

Abstract
Integrins are considered to be an important mechanosensor in cardiac myocytes. To test whether integrins can influence cardiac contractile function, the force–frequency relationships of mouse papillary muscle bundles were measured in the presence or absence of a synthetic integrin-binding peptide, GRGDNP (gly–arg–gly–asp–asn–pro). Results demonstrate that in the presence of an arginine–glycine–aspartic acid (RGD)-containing synthetic peptide, contractile force was depressed significantly by, 28% at 4 Hz, 37.7% at 5 Hz and 20% at 10 Hz (n = 6, P < 0.01). Treatment of myofibres with either protease-generated fragments of denatured collagen (Type I) or denatured collagen that contain the RGD motif, also reduced force production significantly. An integrin-activating antibody for β1 integrin inhibited the force similar to synthetic RGD peptide. Function-blocking integrin antibodies for α5 and β1 integrins reversed the effect of the RGD-containing peptide, and α5 integrin also reversed the effect of proteolytic fragments of denatured collagen on contractile force, whereas experiments with function-blocking antibody for β3 integrin did not reverse the effect of RGD peptide. Force–[Ca2+]i measurements showed that the depressed rate of force generation observed in the presence of the RGD-containing peptide was associated with reduced [Ca2+]i. Data analyses further demonstrated that force per unit of Ca2+ was reduced, suggesting that the myofilament activation process was altered. In addition, inhibition of PKC enzyme using the selective, cell-permeable inhibitor Ro-32-0432, reversed the activity of RGD peptide on papillary muscle bundles. In conclusion, these data indicate that RGD peptide, acting via α5β1 integrin, depresses the force production from papillary muscle bundles, partly associated with changes in [Ca2+]i and the myofilament activation processes, that is modulated by PKCε.
Cardiac dysfunction is closely linked to the disruption of the extracellular matrix (ECM) proteins. ECM in the heart is made up of type 1 collagen (up to 80%) and the remaining 20% consists of type 3 collagen, vitronectin, fibronectin and with minor contributions from other components of ECM. It is well documented that within minutes of the onset of myocardial infarction, there is a major loss of intact collagen associated with acute dysregulation of cardiac function (Nishikawa et al. 2003). It is also hypothesized that many chronic pathophysiological conditions associated with impaired myocardial function, are directly related to altered collagen alignment, structure and tissue support. Examples include progressive left ventricular dilatation and remodeling during decompensatory phase of congestive heart failure. Collectively, there is ample evidence to suggest an important role for ECM in the regulation of cardiac myocyte function.
A number of ECM-degrading enzymes have been identified in heart tissue. The ECM-degrading enzymes that are activated following myocardial infarction belong to the families of serine proteinases (plasmin, plasminogen activator, tissue-type plasminogen activator, thrombin, elastase, cathepsin G) and matrix metalloproteinases (MMPs: collagenases, gelatinases, stromelysins, metaloelastase, and membrane-type matrix metalloproteinase). It has been reported that MMPs are secreted by a number of cell types that include fibroblasts, vascular smooth muscle cells, endothelial cells and also recently demonstrated by adult mammalian cardiac myocytes (D'Armiento, 2002). Other candidate cell types responsible for degradation of ECM are the inflammatory cells, such as neutrophils, that release a number of ECM-degrading enzymes. Neutrophils often represent the first inflammatory cell type that migrate to the site of injury and releases neutrophil elastase enzyme that is essential for phagocytosis. Human neutrophil elastase enzyme cleaves the peptide bonds on the carboxyl-terminal side of hydrophobic or aromatic amino acids with a preference for amino acids with longer aliphatic chains at this position. These enzymes act on ECM proteins and reportedly expose putative sites known as ‘matricryptic sites’ consisting of consensus arginine–glycine–aspartic acid (RGD) sequences that can interact with integrins (Davis et al. 2000). Further degradation of ECM proteins results in release of these sequences as soluble peptide fragments or ‘matricryptins’ which have been implicated to play a role as ‘wound signals’ in the injury-inflammation process through an interaction with integrins.
We have previously shown that vasomotor tone in isolated rat cremaster arterioles is altered by integrin-binding peptides that contain the RGD sequence by interacting with the αvβ3 or α5β1 integrins or the leucine–aspartic acid–valine (LDV) sequence by interacting with α4β1 (Mogford et al. 1996, 1997; Waitkus-Edwards et al. 2002; Martinez-Lemus et al. 2003). These changes are associated with an inhibition or enhancement, respectively, of current through L type Ca2+ channels in vascular smooth muscle cells (Wu et al. 1998). Furthermore, Mogford et al. (1996) reported that neutrophil elastase-generated fragments of type I collagen also caused vasodilatation through RGD-dependent interaction with αvβ3 integrin. Taken together, these studies support the general hypothesis that integrins may play an important role in altering contractile function of vascular smooth muscle cell. In addition, these findings have led us to speculate whether integrin-binding peptides that contain the RGD sequence can modulate force development in the cardiac myocytes, consequences of which could have important pathophysiological implications.
Studies have shown that in cardiac muscle differential expression of the α subunit of integrins (α1, α3 and α5) as well as the β subunit (β1 and β3) occurs following myocardial infarction (Nawata et al. 1999; Sun et al. 2003). Hilenski et al. (1992) has reported on the importance of ECM proteins on myofibrillar and cytoskeletal assembly in neonatal cardiac myocytes. The significance of α5β1 and α3β1 integrins in fetal cardiac myocyte proliferation was demonstrated by Hornberger et al. (2000). Transgenic mice expressing a dominant-negative form of β1 integrin showed hypertrophic changes in the heart and cardiac-specific knockout of β1 integrin led to myocardial fibrosis and cardiac failure (Shai et al. 2002). β3 integrin is shown to be involved in endothelial cell migration and blood vessel formation in granulation tissue. In rat hearts, β3 integrin was up-regulated at the site of myocardial infarction during inflammatory and fibrogenic stages of healing (Sun et al. 2003). Thus, integrins and ECM proteins appear to be key elements associated with myocardial injury and subsequent remodelling events involving cardiac myocytes. The purpose of our study was to test the hypothesis that interaction of integrins with RGD-containing peptides would alter myofilament contractile function.
Methods
Force measurements in intact papillary muscle fibres
Adult male mice (FVB/N strain, 15–20 weeks, 25–35 g, Harlan Houston, TX, and Charles River, USA) were anaesthetized by an intraperitoneal injection of sodium pentobarbital (∼ 10–20 mg) and hearts were excised rapidly. The hearts were placed in cold (4°C) Krebs-Henseleit (KH) buffer containing 30 mm BDM (2,3-butanedione monoxime). KH buffer was composed of (mm): 119.0 NaCl, 11.0 glucose, 4.6 KCl, 25.0 NaHCO3, 1.2 KH2PO4, 1.2 MgSO4 and 1.8 CaCl2. The buffer solution was gassed with 95% O2–5% CO2 to maintain the pH (7.35–7.40). The right ventricular papillary muscle (mm: 1.2 ± 0.22 in length, 0.408 ± 0.30 in width and 0.24 ± 0.10 in thickness) was isolated by dissection with a segment of the tricuspid valve at one end and a portion of the myocardial septum at the other. The muscle tapered from the ventricular wall toward the attachment to the tricuspid valve such that the shape approximated that of a triangle with length and with an oval base width and a base depth. The force was normalized per cross-sectional area at the base using the approximation area = 0.75 × width × depth (Li et al. 2000). The papillary muscle bundles were mounted between a force transducer and a voltage-controlled motor positioner within a muscle measurement suite (Scientific Instruments, Germany). Stimulation pulse duration was 3.5 ms with an initial rate of 0.5 Hz. The papillary bundle was continuously superfused with KH maintained at 34°C. Stimulation voltage and bundle length were adjusted until maximum force was reached. The fibre bundle was then stimulated at 0.5 Hz for 20–30 min before executing the experimental protocol. A digital phosphor oscilloscope suite (Tektronix TDS 3014 with IEEE-488 communication module and Wavestar software) measured stimulation frequency, twitch force amplitude, averaged force amplitude within preset time windows, and continuously logged the data into the computer.
The experimental protocol consisted of: (1) increasing stimulation frequency from 0.5 to 1.0 Hz, then up to 10.0 Hz, by 1.0 Hz increments (duration of 2 min or until a steady force value had been reached); (2) decreasing stimulation frequency to a randomly selected frequency (4, 5, or 10 Hz) and superfusing with KH solution containing either 1 mm GRGDNP peptide or the control peptides GRADSP or GRGESP (Biomol) for 10 min; (3) integrin-activating antibody for β1 integrin (9EG7, 40 μg ml−1) replaced RGD peptide in the previous protocol. (The mAb 9EG7 recognizes an extracellular conformation-sensitive epitope, whose exposure correlates with active or ligand binding state of β1 integrins (Lenter et al. 1993; Bazzoni et al. 1998; Wennerberg et al. 1998; Armulik et al. 2000, 2004). Binding of this antibody has also been linked to activation of β1 integrins (Faull et al. 1996).); (4) in experiments with function-blocking antibodies for α5 (HMα5-1, 20 μg ml−1), β1 (HMβ1-1, 100 μg ml−1) and β3 integrins (HMβ3-1, 20 μg ml−1) (Pharmingen), the preparations were incubated for 10 min with the antibody prior to perfusion with 1 mm GRGDNP peptide or with proteolytic fragments of type 1 collagen. The papillary muscle was kept at minimal flow without stimulation while it was superfused with antibody-containing solution. Then the stimulation frequency was increased to 5 Hz and superfused with 1 mm GRGDNP peptide or with proteolytic fragments of type 1 collage; and (5) for PKC inhibitor studies, the papillary muscle fibres were incubated with 8 μm Ro-32-0432 (Calbiochem, USA) compound, a selective, cell-permeable inhibitor of PKC for 30 min at 0.5 Hz. The stimulation frequency was increased to 5 Hz and 1 mm RGD peptide solution reconstituted in KH solution was started in the flow.
Preparations of native, denatured and digested fragments of type 1 collagen
Type 1 collagen was isolated from mice tails using the acetic acid method (Ehrmann & Gey, 1956; Bornstein, 1958). Purified collagen was dialysed in physiological saline solution (mm: 145.0 NaCl, 4.7 KCl, 2.0 CaCl2, 1.0 MgSO4, 1.2 NaH2PO4, 5.0 glucose, 3.0 Mops, 0.02 EDTA) and a fraction was heat denatured by boiling at 100°C for 15 min. Denatured type 1 collagen was digested with sepharose-coupled human neutrophil elastase enzyme (Calbiochem) (Mogford et al. 1996). The digested fragments of collagen or the denatured collagen were suspended in KH solution. Purified and dialysed ‘native’ type 1 collagen was also used in the experiments. The preparations of native, denatured and digested fragments of collagen were confirmed by running a 7.5% PAGE under non-reducing and non-denaturing conditions. Sample buffer (10 ×) was composed of 1 m Tris-HCl pH 6.8, SDS 1%, EDTA 0.003%, sodium azide 0.01%, glycerol 10%, and bromophenol blue 0.1%. The protein samples (10 μg) were heated at 37°C for 30 min. The electrophoresis was done at 4°C at 160 V. The gel was then stained with Coomasie Blue.
A concentration of 10−6m was selected for all the functional experiments with native, denatured and digested fragments of type 1 collagen. All the experiments were performed while the papillary bundle preparation was stimulated at 5 Hz. The force measurements were collected for 3 min after the start of perfusion. The protocol for experiments with function-blocking antibody for integrin was similar as mentioned before. Briefly, papillary muscles were pre-incubated with antibody for α5 integrin (HMα5-1, 20 μg ml−1), 10 min prior to addition of proteolytic fragments of type 1 collagen (10−6m) digested with neutrophil elastase enzyme.
Force–[Ca2+] measurements in intact papillary muscles
Force–[Ca2+]i measurements were performed as we have previously described (Tong et al. 2004; Gaffin et al. 2004). Isolated papillary muscle preparations were stabilized in the same apparatus mentioned previously, and except for calcium measurements the experiments were performed at room temperature. Measurements were collected using a National Instruments A/D board and Labview software with the digital oscilloscope suite providing continuous monitoring. A mercury lamp and filter-wheel provided alternating ultraviolet (UV) pulses of 340 and 380 nm at 250 Hz with a pulse duration of 1.5 ms to illuminate the bundle. A combination of microscope, dichroic mirror, filter and photomultiplier tube (PMT) collected the fura-2 fluorescence. The loading solution was made up of 10 μm fura-2 AM in KH solution and 5.0 g l−1 cremophor. The solution was loaded for a duration of 1.5 h followed by de-estrification for 20 min. Under these conditions a fluorescence signal of 3-fold more than background was obtained. The ratio of fluorescence from 340 nm excitation to fluorescence from 380 nm excitation was calculated after subtracting the background fluorescence. Intracellular concentration of calcium was obtained by using the following equation with Kd,apparent equating to Kd×β after subtracting the background fluorescence (Grynkiewicz et al. 1985). β is the fluorescence of free dye versus fluorescence of bound dye at 380 nm and can be measured as the ratio of fluorescence from 380 nm excitation at no calcium and 380 nm excitation at saturating calcium.
The in vitro calibrations utilizing 25 μm Fura-2 simulated in vivo conditions and produced the following values: Rmin = 0.106, Rmax = 2.9066 and Kd,apparent = 0.284 μm. The experimental protocol was the same as described in the earlier section, except that stimulation frequency was increased only up to 3.0 Hz. The measurements were obtained for fluorescence before and after the addition of 1 mm RGD peptide. RGD peptide by itself does not affect the fluorescence signal as we have shown previously (D'Angelo et al. 1997) in rat cremaster arterioles.
Data analyses of the force–calcium loop
From the calcium and force values, first, time-to-peak Ca2+ amplitude and time-to-peak tension (TPT), decay time for Ca2+ amplitude and relaxation time for force were calculated. The data were fitted to a sigmoidal dose–response equation with a non-linear curve fit using Prism 3.0 to derive the EC50 value. Second, data analyses were done as described recently (Tong et al. 2004; Gaffin et al. 2004). We analysed the measured force–calcium values at specific points during a twitch cycle. The points consisted of: (1) the resting point ‘A’, (2) maximum calcium point, ‘B’, and (3) maximum force point, ‘C’. Delta gain (DG) is defined as the active force/area versus the change in calcium concentration from the point A. This formula was also used to calculate DG at points B and C.
[Ca2+]NState represents [Ca2+] as either ‘B’ or ‘C’ state. The ratio of DG to the maximum DG obtained at 3 Hz was calculated to provide the normalized delta gain (NDG). This provided a value between 0 and 1.
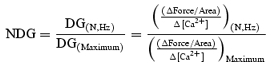 |
Rate of isometric tension development and relaxation
The maximum rate of isometric tension development (+dF/dt) and rate of relaxation (−dF/dt) was calculated using the following equation, where F(t) is the measured force at a particular time t and Δt is the sampling period, 1 ms:
A three data points averaging window was used as a filter for each dF/dt calculation to minimize the noise.
Reverse transcriptase polymerase chain reaction
RNA was isolated from the pooled papillary muscles using Trizol reagent (Invitrogen) according to manufacturer's recommendations. Total RNA (∼ 1 μg) was reverse transcribed into cDNA using 15 U of Thermoscript RT enzyme (Invitrogen) and gene-specific primers for α5 and β1 integrins. PCR assays were performed in a total volume of 50 μl using 5 μl of the RT product in a PCR buffer containing 1.5 mm MgCl2, 10 pmol forward primer, 10 pmol reverse primer and 0.75 U Taq DNA polymerase enzyme. The PCR reaction involved initial incubation at 94°C for 3 min, denaturation at 94°C for 1 min, then annealing at 60°C for 1 min, and extension at 72°C for 1 min. To demonstrate the linearity of RT-PCR conditions, we amplified GAPDH transcripts from papillary muscle as we have done previously (Hein et al. 2001). Since GAPDH, an abundantly expressed message, was not saturated at 30 cycles (data not shown), we normalized the expression of integrin transcripts to GAPDH at 30 cycles. The DNA sequence of the primers used for the amplification of mouse α5 transcript were: forward primer, 5′ AAA AGA AAC TTC AGG TGG CCA 3′; reverse primer, 5′ GAG TCT GAG ATC AGG AGG ACT 3′ obtaining a 241 bp PCR product; for the β1 transcript: forward primer, 5′ ACT GTC CTA GTC CCG ACA 3′; reverse primer, 5′ CGG AAA CTC AGA GAC CAG CTT 3′ yielding a 249 bp PCR product; and for the β3 transcript were: forward primer, 5′ GTC AGA TTC CAG TAC TAC GAA G 3′; reverse primer, 5′ CAT TAA GTC CCC CGG TAG GT 3′ obtaining a 300 bp PCR product.
Western blot analysis
Heart isolated from mouse was rinsed well with physiological saline solution before homogenizing it with lysis buffer (0.06 m KCl, 0.025 m Mops, 0.0025 m MgCl2) containing 1 μg μl−1 leupeptin and 1 mm phenylmethylsulphonyl fluoride. The homogenate was centrifuged at 12 000 g for 15 min at 4°C and the supernatant was collected. Protein concentration was determined using a BCA protein estimation kit (Pierce). Proteins were separated by 10% SDS polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane according to the method of Towbin et al. (1979) for Western blot analysis. Primary antibody incubation in 5% non-fat dry milk was performed with α5 integrin (1: 100) for 1.5 h at room temperature, and overnight for β1 integrin (1: 200) at 4°C. Chemiluminescence (SuperSignal West Dura; Pierce) was used as a detection agent. Antibodies for α5, β1 and β3 (sc-10729, sc-8978 and sc-14009, respectively) integrins were bought from Santa Cruz Biotechnology Inc. (CA, USA).
To verify the equal loading of the protein samples, the nitrocellulose membrane was stripped with Restore Western Blot Stripping Buffer (Pierce) and probed for a second time with anti-sarcomeric actin antibody (1: 5000; Sigma) followed by HRP conjugated goat anti-mouse IgM (1: 10 000 from a 0.6 mg ml−1 stock; Sigma) as the secondary antibody.
Data analyses and statistics
Data are presented as mean ± s.e.m. The significance of differences between means for experiments with RGD, control peptides, and for the calcium study was evaluated using paired Student's t test and values of P < 0.05 were considered statistically significant. Completely randomized design ANOVA with Duncan's multiple range test was used to compare significance of difference in experiments with function blocking antibodies for integrins, and with collagen experiments; values of P < 0.05 were considered statistically significant.
Results
Expression of α5, β1 and β3 integrins in the papillary muscle
To determine the presence of integrins, we conducted RT-PCR analyses for α5, β1 and β3 integrin transcripts in the total RNA isolated from mouse papillary muscle. Figure 1A shows that both α5 and β1 integrins are easily detected in the papillary muscle. In addition, the relative level of β3 integrin is lower in the sample. To confirm our analyses at the protein level, the total homogenate preparations from the mice hearts were analysed using specific monoclonal antibodies for α5, β1 and β3 integrins. The results show the presence of α5, β1 and β3 integrins in the mouse heart (Fig. 1B).
Figure 1. Expression of integrins in mouse papillary fibres.

A, RT-PCR analyses of α5, β1 and β3, integrins and GAPDH gene in papillary muscle of mouse hearts. Electrophoresis of PCR gene products on a 1.8% agarose gel were carried out and visualized with ethidium bromide staining. Molecular weight markers represent Hae III-digested φX-174 DNA. These data are representative of three independent experiments. B, representative Western blot analysis of α5, β1 and β3 integrins (n = 3). Ten and 20 μg of total heart homogenates were run on a 10% SDS-PAGE and probed with specific antibodies as detailed in Methods. The blots were re-probed with sarcomeric actin.
Effect of RGD-containing peptide
A typical force–frequency response of the papillary fibre bundle is shown in Fig. 2A. The fibres demonstrating a positive force–frequency response from 2 to 5 Hz were selected for further experimental protocols as described in Methods. An example trace for force, before and after RGD peptide perfusion, is shown in Fig. 2B. Result shows that perfusion of the RGD peptides causes a significant inhibition of force over a duration of 1–2 min, and then the force gradually returns to its previous state. Figure 2C shows that the inhibition of force by the RGD peptide was maximal at 1 mm concentration.
Figure 2. Effects of RGD peptide on force development.

A, representative graph of force versus frequency relationship. B, acute effect of 1 mm RGD peptide on the active force generated by papillary muscle from the right ventricle of a mouse heart. A significant inhibition of force is observed. C, effect of varying concentrations of RGD on force development. Note that 1 mm GRGDNP peptide causes maximal inhibition of force development. The 100% value of force/cross-sectional area represents the control measurement prior to addition of the RGD peptide. Data are presented as mean ± s.e.m., *P < 0.05, paired t test.
To further determine the specificity of the effect of RGD-containing peptides, the papillary fibre preparations were superfused with KH solution containing control peptides, such as GRADSP or GRGENP. As seen in Fig. 3 A and B, a significant depression of force is seen only with RGD-containing peptides at all stimulation frequencies examined. Figure 3A shows that after the fibres are superfused with the RGD-containing peptides, the force is reduced by 28, 37.7 and 20%, at 4 Hz (control, F= 20.55 ± 2.74 mN mm−2; with RGD, F= 14.55 ± 2.03 mN mm−2, n = 6, P = 0.003), 5 Hz (control, F= 20.78 ± 4.18 mN mm−2; with RGD, F= 12.79 ± 3.08 mN mm−2, n = 6, P = 0.01), and 10 Hz (control, F= 21.64 ± 2.61 mN mm−2; with RGD, F= 17.18 ± 1.92 mN mm−2, n = 6, P = 0.003), respectively. As seen in Fig. 3B, neither the GRGENP peptide nor GRADSP peptide inhibited the force production of the papillary fibres at any stimulation frequencies tested (n = 6, P > 0.1 in all cases). These results indicate that the force production by the papillary fibres was specifically inhibited by the peptide containing the RGD sequence.
Figure 3. Effects of RGD and control peptides on force development at different frequencies.

A, effect of 1 mm RGD peptide at different frequencies: 4, 5 and 10 Hz. Columns with RGD represent the reduced force produced in the presence of peptide. B, effects of control peptides, GRADSP (at 4 and 5 Hz) and GRGESP (at 10 Hz). The force in the presence of peptides is shown as a percentage of the normalized force (100%) before the addition of peptides. Data are presented as mean ± s.e.m., *P < 0.01, paired t test.
Effects of native, denatured and digested fragments of Type I collagen on the force development
To verify that the collagen underwent denaturation and enzymatic digestion, we examined the samples of native, denatured and digested collagen fragments in a native PAGE preparation (Fig. 4A) as described in Methods. Results depict several low molecular weight bands in the elastase enzyme-digested type I collagen sample and some higher molecular weight bands in heat-denatured collagen. The native collagen preparation shows an intact collagen molecule, showing both α1 and α2 chains of collagen, also present in the denatured collagen sample. To determine the effect of type 1 collagen, the force measurements in the papillary muscle preparations were conducted before and after incubation with 10−6m denatured, digested fragments or native type 1 collagen. Figure 4B summarizes data showing that in the presence of either denatured or digested fragments of collagen the force was inhibited by 11.28% (n = 6, P = 0.01) or 14.32% (n = 6, P = 0.001), respectively. In comparison, the force produced by the papillary muscle preparations was not reduced by native type 1 collagen.
Figure 4. Effects of native, denatured and digested fragments of collagen on force development.

A, 7.5%PAGE analysis of collagen samples under non-denaturing and non-reducing conditions. NC, native collagen; DEN, denatured collagen; DIGEST, collagen digested with human neutrophil elastase enzyme. B, effect of native type 1 collagen versus denatured and digested fragments of collagen on papillary muscle force development. Denatured and digested fragments of collagen significantly inhibit the force production. Data are presented as mean ± s.e.m., *P < 0.05 (ANOVA with Duncan's multiple range test).
Effects of function blocking and activating antibody for integrins
To investigate the role of integrins on the inhibition effect on force development in RGD peptides, we pretreated the papillary fibre bundles with function-blocking antibodies for α5, β1 or β3 integrins and measured the force development as described in Methods. Results show that the effect of RGD peptide was significantly attenuated in the presence of antibodies for integrins (Fig. 5A). The data show that the inhibition effect (37.7%) of RGD peptide alone on the force development of papillary muscle preparations at 5 Hz (n = 6, P = 0.01) was significantly reduced to 13.54% (n = 6, P < 0.05) in the presence of α5 and 17.23% (n = 3, P < 0.05) in the presence of β1 integrin function-blocking antibody. However, the depressive effect of RGD peptide was not reversed significantly in the presence of β3 integrin function-blocking antibody (25.03% (n = 6, P < 0.05)). Function-blocking antibodies alone or the whole IgG molecule had no effect on force development (data not shown).
Figure 5. Effects of function-blocking integrin antibodies.

A, effect of function-blocking antibodies for α5, β1 and β3 integrins. Force obtained before the addition of peptide or fragments of denatured collagen is normalized to 100%. α5 and β1 integrin antibodies significantly reverse the effect of RGD peptide. Data are presented as mean ± S.E.M, *P < 0.01 paired t test, #P < 0.05 (ANOVA with Duncan's multiple range test). B, effect of 10−6m digested fragments of type 1 collagen in the presence of function-blocking antibody for α5 integrin. α5 antibody significantly reverses the effect of digested fragments of collagen. C, a representative trace showing the effect of mAb 9EG7 an integrin-stimulating antibody for β1 integrin on normalized force. D, mAb 9EG7 depresses the force production from papillary muscle fibres similar to RGD peptide. Data are presented as mean ± s.e.m., *P < 0.01 paired t test.
In order to confirm that the depressive effect of digested fragments of collagen was also mediated by RGD-binding integrins, we pretreated the papillary muscle with function blocking antibody for α5 integrin before perfusing with 10−6m of digested fragments of type 1 collagen. Figure 5B shows that 14.32% inhibition of force by fragments alone (n = 6, P = 0.001) was reduced to 5% in fibres pretreated with function blocking for α5 integrin (n = 6, P = 0.0007).
To further investigate whether the effect of RGD peptide on the papillary muscle is due to the alteration in integrin-mediated signalling mechanisms, we decided to use a function-stimulating antibody for β1 integrin. The trace in Fig. 5C shows the effect of β1 integrin antibody (9EG7) on force production by the papillary muscle bundle. Results clearly demonstrate that similar to RGD peptide, treatment with an integrin-activating antibody for β1 leads to 16.58% (n = 6, P = 0.01) depression in force production (Fig. 5D). These results indicate that RGD peptide depresses force production by activation of β1 integrin.
Depressed [Ca2+]i and myofilament activation processes with decrease in force
To investigate the mechanism for the reduction of force observed in the presence of synthetic RGD-containing peptide, we measured the force and [Ca2+]i simultaneously, in the papillary muscle fibres. The Ca2+ transients and force measured at 3 Hz in the absence or presence of RGD peptide are shown in Fig. 6A and B. In the absence or presence of RGD peptide the isolated papillary muscles exhibited no significant change in time-to-peak [Ca2+]i amplitude control, EC50 = 10.7 ± 0.42 ms; RGD, EC50 = 11.1 ± 0.75 ms, n = 9, P = 0.4). After reaching a peak, the Ca2+ transients declined more slowly, in the presence of RGD peptide, but were not statistically significantly different (control, decay time measured as EC50 = 0.7 ± 0.42 ms, RGD, EC50 = 3.21 ± 1.53 ms, n = 9, P = 0.1). Isometric tension measurements on the papillary muscles showed an increase in time-to-peak tension (TPT) in the presence of RGD peptide (control, EC50 = 33.8 ± 0.13 ms, RGD, EC50 = 37.2 ± 0.16 ms, n = 9, P = 0.01) whereas the decrease in peak tension was not statistically significant (control EC50 = 207.9 ± 102.7 ms, RGD, EC50 = 239.2 ± 150.3 ms, n = 9, P = 0.5).
Figure 6. Ca2+ transients and tension.

Shown are the mean traces of the Ca2+ amplitude (A) and tension (B). Time-to-peak Ca2+ amplitude, decay time and time-to-peak tension (TPT) and relaxation time are the times representing EC50 value. Data are presented as mean ± s.e.m., *P < 0.01 paired t test.
To further analyse the force–calcium results, the data are plotted as force versus[Ca2+]i to display a cardiac function loop as we have defined previously (Tong et al. 2004). Figure 7A shows force plotted against calcium at 3 Hz for a papillary muscle preparation in the presence or absence of RGD peptide. The data show that there is a depression in the force generated at a given [Ca2+]i in the presence of RGD peptide. To further analyse these results, three specific points A, B and C were identified on the force versus[Ca2+]i loop: point A, after full relaxation following a previous contraction; point B, maximum calcium concentration; and point C, when maximum force is obtained in a twitch contraction. At point A there are no differences in force and calcium in the presence or absence of RGD peptide (data not shown). At maximum calcium concentration, point B, the active force is reduced in the presence of the peptide-containing RGD sequence (control, F= 13.00 ± 1.2 mN mm−2; with RGD, F= 11.50 ± 1.07 mN mm−2, n = 11, P = 0.005) (Fig. 7B, top left). In addition, a significant decrease in the maximum [Ca2+]i (control, [Ca2+]max = 0.76 ± 0.09 μm; with RGD, [Ca2+]max = 0.72 ± 0.09 μm, n = 10, P = 0.04) (Fig. 7B, top right) is seen when the RGD peptides are present. Furthermore, delta gain (ΔForce/Δ[Ca2+]i) was calculated as described in Methods. Since delta gain quantifies changes in force per unit of calcium, the alteration in this parameter represents changes in the myofilament activation processes. There is no significant change in this ratio at point B (Fig. 7B, bottom panel).
Figure 7. Analyses of the force-calcium loop from control and RGD peptide-superfused.

A, representative force–calcium loop analyses in the absence or presence of RGD peptide. A, B and C points represent the resting, maximum calcium concentration and maximum force points, respectively, in the twitch cycle. B, analyses at maximum calcium B point. Changes in active force, [Ca2+]i and delta gain (ΔForce/Δ[Ca2+]i) in the absence or presence of RGD peptide are shown. C, analyses at maximum force C point. Changes in active force, [Ca2+]i and delta gain (ΔForce/Δ[Ca2+]i) in the absence or presence of RGD peptide are shown. Data are presented as mean ± s.e.m., *P < 0.01 paired t test.
At the maximum force, point C, RGD-containing peptides cause a significant depression of maximum force (control, Fmax = 25.29 ± 1.36 mN mm−2; with RGD, Fmax = 22.31 ± 1.40 mN mm−2, n = 11, P = 0.000007) (Fig. 7C, top left). However, the [Ca2+]i is not changed significantly at point C (control, [Ca2+]i = 0.60 ± 0.07 μm; with RGD, [Ca2+]i = 0.58 ± 0.07 μm, n = 11, P = 0.18) (Fig. 7C, top right). In addition, normalized delta gain (NDG) as explained in Methods, was significantly different at point C (with RGD NDG = 90% of NDG of control) (Fig. 7C, bottom panel). Since, NDG is a calcium-independent parameter, these data demonstrate that the depression of force in the presence of RGD is independent of [Ca2+]i and is the result of changes in myofilament activation processes.
Effects of RGD peptide on the rates of contraction and relaxation
Rates of contraction and relaxation were calculated as described in Methods. RGD peptide caused a significant decrease in maximum +dF/dtmax from 350 ± 25.66 mN mm−2 s−1 in control to 309.94 ± 26.14 mN mm−2 s−1 in papillary muscles perfused with RGD peptide (n = 11, P = 0.00002) (Table 1), suggesting a significant decrease in myocardial systolic performance in the presence of peptide. The maximum rate of relaxation −dF/dt was not significantly different between the two groups (control =−209.93 ± 18.7 mN mm−2 s−1; with RGD =−193.14 ± 11.8 mN mm−2 s−1, n = 11, P = 0.1).
Table 1.
Maximum rates of force generation and relaxation
| Max +dF/dt (mN mm−2 s−1) | Max −dF/dt (mN mm−2 s−1) | ||||||
|---|---|---|---|---|---|---|---|
| Frequency | n | Control | With RGD | P value | Control | With RGD | P value |
| 3 Hz | 11 | 350.28 ± 25.6 | 309.94 ± 26.1 | 0.00005 | −209.93 ± 18.7 | −193.14 ± 11.8 | 0.1 |
n, number of experiments; +dF/dt, rate of force generation; −dF/dt, rate of relaxation.
Inhibitor of PKC enzyme reverses the RGD peptide effect
The depression in cardiac contractility has been linked to phosphorylation of TnI (Ser-43 and Ser-45 residues) by PKCε enzyme (Solaro & Rarick, 1998; Solaro, 1999; Burkart et al. 2003; Kobayashi et al. 2004). We hypothesized that the change in the myofilament activation processes in the presence of RGD peptide could be due to the activation of PKC. To test this hypothesis, papillary muscle fibres were pretreated with Ro-32-0432, an inhibitor of PKCε enzyme prior to perfusion with RGD peptide. Figure 8 clearly demonstrates that the effect of RGD on the force development is greatly reduced in the fibres treated with PKC inhibitor (n = 6, P = 0.01).
Figure 8. Effect of the PKC inhibitor Ro-32-0432.

Pretreatment with PKC inhibitor reversed the effect of RGD peptide significantly. Data are presented as mean ± s.e.m., *P < 0.01 paired t test.
Discussion
The main goal of this study was to determine the function of integrins in cardiac contractility. A significant depression of force was seen in the presence of a synthetic RGD-containing peptide, and in the presence of denatured type 1 collagen or digested fragments of collagen. Furthermore, the force–calcium data analyses demonstrated that depression in force was accompanied by a decrease in calcium concentration and changes in the myofilament activation processes. Thus, treatment of papillary muscle with an integrin-binding RGD peptide leads to altered myocardial systolic performance. This attenuation of force was significantly reversed in the presence of function-blocking antibodies against α5 and β1 integrins. In addition, the results from the integrin-stimulating antibody for β1 integrin have suggested that β1 integrin plays an active role in modulating the force development in papillary muscle fibre bundles. Furthermore, the PKC inhibitor reverses the effects of RGD peptide on muscle fibre force development. Thus, our data provide the first evidence that RGD peptide or digested collagen fragments depress the force development possibly through α5β1 integrins and PKCε plays an important role in mediating this effect.
Role of ECM proteins in cardiac muscle contractility
ECM of heart consists of about 80% of type 1 collagen and based on its primary structure type 1 collagen has seven RGD motifs. In native type 1 collagen the RGD motifs are not accessible to interact with integrins. However, denaturation and proteolysis of collagen, as might occur during injury following a myocardial infarction (MI), leads to exposure of these ‘cryptic sites’ and their accessibility to integrin binding (Davis, 1992; Davis et al. 2000). In order to mimic an in vivo state following MI, we superfused papillary muscle with denatured collagen and with proteolytic fragments of type 1 collagen. Our results show that both digested fragments and denatured collagen significantly depress the force in the myofibres. These data support previous observations that the biological activity of the RGD motifs in collagen is unmasked after proteolysis (Davis et al. 2000). Additionally, the effect of digested fragments of collagen was reversed in the presence of α5 function-blocking antibody suggesting that α5 could play a significant role in regulating cardiac contractility during a pathophysiological state. However, at the present time the effect of other non-RGD motifs in the collagen or other integrins, cannot be ruled out without additional study.
Recent studies have supported the role of the matrix in regulating the function of myocardium (Baicu et al. 2003; Lamberts et al. 2004). In their experimental approaches they used enzymatic methods to digest ECM proteins to study its effect on cardiac function, by incubating isolated papillary muscles with MMPs. Lamberts et al. used a specific type 1 collagenase enzyme and observed an increase in diastolic and developed tension, whereas Baicu et al. mimicked the pathological state by activating the MMP cascade by plasmin (a non-specific enzyme) and observed a decrease in systolic myocardial performance. Our observation of depressed myocardial systolic performance is consistent with the findings of Baicu et al. (2003) Furthermore we have determined that this depressive effect of RGD peptide is mediated via α5β1 integrin and the PKC-mediated myofilament activation process is altered.
We provide evidence that denatured or digested fragments of type 1 collagen, a predominant matrix protein, can regulate force production from papillary muscle. Specifically this effect is similar to that of synthetic RGD-containing peptide, suggesting that it is the RGD sequence of type 1 collagen responsible for regulation of force production. The RGD sequence of matrix proteins acts as a ligand for integrins, an important group of cell receptor molecules involved in normal and pathological processes in the heart (Ross, 2004).
Role of integrins in cardiac muscle contractility
Normal cardiac myocytes express α1, α3, α5, α6, α7, α9, α10 and β1 integrins, with at least four prevalent subtypes, which include α1β1, α3β1, α5β1 and α7β1. Among these, α1β1, α3β1 and α5β1 recognize RGD motifs present in the ECM proteins. However, α5β1 integrin has been reported to play an important role in normal physiology and pathology of the heart (Ross & Borg, 2001; Ross, 2004), which led us to concentrate on this particular integrin. Furthermore, Nagai et al. (1999) have reported expression of αvβ3 integrin in cardiac myocytes, which is an RGD-binding integrin. A transgenic overexpression of the human α5 integrin subunit lacking the cytoplasmic domain in cardiac myocytes leads to perinatal lethality, while an overexpression of α5 integrin complex with an unopposed β1 integrin cytoplasmic domain caused ECG abnormalities, atrial enlargement, cardiac fibrosis and sudden death (Valencik & McDonald, 2001). Cardiac-specific knockout of β1 integrin leads to myocardial fibrosis and cardiac failure, emphasizing its importance in the regulation of cardiac function (Shai et al. 2002). Results presented in this study from function blocking or stimulating integrin antibodies clearly support the specificity of α5β1 integrin in modulating the force development. The function-blocking integrin antibodies bind to the motifs of integrins where the RGD peptide would interact. Binding of these antibodies alone does not have any effect on the force generation. This indicates that disengagement of integrin–ECM contact alone, if it is occurring at all, does not cause any modulation in the force development. Since the antibodies compete with the soluble RGD peptides binding to integrins, the effect of RGD peptide is inhibited in the presence of blocking antibody. On the other hand, binding of β1 stimulating antibody (mAb 9EG7) is associated with activation of β1 integrins (Faull, 1996). However, these data do not rule out the possibility of roles of α1, α3 and α7 integrins in the regulation of force development in mouse papillary muscles.
A number of integrins, such as α11bβ3, α8β1, αvβ3, αvβ5, αvβ6, αvβ8, α3β1, α4β1, α2β1, α1β1 and α5β1, recognize the RGD motif present in many ECM proteins such as fibronectin, collagen, vitronectin, osteopontin and fibrinogen. Roles of α5β1, α4β1 and αvβ3 integrins in vasoregulation during myogenic response and remodelling in pathological and physiological conditions are well studied in isolated blood vessel experiments (Mogford et al. 1996, 1997; Waitkus-Edwards et al. 2002; Martinez-Lemus et al. 2003). These studies elucidate the relative importance of the RGD motif as a functionally active integrin ligand, capable of altering vascular smooth muscle tone. Our data highlight the current understanding of the role of α5β1 integrin in myocytes by linking this receptor to the regulation of myocyte contractile function.
A key issue that needs to be addressed is whether RGD peptide is acting by competing with established ECM–integrin interactions (passive effect) or whether it is acting by ligating with integrin receptors on the healthy cardiac myocytes and thereby affecting the contractility via intracellular signalling mechanism (active effect). We used an integrin-stimulating antibody to differentiate between the two effects. A mAb 9EG7 has been used as a function-stimulating antibody for β1 integrin. In our experiments it mimicked the effect of synthetic RGD-containing peptide, confirming that depression of cardiac contractility is due to the active effect of RGD. In addition, experimental evidence indicates activation and cytoskeletal recruitment of FAK and c-Src in the RGD stimulated, collagen embedded, three-dimensional model of adult feline cardiac myocytes (Balasubramanian & Kuppuswamy, 2003), suggesting a direct effect of RGD peptide on isolated adult cardiac myocytes, that can be linked to contraction in a cardiac myocyte.
RGD-containing peptides affects the myofilament activation mechanisms
Myocardial contraction and relaxation is dependent on the activation of cardiac muscle myofilaments, which consist of thin (actin, tropomyosin (TM) and troponin complex (Tn)) and thick filament (myosin) proteins. In the myofilament activation process, calcium binding to troponin C alters the TM position on actin from a blocked state (‘off’ state) to a closed state. This allows weak attachment of myosin heads to actin. The transition from the weakly bound crossbridges to the strongly bound state then moves TM to an open state (‘on’ state). This state facilitates further crossbridge binding, and the strongly bound crossbridges keep tropomyosin in the ‘on’ state (McKillop & Geeves, 1993; Geeves & Lehrer, 1994; Swartz et al. 1996; Vibert et al. 1997). Thus, for active force production, both calcium activation of the thin filament and positive feedback by strongly bound crossbridges are necessary. In addition, both of these mechanisms make significant contributions to each phase of the contractile cycle. Furthermore, we have recently described the components of the force–calcium loop and have discussed that both calcium activation of the thin filament and positive feedback by strongly bound crossbridges contribute significantly to each phase of the contractile cycle (Tong et al. 2004). Thus, alterations in myofilament activation mechanisms can be determined by analysing the force–calcium data.
The mechanism by which the RGD motif–integrin interaction is affecting contractility is an important question that needs to be addressed. As [Ca2+]i is largely responsible for the contractile state of a cell, inhibition of force can be explained by the observed depression of intracellular Ca2+ concentration in our experiments. Thus, ligation of the integrin receptor with the RGD ligand affects either the influx of Ca2+ via L type Ca2+ channel (Wu et al. 1998), calcium-induced calcium release (CICR) from ryanodine receptors or efflux of Ca2+ via SERCA, or Na+–Ca2+ exchanger. Contraction in a vascular smooth muscle cell has been linked to clustering of integrins and recruitment of non-receptor tyrosine kinases such as focal adhesion kinase (FAK) and c-Src. In addition, we have seen that the NDG (normalized delta gain), a Ca2+-independent parameter, is also decreased in the presence of RGD peptide (Fig. 7C), indicating that the process of strong crossbridges formation between thick and thin filaments is altered, to reduce the force development. In addition, the Ca2+ transient and tension analyses have demonstrated that time-to-peak tension is altered independently of the changes in Ca2+ transients. In conclusion, RGD motif–integrin interaction could depress force production by two mechanisms: (1) Ca2+ dependent, and (2) Ca2+ independent (myofilament activation).
Role of PKC
A potential candidate molecule responsible for intracellular signalling events following RGD motif–integrin interaction is protein kinase C (PKC). Two key molecules involved in cardiac myocyte contraction are myosin ATPase that drives crossbridges cycling, and the myosin light chain (MLC) associated with globular myosin head. MLC phosphorylation is regulated by myosin light chain kinase (MLCK) and MLC phosphatase. Ca2+ binding with calmodulin protein controls the activity of MLCK, while the Rho effector and PKC modulate the activity of MLC phosphatase. Experimental data conclude that integrin binding to their ligands and clustering of integrin receptors can activate both Rho and protein kinase C pathways (Van Der Flier & Sonnenberg, 2001; Polte et al. 2004). Interestingly alpha, epsilon and iota isoforms of PKC are translocated to particulate fractions allowing phosphorylation of PKC-associated proteins in an ischaemic myocardium (Albert & Ford, 1999). In addition, studies have indicated that PKCε-mediated phosphorylation of cardiac troponin I (cTnI) at ser-43 and ser-45 leads to a significant decrease in maximum tension of the myofilaments (Solaro & Rarick, 1998; Solaro, 1999; Burkart et al. 2003; Kobayashi et al. 2004). Our results, showing reversal of the RGD peptide effect on the force development of papillary muscle fibres by the PKC inhibitor indicate that PKC is acting downstream of the RGD–integrin axis to modulate the myofilament activation processes. Our data, showing a decrease in active force at points B and C in the force–calcium loop, indicate that the RGD effect is seen both at the A to B and B to C segments of the contractile cycle. These results suggest that both calcium activation of the thin filament (predominant in the A to B segment) and positive feedback by strongly bound crossbridge (predominant in the B to C segment) mechanisms, as we have previously discussed (McKillop & Geeves, 1993; Geeves & Lehrer, 1994; Swartz et al. 1996; Vibert et al. 1997; Tong et al. 2004) are perturbed in the presence of RGD peptide. Taken together, we suggest that in the presence of RGD α5β1 integrin-mediated signalling changes the characteristics of myofilament proteins by PKC, which further alters the myofilament activation processes, leading to the observed depression of force.
This study identifies an important role for the ECM–integrin pathway in cardiac myocytes relative to myocyte contractile function. This pathway is of potential importance to our understanding of the complexity of pathological processes associated with depressed cardiac function such as myocardial ischaemia (Gwathmey et al. 1995). Reduced contractile function following ischaemia can be explained in two ways. First, digestion of the matrix scaffolding of heart by MMPs could lead to reduced efficiency of cardiac myocyte–myocyte co-ordination; while this loss of geometry plays a significant role during remodelling, it may not explain the depression of contractile function within minutes of ischaemia. Second, and perhaps more acute, is the direct effect of degraded ECM on cardiomyocytes via the ECM–integrin–cytoskeletal protein axis. We believe that this possibility may explain the findings of this study. We speculate that this depression of force would allow the heart to recover from injury, and also decreasing the contractility prevents the perfusion of causative agents into neighbouring healthy tissue. The data presented here provide the first experimental evidence that RGD-containing peptide, either synthetic or digested fragments of collagen, depress the force in the mouse papillary muscle fibres through α5 and β1 integrins. Furthermore, the modulation of force in the presence of RGD-containing peptide is associated with a decrease in [Ca2+]i and changes in the myofilament activation process, partially due to the effects of PKC on myofilament proteins. In conclusion, our results indicate an association between integrins, [Ca2+]i homeostasis and the myofilament activation processes in cardiac myocytes.
Acknowledgments
This study was supported by NIH grants HL-60758 to M.M. and HL-58690 to G.A.M. The authors would like to thank Dr A. Trache for technical assistance with the instrumentation.
References
- Albert CJ, Ford DA. Protein kinase C translocation and PKC-dependent protein phosphorylation during myocardial ischemia. Am J Physiol. 1999;276:H642–650. doi: 10.1152/ajpheart.1999.276.2.H642. [DOI] [PubMed] [Google Scholar]
- Armulik A, Svineng G, Wennerberg K, Fassler R, Johansson S. Expression of integrin subunit beta1B in integrin beta1-deficient GD25 cells does not interfere with alphaVbeta3 functions. Exp Cell Res. 2000;254:55–63. doi: 10.1006/excr.1999.4722. 10.1006/excr.1999.4722. [DOI] [PubMed] [Google Scholar]
- Armulik A, Velling T, Johansson S. The integrin beta1 subunit transmembrane domain regulates phosphatidylinositol 3-kinase-dependent tyrosine phosphorylation of Crk-associated substrate. Mol Biol Cell. 2004;15:2558–2567. doi: 10.1091/mbc.E03-09-0700. 10.1091/mbc.E03-09-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baicu CF, Stroud JD, Livesay VA, Hapke E, Holder J, Spinale FG, Zile MR. Changes in extracellular collagen matrix alter myocardial systolic performance. Am J Physiol Heart Circ Physiol. 2003;284:H122–132. doi: 10.1152/ajpheart.00233.2002. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S, Kuppuswamy D. RGD-containing peptides activate S6K1 through beta3 integrin in adult cardiac muscle cells. J Biol Chem. 2003;278:42214–42224. doi: 10.1074/jbc.M303428200. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Ma L, Blue ML, Hemler ME. Divalent cations and ligands induce conformational changes that are highly divergent among beta1 integrins. J Biol Chem. 1998;273:6670–6678. doi: 10.1074/jbc.273.12.6670. [DOI] [PubMed] [Google Scholar]
- Bornstein MB. Reconstituted rattail collagen used as substrate for tissue cultures on coverslips in Maximow slides and roller tubes. Laboratory Invest. 1958;7:134–137. [PubMed] [Google Scholar]
- Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. Phosphorylation or glutamic acid substitution at Protein Kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:11265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Davis MJ, Meininger GA. Calcium and mechanotransduction of the myogenic response. Am J Physiol. 1997;273:H175–182. doi: 10.1152/ajpheart.1997.273.1.H175. [DOI] [PubMed] [Google Scholar]
- D'Armiento J. Matrix metalloproteinase disruption of the extracellular matrix and cardiac dysfunction. Trends Cardiovasc Med. 2002;12:97–101. doi: 10.1016/s1050-1738(01)00160-8. 10.1016/S1050-1738(01)00160-8. [DOI] [PubMed] [Google Scholar]
- Davis GE. Affinity of integrins for damaged extracellular matrix: alpha v beta 3 binds to denatured collagen type I through RGD sites. Biochem Biophys Res Commun. 1992;182:1025–1031. doi: 10.1016/0006-291x(92)91834-d. 10.1016/0006-291X(92)91834-D. [DOI] [PubMed] [Google Scholar]
- Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol. 2000;156:1489–1498. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrmann RL, Gey GO. The growth of cells on a transparent gel of reconstituted rat-tail collagen. J Natl Cancer Inst. 1956;16:1375–1403. [PubMed] [Google Scholar]
- Faull RJ, Wang J, Leavesley DI, Puzon W, Russ GR, Vestweber D, Takada Y. A novel activating anti-beta1 integrin monoclonal antibody binds to the cysteine-rich repeats in the beta1 chain. J Biol Chem. 1996;271:25099–25106. doi: 10.1074/jbc.271.41.25099. 10.1074/jbc.271.41.25099. [DOI] [PubMed] [Google Scholar]
- Gaffin RD, Tong CW, Zawieja DC, Hewett TE, Klevitsky R, Robbins J, Muthuchamy M. Charged residue alterations in the inner-core domain and carboxy-terminus of α-tropomyosin differentially affect mouse cardiac muscle contractility. J Physiol. 2004;561:777–791. doi: 10.1113/jphysiol.2004.070631. 10.1113/jphysiol.2004.070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeves MA, Lehrer SS. Dynamics of the muscle thin filament regulatory switch: the size of the cooperative unit. Biophys J. 1994;67:273–282. doi: 10.1016/S0006-3495(94)80478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gwathmey JK, Liao R, Helm PA, Thaiyananthan G, Hajjar RJ. Is contractility depressed in the failing human heart? Cardiovasc Drugs Ther. 1995;9:581–587. doi: 10.1007/BF00878090. 10.1007/BF00878090. [DOI] [PubMed] [Google Scholar]
- Hein TW, Wang W, Zoghi B, Muthuchamy M, Kuo L. Functional and molecular characterization of receptor subtypes mediating coronary microvascular dilation to adenosine. J Mol Cell Cardiol. 2001;33:271–282. doi: 10.1006/jmcc.2000.1298. 10.1006/jmcc.2000.1298. [DOI] [PubMed] [Google Scholar]
- Hilenski LL, Ma XH, Vinson N, Terracio L, Borg TK. The role of beta 1 integrin in spreading and myofibrillogenesis in neonatal rat cardiomyocytes in vitro. Cell Motil Cytoskeleton. 1992;21:87–100. doi: 10.1002/cm.970210202. 10.1002/cm.970210202. [DOI] [PubMed] [Google Scholar]
- Hornberger LK, Singhroy S, Cavalle-Garrido T, Tsang W, Keeley F, Rabinovitch M. Synthesis of extracellular matrix and adhesion through beta (1) integrins are critical for fetal ventricular myocyte proliferation. Circ Res. 2000;87:508–515. doi: 10.1161/01.res.87.6.508. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Dong WJ, Burkart EM, Cheung HC, Solaro RJ. Effects of protein kinase C dependent phosphorylation and a familial hypertrophic cardiomyopathy-related mutation of cardiac troponin I on structural transition of troponin C and myofilament activation. Biochemistry. 2004;43:5996–6004. doi: 10.1021/bi036073n. [DOI] [PubMed] [Google Scholar]
- Lamberts RR, Willemsen MJ, Perez NG, Sipkema P, Westerhof N. Acute and specific collagen type I degradation increases diastolic and developed tension in perfused rat papillary muscle. Am J Physiol Heart Circ Physiol. 2004;286:H889–894. doi: 10.1152/ajpheart.00967.2001. 10.1152/ajpheart.00967.2001. [DOI] [PubMed] [Google Scholar]
- Lenter M, Uhlig H, Hamann A, Jeno P, Imhof B, Vestweber D. A monoclonal antibody against an activation epitope on mouse integrin chain beta 1 blocks adhesion of lymphocytes to the endothelial integrin alpha 6 beta 1. Proc Natl Acad Sci U S A. 1993;90:9051–9055. doi: 10.1073/pnas.90.19.9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Desantiago J, Chu G, Kranias EG, Bers DM. Phosphorylation of phospholamban and troponin I in beta-adrenergic-induced acceleration of cardiac relaxation. Am J Physiol Heart Circ Physiol. 2000;278:H769–779. doi: 10.1152/ajpheart.2000.278.3.H769. [DOI] [PubMed] [Google Scholar]
- Martinez-Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ, Meininger GA. Integrins as unique receptors for vascular control. J Vasc Res. 2003;40:211–233. doi: 10.1159/000071886. 10.1159/000071886. [DOI] [PubMed] [Google Scholar]
- McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogford JE, Davis GE, Meininger GA. RGDN peptide interaction with endothelial alpha5beta1 integrin causes sustained endothelin-dependent vasoconstriction of rat skeletal muscle arterioles. J Clin Invest. 1997;100:1647–1653. doi: 10.1172/JCI119689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogford JE, Davis GE, Platts SH, Meininger GA. Vascular smooth muscle alpha v beta 3 integrin mediates arteriolar vasodilation in response to RGD peptides. Circ Res. 1996;79:821–826. doi: 10.1161/01.res.79.4.821. [DOI] [PubMed] [Google Scholar]
- Nagai T, Laser M, Baicu CF, Zile MR, Cooper G, IV, Kuppus D. Beta3-integrin-mediated focal adhesion complex formation: adult cardiocytes embedded in three-dimensional polymer matrices. Am J Cardiol. 1999;83:38–43. doi: 10.1016/s0002-9149(99)00256-8. 10.1016/S0002-9149(99)00256-8. [DOI] [PubMed] [Google Scholar]
- Nawata J, Ohno I, Isoyama S, Suzuki J, Miura S, Ikeda J, Shirato K. Differential expression of alpha 1, alpha 3 and alpha 5 integrin subunits in acute and chronic stages of myocardial infarction in rats. Cardiovasc Res. 1999;43:371–381. doi: 10.1016/s0008-6363(99)00117-0. 10.1016/S0008-6363(99)00117-0. [DOI] [PubMed] [Google Scholar]
- Nishikawa N, Yamamoto K, Sakata Y, Mano T, Yoshida J, Miwa T, Takeda H, Hori M, Masuyama T. Differential activation of matrix metalloproteinases in heart failure with and without ventricular dilatation. Cardiovasc Res. 2003;57:766–774. doi: 10.1016/s0008-6363(02)00792-7. 10.1016/S0008-6363(02)00792-7. [DOI] [PubMed] [Google Scholar]
- Polte TR, Eichler GS, Wang N, Ingber DE. Extracellular matrix controls myosin light chain phosphorylation and cell contractility through modulation of cell shape and cytoskeletal prestress. Am J Physiol Cell Physiol. 2004;286:C518–528. doi: 10.1152/ajpcell.00280.2003. 10.1152/ajpcell.00280.2003. [DOI] [PubMed] [Google Scholar]
- Ross RS. Molecular and mechanical synergy: cross-talk between integrins and growth factor receptors. Cardiovasc Res. 2004;63:381–390. doi: 10.1016/j.cardiores.2004.04.027. 10.1016/j.cardiores.2004.04.027. [DOI] [PubMed] [Google Scholar]
- Ross RS, Borg TK. Integrins and the myocardium. Circ Res. 2001;88:1112–1119. doi: 10.1161/hh1101.091862. [DOI] [PubMed] [Google Scholar]
- Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, Omura M, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, Ross RS. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–464. doi: 10.1161/hh0402.105790. 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- Solaro RJ. Troponin I, stunning, hypertrophy, and failure of the heart. Circ Res. 1999;84:122–124. doi: 10.1161/01.res.84.1.122. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Rarick HM. Troponin and tropomyosin: proteins that switch on and tune in the activity of cardiac myofilaments. Circ Res. 1998;83:471–480. doi: 10.1161/01.res.83.5.471. [DOI] [PubMed] [Google Scholar]
- Sun M, Opavsky MA, Stewart DJ, Rabinovitch M, Dawood F, Wen WH, Liu PP. Temporal response and localization of integrins beta1 and beta3 in the heart after myocardial infarction: regulation by cytokines. Circulation. 2003;107:1046–1052. doi: 10.1161/01.cir.0000051363.86009.3c. [DOI] [PubMed] [Google Scholar]
- Swartz DR, Moss RL, Greaser ML. Calcium alone does not fully activate the thin filament for S1 binding to rigor myofibrils. Biophys J. 1996;71:1891–1904. doi: 10.1016/S0006-3495(96)79388-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong CW, Gaffin RD, Zawieja DC, Muthuchamy M. Roles of phosphorylation of myosin binding protein-C and troponin I in mouse cardiac muscle twitch dynamics. J Physiol. 2004;558:927–941. doi: 10.1113/jphysiol.2004.062539. 10.1113/jphysiol.2004.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencik ML, McDonald JA. Cardiac expression of a gain-of-function alpha(5)-integrin results in perinatal lethality. Am J Physiol Heart Circ Physiol. 2001;280:H361–367. doi: 10.1152/ajpheart.2001.280.1.H361. [DOI] [PubMed] [Google Scholar]
- Van Der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res. 2001;305:285–298. doi: 10.1007/s004410100417. 10.1007/s004410100417. [DOI] [PubMed] [Google Scholar]
- Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- Waitkus-Edwards KR, Martinez-Lemus LA, Wu X, Trzeciakowski JP, Davis MJ, Davis GE, Meininger GA. Alpha(4)beta(1) integrin activation of L-type calcium channels in vascular smooth muscle causes arteriole vasoconstriction. Circ Res. 2002;90:473–480. doi: 10.1161/hh0402.105899. 10.1161/hh0402.105899. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Fassler R, Warmegard B, Johansson S. Mutational analysis of the potential phosphorylation sites in the cytoplasmic domain of integrin beta1A. Requirement for threonines 788–789 in receptor activation. J Cell Sci. 1998;111:1117–1126. doi: 10.1242/jcs.111.8.1117. [DOI] [PubMed] [Google Scholar]
- Wu X, Mogford JE, Platts SH, Davis GE, Meininger GA, Davis MJ. Modulation of calcium current in arteriolar smooth muscle by alphav beta3 and alpha5 beta1 integrin ligands. J Cell Biol. 1998;143:241–252. doi: 10.1083/jcb.143.1.241. 10.1083/jcb.143.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]