

Abstract
At least two functionally distinct transient outward K+ current (Ito) phenotypes can exist across the free wall of the left ventricle (LV). Based upon their voltage-dependent kinetics of recovery from inactivation, these two phenotypes are designated ‘Ito,fast’ (recovery time constants on the order of tens of milliseconds) and ‘Ito,slow’ (recovery time constants on the order of thousands of milliseconds). Depending upon species, either Ito,fast, Ito,slow or both current phenotypes may be expressed in the LV free wall. The expression gradients of these two Ito phenotypes across the LV free wall are typically heterogeneous and, depending upon species, may consist of functional phenotypic gradients of both Ito,fast and Ito,slow and/or density gradients of either phenotype. We review the present evidence (molecular, biophysical, electrophysiological and pharmacological) for Kv4.2/4.3 α subunits underlying LV Ito,fast and Kv1.4 α subunits underlying LV Ito,slow and speculate upon the potential roles of each of these currents in determining frequency-dependent action potential characteristics of LV subepicardial versus subendocardial myocytes in different species. We also review the possible functional implications of (i) ancillary subunits that regulate Kv1.4 and Kv4.2/4.3 (Kvβ subunits, DPPs), (ii) KChIP2 isoforms, (iii) spider toxin-mediated block of Kv4.2/4.3 (Heteropoda toxins, phrixotoxins), and (iv) potential mechanisms of modulation of Ito,fast and Ito,slow by cellular redox state, [Ca2+]i and kinase-mediated phosphorylation. Ito phenotypic activation and state-dependent gating models and molecular structure–function relationships are also discussed.
The diverse nature of cardiac myocyte action potentials arises from the non-linear and finely balanced gating characteristics of many different ionic currents (Carmeliet & Vereecke, 2002; Rudy, 2002; Nerbonne & Kass, 2003; Bondarenko et al. 2004; Puglisi, Wang & Bers, 2004). This electrical diversity arises from the combined effects of spatial tissue gradients of gene expression, biophysical channel gating mechanisms, second messenger systems, intracellular Ca2+ regulatory mechanisms and complex interactions between these multiple systems. With the advent of isolated cardiac myocyte techniques, patch clamp technology and molecular biological protocols our knowledge of many of these currents and their associated regulatory systems has significantly advanced. Nonetheless, it is sobering to realize that we are still unable to explain and quantitatively model (without making unverified assumptions) many fundamental aspects of cardiac myocyte action potentials. This is particularly true in the case of ventricular myocytes.
Due to their primary role in repolarization, and hence potential status as substrates for therapeutic interventions, extensive experimental effort has been devoted to characterizing mechanisms underlying expression and regulation of voltage-dependent potassium (Kv) channels in ventricular myocytes (Archer & Rusch, 2001; Strauss et al. 2001; Nerbonne & Kass, 2003). Patch clamp studies on atrial and ventricular myocytes have revealed a minimum of at least five distinct Kv-mediated native potassium current phenotypes which can be classified into two general functional groups: (i) ‘Ito’, rapidly activating and inactivating transient outward currents; and (ii) ‘IK’ or ‘delayed rectifiers’. In this review we focus on recent advances made in our understanding of the molecular, cellular and biophysical mechanisms underlying generation and regulation of Ito current density and phenotypic expression gradients across the well-studied left ventricular free wall. We also point out areas where there is either significant lack of understanding or major debate. This article can thus be viewed as an ‘update’ of the two excellent reviews on this subject that have previously appeared in this journal (Nerbonne, 2000; Sah et al. 2003). For readers interested in aspects of Ito not covered in this review the following sources may be of value: cell biology of Ito, Birnbaum et al. (2004); pathology, Chien (1999), Oudit et al. (2001), Nerbonne (2004) and Zicha et al. (2004); Ito expression gradients in other regions of the heart (e.g. apical–basal gradients), Brahmajothi et al. (1999), de Bakker & Opthof (2002) and Brunet et al. (2004); and the role of Ito phenotypes in supraventricular disturbances, Chien (1999), Van Wagoner & Nerbonne (2001) and Gussack & Antzelevitch (2003).
Cardiac ventricular Ito phenotypes
General characteristics
Ventricular Ito currents display several shared basic characteristics (Campbell et al. 1995; Strauss et al. 2001). First, all display high K+ selectivity (PNa/PK < 0.1), and hence are repolarizing currents. Second, ventricular Ito currents display similar voltage-dependent gating characteristics in that all (i) activate rapidly (time constants on the order of milliseconds) in a voltage-dependent manner which is independent of previous Ca2+ influx, and (ii) inactivate (either as a single or mutliexponential process) with maintained depolarization (time constants on the order of tens to hundreds of milliseconds). All Ito currents are thus modulators of both early phase 1 repolarization and the excitation–contraction (EC) coupling cycle (Campbell et al. 1995; Bers, 2001; Oudit et al. 2001; Nerbonne & Kass, 2003; Sah et al. 2003) (Fig. 1).
Figure 1. Conventional whole cell patch clamp view of Ito: theoretically predicted kinetic behaviour of Ito,fast during a ventricular action potential.

The illustrated action potential was recorded from a ferret right ventricular myocyte (22°C), while the corresponding Ito,fast waveform was generated using the recorded action potential waveform as the voltage ‘driving function’ for the Ito,fast gating equations developed by Campbell et al. (1993a, b). Such predicted behaviour may only reflect the kinetics of Ito,fast under ‘basal’ conditions, i.e. in the absence of normal [Ca2+]i transients and minimal kinase-mediated regulation. Figure reproduced from Campbell et al. (1995) with permission from Elsevier.
Prominent Ito currents have been recorded in ventricular myocytes isolated from the hearts of many species, including mice, rats, rabbits, cows, cats, dogs, ferrets and humans (Campbell et al. 1995; Strauss et al. 2001; Carmeliet & Vereecke, 2002; Nerbonne & Kass, 2003; Nerbonne, 2004). One notable exception is the guinea pig, where Ito is very small or absent (Varro et al. 1993; Nerbonne & Kass, 2003; Zicha et al. 2003). Thus, in the vast majority of species Ito is a prominent current in ventricular myocytes. The term ‘Ito’ has therefore become synonymous with any rapidly activating and inactivating ventricular K+ current. However, depending upon species and anatomical region, there are at least two distinct Itophenotypes that can be distinguished based upon molecular, biophysical and pharmacological properties. For the left ventricular (LV) free wall, depending upon species, two Itoexpression gradients exist: (i) a gradient of two functionally distinct Ito phenotypes; and (ii) a gradient in peak current density (pA/pF) of a single functional Ito phenotype. In many species, including humans, both gradient types are simultaneously expressed.
Functionally distinct Ito phenotypic gradients
In the LV free walls of rats, ferrets and humans an expression gradient of at least two major functionally distinct Ito phenotypes exists (e.g. Shimoni et al. 1995; Näbauer et al. 1996; Brahmajothi et al. 1999). Multiple Ito phenotypes are also expressed in different anatomical regions of the mouse heart (e.g. Xu et al. 1999; Guo et al. 1999; Brunet et al. 2004).
Inactivation/recovery kinetics
The most distinguishing biophysical characteristics for the two major ventricular Ito phenotypes are differences in the kinetics of inactivation and, in particular, the kinetics of recovery from inactivation (‘recovery’). In LV subepicardial (LV epi) myocytes of humans and ferrets inactivation of native Ito is approximated as a single exponential process, with time constants of ∼30–100 ms (Näbauer et al. 1996; Brahmajothi et al. 1999) (Fig. 2). Inactivation time constants become voltage independent at depolarized potentials, suggesting a voltage-independent rate-limiting step. Voltage-dependent recovery kinetics (∼−100 to −60 mV) of this Ito phenotype are fast, with time constants on the order of ∼20–100 ms (Campbell et al. 1995; Strauss et al. 2001; Nerbonne & Kass, 2003). Because of these fast recovery kinetics, rapid repetitive voltage clamp pulses result in very little to no cumulative inactivation (Aldrich, 1981). This Ito phenotype is therefore designated ‘Ito,fast’, with ‘fast’ describing the recovery kinetics.
Figure 2. Distinct recovery kinetics and frequency-dependent inactivation characteristics of Ito,fast and Ito,slow.

A, recovery waveforms (22°C, HP = −70 mV) for ferret LV epi myoctye Ito,fast (upper panel) and LV endo myocyte Ito,slow (lower panel). B, frequency-dependent recovery characteristics of human LV epi myocyte Ito,fast (upper panel; minimal cumulative inactivation) and LV endo myocyte Ito,slow (lower panel; marked cumulative inactivation). From HP = −80 mV currents were repetitively generated (2 Hz, 250 ms clamp step pulses to +40 mV). Data from: ferret, modified from Brahmajothi et al. (1999) by copyright permission of The Rockefeller University Press; human, Näbauer et al. (1996) with permission of Lippincott Williams & Wilkins.
In many species a second Ito phenotype has been identified which inactivates as a distinct double exponential process. The rapid time constant of inactivation has values similar to those displayed by Ito,fast, while the slower time constant is on the order of hundreds of milliseconds. However, this Ito phenotype is truly distinguished by its very slow recovery kinetics, with voltage-dependent time constants on the order of seconds (e.g. Giles & Imaizumi, 1988; Clark et al. 1988; Näbauer et al. 1996; Brahmajothi et al. 1999; Nerbonne & Kass, 2003). Another unique kinetic characteristic of this current is development of prominent cumulative inactivation during rapid repetitive voltage-clamp pulses (Aldrich, 1981; Giles & Imaizumi, 1988; Näbauer et al. 1996; Brahmajothi et al. 1999; Nerbonne & Kass, 2003) (Fig. 2). This cumulatively inactivating Ito phenotype is therefore designated ‘Ito,slow’, with ‘slow’ describing the recovery kinetics.
Activation/deactivation kinetics
Limited quantitative data exist on the voltage-dependent activation and deactivation kinetics of Ito,fast and Ito,slow in LV myocytes. The most detailed analysis has been conducted on Ito,fast in ferret right ventricular (RV) myocytes (Campbell et al. 1993b), which demonstrated that voltage-dependent activation kinetics were sigmoidal. Sigmoidicity of activation and subsequent inactivation (well-approximated as a single exponential) could be described with a Hodgkin-Huxley-like ‘a3i’ formulation (Hodgkin & Huxley, 1952). While the HH-like kinetic model developed from this analysis was able to successfully reproduce many aspects of RV Ito,fast gating (Campbell et al. 1993a, b), it is unlikely that its basic assumptions, particularly the assumption of independence of activation and inactivation, are correct at the molecular level (for further discussion see: Yellen, 1998, 2002; Bezanilla, 2000; Hille, 2001a; Wang et al. 2004).
Pharmacological properties of Ito,fast versus Ito,slow: 4-aminopyridine and spider toxins
4-Aminopyridine. While 4-aminopyridine (4-AP) is non-specific in that it blocks both Ito,fast and Ito,slow, the underlying mechanisms are quite distinct. Block of Ito,slow displays classic characteristics of open-state block: minimal tonic block, apparent acceleration of inactivation, and use dependence (Campbell et al. 1993a; Hille, 2001b). In contrast, 4-AP block of Ito,fast occurs through closed-state binding and displays ‘reverse use dependence’. These properties have been most thoroughly analysed for ferret RV Ito,fast (Campbell et al. 1993a, b). Closed-state block is manifested not only by a reduction in peak Ito,fast amplitude but also by a slowing in the kinetics of activation and inactivation, ‘crossover’ of the partially blocked current with the unblocked current, relief of block during repetitive voltage clamp pulses, and redevelopment of block upon repolarization (Fig. 3).
Figure 3. 4-Aminopyridine: closed-state ‘reverse use-dependent’ block of native Ito,fast in ferret right ventricular myocytes.

A, steady-state block of Ito,fast elicited at +50 mV by 5 mm 4-AP. 4-AP slows both activation and inactivation kinetics, producing a crossover in the current waveforms. B, relief of 4-AP block with increasing depolarizing pulse duration. Currents were elicited during the P2 pulse to +50 mV as the duration of the preceding P1 pulse (500 ms, +50 mV) was progressively increased. C, relief of block and subsequent reassociation of 4-AP (10 mm) during a conventional 500 ms P1–P2 double pulse protocol to +50 mV (HP = −60 mV). Note relief of block during the P1 pulse, and reassociation of 4-AP to channel closed-states as the interpulse interval duration is increased. Data from Campbell et al. (1993a) by copyright permission of The Rockefeller University Press; protocol details given therein.
Spider toxins. Heteropoda toxins (HPTXs 1–3) are 30–33 amino acid peptides isolated from the venom of the Malaysian spider Heteropoda venatoria (Sanguineti et al. 1997; Bernard et al. 2000; amino acid sequences illustrated in Fig. 14A). HPTX2 at 150 nm has no effect on Ito,slow in ferret LV endo myocytes but inhibits Ito,fast in LV epi myocytes (Brahmajothi et al. 1999). Inhibition is voltage dependent and relieved with depolarization (Fig. 4A). The apparent Kd for inhibition ranges from 105 nm at +20 mV to 559 nm at +70 mV. HPTXs also block Ito,fast but not Ito,slow in mouse ventricular myocytes (Xu et al. 1999; Guo et al. 1999). Consistent with these results, 30 nm HPTX2 lengthens action potentials in rat ventricular myocytes (Sanguinetti et al. 1997; Fig. 4B).
Figure 14. Spider toxins.

A, amino acid sequence alignments of PaTXs and HPTXs. Cysteine residues involved in ICK motif formation are indicated by grey boxes. B, the ICK motif is schematically illustrated in the lower panel. Roman numerals I–VI correspond to the cysteine residues indicated in A. The upper panels illustrate MOLESCRIPT representations of the predicted solution structures of PaTX1 and HPTX2. C, predicted solution structures. The upper panels illustrate putative interaction surfaces, with the orientation of the predicted dipole moments emerging from the hydrophobic patches indicated by red arrows. CPK representations are illustrated in the lower panels in the same orientation. Colour coding: polar uncharged residues, green; basic residues, blue; acidic residues, red; aromatic residues, purple; aliphatic residues, yellow. Data modified from Chagot et al. (2004) (with permission from Cold Spring Harbor Laboratory Press) and the Cyclotide webpage (with permission from David Craik).
Figure 4. Spider toxins: HPTX2 selectively blocks native Ito,fast in a voltage-dependent manner.

A, block of ferret LV epi myocyte Ito,fast by 150 nm HPTX2. Inset: Ito,fast in control solution (continuous lines) and after extracellular application of 150 nm HPTX2 (dashed lines). B, effect of 30 nm HPTX2 on the action potential recorded from a rat ventricular myocyte. Data for ferret from Brahmajothi et al. (1999) by copyright permission of The Rockefeller University Press; for rat reproduced with permission from Sanguinetti et al. (1997).
Another family of spider toxins that selectively interact with Ito,fast are the phrixotoxins (PaTXs; Diochot et al. 1999). PaTXs are 29–31 amino acid peptides isolated from the venom of the Chilean fire tarantula, Phrixotrichus aurata (amino acid sequences given in Fig. 14A). When injected into mice, PaTX1 produces varying degrees of atrioventricular block, premature ventricular beats, tachycardia and prolongation of the QT interval. However, the extent to which these effects are due to inhibition of LV Ito,fast versus Ito,fast in other tissues (particularly neurones) is unclear.
Ito density gradients
In most species there is a marked peak current density gradient (pA/pF) for Ito in LV epi versus LV endo myocytes. Such density gradients have been reported for rat, rabbit, dog, ferret and human (Strauss et al. 2001; Nerbonne & Kass, 2003). However, the phenotypic nature of these density gradients is species specific (Fig. 5). For example, in ferret and human phenotypic Ito gradients are present, with Ito,fast being prominently expressed in LV epi myocytes and Ito,slow in LV endo myocytes (Näbauer et al. 1996; Brahmajothi et al. 1999). In both species the density of LV epi Ito,fast is ∼4–5 times greater than LV endo Ito,slow. In contrast, in dog there is only a density gradient of Ito,fast (∼4–5 times higher in LV epi than LV endo myocytes; Liu et al. 1993; Antzelevitch, Zygmunt & Dumaine, 2003), while in rabbit Ito,slow density is higher in LV epi than LV endo myocytes (Fedida & Giles, 1991).
Figure 5. Comparative Ito cellular physiology.

Representative LV free wall Ito phenotypic and density gradients. Peak Ito current density–voltage relations in dog (A) and ferret (B) LV epi and LV endo myocytes. Also illustrated in A is the peak current–voltage relationship obtained in dog midventricular M-cells. Both species display Ito density gradients. Currents generated as follows: dog, HP = −80 mV, pulses from −20 to +70 mV (overlapping inward currents not blocked); ferret, HP = −70 mV, pulses from 0 to +60 mV (LV epi) and −10 to +70 mV (LV endo). Data from: dog, Liu et al. (1993) by permission of Lippincott Williams & Wilkins; ferret, Brahmajothi et al. (1999) by copyright permission of The Rockefeller University Press.
Molecular basis of ventricular Ito phenotypes
The three candidate K+ channel clones for generating distinct Ito phenotypes are Kv1.4, Kv4.2 and Kv4.3 (Nerbonne, 2000; Archer & Rusch, 2001; Oudit et al. 2001; Strauss et al. 2001; Sah et al. 2003; Nerbonne & Kass, 2003). These clones have been assigned to native Ito phenotypes recorded in LV myocytes based upon their electrophysiological characteristics, sensitivity to HPTXs/PaTXs and 4-AP, molecular expression patterns, and properties when manipulated in transgenic mice.
Kv1.4
Kv1.4 α subunits (Fig. 6) display: (i) rapid sigmoidal activation kinetics; (ii) biexponential inactivation kinetics; (iii) very slow kinetics of recovery (time constants on the order of seconds); (iv) cumulative inactivation during repetitive voltage clamp pulses; (v) open-state block by 4-AP (Fig. 7); and (vi) insensitivity to HPTXs, PaTXs and flecanide (Tseng-Crank et al. 1990; Tamkun et al. 1991; Po et al. 1992; Comer et al. 1994; Rasmusson et al. 1995a, b, 1998; Sanguinetti et al. 1997; Roeper et al. 1997; McKinnon, 1999; Petersen & Nerbonne, 1999; Robertson, 2001; Oudit et al. 2001; Jiang et al. 2003a; Li et al. 2003; Nerbonne & Kass, 2003; Bett & Rasmusson, 2004). Hence, the biophysical, kinetic and pharmacological characteristics of Kv1.4-mediated currents are very similar to those displayed by native LV Ito,slow.
Figure 6. Currents generated by heterologously expressed Kv1.4 and Kv4.2/4.3.

A, while both Kv1.4 and Kv4.2/4.3 (Kv4.2 illustrated) give rise to transient outward K+ currents at depolarized potentials that resemble native LV Ito phenotypes, there is approximately an order of magnitude difference in their kinetics of recovery. B, Kv4.3 recovers with time constants on the order of hundreds of milliseconds, while Kv1.4 recovers with time constants on the order of seconds and displays marked cumulative inactivation during repetitive voltage clamp pulses. Data from: A, Kv1.4, Comer et al. (1994) (used with permission from the American Physiological Society); Kv4.2, Yeola & Snyders (1997) with permission from the European Society of Cardiology; B, Kv1.4, Rasmusson et al. (1995b); Kv4.3, Favre et al. (1999) with permission from the European Society of Cardiology.
Figure 7. 4-Aminopyridine: Kv1.4 versus Kv4.2 – open-state versus closed-state blocking effects.

A, effect of 4-AP on Kv4.2 (20 mm; compare to Fig. 3A) and Kv1.4 (1 mm) peak currents and kinetics. Kv4.2, 250 ms pulses to +60 mV, Kv1.4, 500 ms pulses to +20 mV, both elicited from HP = −80 mV and frequency of 1 pulse min−1. Illustrated are currents before application of 4-AP (C), during the first pulse after application of 4-AP (1st) and steady-state currents (SS). B, effects of 10 mm 4-AP on the ferret Kv1.4 N-terminal deletion mutant NCO (removes rapid N-type inactivation). Step pulses of 500 ms to +50 mV, HP = −90 mV, frequency 0.1 Hz. Note both classic use dependence and development of apparent inactivation-like behaviour in the presence of 4-AP. C, time dependence of 4-AP (10 mm) unblock of Kv4.2 at depolarized potentials. Pulses V1 and V2 were both applied to +60 mV at a frequency of 1 pulse/2 min (HP = −80 mV), while the V1 duration was progressively increased. Compare to Fig. 3B. D, reassociation of 4-AP to Kv4.2 closed states after a depolarizing pulse. V1 and V2 both to +60 mV. Corresponding V2 currents overlaid for the indicated interpulse intervals in seconds. Compare to Fig. 3C. Data for Kv4.2 reproduced with permission from Tseng et al. (1996); for Kv1.4 from Rasmusson et al. (1995b).
Very strong evidence for Kv1.4 underlying the LV Ito,slow phenotype comes from elegant studies on transgenic mice which have shown that interventricular myocytes with a targeted deletion of Kv1.4 display complete loss of Ito,slow but not Ito,fast. In contrast, mice expressing a truncated Kv4.2 display reduced Ito,fast expression, and a Kv4.2 pore mutant (Kv4.2W62F) which acts as a dominant negative results in selective loss of Ito,fast (Barry et al. 1998; Guo et al. 1999; Xu et al. 1999; Wickenden et al. 1999). In these mice with deleted/reduced Ito,fast, Ito,slow becomes more prominent in the LV, indicating that the two current phenotypes are regulated by different potassium channels. Finally, in ferret both the phenotypic and density gradients of Ito,slow closely parallel the Kv1.4 protein expression gradient, with both Ito,slow and Kv1.4 being highly expressed in the LV endo but not LV epi or right ventricle (Brahmajothi et al. 1999).
Kv4.2 and Kv4.3
Kv4.2 and Kv4.3 α subunits (Fig. 6) display: (i) rapid sigmoidal activation kinetics; (ii) multiexponential inactivation kinetics; (iii) rapid kinetics of recovery (time constants on the order of 10 to hundreds of milliseconds); (iv) little to no cumulative inactivation during repetitive voltage clamp pulses; (v) closed-state, reverse use-dependent block by 4-AP (Fig. 7); and (vi) sensitivity to HPTXs, PaTXs and flecanide (Serodio et al. 1996; Tseng et al. 1996; Dixon et al. 1996; Yeola & Snyders, 1997; Sanguinetti et al. 1997; Kääb et al. 1998; Favre et al. 1999; Franqueza et al. 1999; Jerng et al. 1999; Nerbonne, 2000; Archer & Rusch, 2001; Robertson, 2001; Strauss et al. 2001; Beck & Corvarrubias, 2001; Oudit et al. 2001; Bähring et al. 2001a, b; Beck et al. 2002; Guo et al. 2002a; Patel et al. 2002a, b, 2004; Wang et al. 2002, 2004; Shahidulla & Covarrubias, 2003; Sah et al. 2003; Nerbonne & Kass, 2003; Gebauer et al. 2004; Hatano et al. 2004; Jerng, Qian & Pfaffinger, 2004).
Similar to their effects on native LV epi Ito,fast, HPTXs and PaTXs both inhibit Kv4.2/4.3 in a voltage-dependent manner, with inhibition relieved with progressive depolarization (Sanguinetti et al. 1997; Brahmajothi et al. 1999). Assuming a single binding site, apparent Kd values for HPTX2 inhibition (100 nm) of peak Kv4.2 current range from 35 nm at +20 mV to 138 nm at +50 mV (Sanguinetti et al. 1997). Similarly, percentage inhibition of peak Kv4.3 current by PaTX1 (100 nm) ranges from ∼50–60% at −30 mV to ∼20% at +50 mV (Diochot et al. 1999).
While pharmacological properties of Ito,fast are mimicked by Kv4.2/4.3, many physiologically important kinetic properties of Ito,fast are only partially reconstituted by these clones. In particular, while recovery kinetics of Kv4.2/4.3 are ∼10-fold faster than those of both Kv1.4 and native LV Ito,slow, they are still ∼5-fold slower than native LV Ito,fast. This suggests involvement of additional regulatory subunits in LV myocytes. Also, both Kv4.2 and Kv4.3 display little to no cumulative inactivation, either when expressed alone or in the presence of regulatory subunits (discussed below).
Regulatory subunits
Kv1.4-mediated Ito,slow
While the kinetic properties of Kv1.4 α subunits expressed alone closely reproduce the properties of native ‘basal’ LV Ito,slow, Kv1.4 α subunits are also subject to regulation by β subunits. Four homologous β subunits have been identified to date (Kvβ1–β4); however, only Kvβ1, β2 and β3 interact with Kv1 channels. Among these, Kvβ1 (alternative splice variants β1.2 and β1.3) and Kvβ2 RNA are expressed in the heart (e.g. Morales et al. 1995; Deal et al. 1996). These subunits lack transmembrane domains and thus are of putative intracellular topology. Their regulatory properties include chaperone effects, modulation of voltage dependence and alteration of inactivation kinetics (Nerbonne & Kass, 2003). For example, Kvβ1.3 accelerates both the fast and slow components of inactivation, promotes the slower component of inactivation and slows recovery of Kv1.4 (Morales et al. 1995; Castellino et al. 1995; Fig. 8A). β subunits can also act as ‘inactivation balls’– coexpression with N-terminus-deleted Kv1.4 channels that lack fast N-type inactivation results in reestablishment of inactivation (Fig. 8B).
Figure 8. Representative effects of Kvβ subunits on Kv1.4 inactivation kinetics.

A, effects of Kvβ3 on ferret Kv1.4 (originally designated FK1 by Comer et al. 1994). Depolarizing voltage clamp pulses to +50 mV from HP = −90 mV. Kvβ3 induced both a rapid early phase of inactivation and a smaller, slower phase of inactivation. B, in the absence of rapid N-type inactivation (N-terminal amino acids 2–146 deleted from Kv1.4 (FK1Δ2–146)) Kvβ3 can act as a partial ‘inactivation ball’ with minimal effects on activation kinetics. Data in A from Morales et al. (1995) reproduced with permission of the American Society for Biochemistry & Molecular Biology; data in B from Castellino et al. (1995), used with permission from the American Physiological Society.
Kv1-interacting β subunits are non-enzymatic homologues of aldo-keto reductases (McCormack & McCormack, 1994; Gulbis et al. 1999; Campomanes et al. 2002). It is thus possible that β subunits may impart selective cellular redox-state sensitivity to LV Ito,slow. For example, in the LV free walls of ferrets and humans the expression gradients of Type III nitric oxide synthase (eNOS) and sarcolemmal bound superoxide dismutase (ECSOD) closely parallel the phenotypic Ito gradients (Näbauer et al. 1996; Brahmajothi et al. 1999) (Fig. 9). But it is unknown if expression gradients of Kvβ subunits also parallel those of eNOS/ECSOD and the two Ito phenotypes. If this turns out to be the case, could Kvβ subunits provide a ‘missing link’ for selective O2, NO- and/or redox-related modulation of LV repolarization and EC-coupling characteristics under normal and pathological conditions (Ruppersberg et al. 1991b; Campbell et al. 1996; Perez-Garcia et al. 1999; Brahmajothi & Campbell, 1999; Holmqvist et al. 2001)?
Figure 9. Immunofluorescent localization: overlap of expression gradients of Itoα subunit proteins and eNOS and ECSOD proteins in ferret heart.

A, expression gradients of Kv1.4 (blue), Kv4.2 (green) and Kv4.3 (red). CA, coronary artery; Ao, aorta; Sep, septum; RV, right ventricle; LV, left ventricle. In addition to the transmural LV free wall gradients also note the apical to basal gradients. B, expression gradients of eNOS (red) and ECSOD (green). Data from: Itoα subunits, Brahmajothi et al. (1999) by copyright permission of The Rockefeller University Press; eNOS and ECSOD, Brahmajothi & Campbell (1999) by permission of Lippincott Williams & Wilkins.
Kv4.2/4.3-mediatedIto,fast
Kv Channel Interacting Proteins. The discrepancy between recovery kinetics of Kv4.2/4.3 and native LV epi Ito,fast suggests the presence of additional ancillary subunits involved in regulation of Ito,fast gating kinetics. The Kv Channel Interacting Proteins (KChIPs) are one such family of ancillary subunits. KChIPS are members of the neuronal calcium sensors subfamily of Ca2+-binding proteins typically characterized by four Ca2+-binding EF-hands (Linse & Forsén, 1995; Nef, 1996; Ikura, 1996). Additional members of this family include DREAM (calsenilin), frequenin (neuronal calcium sensor-1) and hippocalcin.
An et al. (2000) originally identified three KChIPs (KChIP1, 2 and 3) encoded by different genes. Each KChIP has a distinct N-terminus, but ∼70% amino acid identity over the C-terminus. The C-termini of these KChIPs also share significant homology with frequenin and hippocalcin. Since the original work of An et al. (2000), four KChIP genes have now been identified (KChIP1–4). Members of the KChIP2 family are the predominant isoforms expressed in ventricular muscle. To date, at least eight KChIP2 isoforms have been identified and characterized (An et al. 2000; Kuo et al. 2001; Ohya et al. 2001; Bähring et al. 2001a; Rosati et al. 2001, 2003; Decher et al. 2001, 2004; Patel et al. 2002a, b; Hatano et al. 2004).
KChIPs bind to the N-terminus of Kv4 channels with a stoichiometry of 1 KChIP per Kv4 α subunit (4 KChIPs per tetrameric channel complex; Kim et al. 2003). Biophysically, most KChIP2 isoforms influence Kv4 channels by (Fig. 10): (i) slowing the kinetics of the fast component of inactivation; (ii) accelerating the kinetics of the slow component of inactivation; (iii) favouring gating shifts such that the slow component of inactivation becomes dominant at depolarized potentials; and (iv) accelerating the kinetics of recovery to rates similar to those of native LV epi Ito,fast (An et al. 2000; Kuo et al. 2001; Bähring et al. 2001a; Rosati et al. 2001, 2003; Decher et al. 2001, 2004; Patel et al. 2002a, b, 2004). In contrast to their regulatory effects on inactivation and recovery, KChIP2 isoforms have minimal effects on the kinetics or voltage dependence of Kv4 channel activation; however, they significantly accelerate deactivation kinetics (Patel et al. 2004).
Figure 10. ‘Conventional’ regulatory effects of KChIP isoforms 2 (□), 2a (•) and 2b (▵) on Kv4.3 (○) gating characteristics.

A, steady-state activation relationships. B, inactivation relationships. C, inactivation kinetics at +50 mV (peak currents normalized for comparison). D, recovery kinetics at HP = −100 mV. Data from Patel et al. (2002a).
KChIPs also promote Kv4 channel cell surface expression, functionally manifested by an increase in peak current amplitude/density with no change in single channel conductance (Bähring et al. 2001a; Beck et al. 2002; refer to Birnbaum et al. (2004) for further discussion of KChIP-related trafficking effects). This function of KChIP2 is dramatically illustrated by the absence of Ito,fast in mice lacking the KChIP2 gene (Kuo et al. 2001).
While it is assumed that EF-hands in KChIPs play important regulatory roles, little is actually understood about how KChIPs and Ca2+ binding to them regulate Kv4.2/4.3 gating. In KChIP1, EF-hands 1 and 2 are non-canonical and degenerate (Zhou et al. 2004; see Fig. 15E), leaving EF-hands 3 and 4 as the potential sensors/transducers of Ca2+-dependent regulation. This is further supported by ANS fluorescence studies of the metal binding properties of KChIP1, which suggest that EF-hand 4 has the greatest structural and functional importance (Chang et al. 2003).
Figure 15. KChIP1 crystal structure.

A, KChIP1 ribbon representation. EF-hands designated EF1–4, Ca2+ ions as yellow spheres and α helices as H1–H10. Arrows 3 and 4 point to the Ca2+ ions bound to EF-hands 3 and 4, respectively. B, upper panel: stereo views of cylinder model of KChIP1 monomer, with Kv4.2 N-terminus in orange. Note that the structurally unresolved region between H7 and H8 (indicated as a break in A and a dotted line in B) corresponds to the ‘recovery loop’ region originally mutated by Patel et al. (2002b). Lower panel: stereo view of ribbon model of the KChIP1-Kv4.2 N30 dimeric complex, rotated 90 deg around the horizontal axis from upper panel view. Ca2+ ions bound to EF-hands 3 and 4 indicated as grey spheres. N and C indicate the N- and C-terminus of each individual KChIP. Arrows 3 and 4 designate Ca2+ ions bound to EF-hands 3 and 4, respectively. C, lipophilic (Kv4-binding) and hydrophilic (cytoplasmic-orientated) faces of KChIP1. Surfaces have been colour coded to indicate lipophilicity, with brown being highly lipophilic and blue highly hydrophilic. D, surface map structure of the KChIP1 hydrophobic binding pocket and its interaction with the Kv4.2N30 construct (green ribbon model; see Zhou et al. 2004 for further details). Colour coding of surface residues: polar residues, grey; non-polar residues, yellow. Red residues display sequence homology to other NCS proteins. Figures from: A and C, Scannevin et al. (2004) reproduced with permission from Elsevier; B and D, Zhou et al. (2004) reproduced with permission from Elsevier.
Currently, no quantitative measurements of Ca2+-binding affinities of EF-hands in KChIP2 isoforms are available. However, DREAM (KChIP3) has a high affinity Ca2+-binding site (Kd∼6 × 10−7m) located in either EF hand 3 or 4, and Ca2+ binding to this site produces significant changes in secondary structure (Craig et al. 2002). If the Kd of DREAM is similar to KChIP2 isoforms, then cyclical changes in [Ca2+]i that occur during a normal LV myocyte action potential could dynamically alter Kv4.2/4.3 gating kinetics (Patel et al. 2002b, 2004).
The functional role of EF-hand 4 of the KChIP2 isoforms has been studied utilizing KChIP2d, which corresponds to the last 70 C-terminal amino acids of the other larger isoforms, and thus contains only the fourth EF-hand (Patel et al. 2002b, 2004). Nonetheless, KChIP2d retains both Ca2+-dependent and Ca2+-independent functional effects on Kv4.3 gating kinetics. Ca2+ dependency manifests itself in the promotion of the slower component of inactivation, while the reduction of the faster component of inactivation and acceleration of recovery are both Ca2+ independent (Patel et al. 2002b). The presence of EF-hand 4 alone is thus sufficient for mediating the Ca2+-dependent effects on Kv4.3 gating. The magnitude of these Ca2+-dependent effects is greater in the presence of KChIP2b with its four EF-hands.
Other regions of KChIP molecules that regulate specific Kv.4 functions have also been studied. Truncation of both KChIPs 1 and 2 to the highly conserved 185 amino acid C-terminus sequence (KChIP1ΔN2-31, KChIP2ΔN2-67) resulted in no significant loss of regulatory function on Kv4.2 kinetics (An et al. 2000), results consistent with the regulatory effects displayed by KChIP2d (Patel et al. 2002b, 2004). Analysis of the effects of various KChIP2 chimaeric constructs (specific regions swapped between neuronal Ca2+ sensor-1 and KChIP2) on Kv4.3 gating indicated three important regions of KChIP2: (i) the linker between EF-hands 1 and 2; (ii) the linker between EF-hands 3 and 4; and (iii) the C-terminal peptide after EF-hand 4 (Ren et al. 2003). The linker between EF-hands 3 and 4 corresponds to amino acids 14–21 in KChIP2d found to be involved in regulation of Kv4.3 recovery (Patel et al. 2002b). These data indicate that the N-termini of KChIPs do not significantly contribute to regulatory function, while the C-terminus is essential.
While most KChIPs exert ‘conventional’ regulatory effects on Kv4 gating (promotion of the slow component of inactivation, acceleration of recovery), a few novel KChIPs have very different properties. KChIP4a has a unique N-terminus containing a ‘K+-channel inactivation suppressor’ (KIS) domain which eliminates fast inactivation of Kv4 channels, making them behave as very slowly inactivating delayed rectifiers (Holmqvist et al. 2002). KChIP4a is not expressed in heart. However, three cardiac KChIP2 isoforms with either an alternatively spliced C-terminus (KChIP2e, KChIP2f) or N-terminus (KChIP2g) have been identified (Decher et al. 2004). KChIP2e accelerates Kv4.3 inactivation and slows recovery, while KChIP2f slows inactivation but has no effect on recovery. In contrast to other KChIP2 isoforms, coexpression of Kv4.3 + KChIP2g generates a hyperpolarizing shift in V1/2 of the steady-state inactivation relationship (Decher et al. 2004). Functionally, the unique effects produced by these isoforms may further contribute to heterogeneity of Ito phenotypes in the LV. Experimentally, these ‘unconventional’ isoforms could be exploited in coexpression studies to gain new insights into mechanisms governing Kv4 channel gating and KChIP function.
DPPs. Nadal et al. (2001) originally demonstrated that coexpression of Kv4.2 with high molecular weight RNA isolated from rat cerebellum resulted in acceleration of inactivation kinetics. The unknown regulatory protein factor was designated ‘K+ Channel Accelerating Factor’ (KAF). Subsequent studies (Nadal et al. 2003) identified KAF as the transmembrane glycoprotein DPP6 (dipeptidyl aminopeptidase related protein 6; Wada et al. 1992). Two alternatively spliced isoforms were identified (long, L, and short, S), which differ solely in the length of their cytoplasmic N-termini. Both are integral membrane glycoproteins with a single putative transmembrane domain. The N-terminus is relatively short, while the C-terminus is extensive, consisting of an extracellular matrix binding domain composed of three subdomains: a glycosylation domain, a cysteine-rich domain (possibly conferring targeting and/or cell adhesion properties to Kv4 channels) and an inactive catalytic domain (Nadal et al. 2003).
The regulatory effects of DPP6 on Kv4.2 are very similar to those of KChIP2 isoforms (increased surface expression, acceleration of recovery) except for one fundamental functional difference: DPP6 accelerates Kv4.2 inactivation kinetics, while the majority of KChIP2 isoforms promote the slower component of inactivation. Recently, another dipeptidyl aminopeptidase, DPP10, has also been shown to regulate Kv4.1 and Kv4.2 channels, with effects similar to those produced by DPP6 (Jerng, Qian & Pfaffinger, 2004). This latter work suggests that the intracellular N-terminus of DPP10 is responsible for acceleration of inactivation.
DPP6 and DPP10 are interesting in that they belong to a family of serine proteases but, similar to Kv1-interacting β subunits, lack enzymatic activity (Wada et al. 1992). Both appear to be predominantly expressed in brain. If DPPX is found in LV myocytes, then will the DPPX expression gradients across the LV free wall correlate with the functional gradient of Ito,fast? And how will the effects of DPPXs interact with those of KChIP2 isoforms?
Other potential regulatory subunits. In addition to KChIPs, several other Kv4 regulatory subunits have been studied which have varying effects on Kv4 expression and kinetics. Kvβ1 and β2 increase the expression of Kv4.3 without altering kinetics, while Kvβ1.2 confers redox (O2) sensitivity to Kv4.2 without altering kinetics (Perez-Garcia et al. 1999; Yang et al. 2001). KChAP (K Channel Accessory Protein) also increases Kv4.3 expression without effects on kinetics (Wible et al. 1998; Kuryshev et al. 2000). In contrast, MinK-related peptide 1 (MiRP1) has been reported to alter Kv4.2 gating kinetics (Zhang, Jaing & Tseng, 2001), and the Na+ channel β1 subunit interacts ‘promiscuously’ with Kv4.3, resulting in increased peak current density, accelerated inactivation and slowed recovery (Deschênes & Tomaselli, 2002). Frequenin, another Ca2+-binding protein of the neuronal calcium sensors subfamily, also binds to Kv4 channels and slows inactivation in a Ca2+-dependent manner (Nakamura et al. 2001). However, frequenin has no to minimal effects on recovery (Nakamura et al. 2001; Guo et al. 2002b). Any of these subunits could potentially modulate LV Ito. At present, the best evidence exists for frequenin, which coimmunoprecipates with Kv4.3 α subunits from mouse heart ventricular extracts (Guo et al. 2002b; however, see Ren et al. 2003). The physiological significance of the other subunits in the heart is still unresolved.
Species differences in LV expression of Kv1.4, Kv4.2, Kv4.3 and KChIP2 isoforms
Compelling evidence indicates that Kv4.2 and/or Kv4.3 underlie generation of LV Ito,fast. However, there are significant differences among species in both expression profiles and possible role(s) of these two clones in generation of Ito,fast. The distribution of both K+ channel and KChIP2 mRNA across the LV free wall has been determined in dog, ferret, human and rat, and measurements of Kv mRNA levels in guinea pig heart have been reported (Dixon & McKinnon, 1994; Dixon et al. 1996; Kääb et al. 1998; Guo et al. 1999; Brahmajothi et al. 1999; Rosati et al. 2001, 2003; Patel et al. 2002a, b; Nerbonne, 2004; Zicha et al. 2004). The mRNA expression patterns obtained from these studies reveal significant heterogeneity across the LV free wall as well as differences between species. With the caveat that mRNA expression does not always imply functional protein expression (Brahmajothi et al. 1999; Robertson, 2001), we will summarize the expression data for these six species in relation to the native current distribution(s) observed in LV myocytes isolated from each species.
Human
Kv1.4, Kv4.3 and KChIP2 mRNA are found in the human LV (Kääb et al. 1998; Rosati et al. 2001). Both KChIP2 mRNA and protein are expressed in a parallel gradient across the LV free wall (LV epi > LV endo), while Kv4.3 mRNA is uniformly expressed within a gradient of Kv4.3 protein (LV epi > LV endo) (Rosati et al. 2001; Zicha et al. 2004). Based upon these gradients of protein expression, both KChIP2 and Kv4.3 appear to contribute to the Ito,fast density gradient across the human LV free wall. Rosati et al. (2001, 2003) propose that the KChIP2 mRNA/protein gradient regulates Kv4.3 expression to generate the transmural LV Ito,fast gradient. However, expression gradients of KChIP2 and Kv4.3 cannot account for the presence of the cumulatively inactivating Ito,slow phenotype in human LV endo myocytes (Näbauer et al. 1996; Fig. 2). Ito,slow may be mediated by Kv1.4. Kv1.4 mRNA is found in the human LV (Kaab et al. 1998), but its distribution has not yet been analysed.
Dog
Kv1.4 mRNA is reported to be present at 16% of the level of Kv4.3 mRNA in the canine LV (Dixon et al. 1996). However, there appears to be no electrophysiological evidence for a slowly recovering cumulatively inactivating Ito,slow phenotype. Thus, the canine LV free wall appears to express only an Ito,fast density gradient. Consistent with this functional observation, the canine LV free wall displays gradients of both KChIP2 and Kv4.3 protein (LV epi > LV endo) that closely parallel the LV transmural Ito,fast gradient (Rosati et al. 2001, 2003; Zicha et al. 2004).
A recent controversy has arisen over the physiological significance of KChIPs in regulation of LV Ito,fast. While the majority of investigators agree on the importance of KChIP2 isoforms in regulation of Ito,fast, using a polyclonal KChIP2 antibody Deschênes et al. (2002) failed to detect an expression gradient across the canine LV free wall. As suggested by both Rosati et al. (2003) and Zicha et al. (2004), a plausible explanation is that the antibody employed by Deschênes et al. (2002) possessed insufficient selectivity for KChIP2 protein, resulting in non-specific binding that obscured the gradient. We agree with this explanation.
Ferret
Similar to human, ferret LV free wall exhibits both density and phenotypic gradients of Ito,fast and Ito,slow. Kv4.2 protein is mainly localized to LV epi while Kv4.3 protein is transmurally expressed across the LV free wall. However, while Kv1.4 mRNA is expressed uniformly across LV free wall Kv1.4 protein is localized almost exclusively to the LV endo (Brahmajothi et al. 1999; Fig. 9A).
These results raise the question: given the presence of Kv1.4 and Kv4.3 protein why is Ito,fast, or a mixed Ito,fast/Ito,slow phenotype, not observed in the majority of ferret LV endo myocytes (Brahmajothi et al. 1999)? An answer likely resides in the fact that, similar to human and dog, there is an expression gradient of KChIP2 (mRNA and protein) across the ferret LV free wall (LV epi > LV endo; Patel et al. 2002a, b). These results are consistent with the proposal of Rosati et al. (2001) that reduced KChIP2 expression may minimize functional Kv4.3 expression.
The ferret LV free wall also displays a gradient of Kv4.2 protein (LV epi > LV endo) that parallels the Ito,fast gradient (Brahmajothi et al. 1999). It is thus unclear if LV epi Ito,fast is due to Kv4.2, Kv4.3 and/or Kv4.2/Kv4.3 heteromultimers (Nerbonne, 2002). Nonetheless, the distributions of Kv1.4, Kv4.2, Kv4.3 and KChIP2b proteins argue for both Ito density and phenotypic gradients. However, the mechanisms regulating Kv1.4 protein expression in ferret LV endo but not LV epi myocytes in the presence of uniform mRNA expression levels are unknown. Could such mechanisms also be responsible for the lack of Ito,slow in dog LV even though Kv1.4 mRNA can be measured (Dixon et al. 1996)?
Rat
Electrophysiological studies indicate the presence of both Ito density and phenotypic gradients across the rat LV free wall (Clark et al. 1993; Casis et al. 1998). Analyses of Kv4.2 and Kv4.3 mRNA reveal distributions similar to those in ferret LV, with uniform expression of Kv4.3 amid a transmural gradient of Kv4.2 (LV epi > LV endo; Dixon & McKinnon, 1994; Dixon et al. 1996; Rosati et al. 2001). However, expression of KChIP2 mRNA is uniform (Rosati et al. 2001). Rosati et al. (2001) thus concluded that the Kv4.2 gradient is responsible for the Ito,fast density gradient. While plausible, this hypothesis raises the question: why does the uniform distribution of Kv4.3 and KChIP2 mRNA not result in uniform Ito,fast expression? Are there mechanisms preventing Kv4.3 and/or KChIP2 protein expression in rat LV endo myocytes?
Kv1.4 mRNA is also expressed uniformly across the rat LV. If Kv1.4 protein expression in the rat LV is similar to that of the ferret (where Kv1.4 mRNA expression is uniform but Kv1.4 protein localizes to the LV endo; Brahmajothi et al. 1999), then Kv1.4 could account for localization of Ito,slow.
Mouse
Initial studies indicated heterogeneous expression of Ito,fast and Ito,slow in mouse heart, with Ito,fast being expressed in the LV apex and Ito,slow in the interventricular septum (Xu et al. 1999). Another extensive analysis of Ito phenotype expression in the mouse heart has recently been conducted by Brunet et al. (2004). While Ito,fast was observed in all LV myocyte types (apex, base, epi, endo), peak current density was higher in apical and LV epi myocytes than in either basal or LV endo myocytes. No evidence could be found for Ito,slow expression in any LV free wall myocyte type. Thus, in mouse it appears that: (i) Ito,slow is present only in the interventricular septum; and (ii) in the LV there are significant apical-to-basal and epicardial-to-endocardial expression gradients of Ito,fast (similar to that predicted for ferret LV; Brahmajothi et al. 1999; Fig. 9A). At the molecular level, there appears to be a general consensus that the Ito,fast gradient across the normal murine LV free wall is due to heterogeneous expression of Kv4.2 (LV epi > LV endo) amid a background of uniform KChIP2 expression (e.g. Guo et al. 1999; Rosati et al. 2003; Brunet et al. 2004).
Transgenic mouse techniques have further helped dissect the molecular components underlying murine Ito phenotypes (London, 2001; Nerbonne, 2004). Targeted deletion of the Kv1.4 gene, Kv1.4−/−, eliminates Ito,slow in mouse septal myocytes but has no effect on Ito,fast in LV myocytes (London et al. 1998b). LV Ito,fast can be completely eliminated by either targeted deletion of the Kv4.2 gene (Kv4.2−/−; Guo et al. 2000) or expression of the dominant negative (DN) pore mutant Kv4.2W362F (Barry et al. 1998). Mice expressing a dominant-negative truncated Kv4.2 (Kv4.2N) display significantly reduced (but not completely eliminated) Ito,fast (Wickenden et al. 1999). Finally, Ito,fast is eliminated in mice with a targeted deletion of the KChIP2 gene, KChIP2−/− (Kuo et al. 2001).
All four of these genetic manipulations (Kv4.2DN, Kv4/2−/−, Kv4.2N, KChIP2−/−) result in prolonged LV myocyte action potentials. However, although there is a complete loss of Ito,fast in Kv4.2 DN and Kv4.2−/− mice, these mice do not display increased susceptibility to arrhythmias (Barry et al. 1998; Guo et al. 2000; Brunner et al. 2001). In contrast, Kv4.2N mice display several serious pathologies, including cardiac hypertrophy, fibrosis and sudden death (Wickenden et al. 1999). KChIP2−/− mice also display increased susceptibility to ventricular tachycardia and sudden death (Kuo et al. 2001). Thus, functional conclusions on the role of Ito,fast in generation of LV pathologies, as well as its contribution to the ECG, appear to be highly dependent upon the particular mouse model. Reasons underlying these functional discrepancies have yet to be clarified. A further discussion of potential limitations associated with transgenic approaches can be found in Nerbonne (2004).
Guinea pig
No measurable Ito phenotype (Ito,fast or Ito,slow) exists in guinea pig LV myocytes. Consistent with this observation Kv1.4, Kv4.2 and Kv4.3 mRNA are all undetectable in guinea pig heart (Zicha et al. 2003).
Conclusion
To conclude this section, it should be noted that use of the relative terms ‘fast’versus‘slow’ is not always consistent among different laboratories. We thus feel it is important to emphasize that in the absence of KChIP2 isoforms, Kv4.2/4.3 recovery time constants are still approximately an order of magnitude faster than those of Kv1.4/Ito,slow, and neither Kv4.2 nor Kv4.3 displays cumulative inactivation, in either the absence or the presence of KChIP2 isoforms. We propose that ‘fast’ be used to denote recovery kinetics with time constants on the order of tens of milliseconds (Kv4.2/4.3 + KChIP2 isoforms), ‘intermediate’ recovery kinetics with time constants on the order of hundreds of milliseconds (Kv4.2/4.3 with reduced to no KChIP expression) and ‘slow’ recovery kinetics with time constants on the order of thousands of milliseconds (Kv1.4). If such a nomenclature is valid, then, in contrast to Ito,slow, both Ito,fast and Ito,intermediate would display HPTX/PaTX sensitivity, closed-state reverse use-dependent block by 4-AP and little to no cumulative inactivation. Of course, this nomenclature is based upon the predicted effects of KChIP2 isoform gradients. If additional regulatory subunits are expressed in LV myocytes it is unclear how they may alter this scenario.
Ito phenotype expression in Purkinje fibres
While the first cardiac Ito phenotype appears to have been recorded in sheep Purkinje fibres (Deck & Trautwein, 1964), very limited data exist at present on the molecular and biophysical bases of Purkinje fibre Ito phenotypes. Recent studies have been conducted on single Purkinje fibres isolated from free-running endocardial false tendons of canine and human hearts (Han et al. 2000, 2002a, b). In both species, an Ito can be recorded that displays: (i) biexponential inactivation kinetics (fast time constants on the order of milliseconds, slow time constants on the order of tens of milliseconds); (ii) biexponential recovery kinetics (fast time constants on the order of tens of milliseconds, slow time constants on the order of thousands of milliseconds); and (iii) prominent cumulative inactivation. These observations suggest a mixture of Ito,fast and Ito,slow phenotypes; however, the Purkinje fibre Ito phenotype displays significant differences from both Kv4.2/4.3-mediated Ito,fast and Kv1.4-mediated Ito,slow in its sensitivity to 4-AP, tetraethlylammonium+ and cellular redox state.
At the molecular level (both mRNA and protein), Han et al. (2002a) report that canine Purkinje fibres express extremely low levels of Kv1.4, no detectable levels of Kv4.2, and levels of Kv4.3 comparable to LV midmyocardial myocytes. KChIP2 mRNA was also found to be much less abundant in Purkinje fibres than LV midmyocardial myocytes. Interestingly, Kv3.4 mRNA and protein were reported to be abundantly expressed (see also Brahmajothi et al. 1996). Kv3.4 channels generate a TEA+-sensitive Ito phenotype (e.g. Vega-Saenz de Miera et al. 1992).
The results of Han et al. (2000, 2002a, b) would indicate that the Purkinje fibre Ito phenotype is unique and predominantly due to Kv3.4 subunits. However, Purkinje fibres possess a hyperpolarization-activated cation current (‘If’; Campbell et al. 1992). It is unclear if deactivating If currents may be contributing to the reported Purkinje fibre Ito characteristics (Verkerk & van Ginneken, 2001; Boyett et al. 2001). This is an important issue that needs to be addressed in future Purkinje fibre studies.
Molecular and biophysical mechanisms of activation, inactivation and recovery
Kv1.4
Activation of Kv1.4 is sigmoidal and can be described using a Hodgkin-Huxley-like ‘a4’ formulation (Hodgkin & Huxley, 1952; Comer et al. 1994), implying the presence of multiple closed channel states. With regard to inactivation, classic studies on Shaker channels demonstrated the presence of two inactivation mechanisms, N-type (N-terminal ‘ball-and-chain’ mechanism) and C-type (pore closure mechanism) (Hoshi et al. 1990, 1991; Zagotta et al. 1990; Choi et al. 1991; Wissmann et al. 2003). Both of these mechanisms are present in cardiac Kv1.4 channels. Mutagenesis studies support the hypothesis that these two mechanisms are allosterically coupled, with N-type inactivation controlling the rapid component of inactivation and C-type inactivation controlling cumulative inactivation and the very slow kinetics of recovery (Rasmusson et al. 1995a; Roeper et al. 1997). The detailed molecular and biophysical characteristics of Shaker N- and C-type inactivation mechanisms have been extensively reviewed (Rasmusson et al. 1998; Yellen, 1998, 2002; Wissmann et al. 2003). We direct readers to the following sources for detailed discussion of N-type and C-type inactivation mechanisms and their relevance to cardiac Kv1.4/Ito,slow gating mechanisms: Rasmusson et al. (1995a, b, 1998), Roeper et al. (1997), Oudit et al. (2001), Jiang et al. (2003a), Li et al. (2003) and Bett & Rasmusson (2004).
Kv4.2/4.3
The molecular mechanisms underlying the biophysical gating characteristics of Kv4.2/4.3 are not as clearly delineated as Kv1.4. Nevertheless, significant progress has occurred in this area over the past several years.
Activation. Two recent independent studies have demonstrated that activation of Kv4.3 channels is sigmoidal and can be well-described using a Hodgkin-Huxley-like ‘a4’ formulation (Hodgkin & Huxley, 1952; Patel et al. 2004; Wang et al. 2004). τactivation values display saturation at depolarized potentials, indicating a voltage-independent rate-limiting step(s). Also, since inactivation of Kv4.3 is approximately an order of magnitude slower than activation, fits to the early phases of activation are not seriously distorted by inactivation. To account for saturation of rate constants, Patel et al. (2004) empirically fitted their observed τactivation−Vm curves with Boltzmann-like functions, while Wang et al. (2004) proposed the following discrete state Markov model consisting of five closed states and one open state, with voltage-dependent rate constants (α and β) for transitions between with the first four closed states and voltage-independent rate constants (kco and koc) for transition between the final preactivated closed-state and open-state:
 |
Scheme 1 |
Two interesting additional observations were that: (i) Increasing [K+]o (2–98 mm) stabilized the channel open state, as manifested by a slowing in the kinetics of deactivation; and (ii) the voltage dependence of τactivation was best described as a biexponential process, suggesting two different populations of gating charges (Wang et al. 2004). These two charges (in 2 mm[K+]o) are estimated to be 0.27eo and 2.11eo per subunit. At very depolarized or hyperpolarized potentials movement of the larger charge component is hypothesized to become essentially instantaneous, while movement of the smaller component becomes dominant and rate limiting. Activation of Kv4 channels may thus be more complex than Shaker channels.
The steepness of the steady-state activation curve for Kv4 channels is typically 2–3 times less than that observed for Shaker channels, with slope factor values for a single subunit of ∼19–25 mV (e.g. Bähring et al. 2001a; Beck et al. 2002; Wang et al. 2004). As pointed out by Wang et al. (2004) this observation may have one physical basis in significant differences between the amino acid sequences of S4 in Shaker versus Kv4 channels: (i) Shaker has seven positive charges, while Kv4.3 has only five; and (ii) there are seven differences among uncharged residues.
Inactivation and recovery. In contrast to Kv1.4, the molecular basis of inactivation and recovery of Kv4 channels is unclear. All Kv4 channels inactivate as multiexponential processes, suggesting multiple mechanisms (Bähring et al. 2001a; Beck et al. 2002; Patel et al. 2002a, b; Shahidulla & Covarrubias, 2003; Hatano et al. 2004). However, the fractional contributions of the different components of inactivation vary considerably among family members. For example, under ‘basal’ conditions Kv4.1 inactivation occurs predominantly through the slow component, while Kv4.3 inactivation is dominated by the fast component (Jerng & Corvarrubias, 1997; Jerng et al. 1999; Beck & Corvarrubias, 2001; Patel et al. 2002a, b, 2004). Further appraisal of the literature suggests that multiple channel domains may be involved in these processes, but also reveals conflicting conclusions and debate on underlying molecular mechanisms.
Due to the predominance of early studies on Shaker channels, which demonstrated allosteric coupling between N- and C-type inactivation (Hoshi et al. 1990, 1991; Choi et al. 1991; Rasmusson et al. 1998; Yellen, 1998, 2002), initial studies focused on the potential involvement of these two mechanisms in regulating Kv4 channel inactivation and recovery. In contrast to the criteria established for characterization of ‘classic’ N- and C-type mechanisms, these studies demonstrated that: (i) neither extracellular nor intracellular tetraethylammonium altered inactivation (Jerng & Corvarrubias, 1997); (ii) increasing [K+]oaccelerated inactivation and slowed recovery (Jerng et al. 1999); and (iii) N-terminal deletion did not slow the kinetics of closed-state inactivation or alter the kinetics of recovery (Bähring et al. 2001a). It was thus concluded that Kv4 channels lacked N- and C-type mechanisms as originally defined for Shaker channels, although it was proposed that concerted interactions of unspecified regions of the cytosolic N- and C-termini and regions near the internal mouth of the pore were involved in regulating inactivation (Jerng et al. 1999). However, it has recently been suggested (Gebauer et al. 2004) that the N-terminus of Kv4.2 can act as an N-type inactivation peptide under certain heterologous coexpression conditions. This has been confirmed by Pourrier et al. (2004), who demonstrated that attachment of the Kv4.2 N-terminus to N-terminus-deleted Kv1.4 both restored rapid inactivation and accelerated recovery. The latter result suggests that the Kv4.2 N-terminus may act as an inactivation ball. However, it is less efficient in promoting Kv1.4 N-type inactivation than the native Kv1.4 N-terminus inactivation ball(s) (Rasmusson et al. 1995a; Wissmann et al. 2003). While these results are of high intrinsic molecular and biophysical interest, their physiological significance to LV Ito is unclear.
Additional studies have yielded intriguing results on regions (other than the N-terminus) of Kv4 channels that may also be involved in regulation of inactivation and recovery. The following three observations are of particular interest.
Mutation of a cysteine to a serine in the S4–S5 loop of Kv4.1 (C322S) slowed both inactivation and deactivation (Jerng et al. 1999) with little effect on recovery.
(ii) Amino acids 420–429 in the Kv4.3 C-terminus have been demonstrated to regulate the voltage dependence of recovery (Hatano et al. 2004). When two arginines in this region were both mutated to alanines (R426A, R429A) there was no effect on inactivation but a dramatic slowing of recovery. We are unaware of any other Kv4.3 mutation that has such unique effects on recovery but not inactivation.
(iii) A mutation near the inner mouth of the Kv4.1 pore (V(404,406)I) dramatically slows inactivation (Jerng et al. 1999), while a similar mutation in Kv4.3 (V(399,401)I) results in only modest effects on inactivation (Wang et al. 2002). These results suggest that this mutation targets the slow component of inactivation that is more dominant in Kv4.1 than Kv4.3, and emphasize the important kinetic differences between members of the Kv4 family. Furthermore, since most KChIPs produce gating shifts promoting the slower component of inactivation, it is interesting to note that coexpression of KChIP2b with the Kv4.3(V(399,401)I) pore mutant results in a dramatic slowing of inactivation (Wang et al. 2002), an effect very similar to that produced by the Kv4.1 (V(404,406)I) pore mutant expressed alone (Jerng et al. 1999). Is this indicating that KChIP-mediated promotion of the slower component of Kv4.3 inactivation involves allosteric interactions with the inner vestibule of the channel pore?
While these three sets of results remain to be mechanistically explained, they suggest that regions of the C-termini and inner pore vestibule are involved in regulation of Kv4 channel inactivation and recovery. Different regions of the channel complex may also control inactivation and recovery separately. No proposed Kv4 channel or native phenotypic Ito gating model can account for all of these observations (Campbell et al. 1993a, b; Greenstein et al. 2000; Winslow et al. 2000; Puglisi & Bers, 2001; Bähring et al. 2001a; Rudy, 2002; Beck et al. 2002; Bondarenko et al. 2004; Jerng, Qian & Pfaffinger, 2004; Wang, Puglisi & Bers, 2004; Iyer et al. 2004; Winslow et al. 2005).
Speculation: voltage dependence of Ito,fast recovery kinetics
Kv4.2/4.3 inactivation kinetics are voltage independent at only moderately depolarized potentials, while recovery kinetics are voltage dependent over the entire physiological range of hyperpolarized potentials (e.g. Campbell et al. 1993b; Patel et al. 2004). How can inactivation be voltage independent but recovery voltage dependent? And how can intracellular KChIPs accelerate recovery in a voltage-dependent manner?
Three independent sets of data may be hinting at mechanisms: (i) Kv4.3 activation and inactivation are likely to be coupled (Patel et al. 2004; Wang et al. 2004); (ii) increases in [K+]o slow both Kv4 recovery (Jerng et al. 1999) and deactivation (Wang et al. 2004); and (iii) KChIPs accelerate Kv4.3 recovery and deactivation (Patel et al. 2002a, b, 2004). These results may indicate that channel regions regulating recovery and deactivation are coupled. Could ‘reverse’ movement of S4 impart voltage dependence to recovery, a mechanism similar to that originally proposed for the voltage-gated sodium channel (Patlak, 1991)? Does the fact that recovery and deactivation are both [K+]o and KChIP dependent indicate allosteric coupling between the gondola, pore domain(s) and movement of S4? And could provision of KChIP-mediated allosteric coupling be the primary role of the N- and/or C-termini of Kv4 channels?
Unique properties and mechanisms of regulation of distinct LV Ito phenotypes
Ito,fast versus Ito,slow: regulation of frequency-dependent action potential characteristics
Action potential (AP) recordings from rabbit, rat and ferret LV myocytes demonstrate significant differences in frequency-dependent modulation of AP morphologies. In canine LV epi and LV endo myocytes, increasing stimulation frequency leads to a shortening of AP duration, which is consistent with the transmural gradient of Ito,fast (Fig. 11A). In rabbit, where Ito,slow is expressed in all LV myocyte types, increasing pulse frequency leads to an increase in AP duration (Fig. 11B). In both rat and ferret LV epi myocytes, increasing pulse duration leads to a decease in AP duration; in contrast, in LV endo myocytes increasing pulse frequency leads to an increase in AP duration (Fig. 11C and D). Thus, in some species the combination of Ito,fast and Ito,slow contributes to the generation of opposite frequency-dependent effects on APs in LV epi versus LV endo myocytes: during periods of increased stimulation, the predominance of Ito,fast correlates with decreased AP durations while the predominance of Ito,slow correlates with increased AP durations. While AP morphology is the summed result of all currents flowing across the sarcolemma, these results are consistent with the predicted effects of the distinct inactivation and recovery kinetics of Kv4.2/4.3-mediated versus Kv1.4-mediated Ito phenotypes.
Figure 11. Frequency-dependent characteristics of action potential (AP) morphologies in LV myocytes of different species: predominance of effects of Ito,fast versus Ito,slow.

A, APs of dog LV epi and LV endo myocytes elicited at increasing basic cycle lengths (BCL) of stimulation from 8000 ms to 300 ms. B, rabbit LV myocyte after 15 s of rest and final 20th steady-state (SS) AP elicited at 0.5 Hz. C, rat LV endo myocyte APs at rest, 1 Hz and 2 Hz. D, APs of ferret LV epi and LV endo myocytes with stimulation frequency progressively increased from 0.2 to 1 Hz. Final 20th steady-state AP at end of each pulse train is illustrated. Data from: dog, Liu et al. (1993) with permission of Lippincott Williams & Wilkins; rabbit, Bassani et al. (2004); rat, Shimoni et al. (1995); ferret, D. L. Campbell, unpublished results, recording conditions as described in Campbell et al. (1993b).
Many investigators have hypothesized that differences in action potential morphologies between distinct LV myocyte types would produce significant repolarization gradients across the LV free wall, and that such gradients may be the source of the T-wave of the electrocardiogram (Gussack & Antzelevitch, 2003). However, numerous whole LV tissue and in vivo animal heart studies have consistently failed to demonstrate the predicted transmural repolarization gradients (Taggart et al. 2003). It is unclear whether this discrepancy is due to technical difficulties (use of bipolar electrodes in whole tissue; absence of electrotonic coupling in isolated myocytes). However, in species possessing an LV epi Ito,fast–LV endo Ito,slow phenotypic gradient (e.g. ferrets, humans), the opposite frequency-dependent regulatory effects that these two Ito phenotypes exert on LV epi and LV endo myocyte action potentials would contribute to minimizing transmural repolarization gradients at normal heart rates. Thus, in such species the LV epi Ito,fast–LV endo Ito,slow gradient would be inherently antiarrhythmic under normal conditions. If correct, then in such species the normal T-wave would arise from other more global repolarization gradients (apical–basal, RV–LV; de Bakker & Opthof, 2002; Taggart et al. 2003). Under pathological conditions, alterations in Ito,fast and/or Ito,slow could lead to alterations in conduction and/or generation of transmural LV repolarization gradients contributing to abnormal T-wave activity (Huelsing et al. 2003; Libbus et al. 2004). These considerations also argue for caution when applying LV myocyte computer models that only incorporate differences in peak density of a single Ito phenotype to results obtained from whole LV tissue (Winslow et al. 2005).
Speculation: regulation of Ito,slow and Ito,fast by phosphorylation
Ito,slow. The AP results obtained from rabbit LV and rat and ferret LV endo myocytes (Fig. 11) strongly argue that Ito,slow can be an important repolarizing current at low heart rates. However, due to its very slow recovery kinetics and cumulative inactivation it is assumed by many investigators that Kv1.4 plays no significant role in repolarization at normal heart rates. This may be correct; however, a study on the effects of Ca2+/calmodulin-dependent protein kinase II (CaMKII) on Kv1.4 kinetics raises a significant challenge to this assumption (Roeper et al. 1997). Roeper et al. (1997) demonstrated that CaMKII is capable of phosphorylating Kv1.4 α subunits at serine residue 123. More intriguing were the observations that phosphorylation resulted in marked slowing of inactivation kinetics, pronounced reduction of cumulative inactivation and a 10-fold acceleration of recovery kinetics, with time constants on the order of hundreds of milliseconds (Fig. 12). These CaMKII-mediated effects were dependent upon [Ca2+]i.
Figure 12. Regulation of Kv1.4 inactivation and recovery kinetics by CaMKII-mediated phosphorylation (HEK-293 cells, perforated patch technique).

A, recovery waveforms (HP = −80 mV) for Kv1.4 alone (control), after microinjection with autothiophosphorylated CaMKII (+ CaM KII) and after 30 min preincubation in 10 µm KN-93 (CaMKII blocker). B, cumulative inactivation waveforms in the presence of CamKII, after KN −93 preincubation, and in the presence of the Kv1.4 S123A mutation. Currents were repetitively stimulated at 10 Hz (5 ms depolarizations to +20 mV from HP = −60 mV). Both KN-93 and the S123A mutation prevented CaMKII-mediated reduction of Kv1.4 cumulative inactivation. Data from Roeper et al. (1997); © 1997 by the Society for Neuroscience.
Roeper et al. (1997) propose that phosphorylation of Kv1.4 results in acceleration of recovery from N-type inactivated states, while dephosphorylation results in increased ‘trapping’ in C-type inactivated states, with the latter resulting in cumulative inactivation and slowed recovery (Rasmusson et al. 1995a). If correct, then the slow recovery and cumulative inactivation of Kv1.4 under ‘basal’ conditions (Fig. 6) would be due to dephosphorylated channels and predominance of C-type inactivation. Similarly, due to the presence of Ca2+ chelators in whole cell patch pipettes, slow recovery and cumulative inactivation of LV Ito,slow (Fig. 2) would be due to reduced CaMKII activity and dephosphorylated Kv1.4 channels. These results raise an interesting question for cardiac cellular electrophysiologists: under normal conditions of cyclically changing [Ca2+]i within LV endo myocytes does Ito,slow inactivation decrease and recovery accelerate? And, if so, how does this relate to the regulation of frequency-dependent modulation of action potential morphologies in LV endo myocytes (Fig. 11)?
Ito,fast: obligatory involvement of KChIP2 isoforms in kinase-mediated modulatory effects? Adenylate cylcase, protein kinase A (PKA) and protein kinase C (PKC) isoforms are all potential physiological regulators of native LV Ito,fast. For example, in many species, the distribution of Ito,fast correlates with positive inotropic effects produced by α1-adrenergic receptor stimulation (Fedida et al. 1993). Voltage clamp studies indicate that α1-receptor stimulation inhibits LV Ito,fast and that these effects may be due to activation of PKC (Parker & Fedida, 2001). With regard to PKA, Schrader et al. (2002) demonstrated that PKA-mediated phosphorylation of Kv4.2 expressed alone generated no regulatory effects. However, PKA-mediated effects were ‘rescued’ when Kv4.2 was coexpressed with KChIP3. Regulatory effects of phosphorylation in the presence of KChIP3 included a reduction in peak current amplitude and slowing of inactivation, but no additional effects on recovery. Between the two potential phospholylation sites, Thr 38 and Ser 552 (Anderson et al. 2000), only Ser 552 was found to be essential (Schrader et al. 2002). KChIP3 association is thus an obligatory prerequisite for PKA-mediated phosphorylation of Kv4.2. If similar effects are displayed by LV Ito,fast, then KChIP2 isoforms may be critical determinants for PKA-dependent modulation of distinct LV Ito phenotypes: regulation of Ito,fast would have an obligatory dependence upon KChIP2 isoforms. This prediction awaits experimental verification.
Speculation: [Ca2+]i, KChIP2 isoforms and Ito,fast
Kinetic analysis of Ito,fast in LV myocytes requires block of ICa,L and highly buffered [Ca2+]i solutions, manoeuvres which eliminate the [Ca2+]i transient. Thus, under conventional whole cell patch clamp conditions Kv4.2/4.3-mediated Ito recovery would still be accelerated by KChIP2 isoforms but the slow component of inactivation would be minimized (Patel et al. 2002b, 2004). However, in an intact LV epi myocyte under physiological conditions the situation may be very different: due to the overlap of Ito,fast, ICa,L and the [Ca2+]i transient, the Ca2+-bound KChIP-dependent slow component of inactivation would be promoted, thus making Ito,fast a much more prominent hyperpolarizing current during all phases of the action potential plateau. In addition, if the predictions of the model of Patel et al. (2004) are correct, then Ca2+-bound KChIP-promoted reopening of Kv4.2/4.3 channels would contribute to final phase 3 repolarization. Thus the conventionally predicted behaviour of Ito,fast during an LV myocyte action potential (Fig. 1) may not be correct. These predictions are speculative, and the physiological importance of such ‘cross-talk’ interactions between Ito,fast, ICa,L and the [Ca2+]i transient have yet to be demonstrated. But if these predictions are verified, then KChIP2 isoforms would be prime candidates for the development of selective antiarrhythmic agents (Strauss & Rasmusson, 2002; Sanguinetti & Bennet, 2003).
Biophysical gating models and recent molecular insights
Proposed gating models
Kv1.4/Ito,slow
The classic analysis by Aldrich (1981) on the cumulatively inactivating IA in Archidoris and Anisodoris neurones provided the original gating model for what is now recognized as the LV Ito,slow phenotype. This model consisted of four channel states:
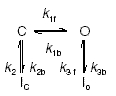 |
Scheme 2 |
Upon depolarization, channels either rapidly inactivate (I) from the resting state (C) or more slowly inactivate from the open state (O). This model was subsequently applied to rabbit atrial myocyte Ito,slow (Clark et al. 1988). During rapid and repetitive voltage clamp pulses, cumulative inactivation results from rapid closed-state inactivation and slower recovery from the open inactivated state, a process now known to be controlled by C-type inactivation (Rasmusson et al. 1995a; Roeper et al. 1997). Although such simple four-state models cannot account for sigmoid activation kinetics, they do successfully reproduce the basic inactivation and recovery properties of Ito,slow. These models also predict reopening currents upon membrane hyperpolarization (Ruppersberg et al. 1991a; Demo & Yellen, 1991; Patel et al. 2004). However, at the molecular level these models cannot account for coupling between N-type and C-type inactivation. Thus, a ‘more realistic’ model of Kv1.4 gating might be:
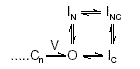 |
Scheme 3 |
where Cn is the final closed state, V denotes the final voltage-sensitive transition, IN is the N-type inactivated state, IC the C-type inactivated state and INC the simultaneously N- and C-type inactivated state (Hoshi et al. 1990, 1991; Rasmusson et al. 1995a, 1998; Yellen, 2002). If the model of Roeper et al. (1997) is applicable, CaMKII-mediated phosphorylation would increase recovery by promoting the IN state.
Kv4.2/4.3/Ito,fast
There are currently several Kv4 gating models. In general, these models agree on the predominance of closed-state inactivation in Kv4 gating; however, they differ on the assignment of kinetic transitions to specific channel conformational states (closed, open, inactivated), number of exponential components of macroscopic inactivation (2 versus 3), regulatory effects of varying [K+]o and KChIPs on gating and presence/predominance of reopening currents upon membrane hyperpolarization (Ruppersberg et al. 1991a; Demo & Yellen, 1991; Bähring et al. 2001a; Beck et al. 2002 Patel et al. 2004).
Kv4 ‘allosteric’ models. These models are based upon triple exponential descriptions of Kv4 channel inactivation kinetics (Bähring et al. 2001a; Beck et al. 2002). Their central proposal is that upon entering the open state (O) Kv4 channels may transiently enter a non-absorbing open-inactivated state (IO) but ultimately accumulate in a closed-inactivated state (IC). It is from the IC state that channels directly recover. Thus, recovery would be an ‘electrically silent’ process due to the absence of reopening currents. In these models an ‘allosteric factor (f)’ is introduced to favour inactivation from preactivated closed-states ‘nearer’ to the open state.
Bähring et al. (2001a) proposed an allosteric model as a descriptor of Kv4.2 channel gating. This model was based upon the observations that Kv4.2 N-terminal deletion did not affect recovery from the IC state and reopening currents were not observed upon repolarization. Beck et al. (2002) expanded upon this model in their analysis of the effects of KChIP1 on Kv4.1 and Kv4.3. The model (Fig. 13A) proposes that Kv4.1/4.3 inactivation is due to a concerted action of the Kv4 α subunit N- and C-termini and interactions between the distal region of S6 and the S4–S5 loop. Two assumptions of this model are: (i) inactivation from the open state, mediated by the N-terminus, kinetically corresponds to the fast time constant, τfast, of inactivation; and (ii) inactivation from the closed state, mediated by conformational changes near the internal mouth of the pore, kinetically corresponds to the slow time constant(s), τslow, of inactivation. It is also proposed that KChIP1 binds and immobilizes the N-terminus leading to a slowing of open-state inactivation. This would result in less steric hindrance around the internal mouth of the pore, favouring closed-state inactivation and lowering the energy barrier for recovery. This model also predicts that minimal to no reopening currents would be generated upon membrane hyperpolarization. While this model could reasonably reproduce Kv4 activation and inactivation kinetics during depolarizing voltage clamp step pulses, it was not demonstrated that it could reproduce recovery kinetics at hyperpolarized potentials.
Figure 13. Proposed Kv4/KChIP gating models.

A, Kv4/KChIP1 allosteric model. C, closed states; I, inactivated states; and O, open states. Rate constants α and β are assumed to be exponentially dependent upon voltage, while the transitions between C4 and O are weakly voltage dependent or voltage independent. The transition from C4 to O is also reversed biased to favour accumulation in inactivated closed states. Channels can inactivate from either open or closed states; however, along with the reverse biasing of the C4 to O transition, the ‘allosteric factor’, f, further biases the voltage dependence of inactivation to progressively favour inactivation from later closed states. Transitions that are hypothesized to be favoured by KChIP1 are indicated by the bolder arrows. B, Kv4.3/KChIP2 model. KChIP2 is hypothesized to slow closed state inactivation and promote the obligatorily coupled open-inactivated state IO, thus resulting in prominent reopening currents upon membrane hyperpolarization. This model also incorporates the effects of varying [Ca2+]i. Models from: A, Beck et al. (2002); B, Patel et al. (2004).
Kv4.3/KChIP2 isoform model. Similar to the allosteric models, it predicts prominent closed state inactivation (Patel et al. 2004). However, it proposes that inactivation, measured as a biexponential process, can occur from both preactivated closed states and the open state by a second, obligatorily coupled inactivation pathway that is at least partially absorbing (Fig. 13B). The assignment of time constants is opposite to that of the allosteric models: closed state inactivation transitions are assigned to τfast and open state inactivation transitions to τslow.
This model predicts that KChIP2 isoforms slow closed state inactivation. They also promote/accelerate the slower component of inactivation via an obligatorily coupled open state inactivation process. As a result, in the presence of KChIP2 isoforms: (i) at depolarized potentials inactivation becomes slowed and approaches single exponential behaviour; and (ii) upon membrane repolarization reopening currents are promoted. The latter prediction is supported by the recent observations of Gebauer et al. (2004) demonstrating reopening of Kv4.2 channels upon membrane repolarization. This Kv4.3/KChIP2 model is also unique in that it incorporates [Ca2+]i dependency: regulation of closed state transitions is [Ca2+]i independent, while regulation of the slower, obligatorily coupled open state transition(s) is [Ca2+]i dependent (Patel et al. 2002b, 2004). The predictions of this model have yet to be quantitatively tested.
Outer pore collapse model. This model (Eghbali et al. 2002) proposes the existence of a high affinity (Kd < 2 mm) K+-selective modulatory site within the extracellular Kv4.x pore vestibule. Due to its high affinity, this site is assumed to be saturated at normal (∼2 mm) [K+]o. This model further proposes that: (i) binding of K+ to the neighbouring high affinity selectivity filter within the pore (Heginbotham et al. 1994; Doyle et al. 1998; Jiang et al. 2003b) promotes electrostatic interactions between K+ ions in the external modulatory site; and (ii) the selectivity filter can bind K+ ions in the closed state. These characteristics would result in destabilization of the modulatory site by electrostatic interactions and allow inactivation from closed states. Thus, this model predicts that raising [K+]o (> 2 mm) would have no effects on Kv4 inactivation or recovery. While this is an interesting proposal, it does not account for the observations that increasing [K+]o well above 2 mm produces concentration-dependent modulatory effects on Kv4 gating kinetics (Jerng et al. 1999; Campbell, Patel & Strauss, 2002; Wang et al. 2004).
Structural–functional insights
In this section we focus on recent advances made in structure–function relationships governing Kv4 channels. For readers interested in similar advances made in Shaker and Kv1.4 channels we suggest the excellent review by Sokolova (2004).
HPTX/PaTX structures
HPTXs and PaTXs inhibit Kv4 channels not by occluding the pore but rather by modifying gating properties. Thus, similar to the effects originally described for hanatoxin on Kv2.1 channels, these toxins should be referred to as Kv4 ‘gating modifiers’ rather than ‘blockers’ (Swartz & MacKinnon, 1997). Gating modification is hypothesized to occur via an external site(s) that is accessible when Kv4 channels are in the closed state(s) (Diochot et al. 1999; Chagot et al. 2004). Depolarization results in reduced accessibility, affinity and/or disruption of the toxin binding site(s) due to conformational changes of the tetrameric α subunit complex.
Insights into the feasibility of this proposal have recently been provided by determination of the solution structures of HPTX2 and PaTX1 (Bernard et al. 2000; Diochot et al. 1999). Both toxins belong to a larger family of peptide toxins that display an ‘inhibitory cysteine knot’ (ICK) motif (Norton & Pallaghy, 1998). The ICK motif consists of a triple-stranded, antiparallel β-sheet ring structure stabilized by a ‘cysteine knot’ composed of six cysteine residues. This produces an embedded ring structure consisting of three β-sheets, the first and second of which are linked by disulphide bonds with the protein backbone and ‘threaded’ with a third disulphide bond between the third sheet and the backbone (Fig. 14B). This motif results in a highly stable structure, with the formation of a hydrophobic core from which only short loops emerge. (A good introduction to cysteine knots and their variations in different peptide toxins can be found at the Cyclotide webpage (http://www.cylcotide.com) and the KNOTTIN website database links (http://knottin.cbs.cnrs.fr/).
Molecular modelling studies suggest that HPTX2 and PaTX1 display very similar interaction surfaces with their Kv4 receptor(s) (Bernard et al. 2000; Shiau et al. 2003; Diochot et al. 1999). Due to overall charge distribution, electrical anisotropy results in a predicted dipole moment emerging through the β-sheet near central Lys 26 in PaTX1 and central Lys 27 in HPTX2 (Fig. 14C, upper panel). Around these central lysines are both a basic residue and an aromatic cluster (Fig. 14C, lower panel). The dipole is thus associated with a ‘hydrophobic patch domain’. Toxin selectivity would arise from a combination of hydrophobic and electrostatic interactions with the Kv4 receptor site(s), the latter presumably being extracellularly accessible regions of S4 (Shiau et al. 2003; Chagot et al. 2004). However, verification of these modelling proposals awaits further experimental evidence (for detailed discussions of this subject please see: Shiau et al. 2003; Lee & MacKinnon, 2004; Ruta & MacKinnon, 2004).
KChIP1 crystal structure
KChIP1 is composed of 10 α helices, with helices H1–H5 forming an N-terminal ‘lobe’ and helices H6–H10 forming a C-terminal ‘lobe’ (Fig. 15). Thus, the overall crystal structure of KChIP1 has two ‘faces’, one hydrophilic (containing functional EF-hands 3 and 4) and the other hydrophobic. The Kv4 N-terminus binds in the groove of the hydrophobic face (Zhou et al. 2004; Scannevin et al. 2004).
Interestingly, the loop domain in KChIP2d (amino acids 14–21), which Patel et al. (2002b) demonstrated to be involved in regulation of Kv4.3 recovery, appears to be a highly flexible structure, as suggested by the fact that Zhou et al. (2004) were unable to structurally resolve the corresponding region in the KChIP1 crystal structure (Fig. 15A and B). Could high conformational flexibility provide a basis for allowing the KChIP loop domain to regulate Kv4.3 recovery kinetics independently of EF-hand- mediated conformational changes (Patel et al. 2002b, 2004)?
Zhou et al. (2004) also propose that Ca2+-binding to EF hands 3 and 4 promotes conformational changes that fully expose the hydrophobic pocket required for KChIP1 binding to the Kv4.2 N-terminus. Without these Ca2+-mediated interactions binding would be much less efficient. However, KChIPs with mutated EF-hands still coimmunoprecipitate with Kv4 α subunits (An et al. 2000) and maintain regulatory effects on Kv4.3 recovery kinetics (Patel et al. 2002b). These results suggest that if the binding interactions proposed by Zhou et al. (2004) are weaker in the absence of Ca2+ or presence of mutated EF-hands, then there must be additional unresolved interactions between KChIPs and Kv4 channel regions to maintain the observed binding and regulatory effects.
Zhou et al. (2004) also reported that under their experimental conditions (introduction of non-physiological covalent bonds between binding domains of KChIP1 and Kv4.2) KChIP1 forms dimers. To account for the 4: 4 stoichiometry of the Kv4.2–KChIP2 complex (Kim et al. 2003), they propose that a pair of KChIP dimers bind to the Kv4 tetrameric complex. As Zhou et al. (2004) point out, this leads to a potential symmetry mismatch between KChIPs and Kv4 channels. However, Chang et al. (2003) have reported that KChIP1 displays a tendency to form dimers in the absence of metal ions and tetramers in the presence of metal ions. Thus, the issues of KChIP dimerization (representative of the native complex within LV myocytes?), dependence upon [Ca2+]i and number of KChIPs bound per Kv4.2/4.3 α subunit are still unclear.
Kv4.2 N-terminus structure
Bähring et al. (2001b) initially demonstrated that a stretch of hydrophobic amino acids in the distal N-terminus of Kv4.2 was involved in KChIP binding. Subsequently, Scannevin et al. (2004) determined the crystal structure of the Kv4.2 T1 (tetramerization) domain. Two regions in the Kv4.2 N-terminus, residues 7–11 and residues 71–90, were found to be involved in KChIP1 interactions. Region 7–11 contains mainly hydrophobic residues, while region 71–90 forms an extended hydrophilic ‘docking loop’. Mutagenesis analysis revealed that the amino acid 71–90 docking loop confers KChIP-binding to Kv1.2 (which does not bind KChIPs under native conditions). In this region, the mutation F74R was most significant in reducing the effects of KChIP1, suggesting that aromatic interactions may be involved.
Three-dimensional (EM) structure of the Kv4.2/KChIP2 channel complex
Using negative stain electron microscopy, Kim et al. (2004) have determined the three-dimensional structure (21 Å resolution) of the Kv4.2/KChIP2 complex. Similar to Shaker (Sokolova et al. 2001), Kv4.2 has a ‘hanging gondola’ held below the transmembrane pore by four N-terminal columns (Fig. 16). Kim et al. (2004) also modelled the structures of Shaker (Sokolova et al. 2001) and frequenin (Bourne et al. 2001) superimposed on the Kv4.2–KChIP2 complex. This modelling revealed that, in marked contrast to Shaker, the Kv4.2–KChIP2 complex has four ‘peripheral columns’ that extend from the membrane-bound Kv4.2 α subunits to the cytosolic regions where KChIP2 molecules bind to the Kv4.2 N-termini. This model predicts that KChIPs extend the gondola platform laterally to meet with the four peripheral columns.
Figure 16. Three dimensional (electron microscopic (EM)) structures of Shaker and Kv4.2/KChIP2.

A, Shaker channel. The left panel illustrates the 3D structure of the homotetrameric Shaker channel. D1 and D2 are subdomains embedded within the membrane and T1 is the tetramerization domain. The right panel is an overlay/docking of the crystal structures of the T1 tetramer (red) and the KcsA channel (blue). B, 3D EM structure of Kv4.2/KChIP2. The left panel illustrates a side view, while the right panel is a side view rotated 45 deg along the central axis of symmetry. C, EM structure of Shaker (white) overlaid on the Kv4.2/KChIP2 complex structure (blue mesh). The upper panel illustrates a side view (only two frequenin molecules (red) are illustrated) while the lower panel shows a ‘bottom’ view. Note the peripheral columns (Kv4.2 C-termini?) in the Kv4.2/KChIP2 complex that are absent in Shaker. Four frequenin molecules will only fit into the structure with their hydrophobic pockets facing the gondola platform thus exposing their hydrophilic EF-hand faces towards the cytosol. A, modified from Sokolova (2004) with permission; B, reproduced from Kim et al. (2004) with permission from Elsevier; C, modified from Kim et al. (2004) with permission from Elsevier.
While the identity of the peripheral columns has yet to be determined, Kim et al. (2004) speculate that they correspond to the C-termini of Kv4.2 α subunits. This is an appealing proposal: could the predicted close proximity of KChIPs and the Kv4 C-termini allow transduction of KChIP-mediated effects between N- and C-termini, and thus transduction of these effects to other physically distant regions of the α subunit? And is the absence of the four peripheral columns in Shaker a physical basis for some of the functional differences between Shaker and Kv4 channels?
It is also interesting to note that the Kv4.2 channel preparation used by Kim et al. (2004) contained no free Ca2+. Might the KChIPs in the derived three dimensional structure have Ca2+-free EF-hands? If this is the case, then could the Kv4 C-terminus serve as another Ca2+-independent binding site for KChIP-mediated interactions, and could these interactions be responsible for Ca2+-independent regulation of Kv4.3 recovery kinetics (Patel et al. 2002b, 2004)? Thus, while the Kv4 N-terminal T1 domain may help maintain Kv4–KChIP interactions, it is possible that there may be even stronger interactions between KChIPs and the Kv4.2/4.3 C-terminus.
Molecular models of Kv channel activation
While no Kv4 α subunit has yet been crystalized, the crystal structure of KvAP, a voltage-dependent K+ channel from the thermolphilic archaebacterium Aeropyrum permix, has been determined (Jiang et al. 2003b, c). However, this pioneering work has led to a debate over the physical mechanisms governing Kv channel activation, with three major models having been proposed. (For readers interested in detailed discussions the following are excellent sources: Bezanilla, 2000, 2002; Gandhi & Isacoff, 2002; Bezanilla & Perozo, 2003; MacKinnon, 2003; Laine et al. 2003, 2004; Blaustein & Miller, 2004; Cuello et al. 2004).
Activation Model 1: ‘sliding helix or corkscrew model’. This is the classic model of voltage-dependent channel gating (Fig. 17A). Recent evidence in its support comes from histidine scanning and LRET and FRET imaging (Starace et al. 1997; Cha et al. 1999; Glauner et al. 1999; Starace & Bezanilla, 2001). The major assumptions of this model are that: (i) the transmembrane segment S4, which contains positively charged arginines at approximately every third residue, is completely surrounded by a proteinaceous ‘activation canal’; (ii) S4 lies close to, and may interact with, the pore domain; and (iii) upon membrane depolarization S4 moves through the activation canal towards the extracellular surface as a rigid rod via either sliding or corkscrewing through the gating pore (Fig. 17Aii).
Figure 17. Kv channel activation models.

A, helical-corkscrew model. S4 is hypothesized to be located next to, and potentially interacting with, the pore domain. i, upper panel: Shaker channel model viewed ‘down’ from the extracellular side. Domains colour coded as follows: S1, light blue; S2, yellow; S3, red; S4, dark blue; pore domain, green. Lower panel: Side view only showing 2 α subunits for clarity. ii, schematic representation of S4 movement in response to membrane depolarization. B, voltage-sensor paddle model. i, KvAP crystal structure (1 α subunit), channel closed state. The S3(b)–S4 voltage-sensor paddle lies parallel to the intracellular membrane surface and is located on the periphery of the channel. ii, upon membrane depolarization the paddles move ∼20 Å perpendicular into the membrane, thus exposing to the extracellular environment amino acids in S3(b)–S4 that are either intracellular or embedded in the membrane in the closed state. C, carrier-like model: hypothesized Shaker S4 gating pore. i, schematic of proton conduction through the S4 gating pore based upon mutagenic replacement of S4 R365 by histidine. Upper panel: R365H mutant (indicated as grey sphere). Conformational changes underlying proton transport. In the presence of an inward proton gradient the R365H channel transports one proton per channel open-to-closed cycle. Lower panel: R362H mutant (indicated as grey sphere). Internal and external protons have simultaneous access to the histidine only when the channel is in the closed conformation allowing proton current flow only when the channel is closed. ii, schematic representation. The R362H mutation is indicated in red. In the closed conformation, a very narrow gate is formed that separates the extracellular and intracellular surfaces. This ‘focuses’ the transmembrane electric field over a very short distance of essentially one amino acid residue. Ai, fromLaine et al. (2003) with permission from Elsevier; Bi, modified from Jiang et al. (2003b) reprinted by permission from Nature, © 2003 Macmillan Publishers Ltd (http://www.nature.com/); Ci, from Starace & Bezanilla (2004) reprinted by permission from Nature, © 2004 Macmillan Publishers Ltd (http://www.nature.com/); Aii, Bii and Cii, Blaustein & Miller (2004) reprinted by permission from Nature, © 2004 Macmillan Publishers Ltd (http://www.nature.com/).
Activation Model 2: ‘voltage-sensor paddle model’. This model (Fig. 17B) is based upon the crystal structure of KvAP (Jiang et al. 2003b, c). The structure of KvAP segments S5 and S6, which form the pore and selectivity filter, were found to be very similar to the crystal structure of KcsA, a two transmembrane domain K+ channel from Streptomyces lividans (Doyle et al. 1988). However, Jiang et al. (2003b) concluded that the structure of the voltage sensor was very different from that predicted by the helical corkscrew model: they proposed that segments S3 and S4 were located at the channel's outer perimeter. The second helix in S3, S3(b), and the amino terminal half of the traditionally defined S4 segment were found to form a hydrophobic helix-turn-helix structure termed the ‘voltage-sensor paddle’. The S3 loop and S4–S5 linker provide flexibility, allowing the paddle to adopt more than one conformation. In the closed state, the S4 helices of the paddle of each α subunit are near the intracellular membrane surface, perpendicular to the pore axis, and are loosely attached to the pore domain. Upon membrane depolarization, the paddles move ∼20 Å perpendicular into the membrane, opening the channel by pulling on the S4–S5 linkers.
The major point of debate in this model is the proposal of S3b–S4 paddles existing on the outer perimeter of the channel and that the corresponding arginines would be exposed to the lipid environment of the membrane. No S4 gating pore is predicted. Arguments have been made that the KvAP structure may have been altered by detergents employed in its isolation, use of FAB fragments and/or crystal-packing forces (see discussions in Laine et al. 2003, 2004; Blaustein & Miller, 2004; Birnbaum et al. 2004). But the magnitude of such distortions is unclear. Furthermore, electron microscopic analysis (10.5 Å resolution) of KvAP in the open conformation suggests that the voltage-sensor paddles are near the extracellular surface and located at the protein–lipid interface (Jiang, Wang & MacKinnon, 2004).
Activation Model 3: ‘gating pore’ or ‘transporter models’. These models do not incorporate a gating paddle but do incorporate many of aspects of the sliding/corkscrew helix model. However, these models propose that S4 is located at the periphery of the channel and is only partially surrounded by a proteinaceous ‘gating pore’. The polar protein walls of the gating pore (provided by transmembrane helices S2 and S3 and the pore domain helices) are hypothesized to insulate the S4 arginines from lipids. Accessibility scanning measurements indicate that, upon depolarization, nine residues in S4 move from an inaccessible location, presumably within the gating pore, to a position that is accessible to the extracellular solution, a movement that corresponds to 13.5 Å of axial length of an α helix (Larsson et al. 1996; Mannuzzu et al. 1996; Baker et al. 1998; Schönherr et al. 2002). FRET measurements further suggest a ∼180 deg helical rotation (Cha et al. 1999; Glauner et al. 1999). The gating pore would thus be ∼13.5 Å long, contain water-filled vestibules at both of its ends, and would focus the transmembrane electric field on a small sequence of S4 within the narrow part of the gating pore (Fig. 18A).
Figure 18. Shaker channels: hypothesized S4 gating pore.

A, schematic of voltage-dependent S4 movement within the gating pore. B, hypothesized locations of S4, gating pore domains (ω) and the channel K+ pore selectivity/conduction domain (α). S4 arginines are hypothesized to face the polar proteinaceous walls of the gating pore formed by S2, S3 and the K+ pore helices. C, proposed ‘ratcheting’ interactions between S4 arginines and S2 E283. Cartoons illustrate relative positions of S4 and S2 in the resting and activated states. At rest, R1 blocks ion conduction through the gating pore. Upon depolarization, R1, R2, R3 and R4 each successively ‘ratchet’ through the gating pore, with each intermediate state blocking ion conduction. PD, channel pore domain; α, K+ current through the channel pore; ω, guanidium current flow through the gating pore in the presence of mutation R1C. D, mutation of R1 to smaller amino acids (guanidium group removal) unblocks the gating pore at rest, leading to flow of a cation-selective ‘omega current’ (blue arrow). Figures from: A, Gandhi & Isacoff (2002) by copyright permission of The Rockefeller University Press; B, C and D, Tombola et al. (2005) reproduced with permission from Elsevier.
Original support for this model (Fig. 17C) came from fluorescent measurements on Shaker channels indicating that S4 moved less than 2 Å in response to membrane depolarization (Cha et al. 1999). This model (Starace & Bezanilla, 2004) differs from the previous two models because it proposes that Kv channel activation occurs through a transporter-like mechanism. The model is based upon results obtained from Shaker channels where the first outermost arginine in S4 was mutated to histidine. This mutation results in production of a steady transmembrane proton current, conducted through the S4 gating pore, when the channel is in the closed-state (Starace & Bezanilla, 2004). Consistent with the gating pore hypothesis, in the closed-state protons can access the single histidine in S4 from both the extracellular and intracellular solutions. This implies that the gating pore focuses the transmembrane voltage gradient across the distance of a single S4 side chain residue! As a result, in response to membrane depolarization S4 moves only a few angstroms at most (in contrast to either the helical corkscrew or paddle models, which predict that S4 moves ∼13–20 Å perpendicular to the cell membrane).
Further evidence for the existence of the S4 gating pore in Shaker has been provided by Tombola et al. (2005). These investigators demonstrated that when the first arginine in S4 was replaced with smaller amino acids, a manoeuvre that removed the guanidium group of arginine, a high conductance cation-selective pathway was formed within the putative S4 gating pore when the channel was in the resting state(s). This current was designated the ‘omega current’ to distinguish it from the conventional K+-selective ‘alpha current’ that normally flows through the selectivity filter/pore domain of the open channel. The omega current, which could be blocked by Mg2+, was supported by guanidium ions, consistent with the hypothesis that arginine residues move through the gating pore during channel activation. These results appear to be inconsistent with the gating paddle model. It was thus proposed that under normal conditions activation of S4 involves ‘ratcheting’ movements between intermediate conformational states where each of the arginines successively occupies the gating pore, thereby occluding ion conduction through the gating pore as the channel activates (Tombola et al. 2005; Fig. 18).
In summary, while there is universal consensus on the role of S4 in Kv channel activation, there is considerable debate on the underlying molecular mechanisms. Also, when considering these models, it should be kept in mind that there are differences in the sequences of S3b–S4 of KvAP, Shaker and Kv4.2/4.3. For example, the first arginine in the S4 segment of Shaker is replaced by a valine in Kv4.2. Thus, might there be differences in the gating pores of Shaker verus Kv channels? And, on a more fundamental level, might there also be differences in the basic structural mechanisms underlying activation of Kv channels in prokaryotes versus eukaryotes?
Concluding remarks
We have attempted to summarize what we feel are the major advances that have been made in our understanding of LV Ito phenotypes. We have emphasized important functional differences between Ito,fast and Ito,slow and differences among species in the transmural LV free wall distributions of these two Ito phenotypes. We have also reviewed those areas where at present we lack understanding of underlying mechanisms. At the molecular level, while analysis of Shaker channels has provided insight into molecular mechanisms governing gating of Kv1.4-mediated Ito,slow, it is now clear that these mechanisms are not always applicable to Kv4.2/4.3 channels, particularly with regard to inactivation and recovery. At the cellular level, particular areas that we feel need further investigation are: (i) the role of normal alterations in [Ca2+]i on Ito,fast kinetics (given that KChIPs provide a means for Ca2+-dependent regulation of Kv4 channels) and its predicted behaviour during an LV myocyte action potential; (ii) the role of Ito,slow in species where this phenotype is prominent in LV endo myocytes; (iii) kinase-dependent modulation of Ito,fast (obligatory KChIP2 involvement?) and Ito,slow; and (iv) determination of protein expression patterns of different regulatory subunits across the LV free wall and the functional consequences of such gradients. These considerations then need to be incorporated into mathematical models of LV electrical activity. Finally, the contributions of different LV Ito phenotypes to both repolarization gradients and arrhythmic conditions need to be evaluated.
Note added in proof
During the production process of this review four articles have appeared which have direct relevance to issues that have been discussed: 1) Radicke et al. (2005; J Physiol. 565.3, 751–756) have cloned DPP6 from human ventricle and demonstrated that it regulates Kv4.3 gating kinetics; 2) Long, Campbell & MacKinnon (2005a, b; Science 309, 897–903, 903–908) have determined the crystal structure of the mammalian Shaker family Kv1.2 channel in the open state; and 3) Wang et al. (2005; Biophys. J. [epub ahead of print]) have both independently reported Kv4.3 reopening currents (Patel et al., 2004) and, using mathematical modelling, have quantitatively demonstrated that proposed allosteric Kv4 channel gating models (e.g. Beck et al., 2002; see Figure 13.A) cannot successfully reproduce the overall gating characteristics of Kv4.3 channels.
Acknowledgments
This work was supported by an American Heart Association Established Investigator Award to D.L.C.
References
- Aldrich RW. Inactivation of voltage-gated delayed potassium current in molluscan neurons. A kinetic model. Biophys J. 1981;36:519–532. doi: 10.1016/S0006-3495(81)84750-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KT, Strassle RW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Anderson AE, Adams JP, Qian Y, Cook RG, Pfaffinger PJ, Sweatt JD. Kv4.2 phosphorylation by cyclic AMP-dependent protein kinase. J Biol Chem. 2000;275:5337–5346. doi: 10.1074/jbc.275.8.5337. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C, Zygmunt AC, Dumaine R. Electrophysiology and pharmacology of ventricular repolarization. In: Gussak I, Antzelevitch C, editors. Cardiac Repolarization. Totowa, NJ, USA: Human Press Inc.; 2003. pp. 63–89. [Google Scholar]
- Archer SL, Rusch NJ. Potassium Channels in Cardiovascular Biology. New York: Kluwer Academic/Plenum Publishers; 2001. [Google Scholar]
- Bähring R, Boland LM, Varghese A, Gebauer M, Pongs O. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J Physiol. 2001a;535:65–81. doi: 10.1111/j.1469-7793.2001.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähring R, Dannenberg J, Peters HC, Leicher T, Pongs O, Isbrandt D. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J Biol Chem. 2001b;276:23888–23894. doi: 10.1074/jbc.M101320200. [DOI] [PubMed] [Google Scholar]
- Baker OS, Larrsson HP, Mannuzzu LM, Isacoff EY. Three transmembrane conformations and sequence-dependent displacement of the S4 domain in shaker K+ channel gating. Neuron. 1998;20:1283–1294. doi: 10.1016/s0896-6273(00)80507-3. [DOI] [PubMed] [Google Scholar]
- Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 α subunit. Circ Res. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- Bassani RA, Altamirano J, Puglisi JL, Bers DM. Action potential duration determines sarcoplasmic reticulum Ca2+ reloading in mammalian ventricular myocytes. J Phyisol. 2004;559:593–609. doi: 10.1113/jphysiol.2004.067959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck EJ, Bowlby M, An WF, Rhodes KJ, Covarrubias M. Remodeling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein. J Physiol. 2002;538:691–706. doi: 10.1113/jphysiol.2001.013127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck EJ, Corvarrubias M. Kv4 channels exhibit modulation of closed-state inactivation in inside-out patches. Biophys J. 2001;81:867–883. doi: 10.1016/S0006-3495(01)75747-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Legros C, Ferrat G, Bischoff U, Marquardt A, Pongs O, Darbon H. Solution structure of HPTX2, a toxin from Heteropoda venatoria spider that blocks Kv4.2 potassium channel. Protein Sci. 2000;9:2059–2067. [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- Bett GC, Rasmusson RL. Inactivation and recovery in Kv1.4 K+ channels: lipophilic interactions at the intracellular mouth of the pore. J Physiol. 2004;556:109–120. doi: 10.1113/jphysiol.2003.055012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- Bezanilla F. Voltage sensor movements. J General Physiol. 2002;120:465–473. doi: 10.1085/jgp.20028660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Perozo E. The voltage sensor and the gate in ion channels. Adv Protein Chem. 2003;63:211–241. doi: 10.1016/s0065-3233(03)63009-3. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan L-Y, Anderson AE, Sweatt JD, Schraeder LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Blaustein RO, Miller C. Shake, rattle or roll? Nature. 2004;427:499–500. doi: 10.1038/427499a. [DOI] [PubMed] [Google Scholar]
- Bondarenko VE, Szigeti GP, Bett GC, Kim SJ, Rasmusson RL. Computer model of action potential of mouse ventricular myocytes. Am J Physiol. 2004;287:H1378–H1403. doi: 10.1152/ajpheart.00185.2003. [DOI] [PubMed] [Google Scholar]
- Bourne Y, Dannenberg J, Pollmann V, Marchot P, Pongs O. Immunocytochemical localization and crystal structure of human frequenin (neuronal calcium sensor 1) J Biol Chem. 2001;276:11949–11955. doi: 10.1074/jbc.M009373200. [DOI] [PubMed] [Google Scholar]
- Boyett MR, Honjo H, Lei M, Kodama I. Transient outward K+ current, ito, in the sinoatrial node. Cardiovascular Res. 2001;52:519–520. doi: 10.1016/s0008-6363(00)00036-5. [DOI] [PubMed] [Google Scholar]
- Brahmajothi MV, Campbell DL. Heterogeneous basal expression of nitric oxide synthase and superoxide dismutase isoforms in mammalian heart. Implications for mechanisms governing indirect and direct nitric-oxide-related effects. Circ Res. 1999;85:575–587. doi: 10.1161/01.res.85.7.575. [DOI] [PubMed] [Google Scholar]
- Brahmajothi MV, Campbell DL, Rasmusson RL, Morales MJ, Trimmer JS, Nerbonne JM, Strauss HC. Distinct transient outward potassium current (Ito) phenotypes and distribution of fast-inactivating potassium channel alpha subunits in ferret left ventricular myocytes. J General Physiol. 1999;113:581–600. doi: 10.1085/jgp.113.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmajothi MV, Morales MJ, Liu S, Rasmusson RL, Campbell DL, Strauss HC. In situ hybridization reveals extensive diversity of K+ channel mRNA in isolated ferret cardiac myocytes. Circ Res. 1996;78:1083–1089. doi: 10.1161/01.res.78.6.1083. [DOI] [PubMed] [Google Scholar]
- Brunet S, Aimond F, Li H, Guo W, Eldstrom J, Fedida D, Yamada KA, Nerbonne JM. Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J Physiol. 2004;559:103–120. doi: 10.1113/jphysiol.2004.063347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner M, Guo W, Mitchell GF, Buckett PD, Nerbonne JM, Koren G. Characterization of mice with a combined suppression of I(to) and I(K,slow) Am J Physiol. 2001;281:H1201–H1209. doi: 10.1152/ajpheart.2001.281.3.H1201. [DOI] [PubMed] [Google Scholar]
- Campbell DL, Patel SP, Strauss HC. ‘Anomalous’ effects of varying [K+]o on inactivation and recovery kinetics of ferret left ventricular Ito. Biophys J. 2002;82:1186. [Google Scholar]
- Campbell DL, Qu Y, Rasmusson RL, Strauss HC. The calcium-independent transient outward potassium current in isolated ferret right ventricular myocytes. II. Closed-state reverse use-dependent block by 4-aminopyridine. J General Physiol. 1993a;101:603–626. doi: 10.1085/jgp.101.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DL, Rasmusson RL, Comer MB, Strauss HC. The cardiac calcium-independent transient outward potassium current: kinetics, molecular properties, and role in ventricular repolarization. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: from Cell to Bedside. Philadelphia: Saunders; 1995. pp. 83–96. [Google Scholar]
- Campbell DL, Rasmusson RL, Qu Y, Strauss HC. The calcium-independent transient outward potassium current in isolated ferret right ventricular myocytes. I. Basic characterization and kinetic analysis. J General Physiol. 1993b;101:571–601. doi: 10.1085/jgp.101.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DL, Rasmusson RL, Strauss HC. Ionic current mechanisms generating vertebrate primary cardiac pacemaker activity at the single cell level: An integrative view. Ann Rev Physiol. 1992;54:279–302. doi: 10.1146/annurev.ph.54.030192.001431. [DOI] [PubMed] [Google Scholar]
- Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J General Physiol. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campomanes CR, Carroll KL, Manganas LN, Hershberger ME, Gong B, Antonucci DE, Rhodes KJ, Trimmer JS. Kv β subunit oxidoreductase activity and Kv1 potassium channel trafficking. J Biol Chem. 2002;277:8298–8305. doi: 10.1074/jbc.M110276200. [DOI] [PubMed] [Google Scholar]
- Carmeliet E, Vereecke J. Cellular Cardiac Electrophysiology. Boston: Kluwer Academic Publishers; 2002. Ionic currents and action potentials in cardiac cells; pp. 95–117. [Google Scholar]
- Casis O, Iriarte M, Gallego M, Sanchez-Chapula JA. Differences in regional distribution of K+ current densities in rat ventricle. Life Sci. 1998;63:391–400. doi: 10.1016/s0024-3205(98)00287-2. [DOI] [PubMed] [Google Scholar]
- Castellino RC, Morales MJ, Strauss HC, Rasmusson RL. Time- and voltage-dependent modulation of a Kv1.4 channel by a β-subunit (Kvβ3) cloned from ferret ventricle. Am J Physiol. 1995;269:H385–H391. doi: 10.1152/ajpheart.1995.269.1.H385. [DOI] [PubMed] [Google Scholar]
- Cha A, Snyder GE, Selvin PR, Bezanilla F. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature. 1999;402:809–813. doi: 10.1038/45552. [DOI] [PubMed] [Google Scholar]
- Chagot B, Escoubas P, Villegas E, Bernard C, Ferrat G, Corzo G, Lazdunski M, Darbon H. Solution structure of Phrixotoxin 1, a specific peptide inhibitor of Kv4 potassium channels from the venom of the therapsid spider Phrixotrichus auratus. Protein Sci. 2004;13:1197–1208. doi: 10.1110/ps.03584304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L-S, Chen C-Y, Wu TT. Functional implication with the metal-binding properties of KChIP1. Biochem Biophys Res Commun. 2003;311:258–263. doi: 10.1016/j.bbrc.2003.09.204. [DOI] [PubMed] [Google Scholar]
- Chien KR. Molecular Basis of Cardiovascular Disease. Philadelphia: W. B. Saunders Company; 1999. [Google Scholar]
- Choi KI, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc Natl Acad Sci U S A. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB, Bouchard RA, Salinas-Stefanon E, Sanchez-Chapula J, Giles WR. Heterogeneity of action potential waveforms and potassium currents in rat ventricle. Cardiovasc Res. 1993;27:1795–1799. doi: 10.1093/cvr/27.10.1795. [DOI] [PubMed] [Google Scholar]
- Clark RB, Giles WR, Imaizumi Y. Properties of the transient outward current in rabbit atrial cells. J Physiol. 1988;405:147–168. doi: 10.1113/jphysiol.1988.sp017326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer MB, Campbell DL, Rasmusson RL, Lamson DR, Morales MJ, Zhang Y, Strauss HC. Cloning and characterization of an Ito-like potassium channel from ferret ventricle. Am J Physiol. 1994;267:H1383–H139. doi: 10.1152/ajpheart.1994.267.4.H1383. [DOI] [PubMed] [Google Scholar]
- Craig TA, Benson LM, Venyaminov SY, Klimtchuk ES, Bajzer Z, Prendergast FG, Naylor S, Kumar R. The metal binding properties of DREAM. Evidence for calcium-mediated changes in DREAM structure. J Biol Chem. 2002;277:10955–10966. doi: 10.1074/jbc.M109660200. [DOI] [PubMed] [Google Scholar]
- Cuello LG, Cortes DM, Perozo E. Molecular architecture of the KvAP voltage-dependent K+ channel in a lipid bilayer. Science. 2004;306:491–495. doi: 10.1126/science.1101373. [DOI] [PubMed] [Google Scholar]
- Deal KK, England SK, Tamkum MM. Molecular physiology of cardiac potassium channels. Physiol Rev. 1996;76:49–67. doi: 10.1152/physrev.1996.76.1.49. [DOI] [PubMed] [Google Scholar]
- de Bakker JMT, Opthof T. Is the apico-basal gradient larger than the transmural gradient? J Cardiovas Pharm. 2002;39:328–331. doi: 10.1097/00005344-200203000-00002. [DOI] [PubMed] [Google Scholar]
- Decher N, Barth AS, Gonzalez T, Steinmyer K, Sanguinetti MC. Novel KChIP2isoforms increase functional diversity of transient outward potassium current. J Physiol. 2004;557:761–772. doi: 10.1113/jphysiol.2004.066720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decher N, Uyguner O, Scherer CR, Karaman B, Yüskelapak M, Busch AE, Steinmeyer K, Wollnik B. hKChIP2 is a functional modifier of hKv4.3 potassium channels: cloning and expression of a short hKChIP2 splice variant. Cardiovas Res. 2001;52:255–264. doi: 10.1016/s0008-6363(01)00374-1. [DOI] [PubMed] [Google Scholar]
- Deck KA, Trautwein W. Ionic currents in cardiac excitation. Pflugers Arch. 1964;280:63–80. doi: 10.1007/BF00412616. [DOI] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Deschênes I, DiSilvestre D, Juang GJ, Wu R, An WF, Tomaselli GF. Regulation of Kv4.3 current by KChIP2 splice variants: a component of native cardiac Ito? Circulation. 2002;106:423–429. doi: 10.1161/01.cir.0000025417.65658.b6. [DOI] [PubMed] [Google Scholar]
- Deschênes I, Tomaselli GF. Modulation of Kv4.3 current by accessory subunits. FEBS Lett. 2002;528:183–188. doi: 10.1016/s0014-5793(02)03296-9. [DOI] [PubMed] [Google Scholar]
- Diochot S, Drici MD, Moinier D, Fink M, Lazdunski M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br J Pharmacol. 1999;126:251–263. doi: 10.1038/sj.bjp.0702283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JE, McKinnon D. Quantitative analysis of mRNA expression in atrial and ventricular muscle of rats. Circ Res. 1994;79:659–668. doi: 10.1161/01.res.75.2.252. [DOI] [PubMed] [Google Scholar]
- Dixon JE, Shi W, Wang H-S, MacDonald CYuH, Wymore R, Cohen IS, McKinnon D. Role of the Kv4.3 potassium channel in ventricular muscle: a molecular correlate for the transient outward current. Circ Res. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Eghbali M, Olcese R, Zarei MM, Stefani E. External pore collapse as an inactivation mechanism for Kv4.3 K+ channels. J Memb Biol. 2002;188:73–86. doi: 10.1007/s00232-001-0173-3. [DOI] [PubMed] [Google Scholar]
- Favre J-F, Calmels TPG, Rouanet S, Javré J-L, Cheval B, Bril A. Characterization of Kv4.3 in HEK 293 cells: comparison with the rate ventricular transient outward potassium current. Cardiovasc Res. 1999;41:188–199. doi: 10.1016/s0008-6363(98)00215-6. [DOI] [PubMed] [Google Scholar]
- Fedida D, Braun AP, Giles WR. Alpha-1 adrenoceptors in myocardium: functional aspects and transmembrane signaling mechanisms. Physiol Rev. 1993;73:469–487. doi: 10.1152/physrev.1993.73.2.469. [DOI] [PubMed] [Google Scholar]
- Fedida D, Giles WR. Regional variations in action potentials and transient outward current in myocytes isolated from rabbit left ventricle. J Physiol. 1991;442:191–209. doi: 10.1113/jphysiol.1991.sp018789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franqueza L, Valenzuela C, Eck J, Tamkun MM, Tamargo J, Snyders DJ. Functional expression of an inactivating potassium channel (Kv4.3) in a mammalian cell line. Cardiovasc Res. 1999;41:212–219. doi: 10.1016/s0008-6363(98)00220-x. [DOI] [PubMed] [Google Scholar]
- Gandhi CS, Isacoff EY. Molecular models of voltage sensing. J General Physiol. 2002;120:455–463. doi: 10.1085/jgp.20028678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer M, Isbrandt D, Sauter K, Callsen B, Nolting A, Pongs O, Bähring R. N-type inactivation features of Kv4.2 channel gating. Biophys J. 2004;56:210–223. doi: 10.1016/S0006-3495(04)74097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles W, Imaizumi Y. Comparison of potassium currents in rabbit atrial and ventricular cells. J Physiol. 1988;405:123–145. doi: 10.1113/jphysiol.1988.sp017325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauner KS, Mannuzzu LM, Gandhi CS, Isacoff EY. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 1999;402:813–817. doi: 10.1038/45561. [DOI] [PubMed] [Google Scholar]
- Greenstein JL, Wu R, Po S, Tomaselli GF, Winslow RL. Role of the calcium-independent transient outward current Ito1 in shaping action potential morphology and duration. Circ Res. 2000;87:1026–1033. doi: 10.1161/01.res.87.11.1026. [DOI] [PubMed] [Google Scholar]
- Gulbis JM, Mann S, MacKinnon R. Structure of a voltage-dependent K+ channel beta subunit. Cell. 1999;97:943–952. doi: 10.1016/s0092-8674(00)80805-3. [DOI] [PubMed] [Google Scholar]
- Guo W, Xu H, London B, Nerbonne JM. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J Physiol. 1999;521:587–599. doi: 10.1111/j.1469-7793.1999.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Li H, London B, Nerbonne JM. Functional consequences of elimination of Ito,f and Ito,s: early afterdepolarizations, atrioventricular block, and ventricular arrhythmia in mice lacking Kv1.4 and expressing a dominant-negative Kv4 alpha subunit. Circ Res. 2000;87:73–79. doi: 10.1161/01.res.87.1.73. [DOI] [PubMed] [Google Scholar]
- Guo W, Li H, Aimond F, Johns DC, Rhodes KJ, Trimmer JS, Nerbonne JM. Role of heteromultimers in the generation of myocardial transient outward K+ current. Circ Res. 2002a;90:586–593. doi: 10.1161/01.res.0000012664.05949.e0. [DOI] [PubMed] [Google Scholar]
- Guo W, Malin SA, Johns DC, Jeromin A, Nerbonne JM. Modulation of Kv4-encoded K+ currents in the mammalian myocardium by neuronal calcium sensor-1. J Biol Chem. 2002b;277:26436–26443. doi: 10.1074/jbc.M201431200. [DOI] [PubMed] [Google Scholar]
- Gussack I, Antzelevitch C. Cardiac Repolarization. Totowa, NJ, USA: Humana Press; 2003. [Google Scholar]
- Han W, Bao W, Wang Z, Nattel S. Comparison of ion-channel subunit expression in canine cardiac Purkinje fibers and ventricular muscle. Circ Res. 2002b;91:790–797. doi: 10.1161/01.res.0000039534.18114.d9. [DOI] [PubMed] [Google Scholar]
- Han W, Wang C, Nattel S. A comparison of transient outward currents in canine cardiac Purkinje cells and ventricular myocytes. Am J Physiol. 2000;279:H466–H474. doi: 10.1152/ajpheart.2000.279.2.H466. [DOI] [PubMed] [Google Scholar]
- Han W, Zhang L, Schram G, Nattel S. Properties of potassium currents in Purkinje cells of failing human hearts. Am J Physiol. 2002b;283:H2495–H2503. doi: 10.1152/ajpheart.00389.2002. [DOI] [PubMed] [Google Scholar]
- Hatano N, Ohya S, Muraki K, Clark RB, Giles WR, Imaizumi Y. Two arginines in the cytoplasmic C-terminal domain are essential for voltage-dependent regulation o A-type K+ current in the Kv4 channel subfamily. J Biol Chem. 2004;279:5450–5459. doi: 10.1074/jbc.M302034200. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys J. 1994;66:1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. 3. Sunderland, MA, USA: Sinauer Associates, Inc.; 2001a. Gating mechanisms: kinetic thinking; pp. 575–602. [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. 3. Sunderland, MA, USA: Sinauer Associates, Inc.; 2001b. Classic mechanisms of block; pp. 503–537. [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist MH, Cao J, Hernandez-Pineda R, Jacobson MD, Carroll KI, Sung MA, Betty M, Ge P, Gilbride KJ, Brown ME, Jurman ME, Lawson D, Silos-Santiago I, Xie Y, Covarrubias M, Rhodes KJ, Distefano PS, An WF. Elimination of fast inactivation in Kv4 A-type potassium channels by an auxiliary subunit domain. Proc Natl Acad Sci U S A. 2002;99:1035–1040. doi: 10.1073/pnas.022509299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist MH, Cao J, Knoppers MH, Jurman ME, Distefano PS, Rhodes KJ, Xie Y, An WF. Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. J Neurosci. 2001;21:4154–4161. doi: 10.1523/JNEUROSCI.21-12-04154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Huelsing DJ, Spitzer KW, Pollard AE. Spontaneous activity induced in rabbit Purkinje myocytes during coupling to a depolarized model cell. Cardiovasc Res. 2003;59:620–627. doi: 10.1016/s0008-6363(03)00507-8. [DOI] [PubMed] [Google Scholar]
- Ikura M. Calcium binding and conformational response in EF-hand proteins. Trends Biochem Sci. 1996;21:14–17. [PubMed] [Google Scholar]
- Iyer V, Mazhari R, Winslow RL. A computational model of the human left-ventricular epicardial myocyte. Biophys J. 2004;87:1507–1525. doi: 10.1529/biophysj.104.043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Corvarrubias M. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys J. 1997;72:163–174. doi: 10.1016/S0006-3495(97)78655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng H, Qian Y, Pfaffinger PJ. Modulation of Kv4.2 channel expression and gating by dipeptidyl peptidase 10 (DPP10) Biophys J. 2004;87:2380–2396. doi: 10.1529/biophysj.104.042358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Shahidulla M, Corvarrubias M. Inactivation gating of Kv4 potassium channels. Molecular interactions involving the inner vestibule of the pore. J General Physiol. 1999;113:641–660. doi: 10.1085/jgp.113.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Bett GC, Li X, Bondarenko VE, Rasmusson RL. C-type inactivation involves a significant decrease in the intracellular aqueous Volume of Kv1.4 K+ channels expressed in Xenopus oocytes. J Physiol. 2003a;549:683–695. doi: 10.1113/jphysiol.2002.034660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003b;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003c;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- Jiang QX, Wang DN, MacKinnon R. Electron microscopic analysis of KvAP voltage-dependent K+ channels in an open conformation. Nature. 2004;430:806–810. doi: 10.1038/nature02735. [DOI] [PubMed] [Google Scholar]
- Kääb S, Dixon J, Duc J, Ashen D, Näbaeur M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current down regulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- Kim LA, Furst J, Butler MH, Xu S, Grigorieff N, Goldstein SA. Composition of Ito channels: Kv4.2-KChIP2 complexes carry four subunits of each type. J Biol Chem. 2003;279:5549–5554. doi: 10.1074/jbc.M311332200. [DOI] [PubMed] [Google Scholar]
- Kim LA, Furst J, Gutierrerz D, Butler MH, Xu S, Goldstein SA, Grigorieff N. Three dimensional structure of Ito: Kv4.2-KChIP2 ion channels by electron microscopy at 21 Å resolution. Neuron. 2004;41:513–519. doi: 10.1016/s0896-6273(04)00050-9. [DOI] [PubMed] [Google Scholar]
- Kuo H-C, Cheng C-F, Clark RB, Lin J, Gu Y, Ikeda Y, Chu P-H, Ross J, Giles WR, Chien KR. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of Ito and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- Kuryshev YA, Gudz TI, Brown AM, Wible BA. KChAP as a chaperone for specific K+ channels. Am J Physiol. 2000;278:C931–C941. doi: 10.1152/ajpcell.2000.278.5.C931. [DOI] [PubMed] [Google Scholar]
- Laine M, Lin MA, Bannister JP, Silverman WR, Mock AF, Roux B, Papazian DM. Atomic proximity between S4 segment and pore domain in Shaker potassium channels. Neuron. 2003;39:467–481. doi: 10.1016/s0896-6273(03)00468-9. [DOI] [PubMed] [Google Scholar]
- Laine M, Papazian DM, Roux B. Critical assessment of a proposed model of Shaker. FEBS Lett. 2004;564:257–263. doi: 10.1016/S0014-5793(04)00273-X. [DOI] [PubMed] [Google Scholar]
- Larsson HP, Baker OS, Dhillon DS, Isacoff EY. Transmembrane movement of the shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Lee S-Y, MacKinnon R. A membrane access mechanism for ion channel inhibition by voltage sensor toxins from spider venom. Nature. 2004;430:232–235. doi: 10.1038/nature02632. [DOI] [PubMed] [Google Scholar]
- Li X, Bett GC, Jiang X, Bondarenko VE, Morales MJ, Rasmusson RL. Regulation of N- and C-type inactivation of Kv1.4 by pHo and K+: evidence for transmembrane communication. Am J Physiol. 2003;284:H71–H80. doi: 10.1152/ajpheart.00392.2002. [DOI] [PubMed] [Google Scholar]
- Libbus I, Wan X, Rosenbaum DS. Electrotonic load triggers remodeling of repolarizing current Ito in ventricle. Am J Physiol. 2004;286:H1901–H1909. doi: 10.1152/ajpheart.00581.2003. [DOI] [PubMed] [Google Scholar]
- Linse S, Forsén S. Determinants that govern high-affinity calcium binding. In: Means AR, editor. Calcium Regulation of Cellular Function. New York: Raven Press, Ltd; 1995. pp. 89–151. [DOI] [PubMed] [Google Scholar]
- Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. 1993;72:671–687. doi: 10.1161/01.res.72.3.671. [DOI] [PubMed] [Google Scholar]
- London B. Cardiac arrhythmias: from (transgenic) mice to men. J Cardiovasc Electrophysiol. 2001;12:1089–1092. doi: 10.1046/j.1540-8167.2001.01089.x. [DOI] [PubMed] [Google Scholar]
- London B, Wang DW, Hill JA, Bennet PB. The transient outward current in mice lacking the potassium channel gene Kv1.4. J Physiol. 1998b;509:171–182. doi: 10.1111/j.1469-7793.1998.171bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack T, McCormack K. Shaker K+ channel beta subunits belong to an NAD(P)H-dependent oxidoreductase superfamily. Cell. 1994;79:1133–1135. doi: 10.1016/0092-8674(94)90004-3. [DOI] [PubMed] [Google Scholar]
- McKinnon D. Molecular identity of Ito: Kv1.4 redux. Circ Res. 1999;84:620–622. doi: 10.1161/01.res.84.5.620. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Potassium channels. FEBS Lett. 2003;555:62–65. doi: 10.1016/s0014-5793(03)01104-9. [DOI] [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- Morales MJ, Castellino RC, Crews AL, Rasmusson RL, Strauss HC. A novel β subunit increases rate of inactivation of specific voltage-gated channel α subunits. J Biol Chem. 1995;270:6272–6277. doi: 10.1074/jbc.270.11.6272. [DOI] [PubMed] [Google Scholar]
- Näbauer M, Beuckelmann DJ, Überfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 1996;93:168–177. doi: 10.1161/01.cir.93.1.168. [DOI] [PubMed] [Google Scholar]
- Nadal MS, Amarillo Y, de Miera EV, Rudy B. Evidence for the presence of a novel Kv4-mediated A-type K+ channel-modifying factor. J Physiol. 2001;537:801–809. doi: 10.1111/j.1469-7793.2001.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Vega-Saenz de Miera YE, Ma Y, Mo W, Goldberg EM, Misumi Y, Ikehara Y, Neubert TA, Rudy B. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron. 2003;37:449–461. doi: 10.1016/s0896-6273(02)01185-6. [DOI] [PubMed] [Google Scholar]
- Nakamura TY, Pountney DJ, Ozaita A, Nandi S, Ueda S, Rudy B, Coetzee WA. A role for frequenin, a Ca2+-binding protein, as a regulator of Kv4 K+-currents. Proc Natl Acad Sci U S A. 2001;98:12808–12813. doi: 10.1073/pnas.221168498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nef P. Neuron specific calcium sensors (the NCS subfamily) In: Celio MR, Pauls T, Schwaller B, editors. Guidebook to the Calcium-Binding Proteins. Oxford: Oxford University Press; 1996. pp. 94–98. [Google Scholar]
- Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J Physiol. 2000;525:285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM. Molecular analysis of voltage-gated K+ channel diversity and functioning in the mammalian heart. In: Page E, Fozzard HA, Solaro RJ, editors. Handbook of Physiology, section 2, The Cardiovascular System, vol. I, The Heart. New York: Oxford University Press; 2002. pp. 568–594. [Google Scholar]
- Nerbonne JM. Studying cardiac arrhythmias in the mouse—a reasonable model for probing mechanisms? Trends Cardiovas Med. 2004;14:83–93. doi: 10.1016/j.tcm.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Physiology and molecular biology of ion channels contributing to ventricular repolarization. In: Gussack I, Antzelevitch C, editors. Cardiac Repolarization. Totowa, New Jersey: Humana Press; 2003. pp. 25–62. [Google Scholar]
- Norton RS, Pallaghy PK. The cysteine knot structure of ion channel toxins and related polypeptides. Toxicon. 1998;36:1573–1583. doi: 10.1016/s0041-0101(98)00149-4. [DOI] [PubMed] [Google Scholar]
- Ohya S, Morohashi Y, Muraki K, Tomita T, Watanabe M, Iwatsubo T, Imaizumi Y. Molecular cloning and expression of novel splice variants of K+ channel interacting protein 2. Biochem Biophys Res Com. 2001;282:96–102. doi: 10.1006/bbrc.2001.4558. [DOI] [PubMed] [Google Scholar]
- Oudit GY, Kassiri Z, Sah R, Ramirez RJ, Zobel Z, Backx PH. The molecular physiology of the cardiac transient outward potassium current (Ito) in normal and diseased myocardium. J Mol Cell Cardiol. 2001;33:851–872. doi: 10.1006/jmcc.2001.1376. [DOI] [PubMed] [Google Scholar]
- Parker C, Fedida D. Cholinergic and adrenergic modulation of cardiac K+ channels. In: Archer SL, Rusch NJ, editors. Potassium Channels in Cardiovascular Biology. New York: Kluwer Academic/Plenum Pubishers; 2001. pp. 387–426. [Google Scholar]
- Patel SP, Campbell DL, Strauss HC. Elucidating KChIP effects on Kv4.3 inactivation and recovery kinetics with a minimal KChIP2 isoform. J Physiol 545. 2002b;1:5–11. doi: 10.1113/jphysiol.2002.031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Campbell DL, Morales MJ, Strauss HC. Heterogeneous expression of KChIP2 isoforms in the ferret heart. J Physiol. 2002a;539:649–656. doi: 10.1113/jphysiol.2001.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Parai R, Parai R, Campbell DL. Regulation of Kv4.3 voltage-dependent gating kinetics by KChIP2 isoforms. J Physiol. 2004;557:19–41. doi: 10.1113/jphysiol.2003.058172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak J. Molecular kinetics of voltage-dependent Na+ channels. Physiol Rev. 1991;71:1047–1080. doi: 10.1152/physrev.1991.71.4.1047. [DOI] [PubMed] [Google Scholar]
- Perez-Garcia MT, López-López JR, Gonzalez C. Kvβ1.2 subunit coexpression in HEK 293 cells confers O2 sensitivity to Kv4.2 but not Shaker channels. J General Physiol. 1999;113:897–907. doi: 10.1085/jgp.113.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KR, Nerbonne JM. Expression environment determines K+ current phenotypes: Kv1 and Kv4 α subunit-induced K+ currents in mammalian cell lines and cardiac myocytes. Pflugers Arch. 1999;437:381–392. doi: 10.1007/s004240050792. [DOI] [PubMed] [Google Scholar]
- Po S, Snyders DJ, Baker R, Tamkun MM, Bennet PB. Functional expression of an inactivating potassium channel cloned from human heart. Circ Res. 1992;71:732–736. doi: 10.1161/01.res.71.3.732. [DOI] [PubMed] [Google Scholar]
- Pourrier M, Herrera D, Caballero R, Schram G, Wang Z, Nattel S. The Kv4.2 N-terminal restores fast inactivation and confers KChIP2 modulatory effects on N-terminal-deleted Kv1.4 channels. Pflugers Arch. 2004;449:235–247. doi: 10.1007/s00424-004-1328-8. [DOI] [PubMed] [Google Scholar]
- Puglisi JL, Bers DM. LabHEART: an interactive computer model of rabbit ventricular myocyte ion channel and Ca transport. Am J Physiol. 2001;281:C2049–C2060. doi: 10.1152/ajpcell.2001.281.6.C2049. [DOI] [PubMed] [Google Scholar]
- Puglisi JL, Wang F, Bers DM. Modeling the isolated cardiac myocyte. Prog Biophys Mol Biol. 2004;85:28–38. doi: 10.1016/j.pbiomolbio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Rasmusson RL, Morales MJ, Castellino RC, Zhang Y, Campbell DL, Strauss HC. C-type inactivation controls recovery in a fast inactivating cardiac K+ channel (Kv1.4) expressed in Xenopus oocytes. J Physiol. 1995a;489:709–721. doi: 10.1113/jphysiol.1995.sp021085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson RL, Morales MJ, Wang S, Liu S, Campbell DL, Brahmajothi MV, Strauss HC. Inactivation of voltage-gated cardiac K+ channels. Circ Res. 1998;20:739–750. doi: 10.1161/01.res.82.7.739. [DOI] [PubMed] [Google Scholar]
- Rasmusson RL, Zhang Y, Campbell DL, Comer MB, Castellino RC, Liu S, Strauss HC. Bi-stable block by 4-aminopyridine of a transient K+ channel (Kv1.4) cloned from ferret ventricle and expressed in Xenopus oocytes. J Physiol. 1995b;485:59–71. doi: 10.1113/jphysiol.1995.sp020712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Shand SH, Takimoto K. Effective association of Kv channel-interacting proteins with Kv4 channel is mediated with their unique core peptide. J Biol Chem. 2003;278:43564–45570. doi: 10.1074/jbc.M302337200. [DOI] [PubMed] [Google Scholar]
- Robertson B. Pharmacology of voltage-gated K+ channels. In: Archer SL, Rusch SJ, editors. Potassium Channels in Cardiovascular Biology. New York: Kluwer Academic/Plenum Publishers; 2001. pp. 195–217. [Google Scholar]
- Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channel Kv1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J Neurosci. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati B, Grau F, Rodriguez S, Li H, Nerbonne JM, McKinnon D. Concordant expression of KChIP2 mRNA, protein, and transient outward current throughout the canine ventricle. J Physiol. 2003;548:815–822. doi: 10.1113/jphysiol.2002.033704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati B, Pan Z, Lypen S, Wang H-S, Cohen I, Dixon JE, McKinnon D. Regulation of KChIP2 potassium channel β subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol. 2001;533:119–125. doi: 10.1111/j.1469-7793.2001.0119b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy Y. The cardiac ventricular action potential. In: Page E, Fozzard HA, Solaro RJ, editors. Handbook of Physiology, section 2, the Cardiovascular System, vol. I, The Heart. New York: Oxford University Press; 2002. pp. 531–547. [Google Scholar]
- Ruppersberg JP, Frank R, Pongs O, Stocker M. Cloned neuronal IKA channels reopen during recovery from inactivation. Nature. 1991a;353:657–660. doi: 10.1038/353657a0. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Koenen M. Regulation of fast inactivation of cloned mammalian IKA channels by cysteine oxidation. Nature. 1991b;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- Ruta V, MacKinnon R. Localization of the voltage-sensor toxin receptor on KvAP. Biochemistry. 2004;43:10071–100079. doi: 10.1021/bi049463y. [DOI] [PubMed] [Google Scholar]
- Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Triveri MG, Zobel C, Backx P. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (Ito) J Physiol. 2003;546:5–18. doi: 10.1113/jphysiol.2002.026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Bennett PB. Antiarrhythmic drug target choices and screening. Circ Res. 2003;93:491–499. doi: 10.1161/01.RES.0000091829.63501.A8. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Johnson JH, Hammerland LG, Kelbaugh PR, Volkmann RA, Saccomano NA, Mueller AL. Heteropodatoxins: peptides isolated from spider venom that block Kv4.2 potassium channels. Mol Pharmacol. 1997;51:491–498. [PubMed] [Google Scholar]
- Scannevin RH, Wang K-W, Jow F, Megules J, Kopsco DC, Edris W, Carroll KC, Lu Q, Xu W, Xu Z, Katz AH, Olland S, Lin L, Taylor M, Stahl M, Malakian K, Somers W, Mosyak L, Bowlby MR, Chandra P, Rhodes KJ. Two N-terminal domains of Kv4 K+ channels regulate binding to and modulation by KChIP1. Neuron. 2004;41:587–598. doi: 10.1016/s0896-6273(04)00049-2. [DOI] [PubMed] [Google Scholar]
- Schönherr R, Manuzzu LM, Isacoff EY, Heinemann SH. Conformational switch between slow and fast gating modes: allosteric regulation of voltage sensor mobility in the EAG K+ channel. Neuron. 2002;35:935–949. doi: 10.1016/s0896-6273(02)00869-3. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Anderson AE, Mayne A, Pfaffinger PJ, Sweatt JD. PKA modulation of Kv4.2-encoded A-type potassium channels requires formation of a supramolecular complex. J Neurosci. 2002;22:10123–10133. doi: 10.1523/JNEUROSCI.22-23-10123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serodio P, Vega-Saenz DM, Rudy B. Cloning of a novel component of A-type K+ channels operating at subthreshold potentials with unique expression in heart and brain. J Neurophysiol. 1996;75:2174–2179. doi: 10.1152/jn.1996.75.5.2174. [DOI] [PubMed] [Google Scholar]
- Shahidullah M, Covarrubias M. The link between ion permeation and inactivation gating of Kv4 potassium channels. Biophys J. 2003;84:928–941. doi: 10.1016/S0006-3495(03)74910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau Y-S, Haung P-T, Liou H-H, Liaw Y-C, Shiau Y-Y, Lou K-L. Structural basis of binding and inhibition of novel tarantula toxins in mammalian voltage-dependent potassium channels. Chem Res Toxicol. 2003;16:1217–1225. doi: 10.1021/tx0341097. [DOI] [PubMed] [Google Scholar]
- Shimoni Y, Severson D, Giles W. Thyroid status and diabetes modulate regional differences in potassium currents in rat ventricle. J Physiol. 1995;488:673–688. doi: 10.1113/jphysiol.1995.sp020999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova O. Structure of cation channels, revealed by single particle electron microscopy. FEBS Lett. 2004;564:251–256. doi: 10.1016/S0014-5793(04)00254-6. [DOI] [PubMed] [Google Scholar]
- Sokolova O, Kolmakova-Partensky L, Gigorieff N. Three dimensional structure of a voltage-gated potassium channel at 2.5 nm resolution. Structure. 2001;9:215–220. doi: 10.1016/s0969-2126(01)00578-0. [DOI] [PubMed] [Google Scholar]
- Starace DM, Bezanilla F. Histidine scanning mutagenesis of basic residues of the S4 segment of the Shaker K+ channel. J General Physiol. 2001;117:469–490. doi: 10.1085/jgp.117.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace DM, Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 2004;427:548–553. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- Starace DM, Stefani E, Bezanilla F. Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron. 1997;19:1319–1327. doi: 10.1016/s0896-6273(00)80422-5. [DOI] [PubMed] [Google Scholar]
- Strauss HC, Morales MJ, Wang S, Brahmajothi MV, Campbell DL. Voltage-dependent K+ channels. In: Sperelakis N, Kurachi Y, Terzic A, Cohen MV, editors. Heart Physiology and Pathophysiology. 4. San Diego: Academic Press; 2001. pp. 259–280. [Google Scholar]
- Strauss HC, Rasmusson RL. Restitution, ventricular fibrillation, and drugs: where are we now? J Cardiovas Electrophysiol. 2002;13:915–917. doi: 10.1046/j.1540-8167.2002.00915.x. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, MacKinnon R. Mapping the receptor site for Hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron. 1997;18:675–682. doi: 10.1016/s0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- Taggart P, Sutton P, Opthof T, Coronel R, Kallis K. Electrotonic cancellation of transmural electrical gradients in the left ventricle in man. Prog Biophys Mol Bio. 2003;82:243–254. doi: 10.1016/s0079-6107(03)00025-7. [DOI] [PubMed] [Google Scholar]
- Tamkun M, Knoth KM, Walbridge M, Kroemer JA, Roden DM, Glover DM. Molecular cloning and characterization of two voltage-gated K+ channel cDNAs from human ventricle. FASEB J. 1991;5:331–337. doi: 10.1096/fasebj.5.3.2001794. [DOI] [PubMed] [Google Scholar]
- Tombola F, Pathak MM, Isacoff EY. Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron. 2005;45:379–388. doi: 10.1016/j.neuron.2004.12.047. [DOI] [PubMed] [Google Scholar]
- Tseng GN, Jiang M, Yao JA. Reverse use dependence of Kv4.2 blockade by 4-aminopyridine. J Pharmacol Exp Ther. 1996;279:865–876. [PubMed] [Google Scholar]
- Tseng-Crank JCL, Tseng G-N, Schwartz A, Tanouye MA. Molecular cloning and functional expression of a potassium channel cDNA isolated from a rat cardiac library. FEBS Lett. 1990;268:63–68. doi: 10.1016/0014-5793(90)80973-m. [DOI] [PubMed] [Google Scholar]
- Van Wagoner DR, Nerbonne JM. Molecular mechanisms of atrial fibrillation. In: Sperelakis N, Kurachi Y, Terzic A, Cohen MV, editors. Heart Physiology and Pathophysiology. 4. San Diego: Academic Press; 2001. pp. 1107–1124. [Google Scholar]
- Varro A, Lathrop DA, Hester SB, Nanasi PP, Papp JG. Ionic currents and action potentials in rabbit, rat and guinea pig ventricular myocytes. Basic Res Cardiol. 1993;88:93–102. doi: 10.1007/BF00798257. [DOI] [PubMed] [Google Scholar]
- Vega-Saenz de Miera E, Moreno H, Fruhling D, Kentros C, Rudy B. Cloning of Sh III (Shaw-like) cDNAs encoding a novel high-voltage-activated, TEA-sensitive, type-A K+ channel. Proc Biol Sci. 1992;248:9–18. doi: 10.1098/rspb.1992.0036. [DOI] [PubMed] [Google Scholar]
- Verkerk AO, van Ginneken A. Considerations when studying the transient outward K+ current in cells exhibiting the hyperpolarization-activated current. Cardiovascular Res. 2001;52:517–518. doi: 10.1016/s0008-6363(01)00482-5. [DOI] [PubMed] [Google Scholar]
- Wada K, Yokotani N, Hunter C, Doi K, Wenthold J, Shimaski S. Differential expression of two distinct forms of mRNA encoding members of a depeptidyl aminopeptidase family. Proc Natl Acad Sci U S A. 1992;89:197–201. doi: 10.1073/pnas.89.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Bondarenko VE, Qu Y, Morales MJ, Rasmusson RL, Strauss HC. Activation properties of Kv4.3 channels: time, voltage and [K+]o dependence. J Physiol. 2004;557:105–717. doi: 10.1113/jphysiol.2003.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Patel SP, Qu Y, Hua P, Strauss HC, Morales MJ. Kinetic properties of Kv4.3 and their modulation by KChIP2b. Biochem Biophys Res Comm. 2002;295:223–229. doi: 10.1016/s0006-291x(02)00658-7. [DOI] [PubMed] [Google Scholar]
- Wible BA, Yang Q, Kurysshev YA, Accili EA, Brown AM. Cloning and expression of a novel K+ channel regulatory protein, KChAP. J Biol Chem. 1998;273:11745–11751. doi: 10.1074/jbc.273.19.11745. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Lee P, Sah R, Huang Q, Fishman GI, Backx PH. Targeted expression of a dominant-negative Kv4.2 K+ channel subunit in the mouse heart. Circ Res. 1999;85:1067–1076. doi: 10.1161/01.res.85.11.1067. [DOI] [PubMed] [Google Scholar]
- Winslow RL, Contassa S, Greenstein JL. Using models of the myocyte for functional interpretation of cardiac proteomic data. J Physiol. 2005;563:73–81. doi: 10.1113/jphysiol.2004.080457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow RL, Scollan DF, Holmes A, Yung CK, Zhaang J, Jafri MS. Electrophysiological modeling of cardiac ventricular function: from cell to organ. Ann Rev Biomed Eng. 2000;2:119–155. doi: 10.1146/annurev.bioeng.2.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissmann R, Bildl W, Oliver D, Beyermann M, Kalbitzer H-R, Bentrop D, Fakler B. Solution structure and function of the ‘tandem inactivation domain’ of neuronal A-type potassium channel Kv1.4. J Biol Chem. 2003;278:16142–16150. doi: 10.1074/jbc.M210191200. [DOI] [PubMed] [Google Scholar]
- Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. J General Physiol. 1999;113:661–677. doi: 10.1085/jgp.113.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E-K, Alvira MR, Levitan ES, Takimoto K. Kvβ subunits increase expression of Kv4.3 channels by interacting with their C termini. J Biol Chem. 2001;276:4839–4844. doi: 10.1074/jbc.M004768200. [DOI] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Quart Rev Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419:35–42. doi: 10.1038/nature00978. [DOI] [PubMed] [Google Scholar]
- Yeola SW, Snyders DJ. Electrophysiological and pharmacological correspondence between Kv4.2 current and rat cardiac transient outward current. Cardiovas Res. 1997;33:540–547. doi: 10.1016/s0008-6363(96)00221-0. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- Zhang M, Jiang M, Tseng GN. MinK-related peptide associates with Kv4.2 and modulates its function: potential role as beta subunit of cardiac transient outward channel? Circ Res. 2001;88:1012–1019. doi: 10.1161/hh1001.090839. [DOI] [PubMed] [Google Scholar]
- Zhou W, Qian Y, Kunjilar K, Pfaffinger PJ, Choe S. Structural insights into the functional interaction of KChIP1 with Shal-type K+ channels. Neuron. 2004;41:573–586. doi: 10.1016/s0896-6273(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Zicha S, Moss I, Allen B, Varro A, Papp J, Dumaine R, Antzelivitch C, Nattel S. Molecular basis of species-specific expression of repolarizing K+ currents in the heart. Am J Physiol. 2003;285:H1641–H1649. doi: 10.1152/ajpheart.00346.2003. [DOI] [PubMed] [Google Scholar]
- Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, Nattel S. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004;561:735–748. doi: 10.1113/jphysiol.2004.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]