

Abstract
The role of protein kinase A (PKA) in insulin exocytosis was investigated with the use of two-photon excitation imaging of mouse islets of Langerhans. Inhibitors of PKA selectively reduced the number of exocytic events during the initial period (< 250 s) of the first phase of glucose-induced exocytosis (GIE), without affecting the second phase, in intact islets or small clusters of islet cells. The PKA inhibitors did not reduce the extent of the glucose-induced increase in [Ca2+]i. The actions of glucose and PKA in Ca2+-induced insulin exocytosis (CIE) triggered by photolysis of a caged-Ca2+ compound, which resulted in large increases in [Ca2+]i and thereby bypassed the ATP-sensitive K+ channel-dependent mechanism of glucose sensing, were therefore studied. A high concentration (20 mm) of glucose potentiated CIE within 1 min, and this effect was blocked by inhibitors of PKA. This PKA-dependent action of glucose required glucose metabolism, given that increasing the intracellular concentration of cAMP by treatment with forskolin potentiated CIE only at the high glucose concentration. Finally, PKA appeared to reduce the frequency of ‘kiss-and-run’ exocytic events and to promote full-fusion events during GIE. These data indicate that a PKA-dependent mechanism of glucose sensing, which is operative even at the basal level of PKA activity, plays an important role specifically in the first phase of GIE, and they suggest that the action of PKA is mediated at the level of the fusion reaction.
Glucose is the most important physiological regulator of insulin secretion from β cells of the islets of Langerhans. Islet β cells rapidly take up and metabolize glucose, resulting in an increase in the cytosolic concentration of ATP within 1 min (Bennett et al. 1996; Porterfield et al. 2000). Such increases in ATP concentration induce the closure of ATP-sensitive K+ (KATP) channels and consequent depolarization of the cell membrane, again within a few minutes of glucose application (Henquin, 1990). Depolarization of the cell membrane to a voltage of > −50 mV results in activation of voltage-dependent Ca2+ channels and an increase in [Ca2+]i that triggers insulin exocytosis. The KATP channels and Ca2+-dependent mechanism are thought to play a central role in glucose sensing for insulin exocytosis. Although additional mechanisms of glucose sensing have been proposed to coexist (Gembal et al. 1993; Aizawa et al. 1994; Kasai et al. 2002), their relative importance has remained unclear.
Exocytosis in many secretory cell types and neurones is regulated by both Ca2+ and cAMP (Wollheim & Sharp, 1981; Trudeau et al. 1996; Renstrom et al. 1997; Fujita-Yoshigaki et al. 1999; Hilfiker et al. 2001; Sato et al. 2002; Sakaba & Neher, 2003; Kaneko & Takahashi, 2004). We have previously shown that cytosolic cAMP potentiates Ca2+-dependent insulin exocytosis (CIE) in β cells (Takahashi et al. 1999). In these studies, individual β cells were subjected to whole-cell patch clamping and stimulated with large increases in [Ca2+]i induced by photolysis of a caged-Ca2+ compound, thereby bypassing the KATP channel-dependent mechanism. We found that CIE was augmented by cAMP in a manner dependent on protein kinase A (PKA) and cytosolic ATP. It was not possible to study the action of extracellular glucose under the whole-cell clamp conditions, however, and it has remained unknown whether PKA contributes to glucose-induced insulin exocytosis (GIE). Inhibitors of PKA have been shown to have relatively small inhibitory effects on GIE in previous studies, in which exocytosis was measured over a long period without separation into the first and second phases (Persaud et al. 1990; Harris et al. 1997).
We therefore subsequently developed an approach based on two-photon excitation imaging to quantify insulin exocytosis in intact pancreatic islets (Takahashi et al. 2002a). This approach has been designated TEP (two-photon extracellular polar-tracer) imaging and TEPIQ (TEP imaging-based quantification) analysis (Kasai et al. 2005). TEP imaging is able to monitor reliably and with a relatively high time resolution (< 1 s) individual insulin exocytic events in intact islet preparations and also allows analysis of the dynamics of the fusion pore that mediates exocytosis. We have previously applied this methodology to study GIE as well as CIE evoked by photolysis of a caged-Ca2+ compound (Takahashi et al. 2004). With the use of this approach, we have now shown that a PKA-dependent mechanism, operative at the basal level of PKA activity, is important for the initial period of the first phase of GIE in mouse pancreatic islets. Furthermore, we found that PKA mediates rapid enhancement of CIE in islets only in the presence of a high glucose concentration, indicating that PKA plays a glucose sensing role, specifically in the first phase of GIE. Given that the first phase of GIE is reduced in many individuals with type 2 diabetes mellitus from an early stage of the disease (Vaag et al. 1995), impairment of this mechanism may contribute to the pathogenesis of this condition.
Methods
Isolation of mouse pancreatic islets and islet cell clusters
Eight- to 12-week-old ICR mice were killed by cervical dislocation and pancreatic islets were isolated by collagenase digestion. Islets were maintained for 1–12 h under a humidified atmosphere of 5% CO2 at 37°C in Dulbecco's modified Eagle's medium containing glucose (1.0 mg ml−1) and supplemented with 10% fetal bovine serum, penicillin (100 μU ml−1), and streptomycin (100 mg ml−1). They were then transferred with a Pipetman (Gilson, Middleton, WI, USA) to thin (0.1 mm) glass coverslips (Matsunami-glass, Osaka, Japan) in the recording chamber. The standard external bathing solution (Sol A) contained 150 mm NaCl, 5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm Hepes-NaOH (pH 7.4), and 2.8 mm glucose at 310 mosmol l−1. A solution containing 20 mm glucose was prepared from Sol A by adjusting the osmolarity with deionized water to 310 mosmol l−1. Imaging experiments were performed within 20 min after placing an islet in Sol A containing 0.7 mm sulforhodamine B (SRB; Molecular Probes, Eugene, OR, USA) with or without 1.5 mm 10-kDa fluorescein dextran (FD; Molecular Probes). Islet cell clusters were prepared by triturating islets with a glass pipette and were plated directly onto the glass-bottomed recording chamber. Forskolin (Sigma, St Louis, MO, USA), cerulenin (Sigma), bisindolylmaleimide I (Calbiochem, La Jolla, CA, USA), KN-62 (Seikagaku, Tokyo, Japan), ML-9 (Calbiochem), KT5720 (Calbiochem), and H89 (Sigma) were initially dissolved in DMSO at 10–50 mm and subsequently diluted in Sol A. Myristoylated PKA inhibitor amide 14–22 (PKI; Calbiochem), the Rp isomer of adenosine 3′,5′-monophosphorothioate (Rp-cAMPS; Sigma), and 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-monophosphate (8-CPT-2′-O-Me-cAMP; Biolog, Bremen, Germany) were dissolved directly in Sol A.
TEP imaging
Two-photon excitation imaging of islets was performed with an inverted microscope (IX70; Olympus, Tokyo, Japan) and a laser-scanning microscope (FluoView, Olympus) equipped with a water-immersion objective lens (UplanApo60xW/IR; NA, 1.2), as previously described (Liu et al. 2005; Kishimoto et al. 2005; Kasai et al. 2005). The laser power at the specimen was 3–10 mW, and two-photon excitation was effected at 830 nm, with images acquired every 0.3–2 s. The fluorescence of SRB and that of fura-2, fura-2FF, or fura-4F were measured at 570–650 nm and at 400–550 nm, respectively. Twelve-bit images were colour-coded with fall colour codes of FluoView. All experimental procedures were performed under illumination with yellow light (FL40S-Y-F; National, Tokyo, Japan) to prevent unintentional photolysis of the caged-Ca2+ compound. Experiments were performed at room temperature (24–25°C).
The number of exocytic events was expressed per cell per minute on the basis of the following procedures. Exocytic events were counted in a region of interest (ROI) with an area of 3000–5000 μm2. An individual ROI in islets contained many (10–40) cells, sectioned at various distances from their equatorial planes, and therefore included all cell aspects (bottom, top and side views) (Takahashi et al. 2002a). We counted the number of exocytic events whose fluorescence intensity was > 20% of the maximal value; these events would be expected to occur within 1 μm from the focal plane, given the axial resolution of 1.4 μm of our set-up (Kasai et al. 2005). The thickness of the image for detection of exocytosis was therefore ∼2 μm along the z-axis. This value was slightly modified from our previous studies (Takahashi et al. 2002a, 2004). Each area of 4000 μm2 thus represented a total cell volume of 8000 μm3, which corresponds to the volume of five β cells, if we assume a cell diameter of 14 μm and that β cells constitute 90% of islet cells. On the basis on this calculation (1 β cell per 800 μm2), we converted the number of events per ROI to events per cell. Moreover, we express the rate of insulin exocytosis as cell−1 min−1 to allow comparison between GIE and CIE. For the mean values of GIE in Table 1, the total events were measured between 0 and 250 s after the onset of high-glucose application and were divided by 2 min, given that the increase in [Ca2+]i occurred with a lag of about 100–150 s (Table 2). For the mean values of CIE in Table 3, the total events were measured between 0 and 15 s after the onset of UV irradiation and were divided by 0.25 min.
Table 1.
Pharmacology of the initial period (0–250 s) of glucose-induced insulin exocytosis (GIE) in intact islets or clusters of islet cells
| Test agent (μM) | Events cell−1 min−1 | Significance |
|---|---|---|
| Islets | ||
| Control | 6.4 ± 0.8 (9) | — |
| Rp-cAMPS (200) | 2.0 ± 0.5 (8) | P < 0.001 |
| PKI (5) | 3.1 ± 0.6 (9) | P < 0.05 |
| H89 (10–20) | 6.4 ± 2.0 (10) | NS (P= 0.66) |
| KT5720 (1–5) | 7.0 ± 1.4 (3) | NS (P= 0.73) |
| Forskolin (2) | 13.8 ± 3.9 (3) | P < 0.05 |
| Clusters | ||
| Control | 8.9 ± 0.9 (4) | — |
| H89 (20) | 5.0 ± 0.7 (4) | P < 0.05 |
| KT5720 (5) | 1.0 ± 0.2 (4) | P < 0.05 |
| Forskolin (2) | 25.2 ± 3.0 (3) | P < 0.05 |
| Forskolin (2) + H89 (20) | 3.8 ± 0.8 (4) | *P < 0.05 |
| Forskolin (2) + KT5720 (5) | 1.5 ± 0.4 (4) | *P < 0.05 |
Islets or cell clusters were pretreated (or not) for 40 min with PKA inhibitors or for 10 min with forskolin and were then stimulated with 20 mm glucose, as indicated. Data are means ± s.e.m. of values for the number of islets or islet cell clusters indicated in parentheses. Statistical analysis was performed with the Kruskal–Wallis test for all values (P < 0.01) followed by the Mann–Whitney U test for comparison with control values or, in the case of the P values indicated by an asterisk, with the corresponding value for forskolin.
Table 2.
Glucose-induced increases in [Ca2+]i estimated with the Ca2+ indicators fura-2 or fura-4F
| Fura-2 | Fura-4F | ||
|---|---|---|---|
| Condition | Latency (s) | (F0−F)/F0 | (F0−F)/F0 |
| Control | 152.4 ± 5.6 (5) | 0.63 ± 0.01 (5) | 0.25 ± 0.05 (5) |
| Forskolin (2 μm) | 121.3 ± 8.3 (4) | 0.60 ± 0.02 (4) | 0.33 ± 0.05 (3) |
| Rp-cAMPS (200 μm) | 113.1 ± 23.2 (5) | 0.67 ± 0.01 (5) | 0.31 ± 0.02 (5) |
| PKI (20 μm) | 104.9 ± 13.9 (5) | 0.62 ± 0.03 (5) | |
| Kruskal–Wallis test | NS (P= 0.078) | NS (P= 0.10) | NS (P= 0.48) |
Islets were pretreated (or not) for 40 min with PKA inhibitors or for 10 min with forskolin and were then stimulated with 20 mm glucose. Latencies were measured as the time between the onset of glucose stimulation and that of the fura-2 signal. The peak amplitudes of the fluorescence signals, (F0−F)/F0, were obtained within 250 s after the onset of glucose stimulation for both indicators. Data are means ± s.e.m. for the number of cells indicated in parentheses. Statistical comparisons were performed with the Kruskal–Wallis test.
Table 3.
Pharmacology of Ca2+-dependent insulin exocytosis (CIE)
| CIE (events cell−1 min−1) | Significance between | ||
|---|---|---|---|
| Test agent (μM) | 2.8 mm Glucose | 20 mm Glucose | 2.8 and 20 mm glucose |
| Control | 68.1 ± 13.7 (9) | 128.4 ± 11.6 (6) | P < 0.05 |
| Rp-cAMPS (200) | 44.5 ± 11.4 (4) | 40.4 ± 3.6 (6) | NS (P= 0.76) |
| NS (P= 0.40) | P < 0.01 | ||
| PKI (5) | 52.6 ± 6.3 (4) | 55.8 ± 10.7 (6) | NS (P= 0.76) |
| NS (P= 0.70) | P < 0.01 | ||
| Forskolin (2) | 82.8 ± 8.9 (5) | 182.5 ± 16.9 (5) | P < 0.01 |
| NS (P= 0.42) | P < 0.05 | ||
| Forskolin (2) + PKI (5) | 48.6 ± 4.5 (5) | 67.0 ± 7.2 (5) | NS (P= 0.22) |
| NS (P= 0.51) | P < 0.01 | ||
| 8-CPT-2′-O-Me-cAMP (10) | 70.5 ± 5.3 (7) | 131.3 ± 8.7 (7) | P < 0.001 |
| NS (P= 0.40) | NS (P= 0.94) | ||
| 8-CPT-2′-O-Me-cAMP (100) | 94.3 ± 7.1 (4) | 159.3 ± 11.7 (4) | P < 0.05 |
| NS (P= 0.14) | P < 0.05 | ||
| 8-CPT-2′-O-Me-cAMP (100) | 50.7 ± 4.4 (4) | 80.2 ± 13.8 (4) | NS (P= 0.11) |
| + PKI (5) | NS (P= 0.82) | NS (P < 0.05) | |
| Bisindolylmaleimide I (1) | 58.0 ± 10.2 (3) | 142.5 ± 27.8 (3) | P < 0.05 |
| NS (P= 0.85) | NS (P= 0.52) | ||
| KN-62 (10) | 37.4 ± 3.7 (4) | 61.1 ± 5.8 (5) | P < 0.05 |
| NS (P= 0.11) | P < 0.01 | ||
| ML-9 (30) | 52.4 ± 6.9 (4) | 95.9 ± 12.5 (3) | P < 0.05 |
| NS (P= 0.70) | NS (P= 0.16) | ||
| Cerulenin (134) | 49.2 ± 9.0 (4) | 89.7 ± 3.8 (4) | P < 0.05 |
| NS (P= 0.70) | P < 0.05 | ||
Exocytic events were counted between 0 and 15 s after the uncaging of NPE. Inhibitors were applied for 40 min, forskolin and 8-CPT-2′-O-Me-cAMP for 10 min, and 20 mm glucose for 1 min before UV irradiation. Data are means ± s.e.m. for the number of islets indicated in parentheses. The actions of the various compounds were compared first with the two-way Friedman test (P < 0.001), after which the Mann–Whitney U test was performed to compare values in the presence of test agent with the corresponding control or between 2.8 and 20 mm glucose, as indicated.
Our data were consistent with the notion that TEP imaging visualized all the exocytic events in the ROI (Takahashi et al. 2002a; Kasai et al. 2005). The measured rate of GIE of 6.4 vesicles cell−1 min−1 (Table 1) corresponds to 12 vesicles cell−1 min−1 at the higher temperature of 35°C (Takahashi et al. 2004). If we assume that a single insulin granule contains 2.6 fg of insulin (Hutton, 1989) and that an islet with a diameter of 200 μm contains 2600 β cells, the rate of insulin secretion at 35°C is predicted to be 82 pg per islet per minute, which is similar to the value of 67 pg per islet per minute obtained for mouse islets by radioimmunoassay (Eto et al. 1999).
Photolysis of NPE and Ca2+ measurement
The acetoxymethyl ester (AM) forms of fura-2 (Molecular Probes), fura-4F (Molecular Probes), and fura-2FF (Tef Laboratory, Austin, TX, USA) as well as that of o-nitrophenyl-EGTA (NPE; Molecular Probes) were dissolved in DMSO at a concentration of 2–10 mm. Islets were loaded with these compounds by incubation for 30 min at 37°C in serum-free Dulbecco's modified Eagle's medium containing 10 μm fura-2-AM (or fura-4F-AM or fura-2FF-AM), 25 μm NPE-AM, 0.03% cremphor EL (Sigma), and 0.1% bovine serum albumin; they were then washed with Sol A. Photolysis of NPE was induced by brief irradiation (0.2–0.5 s) with a mercury lamp (IX-RFC, Olympus) 1 min after switching the glucose concentration of the extracellular solution. In some experiments, we monitored [Ca2+]i simultaneously during this prephotolysis period.
Glucose-induced increases in [Ca2+]i were measured with either fura-2 or fura-4F, whose Kd values are 0.18 and 1.16 μm, respectively (Wokosin et al. 2004). We estimated Fmin/Fmax (minimal/maximal fluorescence intensity ratio) for fura-2 and fura-4F to be 0.3 and 0.46, respectively, in islet cells loaded with the corresponding AM forms. We expressed increases in [Ca2+]i by the ratio (F0−F)/F0 (Takahashi et al. 2002a), where F0 and F represent the fluorescence intensities of the dye before and after glucose stimulation, respectively. If we assume the resting [Ca2+]i to be 0.1 μm, the ratio for fura-4F of ∼0.3 predicted the peak [Ca2+]i during glucose stimulation to be 1.8 μm. Increases in [Ca2+]i induced by uncaging of NPE were monitored as the decrease in fluorescence intensity of fura-2FF and were calculated as 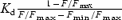 . In vivo calibration of fura-2FF was performed as described (Takahashi et al. 2004); we estimated Fmin/Fmax to be 0.1 and Kd is 31 μm. We assumed that the resting fluorescence was the same as Fmax.
. In vivo calibration of fura-2FF was performed as described (Takahashi et al. 2004); we estimated Fmin/Fmax to be 0.1 and Kd is 31 μm. We assumed that the resting fluorescence was the same as Fmax.
Statistical analysis
Data are presented as means ± s.e.m. Those in Tables 1 and 2 were compared by the one-way Kruskal–Wallis test and those in Table 3 by the two-way Friedman test; further comparisons between two conditions were performed by the Mann–Whitney U test, as indicated. Data in Table 4 were compared by the chi-square test. A P-value of < 0.05 was considered statistically significant. Statistical tests were performed with SPSS 12.0 software (SPSS, Chicago, IL, USA).
Table 4.
Fusion pore dynamics during GIE
| Condition | Transient opening (%) | Latency (s) | T0.5 (s) |
|---|---|---|---|
| Control | 6.5 (367) | 2.1 ± 0.1 (72) | 2.8 ± 0.3 (137) |
| Forskolin (2 μm) | 3.8 (447)* | 2.4 ± 0.1 (101) | 3.0 ± 0.3 (196) |
| Rp-cAMPS (200 μm) | 10.3 (311)* | 2.3 ± 0.1 (71) | 3.2 ± 0.2 (133) |
| PKI (10 μm) | 9.8 (82) | 2.2 ± 0.1 (65) | 2.7 ± 0.3 (71) |
Islets were pretreated (or not) for 40 min with PKA inhibitors or for 10 min with forskolin and were then stimulated with 20 mm glucose. Transient opening of the fusion pore was identified as shown in Fig. 6B. The latency between opening of the fusion pore to 1.4 nm and that to 6 nm was measured as shown in Fig. 6A. The time of flattening of Ω-profiles (T0.5) was measured as shown in Fig. 6C. Latency and T0.5 values are means ± s.e.m. The numbers of events for all data are indicated in parentheses. *
P < 0.05 versus control (chi-square test).
Results
Role of PKA in the first phase of glucose-induced insulin exocytosis
We first examined the participation of PKA in GIE with the use of TEP imaging, in which insulin exocytic events were visualized by two-photon imaging of islets immersed in an extracellular solution containing the polar fluorescent tracer SRB at a concentration of 0.7 mm (see Fig. 4A) (Takahashi et al. 2002a). We imaged mostly the outer second or third layer of cells in the islets because insulin-secreting β cells were abundant in these layers. We detected exocytic events as discrete spots of fluorescence, which reflected diffusion of SRB into individual insulin granules via the fusion pore (Takahashi et al. 2002a). The intensities of the spots of SRB fluorescence were consistent with them reflecting exocytosis of large dense-core vesicles (Kasai et al. 2005).
Figure 4. Effect of glucose on exocytosis induced by uncaging of a caged-Ca2+ compound in islets.

A, insulin exocytosis in a β cell within an islet loaded with NPE and perifused with a solution containing SRB. Exocytosis was triggered by UV-induced photolysis of NPE at time 0. The boxed regions a and b in the micrograph on the left are sites of individual exocytic events after UV irradiation and are shown at higher magnification on the right. B, increases in [Ca2+]i induced by uncaging of NPE at time 0 in islets loaded with the Ca2+ indicator fura-2FF and exposed to glucose at 2.8 or 20 mm. The high-glucose solution was applied 1 min before UV irradiation. Traces represent average time courses obtained from four to six islets. C, latency histograms of insulin exocytosis induced by uncaging of NPE (CIE) in islets exposed to glucose at 2.8 or 20 mm. The bin width is 0.5 s, and data are averages from nine and six islets, respectively, as indicated in Table 3.
Islets were pretreated with either of two inhibitors of PKA, Rp-cAMPS or PKI (Harris et al. 1997), for 40 min and insulin secretion was then stimulated with 20 mm glucose. Figure 1A shows the time courses of averaged exocytic events (cell−1 min−1), with insulin exocytosis first being apparent 1–3 min after the onset of high-glucose application. Rp-cAMPS and PKI each markedly inhibited the initial period (< 250 s) of the first phase of GIE (< 7 min) (Nesher & Cerasi, 2002), with the number of exocytic events within this initial period being reduced in the presence of these agents to one-third or one-half of the control value, respectively (Table 1). The remaining period of the first phase and the second phase of GIE (250–600 s after stimulation) were not affected by Rp-cAMPS (control, 5.3 ± 0.86 events cell−1 min−1, n = 9; Rp-cAMPS, 4.14 ± 1.53 events cell−1 min−1, n = 6, P > 0.1). In contrast, forskolin, which increases the cytosolic concentration of cAMP by activating adenylyl cyclase, increased the extent of secretion both during the first phase of GIE (Table 1) and during the second phase (11.8 ± 0.8 events cell−1 min−1, n = 3, P < 0.05), consistent with previous observations (Takahashi et al. 2002a). These data thus suggested that the initial period of the first phase of GIE, but not the second phase, requires PKA.
Figure 1. Effects of PKA inhibitors on GIE in mouse pancreatic islets.

A, time courses of exocytic events in islets pretreated (or not) with Rp-cAMPS (200 μm) or PKI (5 μm) for 40 min and then stimulated with 20 mm glucose at time 0. B, time courses of exocytic events in islets pretreated (or not) with H89 (10–20 μm) or KT5720 (1–5 μm) for 40 min and then stimulated with 20 mm glucose at time 0. Data in A and B are expressed as the number of exocytic events per cell per minute and are averages from 3–10 islets, as indicated in Table 1. The bin width is 10 s, and the vertical dotted line indicates 250 s after the onset of stimulation with glucose.
In contrast to the effects of Rp-cAMPS and PKI, one of the most widely used inhibitors of PKA, H89 (10–20 μm), did not markedly reduce the extent of GIE in intact islets (Fig. 1B and Table 1). Another inhibitor of PKA, KT5720 (1–5 μm) (Matthies & Reymann, 1993), was similarly ineffective in most of the islets examined (Fig. 1B and Table 1). We noticed, however, that the effects of both H89 and KT5720 were variable, tending to be more pronounced in smaller islets (data not shown), suggesting that their inability to inhibit GIE might be due to their poor penetration into islets. The AM form of the Ca2+ indicator fura-2 is similarly inefficient in its penetration into islets (Takahashi et al. 2002b), with intense staining being limited to the outermost layer of cells.
We therefore tested the effects of H89 and KT5720 in cluster preparations of islets, which consist of groups of only 10–15 cells and would thus be expected to be more accessible to these lipophilic agents (Fig. 2A). Pretreatment of the cluster preparations with H89 or KT5720 resulted in marked inhibition of the first phase of GIE (Fig. 2B and Table 1), with the inhibitory effects again appearing more prominent during the initial period of this phase. Treatment of the clusters with forskolin also greatly increased the extent of the first phase of secretion in a manner sensitive to H89 or KT5720 (Fig. 2C and Table 1). These results thus provided further support for the notion that PKA is required for the initial period of the first phase of GIE.
Figure 2. GIE in cluster preparations of mouse islet cells.

A, a cluster preparation consisting of 10–15 islet cells. The cluster is visualized by SRB fluorescence. B, time courses of exocytic events in cluster preparations pretreated (or not) with 20 μm H89 for 40 min and then stimulated with 20 mm glucose at time 0. C, time courses of exocytic events in cluster preparations pretreated (or not) with 20 μm H89 for 40 min and with 2 μm forskolin for 10 min and then stimulated with 20 mm glucose. Data in B and C are averages from three or four clusters, as indicated in Table 1.
We also measured the possible effects of PKA inhibitors and forskolin on glucose-induced increases in [Ca2+]i in islets. Increases in [Ca2+]i were measured with either the high-affinity Ca2+ indicator fura-2 (Kd= 0.18 μm) (Fig. 3A) or the low-affinity Ca2+ indicator fura-4F (Kd= 1.16 μm) (Fig. 3B); the latter was used in case fura-2 became saturated during the physiological increases in [Ca2+]i (Ito et al. 1999). Indeed, the fractional change in the fluorescence of fura-2 during glucose stimulation of islets was ∼0.63 (Table 2), which is close to the saturation level of ∼0.7 for cells loaded with fura-2-AM (Nemoto et al. 2001), whereas the corresponding fractional change in the fluorescence of fura-4F was ∼0.3 (Table 2). We found that neither the onset (measured by fura-2) nor the maximal value apparent within 250 s (measured by fura-4F) of the glucose-induced increases in [Ca2+]i was affected by forskolin, Rp-cAMPS, or PKI (Fig. 3 and Table 2).
Figure 3. Increases in [Ca2+]i during GIE.

Time courses of glucose-induced increases in the fura-2 (A) or fura-4F (B) fluorescence ratio were determined for β cells in islets pretreated (or not) for 40 min with 200 μm Rp-cAMPS before stimulation with 20 mm glucose at time 0.
These results are consistent with previous observations that cAMP has only a small (if any) effect on glucose-induced increases in [Ca2+]i in islets (Rorsman & Abrahamsson, 1985; Hill et al. 1987; Fournier et al. 1994; Marie & Bailbe, 2000). PKA has been found to modulate [Ca2+]i by affecting voltage-gated Ca2+ channels (Grapengiesser et al. 1991; Yada et al. 1993; Yaekura et al. 1996), Ca2+-induced Ca2+ release (Bugrim, 1999; Dyachok & Gylfe, 2004; Kang et al. 2005), or Ca2+ sequestration (Yaekura & Yada, 1998). Such effects were not evident, however, during glucose-induced increases in [Ca2+]i under our experimental conditions and could not account for the action of PKA in GIE.
Rapid effects of glucose and PKA on Ca2+-induced insulin exocytosis
We next examined whether glucose and PKA might directly potentiate CIE. For these experiments, Ca2+-dependent mechanisms were saturated by large increases in [Ca2+]i generated by photolysis of the caged-Ca2+ compound NPE, which was loaded into the cells of islets in the AM form. Exocytic events were detected as discrete spots of SRB fluorescence (Fig. 4A), as was the case with glucose stimulation (Takahashi et al. 2002a). We confirmed that irradiation with UV light induced an abrupt increase in [Ca2+]i of > 20 μm (Fig. 4B). The latency histogram for the discrete exocytic events was fitted by a probability density function with two exponential components (Fig. 4C), consistent with the characteristics of CIE studied by amperometry (Takahashi et al. 1997, 1999). The second component in the present study was smaller than that detected previously, however, probably as a result of the more rapid recovery of [Ca2+]i (Fig. 4B) compared with that apparent in cells subjected to whole-cell dialysis (Takahashi et al. 1997; Takahashi et al. 1999). It was therefore difficult to compare the effects of glucose on the two components separately, and we counted all exocytic events between 0 and 15 s after the uncaging of NPE for the data shown in Table 3.
Exposure of islets to a high glucose concentration (20 mm) resulted in a marked increase in the extent of CIE (Fig. 4C and Table 3). This facilitatory effect was apparent at a glucose concentration as low as 8 mm (107 ± 25 events cell−1 min−1, n = 6, P < 0.05). Islets were exposed to high glucose for only 1 min before uncaging of NPE, during which time glucose alone did not increase [Ca2+]i (Fig. 3, Table 2). We confirmed that the high-glucose solution did not affect the increase in [Ca2+]i induced by uncaging of NPE (Fig. 4B). Also, the glucose action was not mimicked by 2-deoxy-D-glucose (20 mm) (61.5 ± 5.6 cell−1 min−1, n = 5, P= 0.6), indicating that it required a metabolite of glucose.
The effect of glucose on CIE was abolished by pretreatment of islets (for 40 min) with inhibitors of PKA, including Rp-cAMPS and PKI (Fig. 5A and Table 3). Furthermore, forskolin did not significantly affect CIE at the low glucose concentration of 2.8 mm (Table 3) but potentiated the effect of 20 mm glucose on CIE (Fig. 5B and Table 3). These results suggested that cAMP is necessary but not sufficient for the rapid effect of glucose on CIE, and that a metabolite of glucose, such as ATP, is required for this action of glucose (see Discussion).
Figure 5. Insulin exocytosis induced by uncaging of NPE (CIE) in islets exposed to high glucose, PKA inhibitors, or forskolin.

Rp-cAMPS or PKI was applied for 40 min (A), forskolin for 10 min (B), and high glucose for 1 min (A and B) before UV irradiation. Smooth lines are double-exponential curves, and the bin width is 0.5 s. Data are averages from five to nine islets, as indicated in Table 3.
The effect of glucose on CIE was mediated largely by PKA. Indeed, PKI eliminated the effect of high glucose even in the presence of forskolin (Table 3). The effect of cAMP on insulin exocytosis has been proposed to be mediated also by cAMP-regulated guanine nucleotide exchange factor II (cAMP-GEFII) (Ozaki et al. 2000). We therefore examined the possible role of this protein in our system with the use of 8-CPT-2′-O-Me-cAMP, which selectively activates cAMP-GEFII at 10 μm but also activates PKA at higher concentrations (Enserink et al. 2002). CIE was not affected by 8-CPT-2′-O-Me-cAMP at 10 μm in the presence of low or high glucose, but it was slightly facilitated by this analogue at 100 μm in the presence of high glucose (Table 3), similar to the effect of forskolin. The effect of 20 mm glucose plus 100 μm 8-CPT-2′-O-Me-cAMP was abolished by PKI (Table 3).
We also examined the possible roles of three other protein kinases (protein kinase C, calmodulin-dependent protein kinase II, and myosin light chain kinase) and of protein acylation in CIE with the use of specific inhibitors: bisindolylmaleimide I, KN-62, and ML-9, respectively, for the protein kinases and cerulenin for protein acylation (Yajima et al. 2000; Straub et al. 2002). None of these inhibitors significantly affected CIE in the presence of 2.8 mm glucose; although KN-62 and cerulenin inhibited CIE in the presence of 20 mm glucose, high glucose still had a significant facilitatory effect on CIE in the presence of these inhibitors (Table 3).
Control of the fusion pore by PKA
Finally, we probed the dynamics of the exocytic fusion pore by simultaneous imaging of two fluorescent tracers with different molecular sizes, SRB (∼1.4 nm) and 10-kDa FD (∼6 nm) (Takahashi et al. 2002a). Insulin granules underwent full fusion in most (∼93%) exocytic events during GIE (Fig. 6A), consistent with previous observations (Takahashi et al. 2002a). Transient opening of the initial small pore, as revealed by staining with SRB but not with 10-kDa FD, was detected in 6.5% of events (Fig. 6B). The frequency of such transient opening was reduced to 3.8% in the presence of forskolin and increased to 10.3 or 9.8% in the presence of Rp-cAMPS or PKI (Table 4). These results suggested that PKA affects the fusion pore when its diameter is < 6 nm, and that the effects of the inhibitors on actual insulin secretion are slightly (∼6%) larger than those presented in Table 1, since most insulin secretion occurs after the dilatation of the pore larger than 12 nm (Takahashi et al. 2002a).
Figure 6. Opening of the fusion pore and full flattening of omega (Ω)–shaped exocytic profiles during GIE.

A, typical example of a single exocytic event stained with both SRB and 10-kDa FD in an islet stimulated with 20 mm glucose. The latency between the onset of staining with SRB and that of staining with 10-kDa FD is indicated. AU, arbitrary units. B, example of transient opening of a small fusion pore, as revealed by staining of the event with SRB but not with 10-kDa FD. C, time course of flattening of an Ω-profile stained with SRB. The time (T0.5) required for the fluorescence intensity to decrease to the half-maximal value is indicated.
The latency between the onset of staining of granules with SRB and that of their staining with 10-kDa FD (Fig. 6A), which reflects the dilatation of the pore from 1.4 to 6 nm, was not affected by forskolin, Rp-cAMPS, or PKI (Table 4). Moreover, none of these agents affected the time course of granule flattening, as reflected by the time (T0.5) required for SRB fluorescence to decay to the half-maximal value (Fig. 6C and Table 4). These data suggested that cAMP does not affect the process of exocytosis after dilatation of the fusion pore to > 6 nm.
Discussion
With the use of TEP imaging of pancreatic islets, we found that PKA antagonists markedly and selectively inhibited the initial period (∼250 s) of the first phase (< 7 min) of GIE. These data suggest that the basal activity of PKA in islets is involved in glucose sensing during the initial period of GIE. The action of PKA was found not to be mediated by an effect on Ca2+ signalling, given that the PKA inhibitors did not affect glucose-induced increases in [Ca2+]i under our experimental conditions. Consistently, glucose rapidly potentiated insulin exocytosis induced by the large increases in [Ca2+]i generated by photolysis of a caged-Ca2+ compound (CIE) in a PKA-dependent manner. This action of glucose was not mimicked by increases in the cytosolic concentration of cAMP, suggesting that the glucose action requires a cytosolic factor, which stimulates actions of PKA, and which is generated by glucose, but is not cAMP. Thus, PKA regulates insulin exocytosis in a glucose-dependent manner, and plays a major role in glucose sensing for insulin exocytosis, in addition to the role of the KATP channel-dependent mechanism.
Role of the basal activity of PKA in glucose-induced insulin exocytosis
Previous studies have shown that PKA inhibitors manifest relatively weak inhibitory (−25%) (Persaud et al. 1990; Lester et al. 1997) or insignificant (Harris et al. 1997) effects on GIE. Our data now suggest that the small effects of PKA inhibitors observed in these previous studies might be attributable to several factors. First, these studies relied on radioimmunoassay of insulin secreted from islets during a period of 30–60 min; any effects of the antagonists on the first phase of GIE would thus likely have been masked. Our imaging approach was able to evaluate effects of antagonists on insulin exocytosis with a time resolution of seconds and, consequently, was able to distinguish the first phase of insulin exocytosis from the second. Second, although H89 and KT5720 showed relatively weak effects in intact islets in the present study, our imaging approach revealed that their effects were much greater in small clusters of islet cells, suggesting that their ineffectiveness in whole islets was due to their poor penetration, as has previously been shown to be the case for lipophilic fluorescent tracers (Takahashi et al. 2002b). Finally, we have shown that the effects of PKA inhibitors are dependent on the basal activity of PKA and may be less prominent in preparations in which this basal activity is suppressed, as is the case with the cluster preparations in the present study and with single β cells (Pipeleers et al. 1985). Paracrine regulation, such as that mediated by glucagon secreted from α cells (Pipeleers et al. 1985), might increase the basal activity of PKA in β cells present within intact islets. Furthermore, the PKA-dependent mechanism of glucose sensing might be expected to play a greater role in vivo than in vitro, given that the basal activity of PKA in β cells may also be increased by various hormones, such as incretins, present in the blood.
The PKA-dependent mechanism of glucose sensing
Our finding that a high concentration of glucose enhanced CIE is consistent with the previous demonstration that high glucose facilitated insulin exocytosis induced by a high concentration of K+ (Gembal et al. 1992; Aizawa et al. 1994). These previous studies, however, investigated insulin secretion for longer time periods and under rather complex experimental conditions, in which KATP channels were activated with a channel agonist (diazoxide, 150–250 μm) and insulin secretion was triggered with high K+ (20–50 mm), which induces spatially heterogeneous and moderate increases in [Ca2+]i (Bokvist et al. 1995). In contrast, we quantified CIE beginning 1 min after glucose application in cells subjected to large and spatially homogeneous increases in [Ca2+]i in the absence of a KATP channel agonist. Despite the differences in experimental conditions, certain features of the facilitation by glucose were shared by the present and previous studies. First, forskolin was more effective in enhancing insulin exocytosis in the presence of high glucose (Gembal et al. 1993; Yajima et al. 1999). Second, inhibitors of conventional protein kinase C did not abrogate the glucose action (Sato & Henquin, 1998). Third, the action of glucose was rapid (Gembal et al. 1992) and finally, ATP was implicated in the action of glucose (Detimary et al. 1996; Sato & Henquin, 1998). It is thus possible that the two types of experiments revealed regulation of insulin exocytosis by the same underlying mechanism, whereas the results obtained with KATP channel agonist might also reflect the operation of slower processes activated by glucose, such as protein acylation (Yajima et al. 2000; Straub et al. 2002). Our study has shown that PKA mediates the rapid effect of glucose on CIE, and that this action is operative during the initial period of the first phase of GIE.
The PKA-dependent mechanism does not constitutively regulate exocytosis. Indeed, increases in cytosolic concentrations of cAMP by forskolin were not sufficient for the action of PKA on CIE, and a metabolite of glucose in the cytosol is likely to also be required. One candidate for such a metabolite is ATP, given that glucose induces a rapid increase in the cytosolic concentration of ATP (Henquin, 1990). Moreover, we have previously shown that the fast mode (mode 1) of CIE is augmented by cytosolic ATP in a concentration-dependent (0–5 mm) manner (Takahashi et al. 1999; Kasai et al. 2002) and that this effect requires both cAMP and PKA. Mode-1 exocytosis might thus account for the major part of the first phase of GIE (Fig. 7). An increase in the concentration of ATP can result in the activation of PKA in the absence of an increase in the concentration of cAMP (Takahashi et al. 1999); such an effect might account for glucose action in the absence of cAMP accumulation (Gembal et al. 1993; Yajima et al. 1999).
Figure 7. Scheme for the contribution of two distinct modes of exocytosis (modes 1 and 2) to the two phases of GIE.

The initial period (250 s) of the first phase of GIE requires PKA. It is mediated mostly by mode-1 Ca2+-dependent exocytosis, which is rapid and regulated by both PKA and ATP. Mode-1 exocytic vesicles may be recruited during glucose stimulation in the presence of a high cytosolic concentration of cAMP and can therefore also contribute to the later period of the first phase of GIE. The second phase of GIE is mediated predominantly by mode-2 Ca2+-dependent exocytosis, which is slow and does not require PKA.
The number of insulin vesicles responsible for mode-1 exocytosis is similarly estimated as ∼22 events per cell both from GIE ((6.4 − 2.0) cell−1 min−1× 5 min; Table 1) and from CIE ((128.4 − 40.4) cell−1 min−1× 0.25 min; Table 3), assuming in the latter instance that the large increases in [Ca2+]i rapidly triggered all available mode-1 exocytosis. In the presence of forskolin, however, the number was estimated as 59 events per cell from GIE ((13.8 − 2.0) × 5; Table 1), which is much greater than the value of 35 events per cell estimated from CIE ((182.5 − 40.4) × 0.25; Table 3). It is therefore possible that mode-1 vesicles are recruited when β cells experience a large increase in the cytosolic concentration of cAMP for > 2 min during GIE. Such a scenario may account for the fact that forskolin also augments the second phase of GIE (Takahashi et al. 2002a) (Fig. 7). In the absence of forskolin, the second phase of GIE may be mediated mostly by mode-2 exocytosis (Fig. 7), which is Ca2+ dependent but resistant to antagonists of cAMP and PKA and occurs more slowly than does mode-1 exocytosis (Takahashi et al. 1999).
Our observations that forskolin and inhibitors of PKA affected the fate of the fusion pore suggest that the action of PKA is mediated, at least in part, at the level of the fusion reaction. Several molecules are potential targets of PKA in this regard. One such candidate is SNAP25, a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE); threonine-138 of this protein is phosphorylated by PKA (Nagy et al. 2004) and (Chheda et al. 2001) is implicated in the early phase of Ca2+-triggered exocytosis of large dense-core vesicles in chromaffin cells (Nagy et al. 2004). Snapin, a protein that binds to SNARE complexes, is also a target of PKA in chromaffin cells (Chheda et al. 2001); this protein is also expressed in β cells, but its role in exocytosis of large dense-core vesicles is unknown. Rab-interacting molecule-1 (RIM1) regulates neurotransmitter release at synapses and was shown to be a target of PKA (Lonart et al. 2003); the phosphorylation site (serine-413) of this protein is also conserved in the related molecule RIM2 (Lonart et al. 2003), which is expressed in β cells (Ozaki et al. 2000; Kashima et al. 2001).
In conclusion, we have shown that a PKA-dependent mechanism of glucose sensing operates during the initial period of the first phase of GIE. Impairment of the first phase of GIE has been implicated in the pathogenesis of type 2 diabetes (Ward et al. 1984; Vaag et al. 1995). In addition, the facilitatory effect of glucose on insulin secretion is defective in individuals with this disease (Ward et al. 1984), with correction of this defect having been proposed as a new strategy for diabetes treatment. Indeed, glucagon-like peptide-1 (GLP-1) and exendin, both of which are potential therapeutic agents for individuals with type 2 diabetes, have been found to activate PKA and to enhance insulin secretion in the presence of high plasma concentrations of glucose (Parkes et al. 2001). Further elucidation of the molecular mechanism of PKA-dependent glucose sensing may provide new insight into the aetiology of diabetes mellitus and lead to the development of novel therapies.
Acknowledgments
We thank N. Takahashi, T. Kise, and T. Suzuki for technical assistance. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- Aizawa T, Sato Y, Ishihara F, Taguchi N, Komatsu M, Suzuki N, Hashizume K, Yamada T. ATP-sensitive K+ channel-independent glucose action in rat pancreatic beta-cell. Am J Physiol. 1994;266:C622–C627. doi: 10.1152/ajpcell.1994.266.3.C622. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Jetton TL, Ying G, Magnuson MA, Piston DW. Quantitative subcellular imaging of glucose metabolism within intact pancreatic islets. J Biol Chem. 1996;271:3647–3651. doi: 10.1074/jbc.271.7.3647. [DOI] [PubMed] [Google Scholar]
- Bokvist K, Eliasson L, Ammala C, Renstrom E, Rorsman P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic B-cells. EMBO J. 1995;14:50–57. doi: 10.1002/j.1460-2075.1995.tb06974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugrim AE. Regulation of Ca2+ release by cAMP-dependent protein kinase. A mechanism for agonist-specific calcium signaling? Cell Calcium. 1999;25:219–226. doi: 10.1054/ceca.1999.0027. [DOI] [PubMed] [Google Scholar]
- Chheda MG, Ashery U, Thakur P, Rettig J, Sheng ZH. Phosphorylation of Snapin by PKA modulates its interaction with the SNARE complex. Nat Cell Biol. 2001;3:331–338. doi: 10.1038/35070000. [DOI] [PubMed] [Google Scholar]
- Detimary P, Van den Berghe G, Henquin JC. Concentration dependence and time course of the effects of glucose on adenine and guanine nucleotides in mouse pancreatic islets. J Biol Chem. 1996;271:20559–20565. doi: 10.1074/jbc.271.34.20559. [DOI] [PubMed] [Google Scholar]
- Dyachok O, Gylfe E. Ca2+-induced Ca2+ release via inositol 1,4,5-trisphosphate receptors is amplified by protein kinase A and triggers exocytosis in pancreatic β-cells. J Biol Chem. 2004;279:45455–45461. doi: 10.1074/jbc.M407673200. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Eto K, Tsubamoto Y, Terauchi Y, Sugiyama T, Kishimoto T, Takahashi N, Yamauchi N, Kubota N, Murayama S, Aizawa T, Akanuma Y, Aizawa S, Kasai H, Yazaki Y, Kadowaki T. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science. 1999;283:981–985. doi: 10.1126/science.283.5404.981. [DOI] [PubMed] [Google Scholar]
- Fournier L, Whitfield JF, Schwartz JL, Begin-Heick N. Cyclic AMP triggers large [Ca2+]i oscillations in glucose-stimulated beta-cells from ob/ob mice. J Biol Chem. 1994;269:1120–1124. [PubMed] [Google Scholar]
- Fujita-Yoshigaki J, Dohke Y, Hara-Yokoyama M, Furuyama S, Sugiya H. Presence of a complex containing vesicle-associated membrane protein 2 in rat parotid acinar cells and its disassembly upon activation of cAMP-dependent protein kinase. J Biol Chem. 1999;274:23642–23646. doi: 10.1074/jbc.274.33.23642. [DOI] [PubMed] [Google Scholar]
- Gembal M, Detimary P, Gilon P, Gao ZY, Henquin JC. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J Clin Invest. 1993;91:871–880. doi: 10.1172/JCI116308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gembal M, Gilon P, Henquin JC. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J Clin Invest. 1992;89:1288–1295. doi: 10.1172/JCI115714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grapengiesser E, Gylfe E, Hellman B. Cyclic AMP as a determinant for glucose induction of fast Ca2+ oscillations in isolated pancreatic beta-cells. J Biol Chem. 1991;266:12207–12210. [PubMed] [Google Scholar]
- Harris TE, Persaud SJ, Jones PM. Pseudosubstrate inhibition of cyclic AMP-dependent protein kinase in intact pancreatic islets: effects on cyclic AMP-dependent and glucose-dependent insulin secretion. Biochem Biophys Res Commun. 1997;232:648–651. doi: 10.1006/bbrc.1997.6344. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Cellular mechanisms of the control of insulin secretion. Arch Int Physiol Biochim. 1990;98:A61–A80. [PubMed] [Google Scholar]
- Hilfiker S, Czernik AJ, Greengard P, Augustine GJ. Tonically active protein kinase A regulates neurotransmitter release at the squid giant synapse. J Physiol. 2001;531:141–146. doi: 10.1111/j.1469-7793.2001.0141j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RS, Oberwetter JM, Boyd AE., III Increase in cAMP levels in beta-cell line potentiates insulin secretion without altering cytosolic free-calcium concentration. Diabetes. 1987;36:440–446. doi: 10.2337/diab.36.4.440. [DOI] [PubMed] [Google Scholar]
- Hutton JC. The insulin secretory granule. Diabetologia. 1989;32:271–281. doi: 10.1007/BF00265542. [DOI] [PubMed] [Google Scholar]
- Ito K, Miyashita Y, Kasai H. Kinetic control of multiple forms of Ca2+ spikes by inositol trisphosphate in pancreatic acinar cells. J Cell Biol. 1999;146:405–413. doi: 10.1083/jcb.146.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Takahashi T. Presynaptic mechanism underlying cAMP-dependent synaptic potentiation. J Neurosci. 2004;24:5202–5208. doi: 10.1523/JNEUROSCI.0999-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Chepurny OG, Rindler MJ, Collis L, Chepurny Z, Li WH, Harbeck M, Roe MW, Holz GG. A cAMP and Ca2+ coincidence detector in support of Ca2+-induced Ca2+ release in mouse pancreatic β cells. J Physiol. 2005;566:173–188. doi: 10.1113/jphysiol.2005.087510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Hatakeyama H, Kishimoto T, Liu T-T, Nemoto T, Takahashi N. A new quantitative (two-photon extracellular polar-tracer imaging-based quantification (TEPIQ) analysis for diameters of exocytic vesicles and its application to pancreatic islets. J Physiol. 2005;568:891–903. doi: 10.1113/jphysiol.2005.093047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Suzuki T, Liu TT, Kishimoto T, Takahashi N. Fast and cAMP-sensitive mode of Ca2+-dependent exocytosis in pancreatic beta-cells. Diabetes. 2002;51(Suppl. 1):S19–S24. doi: 10.2337/diabetes.51.2007.s19. [DOI] [PubMed] [Google Scholar]
- Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. Critical role of cAMP-GEFII Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem. 2001;276:46046–46053. doi: 10.1074/jbc.M108378200. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Liu T-T, Hatakeyama H, Nemoto T, Takahashi N, Kasai H. Sequential compound exocytosis of large dense-core vesicles in PC12 cells studied with TEPIQ (two-photon extracellular polar-tracer imaging-based quantification) analysis. J Physiol. 2005;568:905–915. doi: 10.1113/jphysiol.2005.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester LB, Langeberg LK, Scott JD. Anchoring of protein kinase A facilitates hormone-mediated insulin secretion. Proc Natl Acad Sci U S A. 1997;94:14942–14947. doi: 10.1073/pnas.94.26.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T-T, Kishimoto T, Hatakeyama H, Nemoto T, Takahashi N, Kasai H. Exocytosis and endocytosis of small vesicles in PC12 cells studied with TEPIQ (two-photon extracellular polar-tracer imaging-based quantification) analysis. J Physiol. 2005;568:917–929. doi: 10.1113/jphysiol.2005.094011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ. Phosphorylation of RIM1α by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Marie JC, Bailbe D. Cytosolic calcium handling in islets of normal Wistar and diabetic Goto Kakizaki rats in the presence of glucose and truncated glucagon-like peptide 1 (7–36) amide. Ann N Y Acad Sci. 2000;921:464–468. doi: 10.1111/j.1749-6632.2000.tb07016.x. [DOI] [PubMed] [Google Scholar]
- Matthies H, Reymann KG. Protein kinase A inhibitors prevent the maintenance of hippocampal long-term potentiation. Neuroreport. 1993;4:712–714. doi: 10.1097/00001756-199306000-00028. [DOI] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sorensen JB. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron. 2004;41:417–429. doi: 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- Nemoto T, Kimura R, Ito K, Tachikawa A, Miyashita Y, Iino M, Kasai H. Sequential-replenishment mechanism of exocytosis in pancreatic acini. Nat Cell Biol. 2001;3:253–258. doi: 10.1038/35060042. [DOI] [PubMed] [Google Scholar]
- Nesher R, Cerasi E. Modeling phasic insulin release: immediate and time-dependent effects of glucose. Diabetes. 2002;51(Suppl. 1):S53–S59. doi: 10.2337/diabetes.51.2007.s53. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- Parkes DG, Pittner R, Jodka C, Smith P, Young A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism. 2001;50:583–589. doi: 10.1053/meta.2001.22519. [DOI] [PubMed] [Google Scholar]
- Persaud SJ, Jones PM, Howell SL. Glucose-stimulated insulin secretion is not dependent on activation of protein kinase A. Biochem Biophys Res Commun. 1990;173:833–839. doi: 10.1016/s0006-291x(05)80862-9. [DOI] [PubMed] [Google Scholar]
- Pipeleers DG, Schuit FC, in't Veld PA, Maes E, Hooghe-Peters EL, WM, Gepts W. Interplay of nutrients and hormones in the regulation of insulin release. Endocrinology. 1985;117:824–833. doi: 10.1210/endo-117-3-824. [DOI] [PubMed] [Google Scholar]
- Porterfield DM, Corkey RF, Sanger RH, Tornheim K, Smith PJ, Corkey BE. Oxygen consumption oscillates in single clonal pancreatic beta-cells (HIT) Diabetes. 2000;49:1511–1516. doi: 10.2337/diabetes.49.9.1511. [DOI] [PubMed] [Google Scholar]
- Renstrom E, Eliasson L, Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J Physiol. 1997;502:105–118. doi: 10.1111/j.1469-7793.1997.105bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Abrahamsson H. Cyclic AMP potentiates glucose-induced insulin release from mouse pancreatic islets without increasing cytosolic free Ca2+ Acta Physiol Scand. 1985;125:639–647. doi: 10.1111/j.1748-1716.1985.tb07766.x. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Direct modulation of synaptic vesicle priming by GABAB receptor activation at a glutamatergic synapse. Nature. 2003;424:775–778. doi: 10.1038/nature01859. [DOI] [PubMed] [Google Scholar]
- Sato Y, Henquin JC. The K+-ATP channel-independent pathway of regulation of insulin secretion by glucose: in search of the underlying mechanism. Diabetes. 1998;47:1713–1721. doi: 10.2337/diabetes.47.11.1713. [DOI] [PubMed] [Google Scholar]
- Sato K, Ohsaga A, Oshiro T, Ito S, Maruyama Y. Involvement of GTP-binding protein in pancreatic cAMP-mediated exocytosis. Pflugers Arch. 2002;443:394–398. doi: 10.1007/s004240100711. [DOI] [PubMed] [Google Scholar]
- Straub SG, Yajima H, Komatsu M, Aizawa T, Sharp GW. The effects of cerulenin, an inhibitor of protein acylation, on the two phases of glucose-stimulated insulin secretion. Diabetes. 2002;51(Suppl. 1):S91–S95. doi: 10.2337/diabetes.51.2007.s91. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Hatakeyama H, Okado H, Miwa A, Kishimoto T, Kojima T, Abe T, Kasai H. Sequential exocytosis of insulin granules is associated with redistribution of SNAP25. J Cell Biol. 2004;165:255–262. doi: 10.1083/jcb.200312033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Kadowaki T, Yazaki Y, Ellis-Davies GC, Miyashita Y, Kasai H. Post-priming actions of ATP on Ca2+-dependent exocytosis in pancreatic beta cells. Proc Natl Acad Sci U S A. 1999;96:760–765. doi: 10.1073/pnas.96.2.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Kadowaki T, Yazaki Y, Miyashita Y, Kasai H. Multiple exocytotic pathways in pancreatic beta cells. J Cell Biol. 1997;138:55–64. doi: 10.1083/jcb.138.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Kishimoto T, Nemoto T, Kadowaki T, Kasai H. Fusion pore dynamics and insulin granule exocytosis in the pancreatic islet. Science. 2002a;297:1349–1352. doi: 10.1126/science.1073806. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Nemoto T, Kimura R, Tachikawa A, Miwa A, Okado H, Miyashita Y, Iino M, Kadowaki T, Kasai H. Two-photon excitation imaging of pancreatic islets with various fluorescent probes. Diabetes. 2002b;51(Suppl. 1):S25–S28. doi: 10.2337/diabetes.51.2007.s25. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- Vaag A, Henriksen JE, Madsbad S, Holm N, Beck-Nielsen H. Insulin secretion, insulin action, and hepatic glucose production in identical twins discordant for non-insulin-dependent diabetes mellitus. J Clin Invest. 1995;95:690–698. doi: 10.1172/JCI117715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D., Jr Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74:1318–1328. doi: 10.1172/JCI111542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wokosin DL, Loughrey CM, Smith GL. Characterization of a range of fura dyes with two-photon excitation. Biophys J. 2004;86:1726–1738. doi: 10.1016/S0006-3495(04)74241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollheim CB, Sharp GW. Regulation of insulin release by calcium. Physiol Rev. 1981;61:914–973. doi: 10.1152/physrev.1981.61.4.914. [DOI] [PubMed] [Google Scholar]
- Yada T, Itoh K, Nakata M. Glucagon-like peptide-1-(7–36) amide and a rise in cyclic adenosine 3′,5′-monophosphate increase cytosolic free Ca2+ in rat pancreatic beta-cells by enhancing Ca2+ channel activity. Endocrinology. 1993;133:1685–1692. doi: 10.1210/endo.133.4.8404610. [DOI] [PubMed] [Google Scholar]
- Yaekura K, Kakei M, Yada T. cAMP-signaling pathway acts in selective synergism with glucose or tolbutamide to increase cytosolic Ca2+ in rat pancreatic beta-cells. Diabetes. 1996;45:295–301. doi: 10.2337/diab.45.3.295. [DOI] [PubMed] [Google Scholar]
- Yaekura K, Yada T. [Ca2+]i-reducing action of cAMP in rat pancreatic beta-cells: involvement of thapsigargin-sensitive stores. Am J Physiol. 1998;274:C513–C521. doi: 10.1152/ajpcell.1998.274.2.C513. [DOI] [PubMed] [Google Scholar]
- Yajima H, Komatsu M, Schermerhorn T, Aizawa T, Kaneko T, Nagai M, Sharp GW, Hashizume K. cAMP enhances insulin secretion by an action on the ATP-sensitive K+ channel-independent pathway of glucose signaling in rat pancreatic islets. Diabetes. 1999;48:1006–1012. doi: 10.2337/diabetes.48.5.1006. [DOI] [PubMed] [Google Scholar]
- Yajima H, Komatsu M, Yamada S, Straub SG, Kaneko T, Sato Y, Yamauchi K, Hashizume K, Sharp GW, Aizawa T. Cerulenin, an inhibitor of protein acylation, selectively attenuates nutrient stimulation of insulin release: a study in rat pancreatic islets. Diabetes. 2000;49:712–717. doi: 10.2337/diabetes.49.5.712. [DOI] [PubMed] [Google Scholar]