterns employed in the experiments: (1) paired-pulse stimulation with different intervals ((ms) 20, 30, 40, 50, 60, 80, 100, 150, 200, 500), applied in a random sequence and repeated 10 times at 0.1 Hz. The averaged paired-pulse ratio of the amplitudes (PPR = EPSC2/EPSC1) was calculated after recording; (2) short high-frequency trains (5 pulses) at different constant frequencies (5, 10, 20 and 50 Hz), repeated 10 times each at 0.033 Hz. The five-pulse ratio (ratio of the response amplitudes of the fifth versus first pulse, EPSC5/EPSC1) was calculated after recording; and (3) continuous constant frequency stimulation over a range of stimulus frequencies (0.1, 1, 2, 5 and 10 Hz), applied until the EPSC size reached steady state. The steady-state response amplitude was measured for each frequency. In each experiment the stimulus amplitude and duration (0.1 ms) were held constant.
For measurements of the use-dependent block of NDMA responses by MK-801, APV was omitted from the recording solution and 10 μm DNQX was added to block AMPA/kainate receptors. The concentrations of calcium (2.5 mm) and magnesium (1.3 mm) in the recording solution were not changed. NMDA receptor EPSCs were recorded at −40 mV in response to stimulation at 0.1 Hz. After a stable baseline was obtained, 40 μm MK-801 was added and stimulation was turned off for 10 min to allow full wash-in and equilibration of MK-801. Stimulation was resumed at 0.1 Hz, and EPSCs were recorded for at least 120 stimuli. Stimulation at 0.1 Hz was used to avoid causing any short-term plasticity that would alter the release probability. Relative EPSC size was measured by integrating the current in a 40 ms window around the peak (Huang & Stevens, 1997). Averages were made of 10 EPSCs from the baseline before MK-801 perfusion, and for the first 10 EPSCS evoked after the perfusion of MK-801. A four-state kinetic model was fitted to these averages to obtain the fraction of open receptors that were blocked by MK-801 (block fraction) (Huang & Stevens, 1997), which was compared for synapses onto interneurones versus pyramidal cells.
Analysis
Data are presented as mean ± s.e.m. In all figures, stimulus artifacts have been removed for clarity. Except where noted, statistical comparisons were made using Student's t test, and differences are considered significant when P < 0.05. Where noted, statistical comparisons were made using one-way ANOVA, with P < 0.05 considered significant.
Histology
To examine their morphological characteristics, the neurones were labelled with biocytin during recording. After recording, labelled neurones were visualized using an avidin–HRP reaction followed by a peroxidase reaction using diaminobenzidine (DAB). In some slices nickel ammonium sulphate (1%) was added for colour enhancement. Slices containing labelled neurones were stored in a solution of 4% paraformaldehyde in 0.1 m phosphate buffer overnight after the recording, and transferred to the ABC complex (Elite Vectastain ABC Kit, Vector Laboratories, Inc. Burlingame, CA, USA) the next day. The slices were incubated in ABC complex for 4 h, rinsed, and then transferred to the DAB reaction (Peroxidase Substrate Kit, Vector Laboratories, Inc.). After the reaction of 1–5 min, the slices were washed twice in phosphate buffer or water, dehydrated, and mounted on microscope slides. The slices were examined under a microscope and photographs were taken of labelled cells.
Mathematical description
General features
In order to investigate possible mechanisms for the differences in short-term plasticity observed in the present study, a mechanistic mathematical model of short-term plasticity that describes vesicle release from single synapses was developed to incorporate features of several previous models (Dobrunz & Stevens, 1997; Tsodyks et al. 1998; Dittman et al. 2000; Dobrunz, 2002). It is mechanistic in that we attempt to include only variables with physiological counterparts (e.g. readily releasable vesicles) and mechanisms of facilitation and depression based on known physiological processes that govern neurotransmitter release (e.g. depletion of readily releasable vesicles). We therefore also refer to it as a mathematical description of our experimental data.
The model contains three possible states for a synapse: ‘release-ready’, ‘releasing’, and ‘refractory’ (Tsodyks et al. 1998; Dittman et al. 2000). Only synapses in the release-ready state are capable of releasing a vesicle when an action potential arrives, and release of a single vesicle per active synapse occurs with an average release probability P(t). P(t) depends upon the number of readily releasable vesicles and on the release probability per vesicle, both of which are modified by the pattern of activity (Dobrunz & Stevens, 1997; Dobrunz, 2002). Facilitation occurs through a calcium-dependent increase in the release probability per vesicle (Dobrunz & Stevens, 1997; Dobrunz, 2002). Depression results from depletion of the readily releasable vesicle pool (Dobrunz & Stevens, 1997; Dobrunz, 2002) and from synapses becoming refractory after release (Dobrunz et al. 1997; Tsodyks et al. 1998).
One basic assumption in our mathematical analysis is that there is ‘uniquantal release’, which means that at most a single vesicle is released per synapse per action potential (Korn et al. 1994). This assumption has been supported by several previous electrophysiological studies (Redman, 1990; Stevens & Wang, 1995; Dobrunz & Stevens, 1997; Dobrunz et al. 1997; Hanse & Gustafsson, 2001; Dobrunz, 2002; Chen et al. 2004). Some morphological studies have suggested that Schaffer collateral axons can have multiple release sites (active zones) per presynaptic bouton (Harris & Sultan, 1995), although this appears to occur at only a small fraction of synapses. In addition, a small number of Schaffer collateral axons appear to make multiple contacts onto CA1 pyramidal cells (Sorra & Harris, 1993; Schikorski & Stevens, 1997). In consideration of these previous studies, our mathematical model incorporates the following assumptions: (1) if the input (action potential) is delivered to multiple release sites from an axon onto the same postsynaptic neurone, each release site is treated as an independent synapse; (2) one bouton can have multiple release sites (active zones), each of which is treated as an independent single synapse; (3) each synapse (active zone) has uniquantal release. These assumptions work well for the simulation of our experimental data.
Release probability and facilitation
Based on previous studies (Dobrunz & Stevens, 1997; Dobrunz, 2002), the average release probability per release-ready synapse P(t) is determined by the average vesicular release probability α(t) and the average number of readily releasable vesicles n(t). (1 −α) is the average probability that an individual vesicle will fail to release, and therefore (1 −α)n is the average probability that all n vesicles will fail to release, giving the average probability that one vesicle does release as:
| (1) |
For the first action potential at time t=tap1, n equals the initial (maximal) readily releasable vesicle pool size nT, and the vesicle release probability is the initial (baseline) vesicle release probability α1, so that the initial release probability P(tap1) =P1 is given by:
| (2) |
The average readily releasable vesicle pool size n(t) decreases upon each action potential by an amount equal to the average amount of release (the release probability per active synapse × the fraction of active synapses). The empty sites in the readily releasable pool are then refilled, increasing n(t) up to the maximum pool size of nT. For simplicity, we assume that both nT and the refilling rate R are constant. The readily releasable pool size can then be determined by the following equation:
| (3) |
where n≤nT, x(t) is the fraction of synapses in the release-ready state, δ is the Dirac delta function, defined to have units of s−1, and tap is the time of occurrence of an action potential.
The release probability may be enhanced on subsequent action potentials by calcium-dependent facilitation, which increases the vesicle release probability. The enhancement of release probability per vesicle on subsequent pulses is assumed to be directly related to the equilibrium occupancy of the release site by a calcium-bound molecule CaXF with dissociation constant KF, resulting in:
| (4) |
where α1 is the average initial vesicle release probability of an individual synapse. To define the occupancy of the calcium-bound molecule CaXF, it is assumed that CaXF instantaneously rises by ΔF after an action potential at time tap and decays to 0 exponentially with time constant τF (Dittman et al. 2000), giving:
| (5) |
This mathematical description of facilitation (eqns (4) and (5)) is the same as used in the model of Regehr and colleagues (Dittman et al. 2000), except that we use the equation relating facilitation to the calcium-bound molecule CaXF to describe the facilitation of the vesicular release probability α(t) (eqn (4)), while Regehr and colleagues used it for the overall synaptic release probability per active synapse P(t) (which in their model was called F). In our model P(t) also depends upon the readily releasable vesicle pool size n(t) (see above), which is not subject to the same facilitation. As explained in Dittman et al. (2000), this description of facilitation does not directly take into account the time course of intracellular free calcium, but provides an approximation of the magnitude and time course of the resulting facilitation.
Equations for states of the synapse
Release-ready synapses
All synapses are in three cycling states: release-ready state → releasing state → refractory state → release-ready state (Tsodyks et al. 1998). The parameters x, y and z are the fractions of synapses in the release-ready, releasing and refractory states, respectively. Only synapses that are in the release-ready state, which we also refer to as active synapses, are able to release a vesicle when an action potential occurs. The average probability that a vesicle is released by an active synapse is given by P(t) (eqn (1)), and synapses that release a vesicle enter the releasing state. The entry of a synapse into the releasing state is assumed to be nearly instantaneous, and occupancy of the releasing state is very brief. Releasing synapses enter into the refractory state with a time constant τin of a few milliseconds. Refractory synapses recover into the release-ready state with a time constant τrec. The rate of change of synapses in the release-ready state is therefore equal to the increase in synapses recovering from the refractory state minus the decrease due to synapses entering the releasing state. The corresponding kinetic equation for the release-ready state is:
| (6) |
where dx/dt is the rate of change of synapses from the release-ready state, z(t)/τrec is the velocity of recovery of synapses to the release-ready state from the refractory state, x(tap) is the fraction of ready-release synapses at time tap, and P(tap) is the fraction of release-ready synapses that releases a vesicle (i.e. the release probability per active synapse) upon activation by an action potential arriving at time tap.
The rate of recovery of synapses to the release-ready state from the refractory state is assumed to be calcium dependent, and is determined by the equilibrium binding occupancy of a calcium-bound molecule CaXD, such that
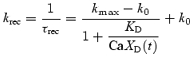 |
(7) |
For CaXD= 0, krec=k0, and z recovers exponentially with time constant τrec= 1/k0. For values of CaXD≫KD, krec=kmax, so z recovers exponentially with τrec≈ 1/kmax. For intermediate values of CaXD, z recovers with both fast and slow kinetic components. In Dittman's model, the same description was used for a ‘depression factor D’ (Dittman et al. 2000).
To define the occupancy of the calcium-bound molecule CaXD, it is assumed that CaXD instantaneously rises by ΔD after an action potential at time tap and decays to 0 exponentially with time constant τD (Dittman et al. 2000), giving:
| (8) |
Releasing synapses
Similarly, the rate of change of synapses in the release state is equal to the increase caused by the activation of an action potential at time tap minus the decrease caused by the synapses entering the refractory state:
| (9) |
where, as above, P(tap) x(tap) is the fraction of synapses that release due to an action potential at time tap, and y(t)/τin is the rate of releasing synapses entering the refractory state. Since the releasing state is very brief, all releasing synapses are assumed to have entered the refractory state before the next action potential occurs. Thus when an action potential occurs at time t=tap,
| (10) |
Refractory synapses
Furthermore, the rate of change of synapses in the refractory state is equal to the velocity of releasing synapses becoming refractory minus the velocity of recovery of refractory synapses to the readily releasable state. Thus:
| (11) |
EPSC amplitude
The amplitude of the postsynaptic current Is(t) for an action potential occurring at time t=tap is:
| (12) |
where A is the amplitude of postsynaptic current induced by one synaptic release (assume one synaptic release is equal to release of one vesicle), Ns is the total number of synapses, and y(tap) is the fraction of synapses that release due to the action potential at time tap.
Paired-pulse ratio
Before the first pulse arrives at time tap1, all synapses are in the release-ready state, such that x(tap1) = 1. When the first action potential arrives, P(tap1) =P1, where P1 is the initial release probability (eqn (2)). The fraction of synapses that release is therefore:
Thus, the amplitude of synaptic current for the first pulse is:
| (13) |
After first pulse, the values of x(t), y(t) and z(t) for t > tap1 can be calculated by solving eqns (3)–(11). The boundary conditions used in the solving of the differential equations are: x(tap1) = 1 −P1, y(tap1) =P1, z(tap1) = 0, n(tap1) =(nT− P1), CaXF(tap1) =ΔF, and CaXD(tap1) =ΔD. If the next pulse arrives at time tap2, the fraction of synapses that release is y(tap2) =P(tap2) x(tap2), where x(tap2) is the fraction of synapses in the release-ready state and P(tap2) is the fraction of release-ready synapses activated by the second pulse, i.e. release probability per active synapse just before the second pulse. Based on eqn (12), when the second pulse arrives, the peak of synaptic current is:
| (14) |
P(tap2) is the average release probability of an individual synapse for the second stimulus, which depends upon the interpulse interval if there is facilitation. If there is no facilitation then α(tap2) =α(tap1) and P(tap2) ≈P1. When facilitation exists, α(tap2) > α(tap1) and P(tap2) can be larger than P1. If the paired-pulse interval is long enough that all of the synapses that released on the first pulse have recovered from the refractory state (x(tap2) = 1), then y(tap2) =P(tap2), otherwise y(tap2) < P(tap2).
The paired-pulse ratio (PPR) measured experimentally is the amplitude of the second EPSC divided by the amplitude of the first EPSC. This gives:
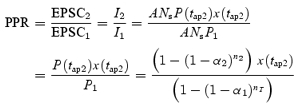 |
(15) |
where α2 and n2 are the values of α(t) and n(t) at time t=tap2. Since both EPSC1 and EPSC2 depend on Ns, the paired-pulse ratio is independent of the number of synapses.
Five-pulse ratio
For regular stimulus trains given at frequency f, eqn (5) has an analytical solution. Combining with eqn (4), the release probability per vesicle just before the ith stimulus can be expressed as:
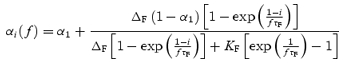 |
(16) |
Similarly, for regular stimulus trains given at frequency f, eqn (3) can be used to give an analytical expression for the size of the readily releasable vesicle pool for the ith stimulus:
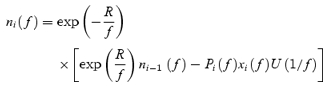 |
(17) |
where U is the unit step function defined to have units of s−1. From eqn (1), the release probability per readily releasable synapse just before the ith stimulus is:
| (18) |
where αi(f) and ni(f) are given by eqns (16) and (17).
In addition, eqns (6)–(11) can be used to yield an analytical expression for the fraction of release-ready synapses for the ith stimulus for regular stimulus trains given at frequency f:
| (19) |
where
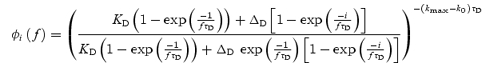 |
(20) |
The five-pulse ratio (FPR), defined as the amplitude of fifth EPSC divided by the first one, can be obtained by using eqns (12) and (18)–(20):
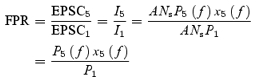 |
(21) |
In the present study, the frequencies 5, 10, 20 and 50 Hz were used in the five-pulse train. Again, because both EPSC1 and EPSC5 depend on Ns, the five-pulse ratio is independent of the number of synapses.
Steady-state EPSCs evoked by constant frequency stimulation
During constant frequency stimulation, the release probability per active synapse reaches steady state (P∞). From eqn (1), at steady state,
| (22) |
Based on eqns (4) and (5), the steady state of the vesicle release probability can be expressed as:
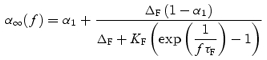 |
(23) |
where ΔF is the incremental increase in CaXF after a stimulus.
During high-frequency stimulation, the readily releasable vesicle pool depletes. For each stimulus frequency, steady state will occur when the rate of vesicle releasing equals the rate of vesicle refilling into the readily releasable pool, resulting in a release probability per active synapse that does not change (P∞). This gives:
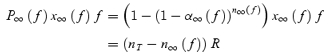 |
(24) |
Using eqns (23) and (24), numerical solutions for n∞(f) can be obtained. During constant frequency stimulation, the number of release-ready synapses reaches a steady-state value that can be expressed as:
| (25) |
where
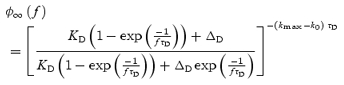 |
(26) |
Using eqns (22)–(26), the steady-state EPSC size can be generated for each stimulus frequency as:
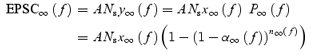 |
(27) |
As this depends upon the number of synapses stimulated, which may differ from cell to cell, we normalize to the initial EPSC size. Since no short-term plasticity occurs when stimulating at 0.1 Hz, we normalize to the value of EPSC∞(0.1), the steady-state EPSC evoked by 0.1 Hz stimulation. The normalized steady-state EPSC size is therefore:
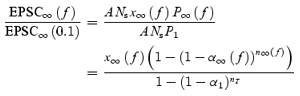 |
(28) |
which is independent of the number of synapses stimulated.
All mathematical calculations were performed using Mathematica software (Fourth Edition, Wolfram Media, Inc. Champaign, IL, USA).
Results
We examined in detail the differences in several forms of short-term plasticity between Schaffer collateral excitatory synapses made by CA3 pyramidal cell axons onto CA1 pyramidal cells and s. radiatum interneurones. CA1 pyramidal cells and interneurones were identified using IR-DIC microscopy and recorded with whole-cell voltage clamp. To allow us to study short-term plasticity of excitatory synapses in isolation, inhibitory (GABAergic) responses and synaptic long-term plasticity were pharmacologically blocked. We compared EPSCs in response to extracellular stimulation of Schaffer collateral axons using three different stimulation protocols that cause different amounts of short-term facilitation versus depression: pairs of pulses at different interpulse intervals, short high-frequency trains of different frequencies, and steady-state responses to constant stimulation at different frequencies. We then used a mechanistic mathematical model to investigate possible mechanisms underlying the differences in short-term plasticity observed between Schaffer collateral inputs to pyramidal cells and s. radiatum interneurones.
Paired-pulse facilitation
We first investigated a simple form of short-term plasticity, paired-pulse facilitation (PPF), at Schaffer collateral excitatory synapses onto CA1 pyramidal cells versus CA1 s. radiatum interneurones across a range of paired-pulse intervals. In response to pairs of pulses close together in time (20–200 ms), synapses onto CA1 pyramidal cells show robust paired-pulse facilitation (example in Fig.1A). In contrast, the synapses onto s. radiatum interneurones are heterogeneous with respect to paired-pulse plasticity. The majority of interneurones have either modest paired-pulse facilitation (example in Fig. 1B) or no paired-pulse plasticity (not shown). A small subset of interneurones, however, showed pronounced paired-pulse depression (PPD) at all intervals tested (example in Fig. 1C). Because there appeared to be two distinct types of responses among the different interneurones with respect to the short-term plasticity of their inputs in response to all three stimulus protocols studied, we divided them into two groups based on their paired-pulse responses. One group had paired-pulse facilitation or no plasticity, referred to from here on as facilitation interneurones (n = 57), and the other group had clear paired-pulse depression (depression interneurones, n = 9). The depression interneurones were rare, and not distinguishable by their morphology or location within s. radiatum (see below), and thus their sample size is relatively small. The average paired-pulse ratios were significantly different between all three groups for paired-pulse intervals between 20 and 200 ms (Fig. 1D, n = 32 pyramidal cells, n = 57 facilitation interneurones, n = 9 depression interneurones, one-way ANOVA, P < 0.01). For all cell types, paired-pulse plasticity had largely disappeared by 500 ms (P > 0.05). We find that paired-pulse facilitation is target cell specific, and that it is reduced at synapses onto interneurones. Because both groups of interneurones had significantly less paired-pulse facilitation than the pyramidal cells, this basic finding also holds true if all interneurones are grouped together instead of being divided. These results show that despite having the same presynaptic input, Schaffer collateral excitatory synapses onto CA1 pyramidal cells and s. radiatum interneurones have different presynaptic properties.
Figure 1. Excitatory synapses onto CA1 s. radiatum inhibitory interneurones have less paired-pulse facilitation than synapses onto CA1 pyramidal cells.

Examples of EPSCS recorded in response to paired-pulse stimulation of Schaffer collateral axons in a pyramidal cell (A), interneurone with facilitation (B) and interneurone with depression (C). Each trace is the average of 10 responses; traces are overlaid for paired-pulse intervals of 30, 50, 80, 100, 150 and 200 ms. D, group results for paired-pulse ratios (mean ± s.e.m.) from pyramidal cells (squares, n = 32), interneurones with facilitation (circles, n = 57) and interneurones with depression (triangles, n = 9). There are significant differences between three groups (one-way ANOVA, P < 0.05).
Facilitation during short trains
The differences between excitatory synapses onto interneurones and pyramidal cells were even more pronounced when we compared short-term plasticity in response to five-pulse trains with different frequencies (5, 10, 20 and 50 Hz). Figure 2 shows examples of average EPSCs evoked by five-pulse stimulation from a CA1 pyramidal cell (Fig. 2A1–A3), a CA1 s. radiatum interneurone with facilitation (Fig. 2B1–B3), and a CA1 s. radiatum interneurone with depression (Fig. 2C1–C3). At all frequencies tested the synapses onto pyramidal cells consistently showed large facilitation that accumulated during the train. In contrast, synapses onto the majority of interneurones showed little facilitation and even had short-term depression at the highest frequency tested (50 Hz, e.g. Fig. 2, 3). Once again, a subgroup of interneurones had short-term depression, which greatly reduced the EPSC size by the end of the trains (Fig. 2C). We quantified short-term plasticity in response to the five-pulse stimulation using the five-pulse ratio (EPSC5/EPSC1). As in the previous section, interneurones were divided into two groups depending on whether they had PPF or PPD. Figure 2D shows the summary of five-pulse ratios for three groups: 16 pyramidal cells, 35 facilitation interneurones and 8 depression interneurones. Pyramidal cells had significantly higher five-pulse ratios compared with both groups of interneurones at all frequencies tested (one-way ANOVA, P < 0.01). In addition, the depression interneurones had significantly lower five-pulse ratios compared with the facilitation interneurones (one-way ANOVA, P < 0.05). These results showed that the differences in short-term plasticity observed with pairs of pulses continue to grow during additional stimuli, resulting in even greater differences between excitatory synapses onto interneurones versus pyramidal cells during short trains of high-frequency stimulation.
Figure 2. Facilitation during short trains is also reduced at excitatory synapses onto interneurones versus pyramidal cells.

Examples of EPSCs recorded in response to five-pulse trains in a pyramidal cell (A), interneurone with facilitation (B) and interneurone with depression (C). Each trace is the average of 10 responses; traces are shown for stimulus trains of 5, 20 and 50 Hz. D, group results for five-pulse ratios (EPSC5/EPSC1, mean ± s.e.m.) for pyramidal cells (squares, n = 16), facilitation interneurones (circles, n = 35) and depression interneurones (triangles, n = 8). There are significant differences between the three groups (one-way ANOVA, P < 0.05).
Figure 3. Steady-state high-frequency depression for excitatory synapses onto CA1 pyramidal cells versus those onto CA1 s. radiatum interneurones.

Examples of EPSCs recorded at steady state during high-frequency stimulation of Schaffer collateral axon at different frequencies in a pyramidal cell (A), facilitation interneurone (B) and depression interneurone (C). Each trace is the average of 10 responses; traces are shown for steady-state responses to stimulation at 0.1, 5 and 10 Hz. D, example of EPSC amplitudes versus time for stimulation at 2, 5 and 10 Hz to show steady state. Data are from a pyramidal cell; curves have been smoothed by 5 point adjacent averaging. E, group results for steady-state high-frequency depression (mean ± s.e.m., normalized to response size at 0.1 Hz) for pyramidal cells (squares, n = 28), facilitation interneurones (circles, n = 42), and depression interneurones (triangles, n = 6). There is no significant difference between pyramidal cells and facilitation interneurones except at 1 Hz (P > 0.5), but there are significant differences between depression interneurones and the other two cell types (one-way ANOVA, P < 0.05).
Steady-state high-frequency depression
Next we examined another form of short-term plasticity, high-frequency depression, by comparing the steady-state EPSC size during continuous constant-frequency stimulation at frequencies of 0.1, 1, 2, 5 and 10 Hz. When stimulated with very long trains of constant-frequency stimulation, Schaffer collateral excitatory synapses onto all cells studied showed a depression of the steady-state EPSC size at high frequencies. Figure 3 shows examples of the average steady-state EPSCs in response to stimulation at 0.1, 5 and 10 Hz for synapses onto a CA1 pyramidal cell (Fig. 3A), and CA1 s. radiatum interneurones with paired-pulse facilitation (Fig. 3B) and paired-pulse depression (Fig. 3C). An example of EPSC amplitudes versus time for three different stimulus frequencies is shown in Fig. 3D to illustrate the change in the EPSC size and attainment of a new steady-state level upon changing the stimulus frequency. Figure 3E shows the summary of the steady-state response size versus stimulus frequency for excitatory synapses onto CA1 pyramidal cells, facilitation interneurones and depression interneurones. For each cell, responses were normalized to the response obtained during 0.1 Hz stimulation, where no short-term plasticity occurred. There is no significant difference between the facilitation interneurones and pyramidal cells in steady-state high-frequency depression at 2, 5 and 10 Hz (n = 42 facilitation interneurones, n = 28 pyramidal cells, P > 0.5). There was a small difference at 1 Hz, where unlike the synapses onto the interneurones, synapses onto the pyramidal cells showed slight steady-state facilitation (P < 0.05). However, the amount of high-frequency depression is significantly greater at synapses onto the depression interneurones (n = 6) compared with each of the other two groups at all frequencies tested (ANOVA, P < 0.05). Thus, although short-term facilitation is greatly reduced at excitatory synapses onto s. radiatum interneurones versus pyramidal cells, high-frequency depression after constant-frequency stimulation is the same in the majority of interneurones, and only slightly greater in the others, as compared with pyramidal cells. This shows that not all forms of presynaptic short-term plasticity are significantly influenced by the target cell at Schaffer collateral excitatory synapses. Using a mathematical model of short-term plasticity, our investigation into the mechanisms that underlie the differences in presynaptic properties of Schaffer collateral synapses gives a possible explanation for this result (see below).
We next considered whether desensitization of postsynaptic receptors might contribute to the short-term depression we observe at steady state. We have previously shown that receptor desensitization does not contribute to short-term depression at Schaffer collateral synapses onto pyramidal cells (Dobrunz et al. 1997; Dobrunz & Stevens, 1997; Dobrunz, 2002); however, it was not known whether desensitization plays a role at synapses onto s. radiatum interneurones. To rule this out, we compared the amount of steady-state high-frequency depression at synapses onto interneurones in control and in the presence of 50 μm cyclothiazide, which inhibits glutamate receptor desensitization. While cyclothiazide prolonged the time course of the EPSC, there was no change in the amount of steady-state high-frequency depression at either 2 Hz (0.82 ± 0.16 versus 0.80 ± 0.017, n = 5, P > 0.2) or 10 Hz (0.34 ± 0.14 versus 0.33 ± 0.08, n = 4, P > 0.8). Because cyclothiazide has been reported to have presynaptic effects at some synapses (Bellingham et al. 1999), we also verified that it did not affect the paired-pulse ratio at synapses onto interneurones at an interpulse interval of 20 ms (2.2 ± 0.5 versus 2.1 ± 0.2, n = 5, P > 0.5) or 40 ms (2.0 ± 0.3 versus 2.0 ± 0.3, n = 5, P > 0.3). Similarly, there was no effect on the five-pulse ratio at either 10 Hz (1.8 ± 0.1 versus 1.7 ± 0.1, n = 3, P > 0.6) or 50 Hz (1.04 ± 0.19 versus 0.95 ± 0.09, n = 3, P > 0.5). These experiments confirm that at Schaffer collateral synapses onto s. radiatum interneurones, receptor desensitization does not play a role in short-term plasticity, as has previously been shown for synapses onto CA1 pyramidal cells (Dobrunz & Stevens, 1997; Hjelmstad et al. 1999).
Heterogeneity of synapses onto interneurones
The results shown above indicate that CA1 s. radiatum interneurones are heterogeneous with respect to the short-term plasticity of their Schaffer collateral inputs. For example, under paired-pulse stimulation, 54 of 66 interneurones tested had moderate facilitation, 9 had depression, and 3 had neither paired-pulse facilitation nor paired-pulse depression. To investigate whether morphological differences between interneurones correlate with differences in short-term plasticity of their inputs, all CA1 s. radiatum interneurones recorded were labelled with biocytin for post hoc morphological analysis. Figure 4 shows two examples of paired-pulse ratios obtained from two s. radiatum interneurones in the same hippocampal slice (Fig. 4A) that displayed different morphological characteristics. However, both interneurones displayed modest paired-pulse facilitation (Fig. 4B), and there is no significant difference between them in paired-pulse ratio at any paired-pulse interval tested (P > 0.6). In addition, other cells showed very similar morphology but markedly different paired-pulse plasticity (data not shown). Overall, we found no correlation between the morphological differences in CA1 s. radiatum interneurones and the short-term plasticity of Schaffer collateral synapses onto them.
Figure 4. Morphology of interneurones does not correlate with the short-term plasticity of their inputs.

Paired-pulse ratios obtained from two CA1 s. radiatum interneurones showing different morphological characteristics. A, two biocytin-filled s. radiatum interneurones (interneurones 1 and 2) recorded in the same slice show different morphological characteristics. S. pyramidale is out of the field of view, off the top of the picture. Scale bar, 100 μm. B, paired-pulse ratios are not different at synapses onto interneurone 1 (▪) and interneurone 2 (◯) (P > 0.6).
Mathematical model of short-term plasticity
To investigate possible mechanisms for the differences between excitatory Schaffer collateral synapses onto s. radiatum interneurones and pyramidal cells, we developed a mechanistic model of presynaptic short-term synaptic plasticity that integrates features of several previous models (Dobrunz & Stevens, 1997; Tsodyks et al. 1998; Dittman et al. 2000; Dobrunz, 2002). Our model assumes that the short-term plasticity we observe is presynaptic, resulting from activity-dependent modulation of the probability of neurotransmitter release from individual synapses. Because there is heterogeneity even among the population of synapses of the same type (e.g. Schaffer collateral synapses onto pyramidal cells (Dobrunz & Stevens, 1997; Dobrunz, 2002)), our model describes the average behaviour of a single synapse for Schaffer collateral inputs onto pyramidal cells, facilitation interneurones and depression interneurones.
The model contains three possible states for a synapse: ‘release-ready’, ‘releasing’ and ‘refractory’. The basic assumptions of the model are: (1) synapses are initially all in the release-ready state. Only synapses in the release ready-state (active synapses) are capable of releasing a vesicle when an action potential arrives; (2) upon a single stimulus, a fraction of the release-ready synapses (P) release one vesicle; (3) a synapse is in the releasing state for a very short time (a few milliseconds), less than the shortest interstimulus interval that is to be considered (20 ms); (4) synapses that release become refractory or inactive; (5) recovery from the refractory state back to the release-ready state is calcium dependent; (6) the fraction of release-ready synapses that release a vesicle depends on the number of readily releasable vesicles (readily releasable pool size, n) and the release probability per vesicle (α), both of which are dynamic; (7) with each stimulus calcium-dependent facilitation increases the release probability per vesicle, which can result in an increase in P on subsequent pulses that occur at short intervals; (8) the readily releasable vesicle pool size decreases when vesicles release and refills up to a maximal size (nT).
Synaptic short-term plasticity depends on the balance between factors causing facilitation and depression. In our model, the overall synaptic release probability for each stimulus is determined by the number of synaptic vesicles available for release, by the release probability per vesicle, and by the fraction of synapses in the release-ready state. All of these factors are in turn dependent upon the release probability, as well as on other model parameters. Facilitation comes from an activity-dependent enhancement of the release probability per vesicle α(t). α(t) increases from its initial value α1 due to the binding of a calcium-bound molecule CaXF, which has a dissociation constant KF. CaXF increases by an amount ΔF after an action potential and decays back to zero with a time constant τF (Dittman et al. 2000). Depression is caused by synapses entering a refractory state after they release and by the depletion of the readily releasable vesicle pool. The fraction of synapses that become inactivated (refractory) depends upon the release probability P(t). In addition, the rate of recovery from refractory depression is accelerated by calcium; this is modelled by having the recovery rate increase from its initial value (k0) up to a maximal value (kmax) depending upon the equilibrium occupancy of a calcium-bound molecule CaXD with a dissociation constant KD (Dittman et al. 2000). Similar to CaXF, the calcium-bound molecule that governs facilitation, CaXD increases by an amount ΔD after each action potential and decays back to zero with a time constant τD. The depletion of the readily releasable vesicle pool is determined by the initial releasable pool size (nT) and the refilling rate (R), as well as by the release probability P(t).
There are a total of 12 independent parameters in the equations (see Tables 1 and 2). We fitted all of the data with 9 of the parameters held constant for all cell types and for all forms of short-term plasticity evoked by using the different stimulation protocols (Table 1). Among them, the values of kmax, k0, KD, τD and ΔD are the same as those used in the study of Dittman et al. (2000). However, the value of ΔF that we used is 4 times higher than that used in the previous study (Dittman et al. 2000). This is due to the difference in the model formulations of facilitation, where facilitation in our model increases the release probability per vesicle rather than the overall release probability. Only two parameters were adjusted as variables for data obtained from the different cell groups, and one additional parameter was a variable for the different stimulus protocols but held the same for all cell groups (Table 2).
Table 1.
Parameters held constant in the model simulations for all cell types and stimulus protocols
| Symbol | Definition | Value | Unit |
|---|---|---|---|
| kmax | Maximum recovery rate from the refractory state | 30a | s−1 |
| k0 | Baseline recovery rate from the refractory state | 2a | s−1 |
| KD | Dissociation constant of CaXD | 2a | N.A. |
| τin | Time constant for entry into refractory state | 3 | ms |
| KF | Dissociation constant of CaXF | 4 | N.A. |
| τD | Decay time constant of CaXD after an action potential | 50a | ms |
| ΔF | Incremental increase in CaXF after a stimulus | 4 | (normalized) |
| ΔD | Incremental increase in CaXD after a stimulus | 1a | (normalized) |
| R | Refilling rate of readily releasable vesicle pool | 0.1 | s−1 |
Parameters are the same of those used by Dittman et al. (2000).
Table 2.
Values of the variables that were adjusted to fit the experimental data
| Simulation object | Initial release probability per vesicle α1 | Initial readily releasable pool size nT | Initial release probability P1 | Decay constant of CaXF τF (s) |
|---|---|---|---|---|
| Pyramidal cells | ||||
| Paired-pulse ratio vs interval | 0.055 | 4.8 | 0.24 | 0.12 |
| Five-pulse ratio versus frequency | 0.055 | 4.8 | 0.24 | 0.16 |
| Steady-state EPSC versus frequency | 0.055 | 4.8 | 0.24 | 0.60 |
| Facilitation interneurones | ||||
| Paired-pulse ratio vs interval | 0.060 | 7.5 | 0.37 | 0.12 |
| Five-pulse ratio vs frequency | 0.060 | 7.5 | 0.37 | 0.16 |
| Steady-state EPSC vs frequency | 0.060 | 7.5 | 0.37 | 0.60 |
| Depression interneurones | ||||
| Paired-pulse ratio vs interval | 0.090 | 10.0 | 0.61 | 0.12 |
| Five-pulse ratio vs frequency | 0.090 | 10.0 | 0.61 | 0.16 |
| Steady-state EPSC vs frequency | 0.090 | 10.0 | 0.61 | 0.60 |
The initial release probability P1 was not an independent variable but was calculated from the values of α1 and nT (eqn (2) in Methods).
Differences in initial release probability cause changes in short-term plasticity
Figure 5 shows the model fits (lines) to the data (symbols) for pyramidal cells, facilitation interneurones and depression interneurones to each of the three stimulus protocols: paired-pulse ratio versus interval (Fig. 5A), five-pulse ratio versus stimulus frequency (Fig. 5B), and steady-state response size versus stimulus frequency (Fig. 5C). All of the differences in short-term plasticity between the three cell groups for all three stimulus protocols could be accounted for by changing only two parameters: the initial release probability per vesicle (α1) and the initial pool size of releasable vesicles (nT) (Table 2). This results in a difference in the initial release probability P1; since P1 is decided by α1 and nT it is not an independent variable. The model simulations therefore indicate that differences in short-term plasticity between cell groups are due to differences in initial release probability that is determined by the initial release probability per vesicle and the initial releasable pool size. From Table 2, the synapses onto interneurones have a larger initial vesicle pool size (nT= 7.5–10.0) than do pyramidal cells (nT= 4.8), and the initial probability per vesicle is different between synapses onto interneurones that facilitate and ones that depress. The heterogeneity in the initial release probability per vesicle accounts for the difference in short-term plasticity between the two groups of interneurones: a larger release probability per vesicle (0.090) leads to short-term depression, while a smaller release probability per vesicle (0.060) leads to short-term facilitation. However, this facilitation is still smaller than that in pyramidal cells because pyramidal cells have a smaller initial readily releasable vesicle pool size, and thus a smaller initial release probability.
Figure 5. Model predicts that differences in initial release probability cause the observed changes in short-term plasticity.

Mathematical simulations (lines) of experimental data (symbols) provide excellent fits to paired-pulse ratios (A), five-pulse ratios (B), and steady-state high-frequency depression (C) obtained from CA1 pyramidal cells (squares), s. radiatum interneurones with facilitation (circles), and with depression (triangles). For each panel continuous curves are from model fits to pyramidal cell data, dashed lines are from model fits to facilitation interneurone data, and dotted lines are from model fits to depression interneurone data. All curves were calculated using equations from Methods with parameters values given in Table 1 and Table 2. Only the values of the initial vesicular release probability α1 and the initial readily releasable pool size nT are different between the three cell groups; for each cell group, values of α1 and nT are the same for all three stimulus protocols.
The amount of paired-pulse facilitation is inversely related to the initial release probability, such that P1 is lowest in the pyramidal cells (0.24), intermediate in the facilitation interneurones (0.37), and highest in the depression interneurones (0.61). With only a difference in initial release probability between the three cell groups, the model provides excellent fits to the paired-pulse ratios across the range of interpulse intervals measured (Fig. 5A). In addition, the same values of α1 and nT (Table 2) fit the data to the five-pulse stimulus protocol for all three cell groups across the range of stimulus frequencies tested (5–50 Hz) (Fig. 5B), with only one other change. To fit the five-pulse data, the variable τF, which is the decay time constant after an action potential of the calcium-bound molecule responsible for facilitation (CaXF), needed to be adjusted to be longer (0.16 versus 0.12). This could be indicative of the saturation of a calcium buffer or of calcium extrusion from the presynaptic terminal during the longer trains, resulting in a slower decay of free intracellular calcium. However, since τF was kept the same for the three cell groups (Table 2), all of the differences between the three groups were determined only by the changes in α1 and nT. Again, the initial release probability is inversely related to the five-pulse ratio. The highest initial release probability (depression interneurones) results in depression of the fifth EPSC in the train, the lowest initial probability (pyramidal cells) results in large facilitation, and the intermediate value of release probability (facilitation interneurones) results in moderate facilitation for some frequencies (5–20 Hz) and depression for the highest frequency tested (50 Hz).
The comparison of the steady-state responses to continuous high-frequency stimulation was more complex. There was no difference in the amount of high-frequency depression of the steady-state responses between the pyramidal cells and the facilitation interneurones, except at 1 Hz where the pyramidal cell responses were slightly facilitated instead of depressed. However, the depression interneurones showed significantly more high-frequency depression than the other two groups at all frequencies tested. As shown in Fig. 5C, the model is able to capture all of these features of the experimental data, using the same values of α1 and nT as used for the paired-pulse and five-pulse data, with a further increase in τF (0.60). Once again, since τF was kept the same for the three cell groups, all of the differences between the three groups were determined only by the changes in α1 and nT. Thus the parameters that govern initial release probability, α1 and nT, are kept the same for each cell group in the simulation of short-term plasticity evoked by the three different stimulation protocols (Table 2).
Comparing the two groups of interneurones suggests that when the initial readily releasable pool size is similar, an increase in the vesicular release probability results in greater steady-state high-frequency depression. Furthermore, there is no difference in the model prediction of steady-state high-frequency depression between pyramidal cells and paired-pulse facilitation interneurones except at 1 Hz, even though the differences in the initial release probability and the initial vesicle pool size are still there (Table 2). Thus the same values of α1 and nT that result in the differences in paired-pulse facilitation and five-pulse facilitation also predict no difference in steady-state high-frequency depression. Even though the initial release probability of the synapses onto the facilitation interneurones is greater than that onto the pyramidal cells (P1= 0.37 versusP1= 0.24), it is caused by increases in both α1 and nT. While increasing α1 alone increases high-frequency depression, increasing nT alone should decrease high-frequency depression. The effects on high-frequency depression due to increasing both α1 and nT in facilitating interneurones versus pyramidal cells offset each other, except at the stimulus frequency of 1 Hz. This suggests that it is not only the overall initial release probability that is important, but that the relative contributions of the readily releasable pool size and the release probability per vesicle to the initial release probability are also critical for determining short-term plasticity.
Release probability is higher at synapses onto interneurones
Our model predicts that the differences in short-term plasticity between synapses onto interneurones versus pyramidal cells are due to a difference in their initial release probability. We tested this experimentally by comparing the rate of block of NMDA receptors by the use-dependent blocker MK-801 (Huang & Stevens, 1997). This is an established method to test for differences in release probability (e.g. Castro-Alamancos & Connors, 1997). Because MK-801 is an irreversible open-channel blocker of NMDA receptors, the rate of block is faster at synapses with a higher average release probability (Huang & Stevens, 1997). We isolated NMDA currents by using DNQX to block AMPA responses and omitting APV from the recording solution. We depolarized the postsynaptic cell to −40 mV to relieve the magnesium block of the NMDA receptors, and left the concentrations of calcium and magnesium unchanged. Figure 6 shows the progressive decrease in the size of the NMDA EPSC in response to 0.1 Hz stimulation in 40 μm MK-801. As predicted, the rate of block was significantly faster at Schaffer collateral synapses onto s. radiatum interneurones (◯, τ = 22.5 ± 3.6, n = 5) as compared with CA1 pyramidal cells (▪, τ = 42.7 ± 11.9, n = 5, P < 0.005). As the depression interneurones are relatively rare, there were none observed in these experiments and thus we have only two cell groups. We also compared the fraction of open NMDA receptors blocked by MK-801 (block fraction) at synapses onto interneurones versus pyramidal cells. We estimated the block fraction by fitting a four-state kinetic model to the average EPSC measured in the absence and presence of MK-801, as described in Huang & Stevens (1997). The block fraction was not different at synapses onto interneurones versus pyramidal cells (0.35 ± 0.05, n = 5 interneurones versus 0.35 ± 0.05, n = 5 pyramidal cells, P > 0.9). The faster rate of block observed at synapses onto interneurones therefore demonstrates that the average release probability is indeed higher at synapses onto interneurones versus pyramidal cells, consistent with our model results and with the lower PPF observed at these synapses.
Figure 6. Synapses onto interneurones have higher initial release probability as shown by a faster MK-801 blocking rate.

Decrease in the NMDA-receptor-mediated EPSC amplitude versus stimulus number in 40 μm MK-801. Rate of block by MK-801 is faster for synapses onto interneurones (◯, mean ± s.e.m., n = 5) versus pyramidal cells (▪, mean ± s.e.m., n = 5, P < 0.005). Inset: examples of average EPSCs in the absence (black line, average of 10 EPSCs from baseline) and presence (grey line, average of first 10 EPSCs in MK-801) of MK-801 for an interneurone. EPSCs have been scaled so their initial peaks match to show the faster decay in the presence of MK-801. Scale bars: 20 ms, 10 pA for control, 5.3 pA in MK-801.
Mathematical model predicts differences in the dynamics of synaptic parameters
Using our model, we further investigated the dynamics of the readily releasable vesicle pool size (n), release probability per vesicle (α), and release probability per active (release-ready) synapse (P), as well as the fraction of synapses that are in the release-ready state (x) during the different stimulus protocols. Figure 7 illustrates the model predictions for these factors that determine the overall neurotransmitter release probability during short-term plasticity for Schaffer collateral synapses onto the three groups of cells: pyramidal cells (continuous lines), interneurones with paired-pulse facilitation (dashed lines) and interneurones with paired-pulse depression (dotted lines). Figure 7A shows the changes in these variables according to time after the first stimulus of a pair. The first stimulus occurs at t= 0, and baseline values of the parameters that determine release on the first stimulus are shown at t < 0. As a result of facilitation, the release probability per vesicle α(t) jumps from its initial value α1 up to a value of α2 at time t= 0, and then decays back to its initial value α1 (Fig. 7A1). As there is no difference in the calcium binding parameters governing facilitation (τF, KF), α(t) increases to a similar degree for synapses onto all three cell groups, and decreases at a similar rate. The differences between these curves are due to the differences in their initial values. Figure 7A2 shows the dynamics of the readily releasable vesicle pool size n(t) after the first stimulus of a pair. Upon the first stimulation, the readily releasable pool size decreases by an amount P1. The refilling rate is quite slow, such that over the time scale shown in Fig. 7A2, n(t) appears to be almost constant for t > 0. In fact, n is not refilled entirely even 1 s after release. Thus, in the case of paired-pulse stimulation, the readily releasable pool has not completely refilled prior to the arrival of the second pulse. However, the decrease in n is very small compared with the initial pool size (nT), and therefore it is likely to play a negligible role in paired-pulse plasticity.
Figure 7. Dynamics of model parameters that govern short-term plasticity for different stimulus patterns.

Dynamics of α, the release probability per vesicle (A1–C1); n, the readily releasable vesicle pool size (A2–C2); P, the release probability per active (release-ready) synapse (A3–C3); and x, the fraction of synapses in release-ready state (A4–C4) for different cell types. For each panel continuous curves are from model fits to pyramidal cell data, dashed lines are from model fits to facilitation interneurone data, and dotted lines are from model fits to depression interneurone data. A shows parameter values for the second pulse of paired-pulse stimulation given at different time intervals after the first pulse, which occurs just prior to t= 0. The initial values of the parameters (which govern release on pulse 1) are shown at t < 0. B shows parameter values for the fifth pulse as a function of the frequency of the five-pulse train. C shows parameter values at steady state during constant frequency stimulation at different frequencies. All curves were calculated using equations from Methods with parameter values given in Table 1 and Table 2.
Figure 7A3 shows changes of the release probability of active synapses (not including synapses in the refractory period) according to time after the first stimulus. Even for the interneurones that have paired-pulse depression, the release probability of active synapses, P(t), increases following the first stimulus, then decays back to its initial value P1. Comparing Fig. 7A1 and 7A3, the differences between the interneurones and pyramidal cells in release probability per active synapse, P(t), are much greater than the differences in release probability per vesicle, α(t), because of the effect of the differences in the releasable vesicle pool size, n(t). Figure 7A4 demonstrates the dynamics of synapses in the release-ready state after the first stimulus (x(t)). The fraction of release-ready (active) synapses decreases at time t= 0 due to synapses entering the releasing state upon the first stimulation. x(t) then increases back towards 1, indicating the recovery to the release ready-state from the refractory state. The initial decrease in release-ready synapses equals the initial release probability, and is therefore largest for the high probability synapses of the paired-pulse depression interneurones. At these high probability synapses, paired-pulse depression is caused by the decrease in the fraction of synapses that are release ready for the second stimulation (x2), due to synapses that have released on the first pulse and are still inactive when the second pulse arrives. The fraction of synapses that releases on the second pulse is y2= (P2x2). So although P2 increases as a result of the increase in α2, (P2x2) is still less than P1, and therefore the paired-pulse ratio is less than 1. While there is also a decrease in the fraction of release-ready synapses (x2) in pyramidal cells and facilitation interneurones, it is smaller and is offset by the larger relative increase of the release probability per active synapse, resulting in net facilitation. In all three cases the fraction of release-ready synapses recovers with a double exponential time course, with time constants of 44 and 511 ms, due to calcium-dependent recovery from inactivation (Dittman et al. 2000).
Figure 7B shows the values of these factors that determine neurotransmitter release probability at the time of the fifth stimulation as a function of stimulus frequency. For all three cell types, the model predicts that release probability per vesicle increases rapidly as a function of frequency between 0.1 and 20 Hz, and increases only a little more at higher frequencies (Fig. 7B1). Figure 7B2 shows that the readily releasable vesicle pool is only slightly depleted at low stimulus frequencies, and that the amount of depletion does not increase any further at higher frequencies. Therefore depletion appears to play only a small role in short-term plasticity for all three cell groups even during five-pulse trains. The combination of large facilitation of the vesicular release probability and only minor depletion of the readily releasable pool results in a very high release probability per release-ready synapse, P5, at frequencies above 10 Hz (Fig. 7B3). The value of P5 is highest for the synapses onto the depression interneurones, where it approaches 1. However, this leads to a decrease in the number of release-ready synapses, as shown in Fig. 7B4. Again, it is the decrease in release-ready synapses due to refractory depression that causes the short-term depression during five-pulse trains in synapses onto the depression interneurones, not a depletion of the readily releasable vesicle pool.
Figure 7C shows the model predictions for the steady-state values as a function of frequency during continuous stimulation. Again, there is a very large increase in the vesicular release probability with stimulus frequency for all three cell groups (Fig. 7C1). For example, for pyramidal cell synapses the steady-state value of α during 10 Hz stimulation is more than 10-fold greater than its initial value. However, the readily releasable vesicle pool becomes significantly depleted during continuous high-frequency stimulation; at steady state the pool is less than 20% full for frequencies above about 2 Hz (Fig. 7C2). Since the vesicle pool is so depleted, the steady-state values of release probability per active synapse, P∞(f), decline at high frequencies (Fig. 7C3), even though release probability per vesicle is greatly facilitated. For depression interneurones P∞(f) decreases for all frequencies above 0.1 Hz; however, for pyramidal cells and facilitation interneurones P∞(f) increases slightly to a peak at about 1 Hz, after which it declines. While the decrease in P∞(f) due to vesicle depletion is one major cause of high-frequency depression of release at steady state, a decrease in the number of active (release-ready) synapses due to inactivation also contributes (Fig. 7C4). The model predicts that although pyramidal cells and facilitation interneurones have almost identical amounts of high-frequency depression at frequencies above 1 Hz, the relative contributions of synapse inactivation (Fig. 7C4) and the decrease in release probability per active synapse (Fig. 7C3) are different. For pyramidal cells steady-state high-frequency depression results almost equally from the decrease in release probability per active synapse and from synapse inactivation, while for facilitation interneurones there is a larger contribution from synapse inactivation, with a smaller effect of the decrease in release probability per active synapse.
In summary, our results demonstrate that Schaffer collateral synapses have target-cell specific short-term plasticity onto neurones in CA1. Compared with excitatory synapses onto pyramidal cells, synapses onto s. radiatum interneurones have less facilitation after paired-pulse stimulation and during short high-frequency trains. Furthermore, excitatory synapses onto interneurones are heterogeneous in their short-term plasticity, unlike those onto pyramidal cells. All synapses onto pyramidal cells showed robust short-term facilitation, while synapses onto most interneurones showed moderate facilitation, but a subset of interneurones had synapses with short-term depression instead. However, when very long trains of stimuli were given at different frequencies, the amount of steady-state high-frequency depression was not significantly different between the synapses onto pyramidal cells versus the majority of interneurones. A theoretical study based on the simulation of experimental data using a mechanistic model of neurotransmitter release predicts that the differences in short-term plasticity are due to differences in the initial release probability. Our model predicts that excitatory synapses onto interneurones have a higher initial release probability, which we confirm experimentally. Our model also predicts that this difference in initial release probability is due to a larger initial vesicle pool size and a higher release probability per vesicle at synapses onto interneurones, and that the variations in short-term plasticity detected from interneurones are primarily due to the variations in the initial release probability per vesicle. A higher release probability per vesicle results in short-term depression, while a lower release probability per vesicle allows for short-term facilitation. However, this facilitation is still smaller than those seen at synapses onto pyramidal cells because synapses onto interneurones have a larger initial readily releasable vesicle pool size, resulting in a larger initial release probability overall. In addition, our model predicts that paired-pulse depression is caused by a large fraction of synapses becoming refractory/inactive following release at high probability synapses, and not by depletion of the readily releasable vesicle pool.
Discussion
Target-cell specificity of short-term plasticity
Our results indicate that synapses made by CA3 pyramidal cell Schaffer collateral axons onto CA1 pyramidal cells and s. radiatum interneurones have target-specific short-term plasticity, and that excitatory synapses onto interneurones have less facilitation in response to both paired-pulse and short train stimulation compared with synapses onto pyramidal cells. Target-cell specificity in paired-pulse plasticity of Schaffer collateral axons was observed in one previous study; however, that study showed that paired-pulse facilitation at Schaffer collateral excitatory synapses onto CA1 interneurones in s. oriens is higher than those onto pyramidal cells (Scanziani et al. 1998). This difference between Schaffer collateral synapses onto CA1 interneurones in s. oriens versus our observations in s. radiatum further emphasizes that short-term plasticity of Schaffer collateral synapses is target-cell specific even for interneurones. This is also shown by the heterogeneity we observe in the properties of inputs to interneurones within s. radiatum.
Another study reported no difference in the synaptic dynamics of excitatory Schaffer collateral inputs onto CA1 pyramidal cells versus CA1 interneurones (Wierenga & Wadman, 2003), specifically in the plateau values of the EPSCs during constant frequency trains. This is in agreement with our result showing no difference in the steady-state responses versus frequency (Fig. 3). However, they also reported that both types of synapses showed similar amounts of paired-pulse facilitation (Wierenga & Wadman, 2003), in contrast to what we report here (Fig. 1). One possible explanation is that they were recording interneurones in s. pyramidale and s. oriens in addition to s. radiatum. Since Schaffer collateral synapses onto s. oriens interneurones have more facilitation than those onto pyramidal cells (Scanziani et al. 1998), while we show that Schaffer collateral synapses onto s. radiatum interneurones have less facilitation, the two effects might average out. Furthermore, in our experiments we attempted to block known forms of postsynaptic short-term plasticity in order to study presynaptic mechanisms in isolation, whereas both pre- and postsynaptic mechanisms may have contributed in their experiments. For example, activity-dependent relief of polyamine block has been shown to postsynaptically cause paired-pulse facilitation at synapses onto interneurones in neocortex that would otherwise show paired-pulse depression (Rozov & Burnashev, 1999). If this also occurs at synapses onto interneurones in CA1, this would suggest the interesting possibility that this postsynaptic mechanism of facilitation is functioning to compensate for the decreased presynaptic short-term facilitation that is a consequence of having high probability synapses driving s. radiatum interneurones.
We found that short-term plasticity recorded from excitatory synapses onto interneurones was heterogeneous in response to all of the stimulus patterns we used. These differences in short-term plasticity did not correlate with any observable morphological differences between the cells. To date, numerous attempts to correlate differences in hippocampal interneurone physiology with morphology have revealed that functional subgroups often do not correspond to distinct morphological types (e.g. McMahon & Kauer, 1997; McMahon et al. 1998; Parra et al. 1998). It has been proposed that interneurones form functional subgroups based on their differential expression of calcium-binding proteins and neuropeptides (Freund & Buzsaki, 1996). For example, in cortex, bitufted interneurones, many of which contain somatostatin, have excitatory inputs that show strong paired-pulse facilitation, in contrast to multipolar interneurones, some of which contain parvalbumin, that show paired-pulse depression (Reyes et al. 1998). However, a recent study of hippocampal interneurones in s. oriens did not find clear correlations between short-term plasticity and the presence of neuropeptides, calcium-binding proteins, or metabotropic glutamate receptors (Losonczy et al. 2002).
A difference in presynaptic properties between synapses onto pyramidal cells versus interneurones could arise in several ways. A subset of CA3 neurones could innervate only pyramidal cells while another subset with different presynaptic properties innervates only interneurones. Alternatively, terminals from the same CA3 cell axon could have different properties when the target cell is an interneurone versus a pyramidal cell. While our experiments do not directly address this question for interneurones in s. radiatum, there is strong evidence for the latter view for Schaffer collateral synapses onto CA1 pyramidal cells versus s. oriens interneurones in organotypic slices (Scanziani et al. 1998). In either case, the dependence of a presynaptic property on the postsynaptic target cell implies the need for a retrograde signal. Possible signals include both secreted factors such as neurotrophins and membrane-bound factors such as cell adhesion molecules (Fitzsimonds & Poo, 1998). Further work will be needed to determine the nature of such a signal, and whether it is present at synaptogenesis or occurs during activity-dependent synaptic maturation.
Mathematical model of presynaptic short-term plasticity
To investigate the biological mechanisms underlying the differences in short-term plasticity, we developed a mechanistic model incorporating features from several existing models that explicitly describes biological factors that govern release probability, such as the number of readily releasable vesicles and the release probability per vesicle. Our model is based on the mechanistic model of Regehr and colleagues in which synaptic facilitation and depression are linked with non-linear Ca2+ binding processes in presynaptic terminals that are related to residual presynaptic calcium (Dittman & Regehr, 1998; Dittman et al. 2000). The basic concepts of this model were employed in our model, as were the equations for the calcium-bound molecules governing facilitation (CaXF) and calcium-dependent recovery from inactivation (CaXD) (eqns (5) and (8) in Methods). We extended their model, however, by directly linking release probability with the readily releasable vesicle pool size and the release probability per vesicle (Dobrunz & Stevens, 1997; Dobrunz, 2002). Furthermore, we modified the mathematical description of facilitation from Dittman et al. (2000) so that it is the vesicular release probability α(t) that is facilitated in a calcium-dependent manner (eqn (4) in Methods), rather than the overall synaptic release probability per active synapse P(t) (which corresponds to the facilitation factor F(t) in their model). In our model P(t) also depends upon the readily releasable vesicle pool size n(t) (see Methods), which is not subject to the same facilitation, but instead can deplete. While we were able to fit the experimental data for the paired-pulse and five-pulse stimulus protocols without making these modifications to their model, they were necessary to enable the model to fit the experimental results for steady-state high-frequency depression. We therefore used the model with these modifications to analyse all of our experimental data. Consistent with this, further analysis of the model as shown in Fig. 7 suggests that depletion of the readily releasable vesicle pool plays very little role during paired-pulse stimulation (Fig. 7A2) and only a minor role during five-pulse stimulation (Fig. 7B2), but is a major source of depression during steady-state high-frequency stimulation (Fig. 7C2).
All of our experimental data for the three different cell groups, each in response to three different stimulus protocols, can be simulated by changing only the values of α1, nT and τF. The best fit of the model to all of the data occurs when all parameters except α1 and nT are kept the same for the three different cell groups. This suggests that there are many identical properties between excitatory synapses onto interneurones and those onto pyramidal cells, and that the differences in short-term plasticity between these two cell types are not due to differences in the mechanisms of facilitation or depression. For each cell group, α1 and nT are kept the same to fit all data obtained for the three different stimulation protocols. Because these parameters reflect the initial state of the synapses, they should be independent of the stimulus protocol.
The third variable τF changes for the different stimulus protocols, but it is the same for all cells recorded whether they were pyramidal cells or interneurones. This variable determines the decay rate after an action potential of the calcium-bound molecule CaXF that governs facilitation. As described in Dittman et al. (2000), this will be dependent upon residual calcium in the presynaptic terminal, although it does not directly reflect the time course of the decay of free calcium. The increases in τF with increasing numbers of stimuli may be due to the saturation of calcium buffers in presynaptic terminals, which could decrease the removal of calcium from the active zone in presynaptic terminals and result in a slower decay of residual calcium and of facilitation. Alternatively, a more complex version of the model that provides a more detailed description of presynaptic calcium dynamics and actually describes the time course of residual free calcium during different stimulation patterns could be developed. Since our model provides a good fit for all of the data if we allow τF to depend upon the stimulus pattern, we chose to use the model in its current, more simplified form. However, since τF is held constant for all three cell groups simulated, the model therefore predicts that differences in short-term plasticity between cell types are only determined by the two parameters that determine the initial release probability of the synapse, α1 and nT.
Difference in initial release probability underlies target-cell specific short-term plasticity
Our model therefore predicts that all of the differences in short-term plasticity between Schaffer collateral synapses onto s. radiatum interneurones versus pyramidal cells result from a difference in initial release probability per vesicle and the initial vesicle pool size. They are both larger at synapses onto interneurones, resulting in a larger synaptic release probability. This is consistent with the observation that the initial release probability per vesicle and the initial vesicle pool size also scale together at individual Schaffer collateral synapses onto CA1 pyramidal cells, contributing to the observed heterogeneity of the overall release probability (Dobrunz, 2002). The values we estimate for α1 (0.055–0.090) for the three cell groups are in the range of what has been previously reported for synapses onto hippocampal neurones in culture (Murthy et al. 1997; Junge et al. 2004) and in hippocampal slices (Dobrunz & Stevens, 1997; Dobrunz, 2002). In particular, the value of α1= 0.055 we obtain for Schaffer collateral synapses onto pyramidal cells is very close to the value of α1= 0.044 estimated by Wesseling & Lo (2002) for Schaffer collateral synapses. Similarly, the values of nT (4.8–10.0) are also comparable to the size of the readily releasable vesicle pool measured electrophysiologically (Stevens & Tsujimoto, 1995; Dobrunz & Stevens, 1997; Dobrunz, 2002) or optically (Murthy et al. 2001) for excitatory synapses onto hippocampal neurones.
Thus the model predicts that the initial release probability is low for inputs to pyramidal cells (P1= 0.24), higher for inputs to the facilitation interneurones (P1= 0.37), and even higher for inputs to the depression interneurones (P1= 0.61). For the pyramidal cells, the value of P1 predicted is in close agreement with previous experimental studies that suggested that the average P1 of their inputs is 0.2–0.3 (Allen & Stevens, 1994; Huang & Stevens, 1997). The release probability of excitatory inputs onto CA1 interneurones has not yet been measured directly, so the values of 0.37–0.61 that the model predicts will need to be confirmed by future experiments. Previous studies have suggested that synaptic connections from CA3 pyramidal cells onto CA3 interneurones are very strong (Miles, 1990) and show only modest paired-pulse facilitation (Miles & Wong, 1986), indicating that they are likely to also be high probability synapses. However, Schaffer collateral synapses onto s. oriens interneurones have large paired-pulse facilitation and thus are likely to be low probability synapses (Scanziani et al. 1998), indicating this is not a feature of all inputs to hippocampal interneurones.
The amount of PPF is a widely used indicator of presynaptic function, and a change in the paired-pulse ratio is often interpreted as indicating a change in the initial release probability in the opposite direction (reviewed in Zucker 1989, 1999). Manipulations that decrease P1, such as lowering the extracellular calcium concentration or activation of presynaptic adenosine receptors, increase the paired-pulse ratio, and vice versa (e.g. Dumas & Foster, 1998). When directly measured at individual synapses of the same type, PPF is inversely proportional to P1 across the population of synapses (Debanne et al. 1996; Dobrunz & Stevens, 1997). Differences in PPF at different types of synapses have been shown to correlate with differences in average P1 (Castro-Alamancos & Connors, 1997). In addition, manipulations that reduce P1 at depressing synapses can unmask PPF (Mennerick & Zorumski, 1995).
Our model results suggest that the lower paired-pulse ratio at excitatory synapses onto CA1 interneurones results from a higher initial release probability at these synapses compared with synapses onto pyramidal cells. It was important to directly test this prediction, because the paired-pulse ratio does not always directly correlate with P1. For example, synapses between layer 5 pyramidal cells all show PPD despite having P1 ranging from 0.1 to 0.95 (Markram et al. 1997; Tsodyks & Markram, 1997). At those synapses, lowering P1 by lowering extracellular calcium does not reveal PPF, suggesting they lack the capacity for facilitation (Markram et al. 1998). Similarly, mice deficient for neurotrophins show reduced PPF with no change in average P1 (Kokaia et al. 1998), and over-expression of the protein neuronal calcium sensor 1 in hippocampal cultures causes an increase in PPF with no change in P1 (Sippy et al. 2003).
We confirmed that synapses onto interneurones have a higher initial release probability by showing a faster rate of block of NMDA EPSCs by the open channel blocker MK-801. While this method does not give a direct measurement of the average P1 (Huang & Stevens, 1997), it is a sensitive indicator for differences in the average P1, provided that there is no difference in the fraction of participating receptors that gets blocked by MK-801. This depends upon the fraction of open NMDA receptors blocked by MK-801 (block fraction) and the fraction of receptors that participate in the current when transmitter is released (Huang & Stevens, 1997). Since the fraction of participating receptors depends upon the NMDA receptor subunit composition (Erreger et al. 2005), it is possible that this fraction could be different at synapses onto interneurones versus pyramidal cells. If so, this could either contribute to or reduce the observed difference in the rate of block by MK-801. However, the time course of NMDA-mediated EPSCS is similar in interneurones and pyramidal cells (Morin et al. 1996), suggesting that their subunit composition is similar and that this is not likely to be a large effect. As the block fraction is not different between synapses onto interneurones and pyramidal cells, the faster rate of block by MK-801 of synapses onto interneurones provides strong support of our model finding that synapses onto interneurones have a higher release probability.
Previous experiments have shown that an increase in P1 can result in an increase in steady-state depression (Markram, 1997), yet for the majority of interneurones we studied (the facilitation interneurones), their inputs had the same amount of steady-state high-frequency depression as pyramidal cell inputs for stimulus frequencies above 1 Hz. However, in that study P1 was raised acutely by increasing extracellular calcium, which our model predicts will result in an increase in the amount of steady-state high-frequency depression due to an increase in α1. Our model also predicts that an increase in P1 due to an increase in the initial readily releasable pool size would decrease high-frequency depression. This is consistent with experiments that have shown that brain-derived neurotrophic factor (BDNF), which increases the number of docked vesicles (Tyler & Pozzo-Miller, 2001) (and presumably also the readily releasable pool size), causes a decrease in high-frequency depression (Gottschalk et al. 1998). Our model suggests that both nT and α1 are greater in synapses onto interneurones as compared with pyramidal cells, and for the facilitation interneurones the effects on high-frequency depression offset each other except at 1 Hz. Both nT and α1 are greater at inputs to the depression interneurones versus the facilitation interneurones, but in this case the increase in α1 has the greater effect, resulting in more high-frequency depression. Therefore, it is not only the initial release probability that determines short-term plasticity; the relative contributions of the readily releasable pool size and release probability per vesicle are also important. Because these two parameters can be modulated independently, this enables differential regulation of the initial synaptic strength and of short-term synaptic dynamics.
Several possible mechanisms could account for the difference in the vesicular release probability α1 between synapses onto interneurones versus pyramidal cells. These include differences in presynaptic calcium influx caused by either variations in the density of calcium channels (Rozov et al. 2001a) or in channel properties (Scheuber et al. 2004), differences in the proximity of calcium channels to vesicles (Bennett et al. 2000; Rozov et al. 2001a; Scheuber et al. 2004), or the differences in effects of presynaptic calcium buffering. In cortex, differences in the density of calcium channels and/or the distance between channels and vesicles are thought to contribute to observed differences in release probability between excitatory synapses onto bitufted versus multipolar interneurones (Rozov et al. 2001a). Thus, this may be a common mechanism governing the target cell specificity of release probability per vesicle at excitatory synapses in neocortex and hippocampus.
Our model also predicts that the initial readily releasable pool size, nT, is larger at synapses onto interneurones. Since the readily releasable pool is the subset of vesicles docked at the active zone that are primed (Sudhof, 2004), this could result from an increase in the number of docked vesicles and/or the fraction of docked vesicles that is primed. At synapses onto pyramidal cells, the number of docked vesicles can be regulated by the neurotrophic factor BDNF (Lu & Chow, 1999; Tyler & Pozzo-Miller, 2001). Similarly, the readily releasable pool size has been shown to be modulated by the second messenger diacylglycerol (Rhee et al. 2002) and by calmodulin (Junge et al. 2004) through interactions with the vesicle priming factor Munc 13 (Augustin et al. 1999; Betz et al. 2001; Rosenmund et al. 2002). The readily releasable vesicle pool size may also be regulated by protein kinase A-dependent phosphorylation of the SNARE protein SNAP-25 (Nagy et al. 2004). However, in cultured hippocampal synapses the readily releasable pool size measured physiologically corresponds closely to the number of anatomically docked vesicles measured with electron microscopy, suggesting that all docked vesicles are normally primed (Murthy et al. 2001; Schikorski & Stevens, 2001). It remains to be determined what mechanisms cause the increases in the readily releasable pool size at excitatory synapses onto interneurones.
Model assumptions
It has been reported that the refilling rate of the readily releasable vesicle pool can be increased by accumulation of intracellular calcium (Ca2+i) during high-frequency stimuli (Neher, 1998; Stevens & Wesseling, 1998; Wang & Kaczmarek, 1998), or by the saturation of calcium buffers at the release site. In our mathematical model the refilling rate of the readily releasable pool was treated as a constant during stimulation and for all of the different cell types. This simplification suggests that there is no difference between different cell types in the refilling rate, rather than requiring no effect of Ca2+i on the refilling rate. A more complex version of the model could include a dependence of the refilling rate, but this simplification worked well for the simulation of our data. Since depletion of the readily releasable vesicle pool appears to play little role in the responses to paired-pulse and five-pulse stimulation, a dependence of the refilling rate on Ca2+i is not likely to have much effect on these simulations. However, during longer trains of stimulation, activity-dependent increases in the refilling rate may help to limit depletion of the readily releasable pool. Unless the refilling rate of synapses onto interneurones is differentially dependent on activity, this is not likely to significantly affect our results.
In our model, we assume all short-term plasticity is due to presynaptic mechanisms by which activity alters the probability of neurotransmitter release. With few exceptions, mechanisms of short-term facilitation have been shown to be presynaptic at all synapses studied (reviewed in Zucker & Regehr, 2002; see also Fisher et al. 1997). Post-synaptic short-term facilitation can be caused by activity-dependent relief of magnesium block of NMDA receptors; we prevent this by blocking NMDA receptors with APV, which also prevents induction of most forms of long-term plasticity. In addition, Ca2+-permeable AMPA receptors show voltage-dependent block by endogenous intracellular polyamines (Koh et al. 1995; Kamboj et al. 1995; Bowie et al. 1998). Relief of this block is use dependent (Bowie et al. 1998; Rozov et al. 1998), and causes a postsynaptic form of short-term facilitation at synapses containing these receptors (McBain, 1998; Rozov & Burnashev, 1999). We included 10 mm ATP in the patch pipette to chelate endogenous polyamines and prevent this postsynaptic form of short-term plasticity (Toth et al. 2000).
Desensitization of postsynaptic receptors can also contribute to short-term depression, although the role of receptor desensitization does not appear to be large at most excitatory synapses (reviewed in Zucker & Regehr, 2002). It has previously been shown that short-term plasticity at Schaffer collateral synapses onto CA1 pyramidal cells occurs presynaptically, with no change in the size of the postsynaptic quantal response (Dobrunz & Stevens, 1997; Dobrunz et al. 1997; Dobrunz, 2002). This indicates that any receptor desensitization that does occur recovers faster than the synapse recovers from the inactive/refractory state, and therefore does not contribute to short-term plasticity. In our model we assume that this is also true for Schaffer collateral synapses onto interneurones. We confirmed this for interneurones with facilitating inputs, which make up the majority of s. radiatum interneurones, by showing no effect of cyclothiazide on short-term facilitation or depression. We also considered the possibility that slow recovery from desensitization may be one cause of the increased short-term depression and the PPD (rather than PPF) that we observed in a small subset of interneurones, as has been observed at synapses onto bipolar interneurones in layer 2/3 of rat neocortex (Rozov et al. 2001b). However, short-term plasticity does not appear to be affected by desensitization at synapses onto layer 2/3 multipolar interneurones, which also have PPD, indicating that this is not a feature of all interneurone synapses with paired-pulse depression. As the depression interneurones are relatively rare in s. radiatum, all the interneurones that we tested with cyclothiazide were facilitating interneurones, and thus we cannot directly rule out this possibility. We think it is unlikely, however, that our depression interneurones contain the same type of AMPA receptors (with the very slow recovery from desensitization) as the layer 2/3 bipolar interneurones, because the time course of recovery from PPD (and from desensitization) is an order of magnitude slower in those cells (4–5 s) (Rozov et al. 2001b) than the time course of PPD in our depression interneurones (approximately 500 ms).
Our model also assumes that the size of the presynaptic calcium influx is the same for every action potential, independent of the stimulus pattern. Presynaptic calcium currents at the calyx of Held have been shown to be able to undergo both activity-dependent facilitation and inactivation (Forsythe et al. 1998; Cuttle et al. 1998; Borst & Sakmann, 1998). However, calcium currents are believed to reliably couple to presynaptic action potentials at synapses in hippocampus (Mackenzie et al. 1996) and cortex (Cox et al. 2000; Koester & Sakmann, 2000), even during bursts of action potentials (Cox et al. 2000), supporting the assumption used in our model. While facilitation is believed to be due to residual calcium in the presynaptic terminal, an activity-dependent increase in calcium influx would also cause facilitation. Activity-dependent inactivation of calcium channels, in contrast, would provide a release-independent form of synaptic depression that would be distinct from the release-dependent inactivation in our model. If conditions are found under which there is facilitation or inactivation of presynaptic calcium currents at hippocampal synapses, our model could be extend to include this mechanism.
Other model result and predictions
We used the mathematical method we developed for theoretical studies of the dynamics of release possibility, vesicle pool size and release-ready state during different stimulus patterns, which are difficult to measure experimentally in intact slices. Previous experiments recording from single synapses onto CA1 pyramidal cells have observed a refractory period after a vesicle release which recovers with a time constant of approximately 20 ms (Dobrunz & Stevens, 1997). From Fig. 7A4, our model predicts that the total recovery of synapses from refractory depression into the release-ready state after a release takes slightly longer than that (τD= 50 ms). However, in the single synapse experiments the measured variable was the overall release probability (called p in that paper, called y here), which includes contributions from both facilitation and refractory depression. The presence of facilitation is likely to have made the recovery from refractory depression appear faster than it actually was.
Paired-pulse depression has traditionally been explained by a depletion of presynaptic vesicles at high probability synapses (Thies, 1965; Betz, 1970), although some studies have suggested that paired-pulse depression may also be evoked by other mechanisms (Brody & Yue, 2000; Caillard et al. 2000; Waldeck et al. 2000; Kirischuk et al. 2002; Munoz-Cuevas et al. 2004), and may be due to a calcium-dependent process (Bellingham et al. 1999). Our results suggest that there are two main factors involved in short-term depression: depletion of the readily releasable pool and the refractory period that occurs after a synapse releases. We find that the depletion of vesicle pool size contributes very little to paired-pulse depression (Fig. 7A2) (Waldeck et al. 2000), and that the paired-pulse depression observed at the high probability synapses onto some interneurones is instead caused by the large fraction of synapses that is refractory following neurotransmitter release (Fig. 7A4). In addition, depletion appears to play only a minor role during short stimulus trains (Fig. 7A3), and synapse inactivation is responsible for the depression seen at synapses onto interneurones. However, depletion of readily releasable vesicles does play a big role in high-frequency depression at steady state (Fig. 7C2), in addition to the accumulation of synapses in the refractory state (Fig. 7C4). Recovery from the refractory state is a calcium-dependent process, which may help to limit depression during periods of high-frequency stimulation (Dittman et al. 2000).
Conclusions
We find that presynaptic short-term plasticity is target-cell specific at Schaffer collateral excitatory synapses onto CA1 pyramidal cells versus s. radiatum interneurones. Since differences in presynaptic function have also been seen between inputs to excitatory versus inhibitory neurones in other parts of the brain, this suggests the possibility that a difference in short-term plasticity may be a general feature of excitatory synapses onto interneurones versus pyramidal cells in the brain. Differences in short-term plasticity between synapses onto interneurones versus pyramidal cells will lead to different frequency dependence in response to complex stimuli, and may result in different output firing rates in the two target cell types even though they receive common input. This will be important in regulating the overall balance of excitation and inhibition in hippocampus, a brain region highly susceptible to epileptiform activity.
By combining electrophysiological recordings with mathematical modelling, we have provided a detailed analysis of the differences in short-term synaptic plasticity at Schaffer collateral excitatory synapses onto CA1 pyramidal cells versus s. radiatum interneurones. Our model predicts that synapses onto interneurones have a higher initial release probability than do synapses onto pyramidal cells, a result confirmed experimentally. The model further predicts that this is caused by increases in both the release probability per vesicle and the readily releasable vesicle pool size. As these two mechanisms for regulating release probability have differing effects on short-term plasticity, this provides a mechanism by which presynaptic terminals can differentially regulate synaptic strength and short-term dynamics to different postsynaptic target cells.
Acknowledgments
The authors thank Brandon Walters, Felecia Hester and Dr Lori McMahon for their technical assistance, as well as Dr McMahon and Dr Smadar Lapidot for their reviews of this manuscript. This work was supported by NIH R01 MH65328 from the National Institute of Mental Health.
References
- Allen C, Stevens CF. An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci U S A. 1994;91:10380–10383. doi: 10.1073/pnas.91.22.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Bahring R, Bowie D, Benveniste M, Mayer ML. Permeation and block of rat GluR6 glutamate receptor channels by internal and external polyamines. J Physiol. 1997;502:575–589. doi: 10.1111/j.1469-7793.1997.575bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC, Lim R, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Farnell L, Gibson WG. The probability of quantal secretion near a single calcium channel of an active zone. Biophys J. 2000;78:2201–2221. doi: 10.1016/S0006-3495(00)76769-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, Brose N. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron. 2001;30:183–196. doi: 10.1016/s0896-6273(01)00272-0. [DOI] [PubMed] [Google Scholar]
- Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol. 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol. 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Lange GD, Mayer ML. Activity-dependent modulation of glutamate receptors by polyamines. J Neurosci. 1998;18:8175–8185. doi: 10.1523/JNEUROSCI.18-20-08175.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. J Neurosci. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillard O, Moreno H, Schwaller B, Llano I, Celio MR, Marty A. Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc Natl Acad Sci U S A. 2000;97:13372–13377. doi: 10.1073/pnas.230362997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Connors BW. Distinct forms of short-term plasticity at excitatory synapses of hippocampus and neocortex. Proc Natl Acad Sci U S A. 1997;94:4161–4166. doi: 10.1073/pnas.94.8.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Harata NC, Tsien RW. Paired-pulse depression of unitary quantal amplitude at single hippocampal synapses. Proc Natl Acad Sci U S A. 2004;101:1063–1068. doi: 10.1073/pnas.0307149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Cox CL, Denk W, Tank DW, Svoboda K. Action potentials reliably invade axonal arbors of rat neocortical neurons. Proc Natl Acad Sci U S A. 2000;97:9724–9728. doi: 10.1073/pnas.170278697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Boudin H. Molecular heterogeneity of central synapses: afferent and target regulation. Nat Neurosci. 2001;4:569–578. doi: 10.1038/88388. [DOI] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Kreitzer AC, Regehr WG. Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci. 2000;20:1374–1385. doi: 10.1523/JNEUROSCI.20-04-01374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE. Release probability is regulated by the size of the readily releasable vesicle pool at excitatory synapses in hippocampus. Intl J Dev Neurosci. 2002;20:225–236. doi: 10.1016/s0736-5748(02)00015-1. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Huang EP, Stevens CF. Very short-term plasticity in hippocampal synapses. Proc Natl Acad Sci U S A. 1997;94:14843–14847. doi: 10.1073/pnas.94.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Response of hippocampal synapses to natural stimulation patterns. Neuron. 1999;22:157–166. doi: 10.1016/s0896-6273(00)80687-x. [DOI] [PubMed] [Google Scholar]
- Dumas TC, Foster TC. Late developmental changes in the ability of adenosine A1 receptors to regulate synaptic transmission in the hippocampus. Brain Res Dev Brain Res. 1998;105:137–139. [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SA, Fischer TM, Carew TJ. Multiple overlapping processes underlying short-term synaptic enhancement. Trends Neurosci. 1997;20:170–177. doi: 10.1016/s0166-2236(96)01001-6. [DOI] [PubMed] [Google Scholar]
- Fitzsimonds RM, Poo MM. Retrograde signaling in the development and modification of synapses. Physiol Rev. 1998;78:143–170. doi: 10.1152/physrev.1998.78.1.143. [DOI] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanse E, Gustafsson B. Quantal variability at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J Physiol. 2001;531:467–480. doi: 10.1111/j.1469-7793.2001.0467i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Sultan P. Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses. Neuropharmacol. 1995;34:1387–1395. doi: 10.1016/0028-3908(95)00142-s. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Isaac JT, Nicoll RA, Malenka RC. Lack of AMPA receptor desensitization during basal synaptic transmission in the hippocampal slice. J Neurophysiol. 1999;81:3096–3099. doi: 10.1152/jn.1999.81.6.3096. [DOI] [PubMed] [Google Scholar]
- Huang EP, Stevens CF. Estimating the distribution of synaptic reliabilities. J Neurophysiol. 1997;78:2870–2880. doi: 10.1152/jn.1997.78.6.2870. [DOI] [PubMed] [Google Scholar]
- Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, Brose N. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell. 2004;118:389–401. doi: 10.1016/j.cell.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol. 1995;486:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Clements JD, Grantyn R. Presynaptic and postsynaptic mechanisms underlie paired pulse depression at single GABAergic boutons in rat collicular cultures. J Physiol. 2002;543:99–116. doi: 10.1113/jphysiol.2002.021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol. 2000;529:625–646. doi: 10.1111/j.1469-7793.2000.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DS, Burnashev N, Jonas P. Block of native Ca2+-permeable AMPA receptors in rat brain by intracellular polyamines generates double rectification. J Physiol. 1995;486:305–312. doi: 10.1113/jphysiol.1995.sp020813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia M, Asztely F, Olofsdotter K, Sindreu CB, Kullmann DM, Lindvall O. Endogenous neurotrophin-3 regulates short-term plasticity at lateral perforant path-granule cell synapses. J Neurosci. 1998;18:8730–8739. doi: 10.1523/JNEUROSCI.18-21-08730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn H, Sur C, Charpier S, Legendre P, Faber D, editors. The One-Vesicle Hypothesis and Multivesicular Release. New York: Raven Press, Ltd; 1994. [DOI] [PubMed] [Google Scholar]
- Laezza F, Doherty JJ, Dingledine R. Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science. 1999;285:1411–1414. doi: 10.1126/science.285.5432.1411. [DOI] [PubMed] [Google Scholar]
- Liaw JS, Berger TW. Dynamic synapse: a new concept of neural representation and computation. Hippocampus. 1996;6:591–600. doi: 10.1002/(SICI)1098-1063(1996)6:6<591::AID-HIPO4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Zhang L, Shigemoto R, Somogyi P, Nusser Z. Cell type dependence and variability in the short-term plasticity of EPSCs in identified mouse hippocampal interneurones. J Physiol. 2002;542:193–210. doi: 10.1113/jphysiol.2002.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res. 1999;58:76–87. [PubMed] [Google Scholar]
- Maass W, Zador AM. Dynamic stochastic synapses as computational units. Neural Comput. 1999;11:903–917. doi: 10.1162/089976699300016494. [DOI] [PubMed] [Google Scholar]
- McBain CJ. A short-term mechanism of plasticity for interneurones? J Physiol. 1998;511:331. doi: 10.1111/j.1469-7793.1998.331bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie PJ, Umemiya M, Murphy TH. Ca2+ imaging of CNS axons in culture indicates reliable coupling between single action potentials and distal functional release sites. Neuron. 1996;16:783–795. doi: 10.1016/s0896-6273(00)80098-7. [DOI] [PubMed] [Google Scholar]
- McMahon LL, Kauer JA. Hippocampal interneurons express a novel form of synaptic plasticity. Neuron. 1997;18:295–305. doi: 10.1016/s0896-6273(00)80269-x. [DOI] [PubMed] [Google Scholar]
- McMahon LL, Williams JH, Kauer JA. Functionally distinct groups of interneurons identified during rhythmic carbachol oscillations in hippocampus in vitro. J Neurosci. 1998;18:5640–5651. doi: 10.1523/JNEUROSCI.18-15-05640.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H. A network of tufted layer 5 pyramidal neurons. Cereb Cortex. 1997;7:523–533. doi: 10.1093/cercor/7.6.523. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Roth A, Sakmann B. Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J Physiol. 1997;500:409–440. doi: 10.1113/jphysiol.1997.sp022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Pikus D, Gupta A, Tsodyks M. Potential for multiple mechanisms, phenomena and algorithms for synaptic plasticity at single synapses. Neuropharmacol. 1998;37:489–500. doi: 10.1016/s0028-3908(98)00049-5. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol. 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R. Synaptic excitation of inhibitory cells by single CA3 hippocampal pyramidal cells of the guinea-pig in vitro. J Physio. 1990;428:61–77. doi: 10.1113/jphysiol.1990.sp018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Wong RK. Excitatory synaptic interactions between CA3 neurones in the guinea-pig hippocampus. J Physiol. 1986;373:397–418. doi: 10.1113/jphysiol.1986.sp016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin F, Beaulieu C, Lacaille JC. Membrane properties and synaptic currents evoked in CA1 interneuron subtypes in rat hippocampal slices. J Neurophysiol. 1996;76:1–16. doi: 10.1152/jn.1996.76.1.1. [DOI] [PubMed] [Google Scholar]
- Munoz-Cuevas J, Vara H, Colino A. Characterization of release-independent short-term depression in the juvenile rat hippocampus. J Physiol. 2004;558:527–548. doi: 10.1113/jphysiol.2004.062133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y. Inactivity produces increases in neurotransmitter release and synapse size. Neuron. 2001;32:673–682. doi: 10.1016/s0896-6273(01)00500-1. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:559–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sorensen JB. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron. 2004;41:417–429. doi: 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Parra P, Gulyas AI, Miles R. How many subtypes of inhibitory cells in the hippocampus? Neuron. 1998;20:983–993. doi: 10.1016/s0896-6273(00)80479-1. [DOI] [PubMed] [Google Scholar]
- Redman SJ. Quantal analysis of synaptic potentials in neurons of the central nervous system. Physiol Rev. 1990;70:165–198. doi: 10.1152/physrev.1990.70.1.165. [DOI] [PubMed] [Google Scholar]
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Sudhof TC, Takahashi M, Rosenmund C, Brose N. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron. 2002;33:411–424. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N. Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature. 1999;401:594–598. doi: 10.1038/44151. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol. 2001a;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Jerecic J, Sakmann B, Burnashev N. AMPA receptor channels with long-lasting desensitization in bipolar interneurons contribute to synaptic depression in a novel feedback circuit in layer 2/3 of rat neocortex. J Neurosci. 2001b;21:8062–8071. doi: 10.1523/JNEUROSCI.21-20-08062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Zilberter Y, Wollmuth LP, Burnashev N. Facilitation of currents through rat Ca2+-permeable AMPA receptor channels by activity-dependent relief from polyamine block. J Physiol. 1998;511:361–377. doi: 10.1111/j.1469-7793.1998.361bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Charpak S. Target cell-specific modulation of transmitter release at terminals from a single axon. Proc Natl Acad Sci U S A. 1998;95:12004–12009. doi: 10.1073/pnas.95.20.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuber A, Miles R, Poncer JC. Presynaptic Cav2.1 and Cav2.2 differentially influence release dynamics at hippocampal excitatory synapses. J Neurosci. 2004;24:10402–10409. doi: 10.1523/JNEUROSCI.1664-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- Sippy T, Cruz-Martin A, Jeromin A, Schweizer FE. Acute changes in short-term plasticity at synapses with elevated levels of neuronal calcium sensor-1. Nat Neurosci. 2003;6:1031–1038. doi: 10.1038/nn1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorra KE, Harris KM. Occurrence and three-dimensional structure of multiple synapses between individual radiatum axons and their target pyramidal cells in hippocampal area CA1. J Neurosci. 1993;13:3736–3748. doi: 10.1523/JNEUROSCI.13-09-03736.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci U S A. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Thies RE. Neuromuscular depression and the apparent depletion of transmitter in mammalian muscle. J Neurophys. 1965;28:427–442. doi: 10.1152/jn.1965.28.3.427. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Molecular frequency filters at central synapses. Prog Neurobiol. 2000;62:159–196. doi: 10.1016/s0301-0082(00)00008-3. [DOI] [PubMed] [Google Scholar]
- Toth K, McBain CJ. Target-specific expression of pre- and postsynaptic mechanisms. J Physiol. 2000;525:41–51. doi: 10.1111/j.1469-7793.2000.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K, Suares G, Lawrence JJ, Philips-Tansey E, McBain CJ. Differential mechanisms of transmission at three types of mossy fiber synapse. J Neurosci. 2000;20:8279–8289. doi: 10.1523/JNEUROSCI.20-22-08279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsodyks MV, Markram H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci U S A. 1997;94:719–723. doi: 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsodyks M, Pawelzik K, Markram H. Neural networks with dynamic synapses. Neural Computation. 1998;10:821–835. doi: 10.1162/089976698300017502. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeck RF, Pereda A, Faber DS. Properties and plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wesseling JF, Lo DC. Limit on the role of activity in controlling the release-ready supply of synaptic vesicles. J Neurosci. 2002;22:9708–9720. doi: 10.1523/JNEUROSCI.22-22-09708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Wadman WJ. Excitatory inputs to CA1 interneurons show selective synaptic dynamics. J Neurophysiol. 2003;90:811–821. doi: 10.1152/jn.00865.2002. [DOI] [PubMed] [Google Scholar]
- Zador AM, Dobrunz LE. Dynamic synapses in the cortex. Neuron. 1997;19:1–4. doi: 10.1016/s0896-6273(00)80341-4. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]