

Abstract
Genes involved in fatty acid catabolism have undergone extensive duplication in the genus Mycobacterium, which includes the etiologic agents of leprosy and tuberculosis. Here, we show that prokaryotic- and eukaryotic-like isoforms of the glyoxylate cycle enzyme isocitrate lyase (ICL) are jointly required for fatty acid catabolism and virulence in Mycobacterium tuberculosis. While deletion of icl1 or icl2 had little effect on bacterial growth in macrophages and mice, deletion of both genes resulted in complete impairment of intracellular replication and rapid elimination from the lungs. The feasibility of targeting ICL1 and ICL2 for chemical inhibition was demonstrated using a dual-specific ICL inhibitor, which blocked growth of M. tuberculosis on fatty acids and in macrophages. The absence of ICL orthologs in mammals should facilitate the development of glyoxylate cycle inhibitors as novel drugs for the treatment of tuberculosis.
INTRODUCTION
Microorganisms that invade, replicate, and persist within mammalian hosts must acquire and assimilate carbon substrates from host tissues. With few exceptions, little is known about the nutrient sources and metabolic pathways utilized by pathogens during infection1. Metabolic adaptations to tissue environments are thought to contribute to the difficulty in eradicating drug-susceptible microbes despite prolonged therapy with appropriate drugs2, a phenomenon known as phenotypic drug tolerance3. In vivo drug tolerance is a major reason why tuberculosis (TB) therapy requires administration of multiple drugs for six months or longer, resulting in high rates of patient non-adherence, treatment failure, and acquired drug resistance4. Elucidation of the metabolic pathways required for growth and persistence of Mycobacterium tuberculosis in the mammalian lung could suggest new avenues of intervention against this recalcitrant pathogen5.
Several lines of evidence suggest that pathogenic mycobacteria primarily utilize fatty acids, rather than carbohydrates, as carbon substrates during infection. Respiration of M. tuberculosis grown in mouse lungs is strongly stimulated by fatty acids but unresponsive to carbohydrates6. Several glycolytic enzymes are apparently dispensable for growth and persistence of M. tuberculosis in mice7, and the terminal step in glycolysis is blocked in the closely related zoonotic pathogen Mycobacterium bovis due to mutation of pykA encoding pyruvate kinase8. Restricted carbohydrate availability in vivo is further suggested by the recent demonstration that phosphoenolpyruvate carboxykinase (PCK) is essential for virulence in M. bovis9,10; in the absence of carbon flux through glycolysis, PCK is required for production of the essential biosynthetic precursor phosphoenolpyruvate (PEP) from TCA cycle intermediates (Fig. 1a). These observations are consistent with the hypothesis that pathogenic mycobacteria chiefly subsist on fatty acid substrates during infection.
Figure 1. Glyoxylate cycle and related metabolic pathways in M. tuberculosis.

(a) The beta-oxidation cycle (β-ox) degrades fatty acids to acetyl-CoA (C2) and propionyl-CoA (C3). The glyoxylate and methylcitrate cycles are required for anaplerosis and for propionyl-CoA metabolism, respectively, in fatty acid-catabolizing cells. Glyoxylate cycle enzymes are ICL1, ICL2, and MLS; ICL1/ICL2 could also function in the methylcitrate cycle with PRPC and PRPD. Enzymes common to the glyoxylate, methylcitrate, and TCA cycles are ACN, SDH, and FUM. Enzymes common to the glyoxylate and TCA cycles are MDH, MQO, GLTA2, and CITA. Pyruvate is produced from malate by MEZ or from oxaloacetate by sequential action of PCK and PYK; coupled decarboxylation of pyruvate by the pyruvate dehydrogenase complex (PDHC) yields acetyl-CoA. Anaplerosis in carbohydrate-catabolizing cells is by carboxylation of pyruvate or phosphoenolpyruvate (PEP) to oxaloacetate by PCA or PPC, respectively. PPC is absent in M. tuberculosis (dashed line). Gene designations are listed in Supplementary Table 1 online. (b) ICL domain organization. Domains I and III are present in all ICLs. Domain I contains the conserved catalytic motif KKCGH. Domain II is present in fungal and plant ICLs and in mycobacterial ICL2, but absent in mycobacterial ICL1. Domain IV is unique to mycobacterial ICL2. (c) Superimposition of in silico-modeled ICL2 monomer (red) and the X-ray crystal structure of ICL1 monomer (blue). Encircled: ICL catalytic signature motif KKCGH (AA 193-203 in ICL1; AA 213-217 in ICL2). Domain II of ICL2 (AA 269-365) was modeled after the X-ray crystal structure of A. nidulans ICL25. Domain IV (not depicted) had no homology to known sequences.
Two pathways are specifically required for utilization of fatty acids: the catabolic beta-oxidation cycle that degrades fatty acids to acetyl-CoA (C2) units, and, if glycolytic substrates are absent or sparse, the anaplerotic glyoxylate cycle. Microorganisms rely on the glyoxylate cycle to replenish TCA cycle intermediates during growth on fatty acid substrates, when concentrations of PEP and pyruvate are insufficient to support anaplerosis via conversion to oxaloacetate by PEP carboxylase (PPC) or pyruvate carboxylase (PCA), respectively (Fig. 1a). The glyoxylate cycle facilitates the biosynthesis of essential cell constituents from C2 units by generating C4 intermediates that serve as biosynthetic precursors11. In conjunction with PCK or malic enzyme12, the glyoxylate cycle can also generate the essential C3 biosynthetic precursors PEP and pyruvate, respectively (Fig. 1a). The extensive duplication of genes encoding beta-oxidation enzymes in M. tuberculosis13,14 poses an obstacle to genetic analysis of this pathway’s role in virulence due to potential functional redundancy of paralogous genes. In contrast, the glyoxylate cycle in M. tuberculosis apparently comprises a single gene encoding malate synthase and two genes encoding isocitrate lyases (Fig. 1a)13,14. The smaller isocitrate lyase gene (icl1) encodes an enzyme closely related to isocitrate lyases in other eubacteria; the larger gene (icl2) encodes a protein more homologous to eukaryotic isocitrate lyases (Fig. 1b). Both recombinant enzymes display isocitrate lyase activity in vitro although ICL1 is more active than ICL215. Expression of both icl116 and icl2 (unpublished) is up-regulated in M. tuberculosis isolated from mouse lungs as compared to bacteria grown in vitro.
Previously we reported that disruption of icl1 impaired the chronic-phase persistence of M. tuberculosis in mice while having little effect on replication during the acute phase of infection17; the role of icl2 was not addressed. Here, we adduce genetic and chemical evidence that icl1 and icl2 are jointly required for growth, survival, and virulence of M. tuberculosis in macrophages and mice. Recent studies have implicated the glyoxylate cycle in the virulence of other bacterial and fungal pathogens18–22; thus, chemical inhibitors of this pathway could have broad therapeutic utility. The development of novel drugs targeting the glyoxylate cycle should be facilitated by the apparent absence of this pathway in mammalian cells23.
RESULTS
ICL isoenzymes in Mycobacterium tuberculosis
The predicted gene products of icl1 (428 AA) and icl2 (766 AA) are 27% identical overall. Bioinformatics analysis revealed three distinct groups of ICL proteins (unpublished), which differed in length and domain organization (Fig. 1b). Group I ICLs comprised small eubacterial ICLs, including M. tuberculosis ICL1. Group II ICLs included medium-length plant and fungal ICLs, which contained a central domain (Domain II) absent in group I. Group III ICLs, so far identified only in Mycobacterium, had a long C-terminal domain absent in groups I and II; they also contained a central region with significant homology to the central domain of the eukaryotic ICLs and were overall ~ 35% identical to them (Fig. 1b). In silico modeling of the three-dimensional structure of ICL2, based on the X-ray crystal structures of the group I ICL1 from M. tuberculosis24 and the group II ICL from Aspergillus nidulans25, revealed a striking level of structural concordance (Fig. 1c). Significantly, the active site loops of ICL1 and ICL2, containing the conserved catalytic motif (KKCGH), were nearly super-imposable (Fig. 1c, encircled). Thus, despite the limited homology between ICL1 and ICL2, conservation of tertiary structure and enzymatic activity suggested that these enzymes might provide overlapping biological functions.
ICL is required for growth on fatty acid substrates
To assess the role of the glyoxylate cycle in fatty acid metabolism, we constructed strains of M. tuberculosis in which icl1, icl2, or both genes were deleted. Deletion of icl1 or icl2 alone had little effect on bacterial replication kinetics in media containing glycerol (Fig. 2a), glucose (Fig. 2b), short-chain fatty acids (Fig. 2c–e), or long-chain fatty acids (Fig. 2f–h) as carbon sources. In contrast, deletion of both icl1 and icl2 eliminated growth on fatty acids but had little effect on utilization of carbohydrates. A slight but reproducible lag was observed for growth of the Δicl1 Δicl2 strain in glucose-containing media (Fig. 2b), indicating some carbon flux through the glyoxylate cycle under these conditions, as reported in Corynebacterium glutamicum26. Complementation of the Δicl1 Δicl2 mutant with plasmids carrying icl1 or icl2 restored growth on fatty acids (Fig. 2c–h), confirming that these genes encode functionally redundant enzymes that are jointly required for fatty acid metabolism.
Figure 2. Overlapping roles of ICL1/ICL2 in fatty acid catabolism.

(a–f) Growth of bacteria in media containing (a) glycerol, (b) glucose, (c) acetate (C2), (d) propionate (C3), (e) butyrate (C4), or (f) polyoxyethylene sorbitan monolaurate (C12). (g) Growth on [14C]-palmitate (C16). (h) Growth on DPPC (C16), an abundant phospholipid in the mammalian lung. (a–h) Bacterial strains: wild-type (filled squares), Δicl1 (open circles), Δicl2 (open triangles), Δicl1 Δicl2 (open diamonds), Δicl1 Δicl2 complemented with pICL1 (filled circles) or pICL2 (filled triangles). Results are representative of at least two experiments.
ICL is required for virulence in mice
We assessed the role of the glyoxylate cycle in replication and persistence of M. tuberculosis in vivo by infecting wild-type C57BL/6 mice with strains lacking icl1, icl2, or both genes (Fig. 3). Deletion of icl1 resulted in modest reduction of the bacterial load in the lungs during the chronic phase but not the acute phase of infection (Fig. 3a) and reduced tissue pathology (Fig. 3g), as reported17, whereas deletion of icl2 had no discernable impact on the kinetics of M. tuberculosis growth and persistence (Fig. 3b) or tissue pathology (Fig. 3g). In contrast, bacteria lacking both icl1 and icl2 were incapable of growth in mice and were rapidly eliminated from the lungs and spleen (Fig. 3c), which remained culture-negative (detection limit: 1 CFU per organ) until the experiment was terminated (2 years post-infection). Mice infected with the Δicl1 Δicl2 mutant did not show any appreciable splenomegaly (Fig. 3e) or lung pathology (Fig. 3g), and remained healthy until the experiment was terminated, whereas mice infected with wild-type bacteria succumbed between 8 and 9 months post-infection. Complementation of the Δicl1 Δicl2 mutant with a plasmid containing icl1 restored growth (Fig. 3d) and tissue pathology in the lungs (Fig. 3g) and spleen (not shown); chronic-phase persistence was largely restored as well (Fig. 3d).
Figure 3. Virulence of ICL-deficient M. tuberculosis in mice.

(a–d) Bacterial loads in lungs of C57BL/6 mice. (e) Spleens of C57BL/6 mice 2 wk post-infection. Left to right: splenic bacterial loads (log10 CFU ± SD) were 6.11 ± 0.15 (wild-type), 5.92 ± 0.12 (Δicl1), 6.24 ± 0.08 (Δicl2), 3.58 ± 0.22 (Δicl1 Δicl2). (f) Bacterial loads in lungs of IFN-γ−/− mice. (g) Lung pathology in C57BL/6 mice 16 wk post-infection. Magnification: 200X. (a–g) Bacterial strains: wild-type (filled squares), Δicl1 (open circles), Δicl2 (open triangles), Δicl1 Δicl2 (open diamonds), Δicl1 Δicl2 transformed with pICL1 (filled circles) or empty vector (cross-marks). Results are representative of two experiments.
TB case fatality rates are increased in HIV-coinfected patients despite appropriate therapy, suggesting that antimicrobials might be less efficacious in immuno-deficient individuals27. We asked what effect ICL deficiency would have on growth and survival of M. tuberculosis in immuno-deficient hosts by infecting highly susceptible mouse strains lacking interferon-γ (IFN-γ) or tumor necrosis factor-α receptor 1 (TNF-R1−/−)28. We found that Δicl1 Δicl2 bacteria were incapable of replicating in IFN-γ−/− mice (Fig. 3f) or TNF-R1−/− mice (not shown), suggesting that ICL inhibitors might have therapeutic value even in patients with compromised immunity. Previously we reported that the attenuation of Δicl1 bacteria in C57BL/6 mice was reversed in IFN-γ−/− mice, suggesting that the in vivo requirement for ICL might be triggered by IFN-γ-mediated macrophage activation17. Our new findings are only partially consistent with this idea because growth of the Δicl1 Δicl2 bacteria was not restored in IFN-γ−/− mice, although clearance was somewhat delayed in IFN-γ−/− mice as compared to wild-type mice (Fig. 3f).
ICL is required for growth and survival in macrophages
The macrophage plays a central and dual role in TB, serving as both a protective immune cell and as a niche for intravacuolar replication and persistence of M. tuberculosis29. Previously we reported that deletion of icl1 impaired survival of M. tuberculosis in IFN-γ-activated murine macrophages but had little effect on survival in non-activated macrophages17. In contrast, survival of Δicl1 Δicl2 bacteria was impaired in both non-activated (Fig. 4a) and IFN-γ-activated (Fig. 4b) murine macrophages, although killing was more rapid in the latter. Complementation of the Δicl1 Δicl2 strain with plasmids containing icl1 (Fig. 4c,d) or icl2 (Fig. 4e,f) restored replication in non-activated macrophages (Fig. 4c,e) and survival in IFN-γ-activated macrophages (Fig. 4d,f). We obtained similar results in assays with human blood monocyte-derived macrophages (Fig. 4g,h), suggesting that M. tuberculosis might also utilize the glyoxylate cycle during human infection.
Figure 4. Survival of ICL-deficient M. tuberculosis in macrophages.

(a–f) Bacterial loads in non-activated (a,c,e) and IFN-γ-activated (b,d,f) mouse bone marrow-derived macrophages. (g,h) Bacterial loads in human blood monocyte-derived macrophages. (a–h) Bacterial strains: wild-type (filled squares), Δicl1 (open circles), Δicl2 (open triangles), Δicl1 Δicl2 (open diamonds), Δicl1 Δicl2 complemented with pICL1 (filled circles) or pICL2 (filled triangles). Results are representative of at least two experiments.
Chemical inhibition of ICL mimics ICL-deficiency
Our demonstration that ICL activity is essential for replication and survival of M. tuberculosis in vivo suggested that chemical inhibition of ICL1 and ICL2 might be an effective therapeutic strategy. We therefore tested the ability of the prototype ICL inhibitor 3-nitropropionate (3-NP)15,23,24 to block growth of M. tuberculosis in media containing specific carbon substrates (Fig. 5) and in macrophages (Fig. 6). Growth of M. tuberculosis on glycerol (Fig. 5a) was not affected by the addition of 0.1 mM 3-NP, and growth on glucose (Fig. 5b) was only slightly affected, comparable to the slightly reduced growth kinetics of Δicl1 Δicl2 bacteria. In contrast, growth of M. tuberculosis on fatty acids (Fig. 5c–d) was eliminated by 3-NP addition, comparable to the phenotype of Δicl1 Δicl2 bacteria. Consistent with our interpretation that 3-NP-mediated growth inhibition was due to inhibition of ICL per se, bacteria that over-expressed ICL1 (6-fold) from a plasmid (Fig. 5f) were less sensitive to 3-NP as compared to wild-type bacteria (Fig. 5e).
Figure 5. Chemical inhibition of ICL blocks M. tuberculosis growth on fatty acids.
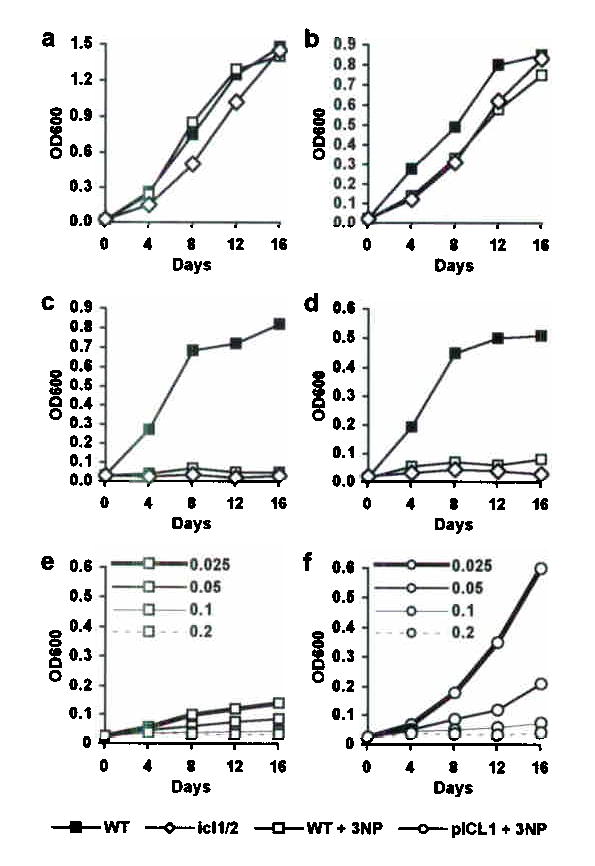
(a–d) Growth of wild-type (squares) and Δicl1 Δicl2 (open diamonds) bacteria in media containing (a) glycerol, (b) glucose, (c) propionate (C3), or (d) polyoxyethylene sorbitan monolaurate (C12), without (filled squares, open diamonds) or with (open squares) addition of 3-NP (0.1 mM). (e,f) Growth of (e) wild-type (open squares) and (f) pICL1-complemented Δicl1 Δicl2 (open circles) bacteria in media containing propionate and 3-NP (0.025, 0.05, 0.1, or 0.2 mM). Results are representative of at least two experiments.
Figure 6. Chemical inhibition of ICL blocks M. tuberculosis growth in macrophages.

(a–e) Bacterial loads in M. tuberculosis-infected (a–d) murine bone marrow-derived macrophages and (e) human blood monocyte-derived macrophages without (filled symbols) or with (open symbols) addition of 3-NP (0.2, 1, 5, or 10 mM). Bacterial strains: wild-type (squares), Δicl1 Δicl2 (diamonds), Δicl1 Δicl2 complemented with pICL1 (circles). Results are representative of two experiments.
Addition of 3-NP to M. tuberculosis-infected murine macrophages (Fig. 6a–d) completely eliminated intracellular growth (Fig. 6b), comparable to the growth inhibition resulting from deletion of icl1 and icl2 (Fig. 6a). 3-NP-mediated inhibition of bacterial growth in murine macrophages was dose-dependent (Fig. 6d), and bacteria that over-expressed ICL1 (6-fold) from a plasmid were more resistant to growth inhibition by 3-NP as compared to wild-type bacteria (Fig. 6c). We did not perform studies in M. tuberculosis-infected mice due to the known neurotoxicity of 3-NP in mice30. Although slightly less effective in human as compared to murine macrophages, 3-NP also inhibited growth of M. tuberculosis in human macrophages in a dose-dependent manner (Fig. 6e). These results provide a proof of concept for the development of ICL inhibitors as anti-tuberculosis drugs.
DISCUSSION
Carbohydrates are thought to comprise the principal carbon source for pathogenic bacteria in such diverse niches as the intestinal lumen31, the host cell cytosol32, and phagocyte vacuoles33. In contrast, pathogenic mycobacteria isolated from the lungs of mice preferentially respire fatty acids rather than carbohydrates6, and mycobacterial fatty acid metabolic genes are up-regulated in macrophages34 and mice16. A central role for fatty acid metabolism is further suggested by the extensive duplication of genes involved in lipid degradation in M. tuberculosis13,14. Homologous gene pairs include icl1 and icl2 encoding isocitrate lyases of the glyoxylate cycle (Fig. 1a). Previously, we reported that icl1 is dispensable for acute-phase growth but essential for chronic-phase persistence in M. tuberculosis-infected mice, and we proposed that M. tuberculosis might switch to fatty acid catabolism at the transition from the acute to the chronic phase of infection17. Here, we adduce evidence that icl1 and icl2 are jointly required for survival of M. tuberculosis in macrophages and mice at early as well as late stages of infection, indicating that fatty acid catabolism has a more fundamental role during infection than we had previously thought. In future, the roles of ICL1 and ICL2 in chronic-phase persistence could be addressed using new technologies for conditional gene expression in mycobacteria35,36. Our studies raise the possibility that drugs capable of inhibiting both ICL isoforms might have potent anti-mycobacterial activity. However, there are significant differences in TB pathology in mice and humans, and it is possible that the in vivo metabolism of M. tuberculosis might be influenced by these factors. Resolution of these issues will require studies on the role of the glyoxylate cycle in human disease.
Primacy of fatty acid metabolism and essentiality of the glyoxylate cycle do not rule out the possibility that M. tuberculosis might also have access to carbohydrates in vivo. During growth on all types of carbon substrates, bacteria must replenish intermediates of central metabolism that are siphoned off to biosynthetic pathways. Nutrient availability and growth rate determine which anaplerotic pathways are employed. Escherichia coli uses PPC to synthesize oxaloacetate during rapid growth with excess glucose, but switches to the glyoxylate cycle during slow growth with limiting glucose37. Similarly, Corynebacterium glutamicum uses PPC or PCA to replenish oxaloacetate during growth on glucose38 (Fig. 1a), but switches to the glyoxylate cycle when growing on mixtures of glucose and fatty acids26.
Adaptation of M. tuberculosis to low concentrations of glycolytic substrates in vivo could involve reduced flux of carbon through glycolysis in order to conserve glycolytic intermediates for essential biosynthetic processes. This idea is consistent with preliminary evidence that glucokinase and a putative carbohydrate transporter are essential for growth of M. tuberculosis in mice, whereas the glycolytic steps catalyzed by triosephosphate isomerase, glyceraldehyde 3-phosphate dehydrogenase, and phosphoglycerate kinase are apparently dispensable7. If correct, these observations suggest that during infection glycolysis is not the source of PEP, an essential biosynthetic precursor of aromatic amino acids and peptidoglycan. Instead, PEP could be generated by the sequential action of the glyoxylate cycle, which converts acetyl-CoA to oxaloacetate, and PCK, which decarboxylates oxaloacetate to PEP (Fig. 1a). Consistent with this idea, pckA (encoding PCK) is up-regulated in the lungs of M. tuberculosis-infected mice16 and is essential for virulence in M. bovis9,10.
During infection, M. tuberculosis could obtain fatty acids from the host cell by hydrolysis of cell membrane lipids39; consistent with this possibility, M. tuberculosis genes encoding exported phospholipases are jointly required for growth in the lungs of mice40. M. tuberculosis might also catabolize internal lipid reserves such as triacylglycerol, particularly during persistence41. Beta-oxidation of odd-chain fatty acids generates both acetyl-CoA (C2) and propionyl-CoA (C3); the latter can be metabolized via the methylcitrate cycle (Fig. 1a), which is required for propionate catabolism in fungi and eubacteria42,43. The M. tuberculosis genome encodes homologs of methylcitrate synthase (prpC) and methylcitrate dehydratase (prpD), which are up-regulated during infection of macrophages34. Methylisocitrate lyase (prpB) is apparently missing in M. tuberculosis; however, ICLs show limited homology to methylisocitrate lyases and possess weak methylisocitrate lysase activity44. These observations, and our finding that ICL1 and ICL2 are jointly required for growth of M. tuberculosis on propionate (Fig. 2d, Fig. 5c), raise the possibility that these enzymes might substitute for PrpB in the methylcitrate cycle.
Our studies demonstrate that the glyoxylate cycle is essential for M. tuberculosis growth and persistence in macrophages and mice. We have presented structural and biological evidence indicating the feasibility of developing dual-specificity ICL1/ICL2 inhibitors as potential therapeutic agents for TB. Mutations in icl1 and icl2 could also be incorporated in the design of live attenuated vaccines that might be safe for use in immuno-suppressed individuals, such as individuals with HIV/AIDS. As a novel target for drug development, the glyoxylate cycle is attractive because it is apparently absent in mammalian cells23 and is essential for virulence in other major pathogens, such as Candida albicans19. There is an urgent need for new and better drugs to treat persistent infections like candidiasis and TB, which require lengthy treatment regimens with potentially toxic drugs, resulting in patient non-adherence, treatment failure, and drug resistance.
METHODS
Bioinformatics
Conceptually translated nucleotide sequences of icl1 (MT0483) and icl2 (MT1966) were aligned with BLAST2 or PSI-BLAST (NCBI). Domains were identified with Pfam and aligned with CDD v1.62 (NCBI). ICL2 was modeled by threading on M. tuberculosis ICL124 and A. nidulans ICL25 using MODELLER.
Bacteriologic media
M. tuberculosis (Erdman) was stored at −80°C in 15% glycerol. Bacteria were grown at 37°C in Middlebrook 7H9 broth (DifCo) + 0.5% albumin, 0.085% NaCl, 0.05% Tween-80, 0.5% glycerol, and 0.2% glucose, or on Middlebrook 7H10 agar (DifCo) + 10% OADC (DifCo) and 0.5% glycerol. For carbon utilization experiments bacteria were grown in 7H9 + 0.5% albumin, 0.085% NaCl, 0.05% Tween-80, and carbon substrate at 0.1% (w/v). Hygromycin (50 μg ml−1), kanamycin (25 μg ml−1), streptomycin (20 μg ml−1), and 3-nitropropionate were from Sigma. Oxidation of [14C]-palmitate to [14C]-CO2 was measured using BACTEC (Becton Dickenson).
Strain construction
A genomic KpnI fragment containing icl2 was sub-cloned into pBSKS (Stratagene) and an internal 1.8 kbp MscI fragment was replaced with an aph kanamycin resistance (Kmr) cassette (pUC4 1.3 kbp BamHI fragment) to generate pEM028. The KpnI fragment containing Δicl2::aph was cloned into the SalI site of pEM025, a shuttle vector containing the pAL5000 ori45, aadA streptomycin-resistance (Smr) marker (pHP45 1.9 kb DraI fragment46), and sacB counter-selection marker to generate pEM029-1. The Δicl2::aph allele was recombined into the M. tuberculosis chromosome by selection of Kmr transformants and counter-selection on 5% sucrose47. The Δicl1 Δicl2 mutant was generated by disrupting icl2 in the chromosome of Δicl1 bacteria17. Replacement of wild-type icl2 with Δicl2::aph was confirmed by PCR and Southern blot. pEM263-4 (pICL1) was constructed by sub-cloning a 1.6 kb SpeI fragment containing icl1 into the HpaI site of pEM262, an episomal vector derived from pMV26148 by replacing the Kmr cassette with the Smr cassette. pEM3E4-8 (pICL2) was constructed by inserting a 6.1 kbp KpnI fragment containing icl2 into the HpaI site of pEM262.
Mouse macrophage infections
Bone marrow was flushed from mouse femurs and incubated (37°C, 5% CO2) 7 days in DMEM (Cellgro) + 0.58 g l−1 L-glutamine, 1 mM sodium pyruvate, 10% C heat-inactivated fetal bovine serum (HIFBS; Hyclone), 10 mM Hepes pH 7.55, 100 U ml−1 penicillin G, 100 μg ml−1 streptomycin, and 20% L929 cell-conditioned medium. Adherent cells were released (1 mM EDTA-PBS), seeded in 12-well plates (2 × 105 cells/well), and incubated 24 h before infection. Where indicated, 50 U ml−1 rIFN-γ (R&D) was added 16 h before infection and thereafter. Bacteria were grown to OD600 0.5 in 7H9, washed in PBS + 0.05% Tween-80 (PBST), and centrifuged 5 min at 130 g to remove aggregates. Macrophage monolayers were infected (MOI 2:1) in DMEM + 0.58 g l−1 L-glutamine, 1 mM sodium pyruvate, 5% HIFBS, and 5% heat-inactivated horse serum (HIHS; Gibco). After 4 h monolayers were washed to remove non-adherent bacteria and incubated with medium changes every 36 h (HIHS omitted). Where indicated, 3-NP was added 4 h post-infection onwards. 3-NP had no effect on macrophage adherence or viability. Infected monolayers were washed and lysed with 0.5% Triton X-100. Lysates were plated on 7H10 and bacterial CFU were enumerated after 3–4 wk at 37°C.
Human macrophage infections
Mononuclear cells were isolated from whole human blood (New York Blood Center) on Ficoll-plaque gradients (Pharmacia Biotech). Cells were seeded in 12-well plates (2.4 × 106 cells/well) and incubated (37°C, 5% CO2) in HIPHS-RPMI = RPMI-1640 (Life Technologies) + 10% heat-inactivated pooled human serum (Labquib, Ltd.), 10 mM Hepes pH 7.55, and 20 μg/ml gentamicin. After 2 h monolayers were washed to remove non-adherent cells and incubated for 7 days. Macrophage monolayers (2 x 105 cells/well) were washed with RPMI and infected with M. tuberculosis per the procedures used to infect mouse macrophages except that cells were cultured in HIPHS-RPMI (gentamicin omitted).
Mouse infections
Male and female C57BL/6, IFN-γ−/−, and TNF-R1−/− mice age 5–8 wk were from Jackson Laboratory. Bacteria were grown to OD600 0.5 in 7H9, collected by centrifugation, washed with PBST, adjusted to 107 CFU ml−1, and cup-sonicated to disrupt aggregates. Mice were infected by injection of 0.1 ml (106 CFU) of the bacterial suspension into a lateral tail vein. Infected mice were euthanized by CO2 overdose. Lung and spleen homogenates were plated on 7H10 and bacterial CFU were enumerated after 3–4 wk at 37°C. Tissues were fixed in phosphate-buffered formalin (10%) and H&E stained for histopathology. Procedures involving vertebrate animals were reviewed and approved by Rockefeller University’s Animal Care and Use Committee.
Statistics
Bacterial CFU plots were compared using Student’s two-tailed t test. Kaplan-Meier plots of mouse survival were compared using Mantel-Haenszel’s test (GraphPad Prism 4.0 software). P values < 0.05 were considered significant.
URLs: TIGR: http://www.tigr.org. NCBI: http://www.ncbi.nlm.nih.gov/. Pfam: http://www.sanger.ac.uk/Software/Pfam/. MODELLER: http://salilab.org/modeller/modeller.html.
Supplementary Material
Acknowledgments
We thank N. Mirkovic and A. Sâli for construction of the ICL2 in silico model, B. Hanna for use of the BACTEC apparatus, J. Timm for assistance with real-time RT-PCR assays, M. Glickman for providing the cosmid containing icl2, S. Ehrt for providing the L-cell line, and W.T. Chan and P. Giannakas for technical assistance. E. Muñoz-Elías was supported by a Robert D. Watkins Graduate Fellowship from the American Society for Microbiology. J. McKinney acknowledges support from the Sequella Global Tuberculosis Foundation, the Ellison Medical Foundation, the Sinsheimer Fund, and the Irma T. Hirschl Trust. This work was funded by grants (J. McKinney) from GlaxoSmithKline and the National Institutes of Health (AI46392).
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Smith H. Questions about the behaviour of bacterial pathogens in vivo. Philos Trans R Soc Lond B Biol Sci. 2000;355:551–564. doi: 10.1098/rstb.2000.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDermott W. Microbial persistence. Yale J Biol Med. 1958;30:257–291. [PMC free article] [PubMed] [Google Scholar]
- 3.Charpentier E, Tuomanen E. Mechanisms of antibiotic resistance and tolerance in Streptococcus pneumoniae. Microbes Infect. 2000;2:1855–1864. doi: 10.1016/s1286-4579(00)01345-9. [DOI] [PubMed] [Google Scholar]
- 4.Gomez JE, McKinney JD. Mycobacterium tuberculosis persistence, latency, and drug tolerance. Tuberculosis. 2004;84:29–44. doi: 10.1016/j.tube.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Boshoff HI, Barry CE., 3rd Tuberculosis - metabolism and respiration in the absence of growth. Nat Rev Microbiol. 2005;3:70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- 6.Segal W, Bloch H. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J Bacteriol. 1956;72:132–141. doi: 10.1128/jb.72.2.132-141.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keating LA, et al. The pyruvate requirement of some members of the Mycobacterium tuberculosis complex is due to an inactive pyruvate kinase: implications for in vivo growth. Mol Microbiol. 2005;56:163–174. doi: 10.1111/j.1365-2958.2005.04524.x. [DOI] [PubMed] [Google Scholar]
- 9.Collins DM, et al. Production of avirulent mutants of Mycobacterium bovis with vaccine properties by the use of illegitimate recombination and screening of stationary-phase cultures. Microbiology. 2002;148:3019–3027. doi: 10.1099/00221287-148-10-3019. [DOI] [PubMed] [Google Scholar]
- 10.Liu K, et al. pckA-deficient Mycobacterium bovis BCG shows attenuated virulence in mice and in macrophages. Microbiology. 2003;149:1829–1835. doi: 10.1099/mic.0.26234-0. [DOI] [PubMed] [Google Scholar]
- 11.Kornberg HL, Krebs HA. Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature. 1957;179:988–991. doi: 10.1038/179988a0. [DOI] [PubMed] [Google Scholar]
- 12.Pertierra AG, Cooper RA. Pyruvate formation during the catabolism of simple hexose sugars by Escherichia coli: studies with pyruvate kinase-negative mutants. J Bacteriol. 1977;129:1208–1214. doi: 10.1128/jb.129.3.1208-1214.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole ST, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 14.Fleischmann RD, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479–5490. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Höner zu Bentrup K, et al. Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J Bacteriol. 1999;181:7161–7167. doi: 10.1128/jb.181.23.7161-7167.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timm J, et al. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc Natl Acad Sci USA. 2003;100:14321–14326. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKinney JD, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 18.Idnurm A, Howlett BJ. Isocitrate lyase is essential for pathogenicity of the fungus Leptosphaeria maculans to canola (Brassica napus) Eukaryot Cell. 2002;1:719–724. doi: 10.1128/EC.1.5.719-724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorenz MC, Fink GR. The glyoxylate cycle is required for fungal virulence. Nature. 2001;412:83–86. doi: 10.1038/35083594. [DOI] [PubMed] [Google Scholar]
- 20.Solomon PS, et al. Pathogenicity of Stagonospora nodorum requires malate synthase. Mol Microbiol. 2004;53:1065–1073. doi: 10.1111/j.1365-2958.2004.04178.x. [DOI] [PubMed] [Google Scholar]
- 21.Vereecke D, et al. Chromosomal locus that affects pathogenicity of Rhodococcus fascians. J Bacteriol. 2002;184:1112–1120. doi: 10.1128/jb.184.4.1112-1120.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang ZY, et al. The glyoxylate cycle is required for temporal regulation of virulence by the plant pathogenic fungus Magnaporthe grisea. Mol Microbiol. 2003;47:1601–1612. doi: 10.1046/j.1365-2958.2003.03412.x. [DOI] [PubMed] [Google Scholar]
- 23.Vanni P, et al. Comparative structure, function and regulation of isocitrate lyase, an important assimilatory enzyme. Comp Biochem Physiol. 1990;95B:431–458. doi: 10.1016/0305-0491(90)90002-b. [DOI] [PubMed] [Google Scholar]
- 24.Sharma V, et al. Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat Struct Biol. 2000;7:663–668. doi: 10.1038/77964. [DOI] [PubMed] [Google Scholar]
- 25.Britton K, et al. The crystal structure and active site location of isocitrate lyase from the fungus Aspergillus nidulans. Structure. 2000;8:349–362. doi: 10.1016/s0969-2126(00)00117-9. [DOI] [PubMed] [Google Scholar]
- 26.Wendisch VF, et al. Quantitative determination of metabolic fluxes during coutilization of two carbon sources: comparative analyses with Corynebacterium glutamicum during growth on acetate and/or glucose. J Bacteriol. 2000;182:3088–3096. doi: 10.1128/jb.182.11.3088-3096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Sadr WM, et al. A review of efficacy studies of 6-month short-course therapy for tuberculosis among patients infected with human immunodeficiency virus: differences in study outcomes. Clin Infect Dis. 2001;32:623–632. doi: 10.1086/318706. [DOI] [PubMed] [Google Scholar]
- 28.Flynn JL, Chan J. Immunology of tuberculosis . Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 29.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 30.Nishino H, et al. Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood-brain barrier: involvement of dopamine toxicity. Neurosci Res. 1997;27:343–355. doi: 10.1016/s0168-0102(97)01170-x. [DOI] [PubMed] [Google Scholar]
- 31.Chang DE, et al. Carbon nutrition of Escherichia coli in the mouse intestine. Proc Natl Acad Sci USA. 2004;101:7427–7432. doi: 10.1073/pnas.0307888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chico-Calero I, et al. Hpt, a bacterial homolog of the microsomal glucose- 6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc Natl Acad Sci USA. 2002;99:431–436. doi: 10.1073/pnas.012363899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eriksson S, et al. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol Microbiol. 2003;47:103–118. doi: 10.1046/j.1365-2958.2003.03313.x. [DOI] [PubMed] [Google Scholar]
- 34.Schnappinger D, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehrt, S. et al. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. [Feb. 01 Epub ahead of print] (2005). [DOI] [PMC free article] [PubMed]
- 36.Blokpoel, M. C. et al. Tetracycline-inducible gene regulation in mycobacteria. Nucleic Acids Res. [Feb. 01 Epub ahead of print] (2005). [DOI] [PMC free article] [PubMed]
- 37.Fischer E, Sauer U. A novel metabolic cycle catalyzes glucose oxidation and anaplerosis in hungry Escherichia coli. J Biol Chem. 2003;278:46446–46451. doi: 10.1074/jbc.M307968200. [DOI] [PubMed] [Google Scholar]
- 38.Peters-Wendisch P, et al. Pyruvate carboxylase from Corynebacterium glutamicum: characterization, expression and inactivation of the pyc gene. Microbiology. 1998;144:915–927. doi: 10.1099/00221287-144-4-915. [DOI] [PubMed] [Google Scholar]
- 39.Kondo E, Kanai K. An attempt to cultivate mycobacteria in simple synthetic liquid medium containing lecithin-cholesterol liposomes. Jpn J Med Sci Biol. 1976;29:109–121. doi: 10.7883/yoken1952.29.109. [DOI] [PubMed] [Google Scholar]
- 40.Raynaud C, et al. Phospholipases C are involved in the virulence of Mycobacterium tuberculosis. Mol Microbiol. 2002;45:203–217. doi: 10.1046/j.1365-2958.2002.03009.x. [DOI] [PubMed] [Google Scholar]
- 41.Daniel J, et al. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J Bacteriol. 2004;186:5017–5030. doi: 10.1128/JB.186.15.5017-5030.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Claes WA, et al. Identification of two prpDBC gene clusters in Corynebacterium glutamicum and their involvement in propionate degradation via the 2-methylcitrate cycle. J Bacteriol. 2002;184:2728–2739. doi: 10.1128/JB.184.10.2728-2739.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabuchi T, Uchiyama H. Methylcitrate condensing and methylisocitrate cleaving enzymes; evidence for the pathway of oxidation of propionyl-CoA to pyruvate via C7-tricarboxylic acids. Agr Biol Chem. 1975;39:2035–2042. [Google Scholar]
- 44.Luttik MA, et al. The Saccharomyces cerevisiae ICL2 gene encodes a mitochondrial 2-methylisocitrate lyase involved in propionyl-coenzyme A metabolism. J Bacteriol. 2000;182:7007–7013. doi: 10.1128/jb.182.24.7007-7013.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ranes M, et al. Functional analysis of pAL5000, a plasmid from Mycobacterium fortuitum: construction of a “Mini” Mycobacterium-Escherichia coli shuttle vector. J Bacteriol. 1990;172:2793–2797. doi: 10.1128/jb.172.5.2793-2797.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prentki P, Krisch HM. In vitro insertional mutagenesis with a selectable DNA fragment. Gene. 1984;29:303–13. doi: 10.1016/0378-1119(84)90059-3. [DOI] [PubMed] [Google Scholar]
- 47.Pavelka MS, Jr, Jacobs WR., Jr Comparison of the construction of unmarked deletion mutations in Mycobacterium smegmatis, Mycobacterium bovis bacillus Calmette-Guerin, and Mycobacterium tuberculosis H37Rv by allelic exchange. J Bacteriol. 1999;181:4780–4789. doi: 10.1128/jb.181.16.4780-4789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stover CK, et al. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.