

Abstract
Mast cells are the major effector cells for immediate hypersensitivity and chronic allergic reactions. These cells accumulate in mucosal tissues of allergic reactions, where immunoglobulin E (IgE) is produced locally. Here we provide evidence that, in addition to antigen that can attract IgE-bound mast cells, the type of IgE molecules that efficiently activate mast cells can promote the migration of mast cells in the absence of antigen. IgEand IgE+Ag-mediated migration involves an autocrine/paracrine secretion of soluble factors including adenosine, leukotriene B4, and several chemokines. Their secretion depends on 2 tyrosine kinases, Lyn and Syk, and they are agonists of G-protein–coupled receptors and signal through phosphatidylinositol 3-kinase γ, leading to mast cell migration. In mouse experiments, naive mast cells are attracted to IgE, and IgE-sensitized mast cells are attracted to antigen. Therefore, IgE and antigen are implicated in mast cell accumulation at allergic tissue sites with local high IgE levels.
Introduction
Mast cells are the major effector cell type in immunoglobulin E (IgE)–mediated immediate hypersensitivity, chronic allergic diseases, and the defense against certain parasites and bacteria. Traditionally, it is thought that mast cells bound to antigen-specific IgE via the high-affinity receptor (Fc∈;RI) encounter multivalent antigen (the stimulation mode hereafter termed IgE+Ag), and then IgE-bound receptors are aggregated, leading to cellular activation.1 Activated mast cells secrete preformed and newly synthesized proinflammatory mediators, such as histamine, proteases, lipids, cytokines, and chemokines.
The mouse Fc∈;RI consists of an IgE-binding α subunit, a β subunit, and 2 molecules of signal-generating γ subunit.2 Fc∈;RI aggregation leads to phosphorylation of the immunoreceptor tyrosine–based activation motifs (ITAMs) of the β and γ subunits by Lyn, and recruitment of Syk to the tyrosine-phosphorylated ITAMs of the γ subunit results in the activation of this protein-tyrosine kinase (PTK).3 Activated Lyn and Syk as well as Fyn eventually lead to the activation of multiple signaling pathways, including phosphatidylinositol 3-kinase (PI3K), phospholipase C-γ/Ca2+/protein kinase C (PKC), and mitogen-activated protein (MAP) kinases.4
Cell–extracellular matrix interactions mediated by integrins play a critical role in multiple cellular functions including cell adhesion and migration. Activation of mast cells by Fc∈;RI aggregation and stem cell factor (SCF) induces adhesion to fibronectin (FN)5 predominantly via integrin α5β1.6-8 Upon Fc∈;RI aggregation and SCF stimulation, FN-adherent cells exhibit stronger effector functions such as histamine release and cytokine production than nonadherent cells.7
We and others recently demonstrated that monomeric IgE can promote mast cell survival.9,10 This observation, together with earlier studies showing that IgE in the absence of antigen can increase the surface expression of Fc∈;RI,11-13 has transformed the traditional view of IgE–mast cell binding as a “sensitization” step prior to receptor aggregation with antigen or other crosslinking reagents into a new one that monomeric IgE can induce survival and “activation” of mast cells.14 We also found that IgE molecules display heterogeneity in that different IgE molecules induce varied levels of activation; at one extreme end of the spectrum, some IgE molecules, termed highly cytokinergic (HC) IgEs, induce the production and secretion of various cytokines and other activation events including degranulation, whereas other IgE molecules, termed poorly cytokinergic (PC), do so very inefficiently.15
Mast cells accumulate at local inflammatory mucosal tissues, as seen in allergic rhinitis and asthma. Interestingly, class switch recombination and somatic hypermutation of the immunoglobulin gene and eventually IgE synthesis and secretion occur at such inflammatory mucosae.16-21 In an allergic individual, local IgE production persists for a long period in the absence of allergen.20 A variety of biologic agents, including growth factors (eg, SCF), chemokines (MCP-1/CCL2, MIP-1α/CCL3, RANTES/CCL5, eotaxin/CCL11, SDF-1α/CXCL12, etc, refer to “Chemokine/chemokine receptor nomenclature”22 for the nomenclature of chemokines), and adenosine nucleotides, are known to attract rodent mast cells.23-27 Consistent with effects of the chemokines on mast cell migration, mast cells express appropriate receptors including CCR1-5, CXCR1-2, and CXCR4.28-32 Antigen can also cause the migration of IgE-sensitized mast cells, which can be suppressed by inhibitors of Rho-kinase/ROCK and p38.33 In this study, we have found that, in addition to IgE+Ag, HC IgEs can attract mast cells. HC IgE- and IgE+Ag-induced mast cell migration involves autocrine/paracrine secretion of soluble factors. The initial phase of migration leading to the release of such factors depends on Lyn and Syk, and the following phase downstream of G-protein–coupled receptor (GPCR) stimulation by these soluble factors requires PI3Kγ. In vivo mouse experiments also suggest that mast cells can be attracted to HC IgEs as well as antigen. Therefore, this study provides a novel mechanism for mast cell accumulation to allergic inflammatory sites.
Materials and methods
Antibodies and other reagents
IgE antibodies used were described previously.15 Antitrinitrophenyl (TNP) IgG and antibodies against integrins were purchased from BD Biosciences Pharmingen (San Diego, CA); MIP-1α, MCP-1, and RANTES were from R&D Systems, Minneapolis, MN; fibronectin, vitronectin, laminin, collagen type IV, dinitrophenyl (DNP)–lysine, adenosine, histamine, serotonin, and MRS 1523 were from Sigma, St Louis, MO; PP2, piceatannol, wortmannin, LY294002, PD98059, SP600125, Gö-6976, PKI, KT5720, Y-27632, cytochalasin D, EGTA [ethylene glycol-bis(beta-aminoethyl ether)–N,N,N′,N′-tetraacetic acid], and leukotriene B4 (LTB4) were from EMD Biosciences (San Diego, CA); N-oleoyl dopamine, U-75302, and LY2552833 were from Cayman Chemical (Ann Arbor, MI). SCF and ER-27319 were gifts from Kirin Brewery (Tokyo, Japan) and Eisai (Tokyo, Japan), respectively. Terreic acid was described previously.34
Cells and stimulation
Bone marrow cells from wild-type and mutant mice were cultured in interleukin 3 (IL-3)–containing medium for 4 to 6 weeks to generate mast cells (BMMCs) with more than 95% purity (c-Kit+ Fc∈;RI+ by flow cytometry). The following mutant mice were used: Fc∈;RIα−/−, lyn−/−, fyn−/−, hck−/−, syk−/−, btk−/−, PKCβ−/−, PKC∈;−/−, PKCθ−/−, and PI3Kγ−/−. For IgE+Ag stimulation, BMMCs were sensitized by overnight incubation with 0.5 μg/mL H1-∈;-DNP-206 (206) IgE. BMMCs washed twice with buffer were used for migration assays. Animal studies were approved by the institutional review board of La Jolla Institute for Allergy and Immunology, San Diego, CA.
In vitro migration assay
BMMC migration for 8 hours was assayed using 24-well Transwell chambers (Corning, Corning, NY) separated by 5-μm polycarbonate filters, the lower surface of which had been coated overnight with 20 μg/mL fibronectin and followed by blocking with 4% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) at 37°C for 1 hour, unless otherwise described. Under standard conditions, upper wells contained 106 cells/well in 0.2 mL medium consisting of RPMI/1% BSA/20 mM HEPES [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid] (pH 7.4) and lower wells contain 0.6 mL medium. Cells migrated into lower wells were counted using a hemocytometer. For inhibition with antibodies, BMMCs were first preincubated with neutralizing antibodies (20 μg/mL-50 μg/mL) for 30 minutes, and then placed in upper wells. Both upper and lower wells contained neutralizing antibodies.
In some experiments, BMMCs were prestained with 10 μM 5- (and 6-) carboxyfluorescein diacetate, succinimidyl ester (CFSE; Molecular Probes, Eugene, OR) in 0.1% BSA/PBS at 37°C for 10 minutes before loading onto upper wells. Cells migrated into lower wells were quantified using flow cytometry.
In vivo mast cell accumulation
Gauze strips (∼7 layers, 10 × 15 mm2) spotted with 50 μL of 10 μg/mL SPE-7 IgE or PBS were applied to the shaved back skin of naive NC/Nga mice for 24 hours. This portion of the back skin was occluded with Tegaderm Transparent Dressing (3M, St. Paul, MN) and BAND-AID (Johnson & Johnson, New Brunswick, NJ). Mice were killed and skin regions were prepared to stain mast cells with toluidine blue. Mast cell numbers were counted under a microscope. In another type of in vivo experiment, mice were first treated similarly with 100 μL of 10 μg/mL anti-DNP (206) IgE or PBS into the back skin, and 1 day later the same area was applied with 50 μL of 100 ng/mL DNP-human serum albumin (HSA) or PBS for 24 hours. For mast cell quantification, dorsal skin samples were fixed in 10% formaldehyde, paraffin embedded, and cut into 6-μm sections. Deparaffinized sections were stained with toluidine blue (pH 4.0) and analyzed by light microscopy. Cells between epithelium and panniculus carnosus were counted at a magnification of ×400. Images were acquired with a Nikon Optiphot photomicroscope (Nikon, Tokyo, Japan) with a Fluor 40×/1.30 NA or Plan 10×/0.25 NA objective, using a DVC-1310 camera (DVC Company, West Austin, TX) and DVC C-View v2.2 software. Immersion oil type FF (Cargille Laboratories Inc, Cedar Grove, NJ) was used for a magnification at 400×.
Data supplements
Four figures are provided as supplemental materials, which are available on the Blood website (see the Supplemental Figures link at the top of the online article): effects of IgE/antigen and IgG/antigen complexes on mast cell migration (Figure S1), IgE- and IgE+Ag-induced migration of mouse mast cell lines (Figure S2), requirements of fibronectin, vitronectin, or laminin (Figure S3), and autocrine/paracrine mechanism of mast cell migration (Figure S4).
Results
HC IgEs can induce mast cell migration
Given the ability of IgEs to induce mast cell activation,15 we examined whether IgEs can promote the migration of mouse BMMCs in the absence of antigen. As shown in Figure 1A, 3 HC IgEs in a lower well induced vigorous migration of BMMCs from an FN-coated upper well after 8 hours of incubation in a transwell assay, whereas typical PC IgEs such as H1-∈;-DNP-206 (206) and IgE-3 did not. Dose-response experiments (Figure 1B) indicated that the extent of migration induced by a typical HC IgE (SPE-7) increases in a dose-dependent manner up to 30 μg/mL. A significant migration was observed at 1 μg/mL IgE, a concentration reachable in a subset of atopic conditions.35,36 Kinetic experiments (Figure 1C) showed that HC IgE–induced migration is smaller in magnitude than that induced by SCF, and plateaus within 10 to 15 hours. Consistent with previous studies,33,37 migration with similar levels and kinetics was also observed with BMMCs in an upper well that had been sensitized with anti-DNP IgE (206) and moved toward antigen, DNP-HSA (Figure 1A-C).
Figure 1.
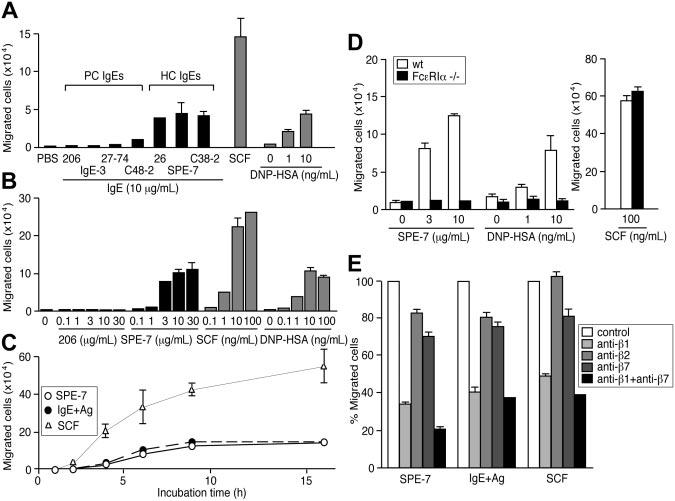
HC IgEs and IgE+Ag induce the migration of BMMCs through Fc∈;RI and β1 integrin. (A) HC, but not PC, IgEs induce BMMC migration efficiently in an 8-hour transwell assay. BMMCs, sensitized overnight with anti-DNP IgE (206) and washed, were also attracted to a lower well containing DNP-HSA. For comparison, SCF (100 ng/ml) was also tested in the same assay. Also shown are dose-response (B) and time-course (C) experiments using 10 μg/mL 206 IgE, 10 μg/mL SPE-7 IgE, 10 ng/mL DNP-HAS, and 100 ng/mL SCF. (D) Migration assays were performed with Fc∈;RIα−/− and control BMMCs. (E) Migration of wild-type BMMCs was inhibited by neutralizing antibodies against integrins. Experiments were performed in triplicate. Representative data (± SD) are shown out of 2 or more experiments.
HC IgE–induced migration signals through the Fc∈;RI and integrins
We next confirmed that IgE-induced migration is mediated through Fc∈;RI. BMMCs from Fc∈;RIα−/− mice38 failed to migrate in response to HC IgEs or IgE+Ag, whereas the mutant cells were attracted to SCF as vigorously as wild-type cells (Figure 1D). Consistent with the notion that these IgE-dependent migrations are triggered by Fc∈;RI aggregation, these migrations were inhibited by monovalent hapten, DNP-lysine (Table 1). Further, BMMC migration was induced by incubation with PC IgE plus DNP-HSA, conditions that mimic acute allergic situations in that antigens are present around IgE-bound mast cells and are different from IgE+Ag in that unbound IgE molecules were removed (IgE+Ag) or not (PC IgE plus DNP-HSA) (Figure S1A). By contrast, BMMC migration was not induced by IgG, IgM (J.K. and T.K., unpublished data, February 2004), or anti-TNP IgG plus TNP-BSA (Figure S1B). Efficient HC IgE–dependent migration was seen with BMMCs and mouse mast cell lines (MC/9 and MCP-5) (Figure S2) incubated in upper wells coated with FN, vitronectin, or laminin (Figure S3). However, collagen or BSA coating did not promote BMMC migration efficiently. These results implicated the involvement of integrins in HC IgE– and IgE+Ag-mediated mast cell migration. This was confirmed by the strong inhibition of migration with a neutralizing antibody against integrin β1. β2 and β7 integrins may play a minor, if any, role in IgE-dependent mast cell migration (Figure 1E).
Table 1.
Inhibition of BMMC migration induced by SPE-7 IgE, IgE + Ag, or SCF by various pharmacologic inhibitors
| Target | Inhibitor | SPE-7 IgE | IgE+Ag | SCF |
|---|---|---|---|---|
| Src PTKs | PP2 | 2 μM | 10 μM | 0.5 μM |
| Syk | Piceatannol | 100 μM | 120 μM | > 200 μM |
| ER-27319 | 20 μM | 20 μM | > 100 μM | |
| Btk | Terreic acid | > 20 μM | > 20 μM | > 20 μM |
| PI3K | Wortmannin | 5 nM | 40 nM | 30 nM |
| LY294002 | 7 μM | 10 μM | > 50 μM | |
| MEK | PD98059 | > 50 μM | > 50 μM | > 50 μM |
| JNK | SP600125 | 30 μM | 40 μM | 40 μM |
| p38 | SB203380 | 5 μM | 2 μM | 20 μM |
| PKC | Ro31-8425 | 4 μM | 8 μM | 20 μM |
| cPKC | Goö-6976 | 2 μM | 0.5 μM | 2 μM |
| PKA | PKI | > 100 nM | > 100 nM | > 100 nM |
| KT5720 | > 100 nM | > 100 nM | > 100 nM | |
| Rho-K | Y-27632 | > 10 μM* | > 10 μM* | > 10 μM* |
| F-actin | Cytochalasin D | 0.02 μM | 0.02 μM | 0.08 μM |
| Ca2+ | EGTA | 1 mM | 1 mM | 2 mM |
| IgV | DNP-lysine | 0.4 μM | 0.4 μM | > 100 μM |
BMMCs were preincubated with pharmacologic reagents for 30 minutes before being placed in upper wells at the time of addition of 10 μg/mL SPE-7 IgE or 100 ng/mL SCF to lower wells. For IgE+Ag stimulation, BMMCs were sensitized by overnight incubation with 0.5 μg/mL of 206 IgE. BMMCs washed with buffer were preincubated with inhibitors for 30 minutes before being placed in upper wells at the time of addition of 10 ng/mL DNP-HSA. Concentrations for 50% inhibition (IC50) are indicated.
Although BMMCs were insensitive to Y-27632, MC/9 mouse mast cells were sensitive with IC50s of 5 μM to 10 μM for SPE-7-, IgE + Ag-, and SCF-induced migration.
Comparison between HC IgE– and chemokine-induced migration
Several chemokines are known to attract mast cells.26,39,40 Remarkably, migration induced by 10 μg/mL SPE-7 IgE was more pronounced than that induced by mast cell–attracting chemokines, RANTES, MCP-1, and MIP-1α (Figure 2A). When used in combinations, HC IgEs did not synergize to induce mast cell migration with SCF or any of the tested chemokines, and HC IgEs determined the extent of migration (Figure 2B-C), suggesting a hierarchically dominant role for HC IgEs in complex situations where various chemoattractants are present. Signaling hierarchy was well known among several chemoattractants of neutrophils.41 An alternative possibility is that the mediators released by HC IgE–activated mast cells (for example, proteases) inactivate SCF, chemokines, or their receptors. Unlike SCF or the chemokines, which induce the movement of mast cells toward the increasing gradient of chemoattractant concentrations (chemotaxis), checkerboard analysis indicated that HC IgEs induce a mixture of directional and nondirectional (chemokinesis) movements (Figure 2D-E).
Figure 2.
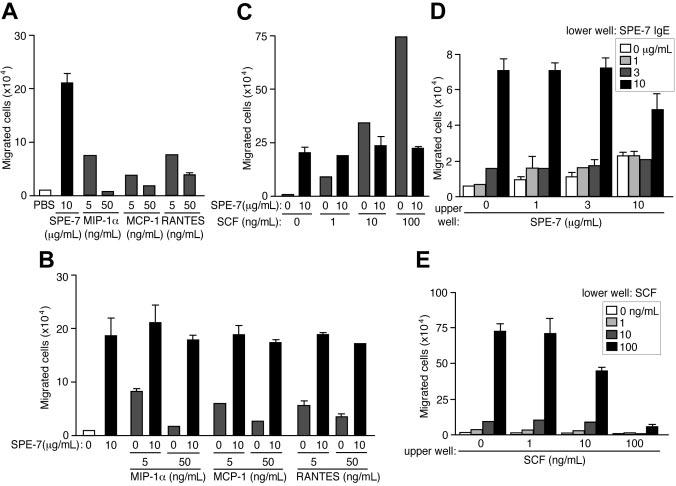
Comparison between HC IgE– and chemokine-induced migration of BMMCs and checkerboard analysis of HC IgE–induced mast cell migration. (A) SPE-7 IgE–induced migration of BMMCs was compared with those induced by mast cell–attracting chemokines, MIP-1α, MCP-1, and RANTES. (B-C) BMMC migration was induced by SPE-7 IgE in combination with chemokines or SCF. (D) BMMCs in upper wells containing various concentrations of SPE-7 IgE were incubated with various concentrations of SPE-7 IgE in lower wells. (E) BMMCs in upper wells containing various concentrations of SCF were incubated with various concentrations of SCF in lower wells. Values (mean ± SD) are shown out of 2 independent experiments.
Signaling requirements for HC IgE– and IgE+Ag-induced migration
Fc∈;RI stimulation by IgE+Ag triggers activation of nonreceptor PTKs of Src, Syk, and Tec families and several signaling pathways.3,4,14 Pharmacologic inhibition suggested that HC IgE– and IgE+Ag-induced migration requires Src and Syk kinases, but not Tec kinases (Table 1). Consistent with these data, IgE-dependent migration was abrogated in syk−/− cells (Figure 3B), but was not affected by Btk deficiency (Figure 3C). Among the tested Src PTKs, Lyn and Fyn played major and minor roles, respectively, in IgE- and IgE+Ag-dependent migration, whereas Hck played no part in this function (Figure 3A). These pharmacologic experiments (Table 1) and previous genetic and biochemical experiments10,15 indicate that signaling events induced by HC IgEs are almost the same as those induced by IgE+Ag. Downstream of these kinases, PI3K and some MAP kinases (p38 and JNK) appeared essential from pharmacologic experiments. A general inhibitor (Ro31-8425) of PKC, an inhibitor (Gö-6976) of Ca2+-dependent PKC isoforms and a Ca2+ chelator (EGTA) all inhibited IgE-dependent migration whereas inhibitors of protein kinase A or Rho-kinase did not. Consistent with these results, deficiency of PKCβ, PKC∈;,or PKCθ modestly reduced IgE-dependent migration (Figure 3D).
Figure 3.
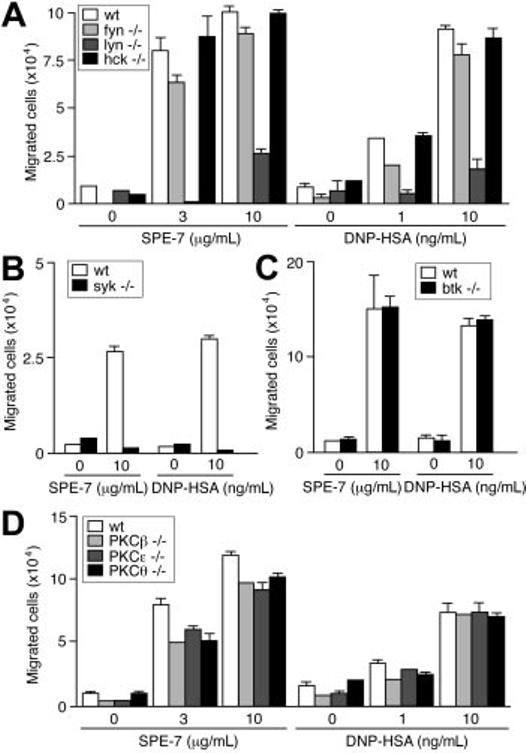
Effects of Src, Syk, Btk, and PKC deficiencies on IgE-induced migration. IgE- and IgE+Ag-induced migration assays were performed using Src family PTKdeficient (A), Syk-deficient (B), Btk-deficient (C), and PKC-deficient (D) BMMCs. Values (mean ± SD) are shown out of 2 independent experiments.
Involvement of autocrine/paracrine soluble factors, GPCRs, and PI3Kγ in HC IgE– and IgE+Ag-induced mast cell migration
To test potential involvement of autocrine/paracrine secretion in migration, we examined whether Fc∈;RIα−/− mast cells that cannot directly respond to IgE can migrate in the presence of wild-type cells. A significant proportion of CFSE-labeled Fc∈;RIα−/− mast cells mixed at a ratio of 1:1 with nonlabeled wild-type cells in upper wells migrated to lower wells in response to HC IgE and IgE+Ag (Figure S4). Further, Fc∈;RIα−/− cells in upper wells were attracted to lower wells that contained wild-type cells in the presence of HC IgE or anti-DNP IgE-sensitized wild-type cells in the presence of DNP-HSA (Figure 4A). More directly, supernatants of wild-type mast cells cultured with HC IgE or IgE+Ag for 0.5, 1, 2, or 6 hours could attract Fc∈;RIα−/− mast cells with similar efficiency (Figure 4B). These results strongly indicate that Fc∈;RIα−/− cells alone or in the mixed cultures with wild-type cells migrated to lower wells that contained HC IgE– or IgE+Ag-induced soluble factors secreted from wild-type cells.
Figure 4.
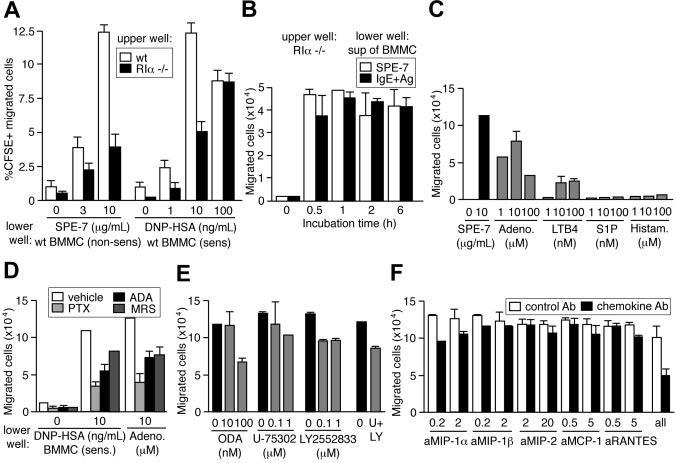
Involvement of adenosine, LTB4, and chemokines in an autocrine/paracrine mechanism of IgEand IgE+Ag-induced mast cell migration. (A) CFSE-labeled wild-type and Fc∈;RIα−/− BMMCs in upper wells were attracted to lower wells containing wild-type cells and SPE-7 or 206 IgE–sensitized wild-type cells and antigen. (B) Supernatants of wild-type BMMCs incubated with SPE-7 for the indicated periods attracted Fc∈;RIα−/− cells. Supernatants of PC IgE (206)–sensitized wild-type cells incubated with antigen for the indicated periods also attracted Fc∈;RIα−/− cells. (C) Migration was induced by adenosine and LTB4, but not by sphingosine 1–phosphate (S1P) or histamine. (D) Pertussis toxin (PTX), adenosine deaminase (ADA), and adenosine A3 receptor inhibitor MRS 1523 inhibit the migration of CFSE-labeled wild-type BMMCs from upper wells to lower wells containing IgE-sensitized wild-type cells and antigen or adenosine. (E) 5-Lipoxygenase inhibitor (N-oleoyl dopamine [ODA]), BLT1 receptor inhibitor (U-75302), and BLT2 inhibitor (LY2552833) inhibit the migration of CFSE-labeled wild-type BMMCs from upper wells to lower wells containing IgE-sensitized wild-type cells and antigen. U-75302 and LY2552833 at 1 μM each were used in a combination (U+LY) as well. (F) Neutralizing antibodies to several chemokines inhibit the migration of CFSE-labeled wild-type BMMCs from upper wells to lower wells containing IgE-sensitized wild-type cells and antigen. All the antibodies at the higher concentrations each were used in a combination (all) as well. Mean values ± SD are shown out of 2 independent experiments.
A variety of chemicals and peptides are released from IgE+Ag- and HC IgE–stimulated mast cells within 30 minutes and exert their functions on multiple cell types including mast cells themselves.10,15,42 For example, histamine (and serotonin) mediates increased vascular permeability in passive cutaneous anaphylaxis, and LTB4 and sphingosine 1–phosphate induce the migration of various leukocytes.43-46 Several chemokines such as MCP-1, MIP-1α, MIP-1β/CCL4, and MIP-2 are produced by BMMCs stimulated weakly by IgE+Ag,47 and MCP-1 secretion can be induced by fast dissociating antigens and characterized as an exception of the kinetic proofreading regimen.48 Like mast cell–produced chemokines such as MCP-1 and MIP-1α (Figure 2), adenosine and LTB4 vigorously induced migration of BMMCs whereas histamine, serotonin, or sphingosine 1–phosphate did not (Figure 4C and J.K., T.K., and T.K., unpublished data, June 2004). Adenosine and LTB4 as well as chemokines are ligands of GPCRs. Consistent with this, migration of Fc∈;RIα−/− BMMCs to supernatants of wild-type mast cells that had been cultured with HC IgE or IgE+Ag was blocked by approximately 60% by pertussis toxin, indicating that some soluble factors use Gαi-coupled receptors (Figure 4D). The role of adenosine in the migration was confirmed by the inhibition with adenosine deaminase that converts adenosine to inosine and an adenosine A3 receptor inhibitor, MRS 1523 (Figure 4D). The involvement of LTB4 was indicated by the inhibition with a 5-lipoxygenase inhibitor (N-oleoyl dopamine) and inhibitors of LTB4 receptors BLT1 (U-75 302) and BLT2 (LY2 552 833) (Figure 4E). Parenthetically, these inhibitors of LTB4 synthesis or receptors did not affect SCF-induced migration (T.K. and T.K., unpublished data, June 2004). Furthermore, roles of mast cell–produced chemokines in the migration were shown by the inhibition by neutralizing antibodies. Antibodies to MIP-1α, MIP-1β, and RANTES individually inhibited weakly (10%-25%) but in a combination inhibited HC IgE– and IgE+Ag-induced mast cell migration more strongly (∼50%; Figure 4F). Adenosine, LTB4, and chemokines are all agonists of GPCRs and signal through PI3Kγ.49 Strikingly, HC IgE– and IgE+Ag-induced mast cell migration was drastically reduced in PI3Kγ-deficient cells (Figure 5A). Therefore, it appears that IgE- and IgE+Ag-dependent migration results from the signals originated from Fc∈;RI aggregation that lead to a rapid release of multiple GPCR agonists including adenosine, LTB4, and several chemokines. Thus, activation of the GPCRs and their downstream signaling molecule, PI3Kγ, is crucial for mast cell migration.
Figure 5.
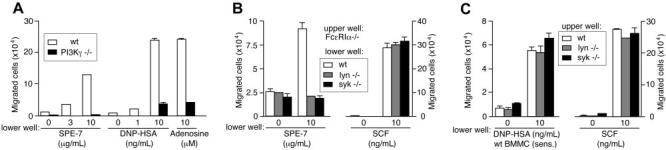
IgE- and IgE+Ag-induced mast cell migration can be divided into the early phase of Lyn/Syk-dependent release of soluble factors and the later PI3Kγ-dependent phase. (A) PI3Kγ−/− BMMCs were defective in migration in responses to SPE-7 IgE, IgE+Ag, or adenosine. (B) Fc∈;RIα−/− cells in upper wells were incubated with lower wells containing wild-type, lyn−/−, or syk−/− cells in the presence of SPE-7 or SCF. (C) Wild-type, lyn−/−, or syk−/− cells in upper wells were incubated with lower wells containing 206 IgE–sensitized wild-type cells in the presence of DNP-HSA or SCF. Mean values ± SD are shown from 2 (A) or 3 (B,C) independent experiments.
We next examined which step, Fc∈;RI stimulation-induced release of GPCR agonists or downstream signaling from GPCRs, is dependent on Lyn and Syk. For this purpose, we tested whether 5-(and -6)-carboxyfluorescein diacetate, succinimidyl ester (CFSE)– labeled Fc∈;RIα−/− BMMCs in upper wells migrate to lower wells containing wild-type, lyn−/−, or syk−/− BMMCs in the presence of SPE-7 IgE or SCF. In the presence of SPE-7 IgE, wild-type BMMCs attracted Fc∈;RIα−/− cells vigorously, but the migration of the latter cells was abrogated toward SPE-7 IgE–incubated lyn−/− and syk−/− cells, although Fc∈;RIα−/− cells migrated vigorously to SCF irrespective of the genotype of mast cells present in the lower wells (Figure 5B). Similar results were obtained when CFSE-labeled Fc∈;RIα−/− cells in upper wells were cultured with lower wells containing 206 IgE–sensitized mutant cells in the presence of antigen or supernatants of 206 IgE–sensitized mutant cells that had been cultured in the presence of antigen (J.K. and T.K., unpublished data, January 2004). Therefore, these results indicate that Lyn and Syk are required for HC IgE– and IgE+Ag-induced release of soluble factors.
We next examined whether Lyn and Syk are required for signaling downstream of GPCRs as well. For this purpose, we tested whether wild-type, lyn−/−, and syk[unk]−/− BMMCs in upper wells are attracted to lower wells containing 206 IgE–sensitized wild-type cells in the presence of antigen or supernatants of 206 IgE–sensitized wild-type cells cultured in the presence of antigen. The mutant cells migrated as vigorously as wild-type cells under these conditions (Figure 5C and J.K. and T.K., unpublished data, December 2003). Furthermore, the mutant cells were also attracted to adenosine, LTB4, MCP-1, and MIP-1α as efficiently as wild-type cells (J.K. and T.K., unpublished data, January 2004), consistent with our previous study showing that Syk is not required for mast cell migratory responses to GPCR stimuli.50 Therefore, we conclude that Lyn and Syk are required for the initial phase of mast cell activation leading to the release of GPCR agonists, but not for the late phase of GPCR activation and further downstream events, in HC IgE– and IgE+Ag-induced migration.
In vivo mouse models for IgE- and IgE+Ag-induced mast cell accumulation
To evaluate the pathophysiologic relevance of our in vitro observations on IgE- and IgE/Ag-induced mast cell migration, we performed in vivo mouse experiments. First, we tested whether epicutaneous application of IgE on the back skin of naive mice can attract mast cells. Microscopic analysis of toluidine blue–stained samples showed that the local density of mast cells in SPE-7 IgE–applied areas was significantly increased after 24 hours compared with that in PBS-applied areas, whereas a modest increase was observed in 206 IgE–treated areas (Figure 6A-B). Most mast cells were localized in perivascular regions of the dermis. SPE-7 IgE–applied areas contained approximately 10% mast cells that had degranulated whereas such degranulating mast cells were very rare in PBS- or 206 IgE–applied areas. Another type of in vivo experiment was designed to test whether antigen can induce mast cell migration in vivo. Mice were epicutaneously treated with anti-DNP (206) IgE or PBS into the back skin, and 1 day later the same area was applied with DNP-HSA or PBS. Microscopic analysis showed that mast cell numbers in IgE/antigen-applied areas are much higher after 24 hours of treatment than those in PBS/PBS-, PBS/antigen-, or IgE/PBS-applied areas (Figure 6C). Similar to the effects of SPE-7 IgE on mast cell accumulation, antigen-treated areas had degranulating mast cells. Although these in vivo experiments do not reveal the mechanism of mast cell accumulation, the perivascular location of these cells suggests that mast cells or their precursors were recruited from the circulation rather than from nearby tissues. Alternatively, mast cells might have proliferated in situ, although this is less likely because of the short assay time and the low capacity of mast cells to proliferate in response to HC IgE and IgE+Ag.
Figure 6.
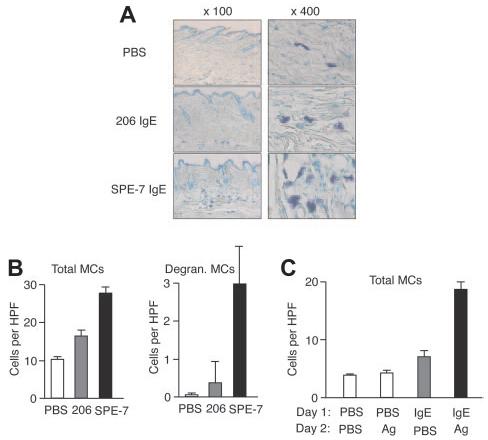
In vivo models for IgE- and IgE+Ag-induced mast cell accumulation. (A) Gauze strips spotted with 10 μg/mL 206 or SPE-7 IgE or PBS were applied to the shaved back skin of naive NC/Nga mice for 24 hours. The mice were killed and skin samples were prepared to stain mast cells. Representative photomicrographs are shown. (B) Total and degranulating mast cell numbers per high-power field were counted. (C) NC/Nga mice were first treated with anti-DNP (206) IgE or PBS on the back skin, and 1 day later the same area was applied with gauze strips spotted with DNP-HSA or PBS for 24 hours. Total mast cell numbers per high-power field were counted. Data (mean values ± SD) are shown representative of 3 similar experiments.
Discussion
This study provides evidence that HC IgEs in addition to IgE+Ag can promote the migration of mast cells. This migration is mediated mainly through integrin β1 and is more potent than some chemokines, and involves 2 phases: an early phase of Lyn- and Syk-dependent release of multiple soluble factors including adenosine, LTB4, and chemokines, and a later phase of PI3Kγ-dependent signaling following the activation of GPCRs by these factors. These in vitro observations are consistent with in vivo mouse experiments in which mast cell accumulation was induced by epicutaneous application of HC IgE and IgE+Ag.
The in vivo results suggest that HC IgE and IgE+Ag can induce accumulation of mast cells without prior inflammation. However, it is conceivable that, in allergic individuals, some inflammatory reactions such as the infiltration of helper T cells have occurred when IgE synthesis in B cells takes places at mucosal sites in the nasal cavity and lung in response to antigen exposure.16-21 Given the vast variety of proinflammatory mediators secreted from activated mast cells,51 IgE- and IgE+Ag-induced mast cell accumulation would amplify inflammatory reactions by recruiting other cells such as T cells, eosinophils, monocytes, and neutrophils. For instance, histamine plays an important role in the pathogenesis of atopic asthma by enhancing the secretion of Th2 cytokines and inhibiting the production of Th1 cytokines.52 LTB4 recruits T cells and myeloid cells,43-45 and mast cell–produced cytokines and chemokines can recruit T cells, eosinophils, monocytes, and neutrophils. CC chemokine transcripts coding for I-309/CCL1, MIP-1α, MIP-1β, and MCP-3/CCL7 are among the most dramatically enhanced ones in IgE+Ag-stimulated mast cells.53 The ability of HC IgEs to attract mast cells suggests that this amplification of inflammation can last as long as local IgE synthesis continues, even after the elimination of antigen. Overall, IgEs in the absence as well as presence of allergen are implicated in mast cell accumulation at allergic tissue sites with local high IgE levels.
This and previous studies10,15 have shown that intracellular signaling events induced by HC IgEs and IgE+Ag are very similar, if not identical: Src and Syk family PTKs are activated, intracellular Ca2+ concentrations are increased, and several serine/threonine kinases such as MAP kinases, PKCs, and Akt are also activated. These signaling events induced by Fc∈;RI aggregation by either HC IgEs or IgE+Ag (Table 1 and Kitaura et al15) result in a variety of biologic outcomes such as degranulation, histamine synthesis, leukotrienes release, receptor internalization, cytokine production, migration, and survival.9,10,15,54 Consistent with the similarities in signaling between HC IgE– and IgE+Ag-stimulated cells, migrations induced by these 2 modes of Fc∈;RI stimulation require identical signaling molecules such as Lyn, Syk, and PI3Kγ. Importantly, both migrations use soluble autocrine factors as a part of the migratory mechanism. Among numerous chemical and peptide agents rapidly secreted from activated mast cells, adenosine, LTB4, and several chemokines were identified as the mediators for mast cell migration. Unlike RBL-2H3 rat mast cells,55 sphingosine 1–phosphate did not substantially induce the migration of BMMCs (Figure 4C). As discussed in the preceding paragraph, these mediators influence an in vivo inflammatory process by recruiting not only mast cells but also other types of inflammatory cells. Assuming that PI3Kγ is involved only in signal transduction of GPCRs,49 this autocrine/paracrine mechanism seems essential for mast cell migration. Thus, only residual levels of migration were observed with PI3K γ−/− mast cells (Figure 5A). Notably, pertussis toxin inhibits HC IgE–, IgE+Ag-, and adenosine-induced migrations by approximately 60%. This indicates that these migrations are mediated mainly via Gαi. However, other Gα proteins or G protein–independent signals may also be involved; for example, integrins might supply additional signals for migration. In line with this possibility, Lyn is required for FN-mediated migration in RBL-2H3 cells.56 In any event, this study has extended the previous findings that soluble factors including adenosine are used to amplify Ca2+ and degranulation responses in IgE+Ag-stimulated mast cells.49,55 LTB4 or chemokines do not induce degranulation. It will be interesting to investigate whether other biologic responses of mast cell activation depend on an autocrine/paracrine mechanism and what molecules mediate such responses. Our knowledge on this mechanism will expand our choices of anti-inflammatory drugs beyond currently used antihistamine and antileukotriene drugs that target this process.
Acknowledgments
We thank Wasif Khan, Michael Leitges, Fu-Tong Liu, Robert O. Messing, and Dan R. Littman for donating mutant mice. We are grateful to Fu-Tong Liu for critical reading of the manuscript. This article is Publication 637 from the La Jolla Institute for Allergy and Immunology.
Footnotes
Supported by grants from the National Institutes of Health, nos. AI50209 and AI/GM38348 (T.K.).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Metzger H. The receptor with high affinity for IgE. Immunol Rev. 1992;125:37–48. doi: 10.1111/j.1600-065x.1992.tb00624.x. [DOI] [PubMed] [Google Scholar]
- 2.Kinet JP. The high-affinity IgE receptor (Fc∈;RI): from physiology to pathology. Annu Rev Immunol. 1999;17:931–972. doi: 10.1146/annurev.immunol.17.1.931. [DOI] [PubMed] [Google Scholar]
- 3.Turner H, Kinet JP. Signalling through the high-affinity IgE receptor Fc∈;RI. Nature. 1999;402:B24–30. doi: 10.1038/35037021. [DOI] [PubMed] [Google Scholar]
- 4.Rivera J. Molecular adapters in Fc∈;RI signaling and the allergic response. Curr Opin Immunol. 2002;14:688–693. doi: 10.1016/s0952-7915(02)00396-5. [DOI] [PubMed] [Google Scholar]
- 5.Dastych J, Costa JJ, Thompson HL, Metcalfe DD. Mast cell adhesion to fibronectin. Immunology. 1991;73:478–484. [PMC free article] [PubMed] [Google Scholar]
- 6.Kinashi T, Springer TA. Steel factor and c-kit regulate cell-matrix adhesion. Blood. 1994;83:1033–1038. [PubMed] [Google Scholar]
- 7.Ra C, Yasuda M, Yagita H, Okumura K. Fibronectin receptor integrins are involved in mast cell activation. J Allergy Clin Immunol. 1994;94:625–628. doi: 10.1016/0091-6749(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 8.Fehlner-Gardiner CC, Uniyal S, von Ballestrem CG, Chan BM. Differential utilization of VLA-4 (α4β1) and -5 (α5β1) integrins during the development of mouse bone marrow-derived mast cells. Differentiation. 1996;60:317–325. doi: 10.1046/j.1432-0436.1996.6050317.x. [DOI] [PubMed] [Google Scholar]
- 9.Asai K, Kitaura J, Kawakami Y, et al. Regulation of mast cell survival by IgE. Immunity. 2001;14:791–800. doi: 10.1016/s1074-7613(01)00157-1. [DOI] [PubMed] [Google Scholar]
- 10.Kalesnikoff J, Huber M, Lam V, et al. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. 2001;14:801–811. doi: 10.1016/s1074-7613(01)00159-5. [DOI] [PubMed] [Google Scholar]
- 11.Furuichi K, Rivera J, Isersky C. The receptor for immunoglobulin E on rat basophilic leukemia cells: effect of ligand binding on receptor expression. Proc Natl Acad Sci U S A. 1985;82:1522–1525. doi: 10.1073/pnas.82.5.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu C, MacGlashan D., Jr. IgE antibody up-regulates high affinity IgE binding on murine bone marrow-derived mast cells. Immunol Lett. 1996;52:129–134. doi: 10.1016/0165-2478(96)02599-0. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi M, Lantz CS, Oettgen HC, et al. IgE enhances mouse mast cell Fc∈;RI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE-dependent reactions. J Exp Med. 1997;185:663–672. doi: 10.1084/jem.185.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawakami T, Galli SJ. Regulation of mast-cell and basophil function and survival by IgE. Nat Rev Immunol. 2002;2:773–786. doi: 10.1038/nri914. [DOI] [PubMed] [Google Scholar]
- 15.Kitaura J, Song J, Tsai M, et al. Evidence that IgE molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the Fc∈;RI. Proc Natl Acad Sci U S A. 2003;100:12911–12916. doi: 10.1073/pnas.1735525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durham SR, Gould HJ, Thienes CP, et al. Expression of ∈; germ-line gene transcripts and mRNA for the ∈; heavy chain of IgE in nasal B cells and the effects of topical corticosteroid. Eur J Immunol. 1997;27:2899–2906. doi: 10.1002/eji.1830271123. [DOI] [PubMed] [Google Scholar]
- 17.Cameron LA, Durham SR, Jacobson MR, et al. Expression of IL-4, C∈; RNA, and I∈; RNA in the nasal mucosa of patients with seasonal rhinitis: effect of topical corticosteroids. J Allergy Clin Immunol. 1998;101:330–336. doi: 10.1016/s0091-6749(98)70244-1. [DOI] [PubMed] [Google Scholar]
- 18.Ying S, Humbert M, Meng Q, et al. Local expression of ∈; germline gene transcripts and RNA for the ∈; heavy chain of IgE in the bronchial mucosa in atopic and nonatopic asthma. J Allergy Clin Immunol. 2001;107:686–692. doi: 10.1067/mai.2001.114339. [DOI] [PubMed] [Google Scholar]
- 19.Snow RE, Djukanovic R, Stevenson FK. Analysis of immunoglobulin E VH transcripts in a bronchial biopsy of an asthmatic patient confirms bias towards VH5, and indicates local clonal expansion, somatic mutation and isotype switch events. Immunology. 1999;98:646–651. doi: 10.1046/j.1365-2567.1999.00910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smurthwaite L, Walker SN, Wilson DR, et al. Persistent IgE synthesis in the nasal mucosa of hay fever patients. Eur J Immunol. 2001;31:3422–3431. doi: 10.1002/1521-4141(200112)31:12<3422::aid-immu3422>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 21.Coker HA, Durham SR, Gould HJ. Local somatic hypermutation and class switch recombination in the nasal mucosa of allergic rhinitis patients. J Immunol. 2003;171:5602–5610. doi: 10.4049/jimmunol.171.10.5602. [DOI] [PubMed] [Google Scholar]
- 22.IUIS/WHO Subcommittee on Chemokine Nomenclature Chemokine/chemokine receptor nomenclature. Cytokine. 2003;21:48–49. [Google Scholar]
- 23.Meininger CJ, Yano H, Rottapel R, Bernstein A, Zsebo KM, Zetter BR. The c-kit receptor ligand functions as a mast cell chemoattractant. Blood. 1992;79:958–963. [PubMed] [Google Scholar]
- 24.Gruber BL, Marchese MJ, Kew RR. Transforming growth factor-β 1 mediates mast cell chemotaxis. J Immunol. 1994;152:5860–5867. [PubMed] [Google Scholar]
- 25.Gruber BL, Marchese MJ, Kew R. Angiogenic factors stimulate mast-cell migration. Blood. 1995;86:2488–2493. [PubMed] [Google Scholar]
- 26.Taub D, Dastych J, Inamura N, et al. Bone marrow-derived murine mast cells migrate, but do not degranulate, in response to chemokines. J Immunol. 1995;154:2393–2402. [PubMed] [Google Scholar]
- 27.McCloskey MA, Fan Y, Luther S. Chemotaxis of rat mast cells toward adenine nucleotides. J Immunol. 1999;163:970–977. [PubMed] [Google Scholar]
- 28.Lippert U, Artuc M, Grutzkau A, et al. Expression and functional activity of the IL-8 receptor type CXCR1 and CXCR2 on human mast cells. J Immunol. 1998;161:2600–2608. [PubMed] [Google Scholar]
- 29.Nilsson G, Mikovits JA, Metcalfe DD, Taub DD. Mast cell migratory response to interleukin-8 is mediated through interaction with chemokine receptor CXCR2/interleukin-8RB. Blood. 1999;93:2791–2797. [PubMed] [Google Scholar]
- 30.Juremalm M, Hjertson M, Olsson N, Harvima I, Nilsson K, Nilsson G. The chemokine receptor CXCR4 is expressed within the mast cell lineage and its ligand stromal cell-derived factor-1α acts as a mast cell chemotaxin. Eur J Immunol. 2000;30:3614–3622. doi: 10.1002/1521-4141(200012)30:12<3614::AID-IMMU3614>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 31.Oliveira SH, Lukacs NW. Stem cell factor and IgE-stimulated murine mast cells produce chemokines (CCL2, CCL17, CCL22) and express chemokine receptors. Inflamm Res. 2001;50:168–174. doi: 10.1007/s000110050741. [DOI] [PubMed] [Google Scholar]
- 32.Juremalm M, Olsson N, Nilsson G. Selective CCL5/RANTES-induced mast cell migration through interactions with chemokine receptors CCR1 and CCR4. Biochem Biophys Res Commun. 2002;297:480–485. doi: 10.1016/s0006-291x(02)02244-1. [DOI] [PubMed] [Google Scholar]
- 33.Ishizuka T, Okajima F, Ishiwara M, et al. Sensitized mast cells migrate toward the antigen: a response regulated by p38 mitogen-activated protein kinase and Rho-associated coiled-coil-forming protein kinase. J Immunol. 2001;167:2298–2304. doi: 10.4049/jimmunol.167.4.2298. [DOI] [PubMed] [Google Scholar]
- 34.Kawakami Y, Hartman SE, Kinoshita E, et al. Terreic acid, a quinone epoxide inhibitor of Bruton's tyrosine kinase. Proc Natl Acad Sci U S A. 1999;96:2227–2232. doi: 10.1073/pnas.96.5.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knauer KA, Adkinson NF., Jr. Clinical significance of IgE. In: Middleton E Jr, Reed CE, Ellis EF, editors. Allergy Principles and Practice. C.V. Mosby Company; St Louis, MO: 1983. pp. 673–688. [Google Scholar]
- 36.Matsuda H, Watanabe N, Geba GP, et al. Development of atopic dermatitis-like skin lesion with IgE hyperproduction in NC/Nga mice. Int Immunol. 1997;9:461–466. doi: 10.1093/intimm/9.3.461. [DOI] [PubMed] [Google Scholar]
- 37.Shimada Y, Hasegawa M, Kaburagi Y, et al. L-selectin or ICAM-1 deficiency reduces an immediate-type hypersensitivity response by preventing mast cell recruitment in repeated elicitation of contact hypersensitivity. J Immunol. 2003;170:4325–4334. doi: 10.4049/jimmunol.170.8.4325. [DOI] [PubMed] [Google Scholar]
- 38.Mayr SI, Zuberi RI, Zhang M, et al. IgE-dependent mast cell activation potentiates airway responses in murine asthma models. J Immunol. 2002;169:2061–2068. doi: 10.4049/jimmunol.169.4.2061. [DOI] [PubMed] [Google Scholar]
- 39.Papadopoulos EJ, Fitzhugh DJ, Tkaczyk C, et al. Mast cells migrate, but do not degranulate, in response to fractalkine, a membrane-bound chemokine expressed constitutively in diverse cells of the skin. Eur J Immunol. 2000;30:2355–2361. doi: 10.1002/1521-4141(2000)30:8<2355::AID-IMMU2355>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 40.Woo CH, Jeong DT, Yoon SB, et al. Eotaxin induces migration of RBL-2H3 mast cells via a RacERK-dependent pathway. Biochem Biophys Res Commun. 2002;298:392–397. doi: 10.1016/s0006-291x(02)02432-4. [DOI] [PubMed] [Google Scholar]
- 41.Heit B, Tavener S, Raharjo E, Kubes P. An intra-cellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galli SJ, Lantz CS. Allergy. In: Paul WE, editor. Fundamental Immunology. 4th Lippincott-Raven Press; Philadelphia, PA: 1999. pp. 1137–1184. [Google Scholar]
- 43.Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat Immunol. 2003;4:974–981. doi: 10.1038/ni971. [DOI] [PubMed] [Google Scholar]
- 44.Goodarzi K, Goodarzi M, Tager AM, Luster AD, von Andrian UH. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat Immunol. 2003;4:965–973. doi: 10.1038/ni972. [DOI] [PubMed] [Google Scholar]
- 45.Tager AM, Bromley SK, Medoff BD, et al. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. 2003;4:982–990. doi: 10.1038/ni970. [DOI] [PubMed] [Google Scholar]
- 46.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalez-Espinosa C, Odom S, Olivera A, et al. Preferential signaling and induction of allergy-promoting lymphokines upon weak stimulation of the high affinity IgE receptor on mast cells. J Exp Med. 2003;197:1453–1465. doi: 10.1084/jem.20021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu ZJ, Haleem-Smith H, Chen H, Metzger H. Unexpected signals in a system subject to kinetic proofreading. Proc Natl Acad Sci U S A. 2001;98:7289–7294. doi: 10.1073/pnas.121171998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laffargue M, Calvez R, Finan P, et al. Phosphoinositide 3-kinase γ is an essential amplifier of mast cell function. Immunity. 2002;16:441–451. doi: 10.1016/s1074-7613(02)00282-0. [DOI] [PubMed] [Google Scholar]
- 50.Mocsai A, Zhang H, Jakus Z, Kitaura J, Kawakami T, Lowell CA. G-protein-coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood. 2003;101:4155–4163. doi: 10.1182/blood-2002-07-2346. [DOI] [PubMed] [Google Scholar]
- 51.Galli SJ, Costa JJ. Mast-cell-leukocyte cytokine cascades in allergic inflammation. Allergy. 1995;50:851–862. doi: 10.1111/j.1398-9995.1995.tb02490.x. [DOI] [PubMed] [Google Scholar]
- 52.Packard KA, Khan MM. Effects of histamine on Th1/Th2 cytokine balance. Int Immunopharmacol. 2003;3:909–920. doi: 10.1016/S1567-5769(02)00235-7. [DOI] [PubMed] [Google Scholar]
- 53.Nakajima T, Inagaki N, Tanaka H, et al. Marked increase in CC chemokine gene expression in both human and mouse mast cell transcriptomes following Fc∈; receptor I cross-linking: an inter-species comparison. Blood. 2002;100:3861–3868. doi: 10.1182/blood-2002-02-0602. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka S, Takasu Y, Mikura S, Satoh N, Ichikawa A. Antigen-independent induction of histamine synthesis by immunoglobulin E in mouse bone marrow-derived mast cells. J Exp Med. 2002;196:229–235. doi: 10.1084/jem.20012037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jolly PS, Bektas M, Olivera A, et al. Transactivation of sphingosine-1-phosphate receptors by Fc∈;RI triggering is required for normal mast cell degranulation and chemotaxis. J Exp Med. 2004199:959–970. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki T, Shoji S, Yamamoto K, et al. Essential roles of Lyn in fibronectin-mediated filamentous actin assembly and cell motility in mast cells. J Immunol. 1998;161:3694–3701. [PubMed] [Google Scholar]