

Abstract
Fc receptor (FcR)-mediated phagocytosis requires activation of the Rho GTPases Cdc42 and Rac1, but how they are recruited to the FcR is unknown. Here we show that the Ca2+-promoted Ras inactivator (CAPRI), a Ras GTPase-activating protein, functions as an adaptor for Cdc42 and Rac1 during FcR-mediated phagocytosis. CAPRI-deficient macrophages exhibit impaired FcγR-mediated phagocytosis and oxidative burst, as well as a defective activation of Cdc42 and Rac1. CAPRI interacts constitutively with both Cdc42 and Rac1, and translocates to phagocytic cups during FcγR-mediated phagocytosis. CAPRI-deficient mice exhibited an impaired innate immune response to bacterial infection. These results suggest that CAPRI provides a link between FcγR and Cdc42 and Rac1 and is essential for innate immune response.
Phagocytosis by specialized phagocytes such as macrophages and neutrophils mediates an essential role in host innate immune responses to microbial infection. Phagocytes utilize a variety of phagocytic receptors to internalize microbes. These include IgG Fc receptors (FcγR), complement receptors, scavenger receptors, integrins, and Toll-like receptors (TLR)1. The molecular mechanisms underlying FcγR-mediated phagocytosis have been extensively investigated2,3. Three types of FcγR, FcγRI, FcγRII, and FcγRIII, are expressed on mouse macrophages4,5. FcγRI and FcγRIII are associated with a γ subunit containing immunoreceptor tyrosine-based activation motifs (ITAM) and function as activating receptors. FcγRII contains tyrosine-based inhibitory motifs (ITIM) and functions as an inhibitory receptor. IgG-opsonized particles initiate phagocytosis by clustering FcγRs, which triggers intracellular signaling, cytoskeletal rearrangement and membrane trafficking2,3. The aggregated FcγRs, as well as the associated γ chain, are then phosphorylated by members of the Src family of protein tyrosine kinases (PTKs)6 and serve as docking sites for downstream signaling molecules. Syk, a tyrosine kinase that plays an essential role in FcγR-mediated phagocytosis is recruited and subsequently activated7,8. Activated Syk induces a downstream signaling process comprised of many signaling molecules such as LAT, SLP-76, BLNK, Nck, Crk1, protein kinase C, phospholipase Cγ, phosphatidylinositol 3-kinase (PI3K), phospholipase D, phospholipase A2, and extracellular signal-regulated kinase (ERK)2,3. However, the precise roles of these signaling molecules and their order in FcγR-mediated signaling remain to be defined.
An important feature of phagocytosis is the rapid reorganization of the actin cytoskeleton with the accumulation of F-actin underneath the periphagosomal region9,10. This process is termed phagocytic cup formation. Members of the Rho GTPases provide signals for this actin reorganization during FcR-mediated phagocytosis. Several reports have demonstrated an essential role for Rac1 and Cdc42 in actin assembly during FcR-mediated phagocytosis11-13. Inhibition of either enzyme in macrophages results in a complete blockade of FcR-mediated phagocytosis due to defective actin assembly at nascent phagosomes11,12. In addition, membrane targeting of active Cdc42 or Rac1 triggers actin polymerization and phagocytosis14,15. Activation of Cdc42 and Rac1 involves a transition from an inactive guanosine 5'-diphosphate (GDP)-bound to an active guanosine 5'-triphosphate (GTP)-bound form and is catalyzed by guanine nucleotide-exchange factors (GEFs)16. In contrast, GTPase-activating proteins (GAPs) function to accelerate the transition of active GTPase, that are GTP-bound, to the inactive GDP-bound form. Vav, a Rho GTPase GEF, activates Rac1 during FcγR-mediated phagocytosis17. However, Rac1 is recruited to the sites of particle attachment in its inactive, GDP-bound state and the recruitment is independent of Vav activity. These data suggest that other unidentified protein(s) plays a role in the localized recruitment of Rac1 and Cdc42 to the FcR.
CAPRI is a member of the GAP1 family of Ras-specific GAPs18. Other members of the GAP1 family of Ras-GAPs include GAP11P4BP, GAP1m, and RASAL19-23. These proteins share four conserved structural domains: two tandem C2 domains at the N-terminus, a GAP-related domain (GRD) in the middle, and both a pleckstrin homology (PH) domain and a Bruton's tyrosine kinase (Btk) motif at the C-terminus. Human CAPRI was cloned and characterized as a Ras-GAP18. It localizes to the cytosol of unstimulated cells and rapidly translocates to the plasma membrane upon Ca2+ mobilization. Finally, CAPRI exhibits Ras-GAP activity only when expressed in cells but not in its purified form18. Here we show that CAPRI plays a critical role in FcγR-mediated phagocytosis and innate immune response to bacterial infection. Our results suggest that CAPRI functions as an adaptor protein that links FcγR to Cdc42 and Rac1.
Results
Generation of CAPRI-deficient mice. We cloned mouse Rasa4(CAPRI) in a gene differential analysis. Full-length mouse CAPRI protein contains 802 amino acids with 2 C2 domains, a GRD and a PH/Btk domain (Supplemental Fig. 1). Mouse CAPRI shares 95% and 87% amino acid identity with rat and human CAPRI respectively. To determine Rasa4 expression, we probed a mouse tissue mRNA blot with a Rasa4 cDNA fragment. Rasa4 mRNA is expressed at high levels in spleen, LN, and muscle and at low levels in brain, thymus, kidney and lung (Fig. 1a). CAPRI protein expression was readily detected in peritoneal macrophages (Fig. 1d).
Figure 1.
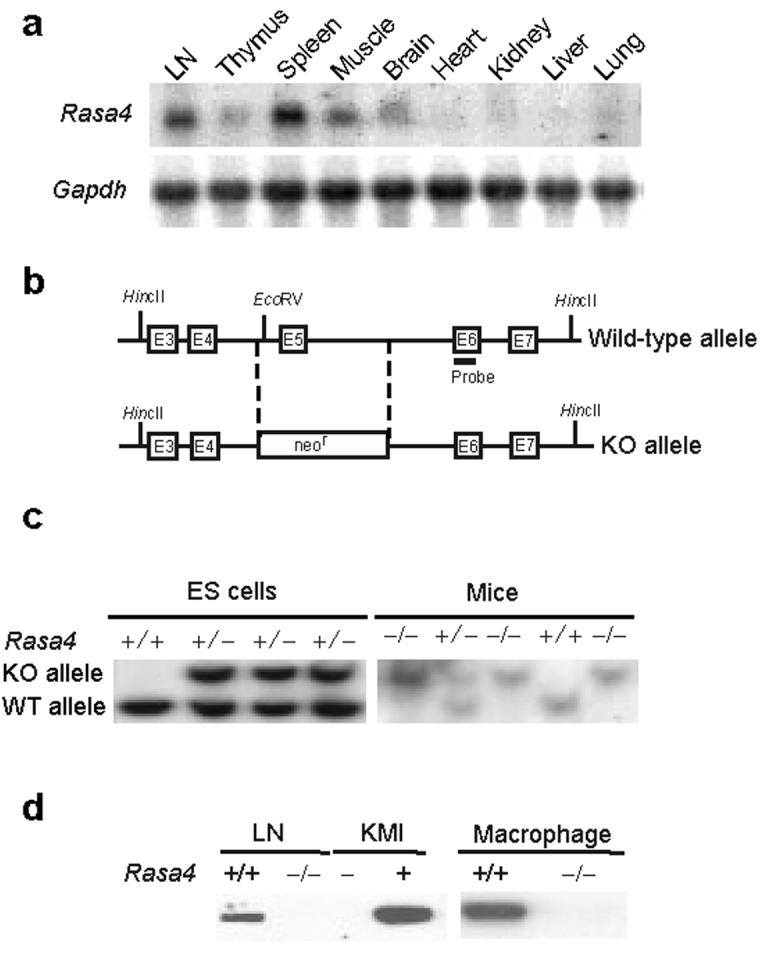
Generation of CAPRI-deficient mice. (a) RNA blot analysis of Rasa4 expression in mouse tissues. Gapdh, glyceraldehyde-3-phosphate dehydrogenase as a loading control. (b) Schematic diagram of the genomic structure of the Rasa4 gene and the targeted allele. Exons are labeled as E3-E7. Restriction sites for EcoRV and HincII and the probe used in DNA blot analysis are indicated. (c) DNA blot analysis of Rasa4-targeted ES cells and mutant mice. Genomic DNA from ES cells or the tails of mutant mice were digested with EcoRV and HincII and hybridized with a probe shown in (b). +/+, +/−, −/− represent wild-type, heterozygous or homozygous alleles of Rasa4, respectively. (d) Immunoblot analysis of CAPRI in lymph nodes and macrophages. Whole cell lysates (10 μg/lane) of lymph node cells and macrophages from wild-type (+/+) and CAPRI-deficient (−/−) mice were blotted with polyclonal anti-CAPRI. Blotting of lysates of KMI T cell hybridoma cells with (+) or without (−) ecotopic CAPRI expression demonstrated the specificity of the anti-CAPRI.
To investigate the function of CAPRI in the immune system, we generated mice lacking CAPRI expression by targeted gene deletion. We replaced exon 5 encoding amino acids 101 to 143 of Rasa4 with a neomycin-resistance cassette (Fig. 1b) and confirmed homologous recombination of the targeting construct in ES cells by DNA blot analysis (Fig. 1c). Three individual ES clones with correct targeting events were used to generate chimeric mice. The initial heterozygous mice on a 129/B6 mixed background were back-crossed with C57BL/6 for 7 generations before phenotypic and functional analyses were performed. CAPRI protein expression was examined with a polyclonal antibody against mouse CAPRI. The antibody specifically detected mouse CAPRI in KMI T hybridoma cells ectopically expressing mouse CAPRI but not in parental cells (Fig. 1d). CAPRI protein was not detected in LN and macrophages of CAPRI-deficient mice in immunoblot analysis (Fig. 1d), indicating a successful deletion of Rasa4 gene in vivo.
Bacterial clearance in CAPRI-deficient mice. CAPRI-deficient mice had normal appearance and lifespan in a specific pathogen free facility. Lymphocyte and myeloid cell development in the thymus, spleen, LN and bone marrow was normal as determined by FACS analysis (data not shown). Given the expression of CAPRI in macrophages, we examined the innate immune response of CAPRI-deficient mice by infecting mutant and control mice with Salmonella typhimurium, a Gram-negative bacterial pathogen. Remarkably, 80% of CAPRI-deficient mice died within 2 days of intraperitoneal injection of S. typhimurium at a dose of 1×105 CFU/mouse whereas none of the controls succumbed (Fig. 2a), indicating that CAPRI-deficient mice are more susceptible to Salmonella infection. We then examined the innate immune response of CAPRI-deficient mice to a Gram-positive bacterial pathogen Streptococcus pneumoniae, a leading cause of community-acquired pneumonia in humans. CAPRI-deficient and control mice were infected with S. pneumoniae by intratracheal injection and observed for 3 days. A majority (60%) of the CAPRI-deficient mice died within 3 days of S. pneumoniae infection. In contrast, all the control mice survived the same bacterial challenge (Fig. 2b). Correlating with their impaired innate immune response to S. pneumoniae infection, histological analysis of lungs from the infected CAPRI-deficient mice showed massive inflammation, with a dense infiltration of neutrophils and monocytes (Fig 2c). In contrast, lungs from infected control mice had normal alveolar architecture and no signs of inflammation (Fig 2c). Furthermore, we did not detect any S. pneumoniae bacteria in lungs of the control mice 3 days after infection. However, an average of 5.3 × 104 S. pneumoniae/g tissue were found in lungs of CAPRI-deficient mice, indicating defective bacterial clearance in lungs of the mutant mice (Fig. 2d). We also infected CAPRI-deficient mice with Haemophilus influenzae and group B streptococcus (GBS). CAPRI-deficient mice showed a dramatically reduced capability to clear these two bacterial strains from their lungs 3 days after intratracheal infection (Fig. 2d). Taken together, these results demonstrate that CAPRI-deficient mice have defective innate immune response to bacterial infection.
Figure 2.
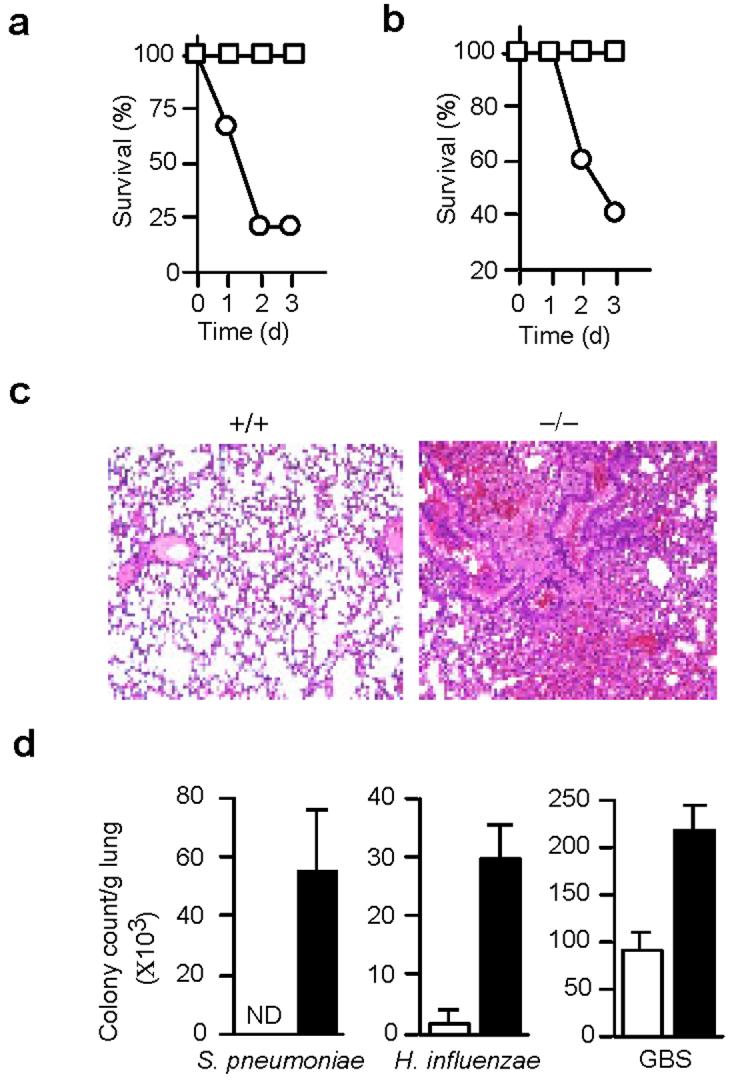
Impaired bacterial clearance in CAPRI-deficient mice. (a) Survival of mice after S. typhimurium infection. Age-matched CAPRI-deficient (circle, n=10) and wild-type control (square, n=10) mice were infected by i.p injection of 1×105 CFU/mouse S. typhimurium and observed for 3 days. (b) Survival of mice after S. pneumoniae infection. Age-matched CAPRI-deficient (circle, n=5) and wild-type control (square, n=5) mice were infected by intratracheal injection of 1×108 CFU/mouse S. pneumoniae and observed for 3 days. (c) Histology of lungs from control (+/+) and CAPRI-deficient (−/−) mice infected with S. pneumoniae (1×105 CFU/mouse). Lungs were removed at day 3 after infection, sectioned and stained by hematoxylin and eosin. Shown are the alveolar architecture of the infected lungs. Representative of 5 mice for each group. (d) Bacterial clearance in the lungs of CAPRI-deficient (filled column) and wild-type control (empty column) mice. Bacteria were inoculated intratracheally (S. pnuemoniae, 105 CFU/mouse; H. influenzae, 2×108 CFU/mouse; GBS, 1.6×105/mouse) into CAPRI-deficient (n=5) and control mice (n=5). 3 days after infection, lungs were removed, homogenized, and plated for colony counts. Shown are mean+SD. ND: not detectable. Data are representative of 2-4 independent experiments.
FcγR-mediated phagocytosis. Macrophages are the major innate immune cell population. Defective bacterial clearance in CAPRI-deficient mice may be the result of impaired phagocytosis and/or activation of macrophages in response to microbial pathogens. To test this, we analyzed bacterial phagocytosis by peritoneal macrophages from CAPRI-deficient and control mice. Compared to controls, CAPRI-deficient macrophages exhibited a 70% reduction in the phagocytosis of S. pneumoniae and a slight decrease in the phagocytosis of H. influenzae. However, phagocytosis of Staphylococcus aureus and GBS was comparable to controls (Fig. 3a). Because the bacteria that were opsonized with normal mouse serum might be phagocytosed by a variety of phagocytic receptors on macrophages, we directly assessed FcγR-mediated phagocytosis using IgG-opsonized sheep red blood cells (SRBC). Compared to control cells, peritoneal macrophages from CAPRI-deficient mice showed an 80% reduction in the phagocytosis of IgG opsonized SRBCs (Fig. 3b). Furthermore, BM-derived macrophages also exhibited similar defect in FcγR-mediated phagocytosis (Fig. 3c). In contrast, CAPRI-deficient macrophages phagocytosed complement-opsonized zymosan particles at amounts comparable to that of control cells (Fig. 3d). Defective FcγR-mediated phagocytosis in CAPRI-deficient macrophages was not due to differences in FcγRII/III expression amounts (Fig. 3e). In addition, the binding of IgG-opsonized SRBCs to CAPRI-deficient macrophages at 4°C was comparable to that of control cells (data not shown). These results suggest that CAPRI may regulate FcγR-mediated phagocytosis downstream of the FcγR.
Figure 3.

Defective FcγR-mediated phagocytosis by CAPRI-deficient macrophages. (a) Phagocytosis of bacteria by CAPRI-deficient macrophages. Elicited peritoneal macrophages from CAPRI-deficient (filled column, throughout a-d) and wild-type control (empty column, through a-d) mice and bacteria opsonized with normal mouse serum were mixed in suspension at a ratio of 1:10 (cell:bacteria) for 20-25 min at 37°C and examined for phagocytosis. (b) FcγR-mediated phagocytosis by CAPRI-deficient peritoneal macrophages. IgG opsonized SRBCs were mixed with macrophages at 10:1 and incubated at 37°C for 15 min before counting. (c) FcγR-mediated phagocytosis by CAPRI-deficient BM-derived macrophages. BM macrophages were mixed with IgG opsonized SRBCs and examined as in (b). (d) Complement receptor-mediated phagocytosis. C3-opsonized FITC-labeled zymosan particles were mixed with CAPRI-deficient and control macrophages at 37°C for 15 min and quenched by trypan blue. The phagocytosed particles were counted under a fluorescence microscope. Data in (a-d) are mean+SD from triplicate determination. Data are representative of 2-4 independent experiments. (e) FcγR expression on CAPRI-deficient (−/−) and control (+/+) macrophages. Elicited peritoneal macrophages were stained with mAb 2.4G2, followed by biotin-anti-rat IgG and PE-avidin. Thick line: 2.4G2 staining. Thin line: cells stained with 2.4G2 plus PE-avidin as background controls.
An early event in FcγR-mediated phagocytosis is actin polymerization underlying the membrane associated with FcγR and phagocytic cup formation. We examined actin polymerization and phagocytic cup formation in CAPRI-deficient and control macrophages using phallacidin to visualize F-actin. Incubation of IgG-opsonized SRBCs with control macrophages readily induced actin polymerization and the formation of multiple phagocytic cups (Fig. 4a). However, a majority of CAPRI-deficient macrophages did not form any visible phagocytic cups (Fig. 4a). The percent of macrophages that contained phagocytic cups is shown (Fig. 4b). Although 92% of the control macrophages formed two or more phagocytic cups upon incubation with IgGSRBCs, phagocytic cup formation was observed in less than 25% of the CAPRI-deficient macrophages, indicating that CAPRI-deficient macrophages are defective in actin polymerization and phagocytic cup formation.
Figure 4.

Defective phagocytic cup formation and oxidative burst in CAPRI-deficient macrophages. (a) Phagocytic cup formation and localization of CAPRI. Elicited peritoneal macrophages from CAPRI-deficient (−/−) and control (+/+) mice were incubated with IgG-opsonized SRBCs at 4°C for 30 min, fixed, permeabilized and stained with phallacidin followed by biotin-anti-CAPRI and Texas-red-avidin. (b) Percent of macrophages from CAPRI-deficient (−/−) and control (+/+) mice with phagocytic cups after incubation with IgG-opsonized SRBCs. A total of 500 macrophages were counted under a fluorescence microscope in each of 3 separate experiments to determine the number of cells with at least 2 or more phagocytic cups. Data are mean+SD. (c) FcγR-mediated respiratory burst in CARPI-deficient (circle) and wild-type control (square) macrophages. Macrophages in suspension were incubated with 120 μg/ml of Fc OxyBURST immune complexes at 37°C and immediately tested for fluorescence by FACS for 10 min. (d) Macrophage production of superoxide anion. Peritoneal macrophages from CAPRI-deficient (−/−) and control (+/+) mice were stimulated with PMA for 30 min and measured for O2− production by chemiluminescent detection.
CAPRI is localized in the cytosol when expressed in COS cells and rapidly translocates to the plasma membrane following increases in intracellular calcium18. Therefore, we examined the localization of CAPRI during FcγR-mediated phagocytosis. CAPRI was clearly enriched in the phagocytic cups within control macrophages after FcγR engagement with IgG-SRBCs (Fig. 4a). In contrast, CAPRI-deficient macrophages displayed only a low amount of background staining and functions as a negative control for the antibody (Fig. 4a). These results suggest that CAPRI translocates to plasma membranes and associates with phagocytic cups during FcγR-mediated phagocytosis.
FcγR-mediated internalization of immune complexes in macrophages induces an oxidative burst that plays an important role in pathogen clearance1. We examined the FcγR-mediated oxidative burst in CAPRI-deficient and control macrophages using insoluble BSA-anti-BSA immune complexes in which the BSA is covalently labeled with dichlorodihydrofluorescein (H2DCF). Oxidation of the H2DCF to DCF results in green fluoresecence that can be monitored by flow cytometry. CAPRI-deficient macrophages exhibited a drastically reduced oxidative burst when compared to control macrophages (Fig. 4c). The impaired FcγR-induced oxidative burst in the mutant macrophages was not due to a general defect in the production of superoxide anion since phorbol-12-myristate-13-acetate (PMA) stimulation of control and mutant macrophages produced similar amounts of superoxide anion (Fig. 4d). Taken together, these results demonstrate that CAPRI plays a critical role in FcγR-mediated phagocytosis and oxidative burst.
CAPRI links FcγR to Cdc42 and Rac1. Activation of Cdc42 and Rac1 promotes actin polymerization and phagocytic cup formation14,15 and is essential for FcγR-mediated phagocytosis11-13. We hypothesize that CAPRI may play a role in the activation of these two Rho GTPases. To assess the activation of Cdc42 and Rac1 in FcγR-stimulated macrophages, we performed an affinity-precipitation assay using the p21-binding domain (PBD) of p21-activated kinase 1 (PAK1) as a binding partner for Cdc42-GTP and Rac1-GTP. Elicited peritoneal macrophages from CAPRI-deficient and control mice were incubated with mAb 2.4G2 recognizing mouse FcγRII and FcγRIII, followed by crosslinking with anti-rat IgG. Cdc42-GTP and Rac1-GTP were readily detected in elicited peritoneal macrophages from control mice and up-regulated upon crosslinking of FcγRII/III by 2.4G2 mAb (Fig. 5a). In contrast, Cdc42-GTP was not detected in freshly isolated peritoneal macrophages from CAPRI-deficient mice and only increased slightly after FcγR stimulation (Fig. 5a). Rac1-GTP levels were also dramatically reduced in resting and FcγR-stimulated CAPRI-deficient peritoneal macrophages (Fig. 5a). We then examined Cdc42 and Rac1 activation in BM-derived macrophages. The amounts of Cdc42-GTP and Rac1-GTP in unstimulated BM macrophages from CAPRI-deficient and control mice were similar (Fig. 5a). However, upregulation of these activated Rho GTPases in CAPRI-deficient BM macrophages after FcγR stimulation was impaired (Fig. 5a). These results demonstrate that CAPRI is critical for the activation of both Cdc42 and Rac1 after FcγR engagement.
Figure 5.

Regulation of Cdc42 and Rac1 activity by CAPRI. (a) FcγR-mediated activation of Cdc42 and Rac1 in CAPRI-deficient (−/−) and control (+/+) macrophages. Equal number of elicited peritoneal or BM-derived macrophages were incubated with anti-FcγRII/III mAb 2.4G2, crosslinked with anti-rat IgG for the indicated times and assayed for Cdc42-GTP and Rac1-GTP with GST-PAK-1 PBD. Total Cdc42 and Rac1 from 10 μl of the lysates were blotted. (b) Amount of Cdc42-GTP and Rac1-GTP in RAW264.7 cells over-expressing CAPRI. RAW264.7 cells expressing hCD2 or CAPRI were treated as in (a) and assayed for Cdc42-GTP and Rac1-GTP with GST-PAK-1 PBD. 10 μl of the whole cell lysate for each sample was blotted with anti-Cdc42, Rac1 and CAPRI antibodies. (c) FcγR-mediated phagocytosis by RAW264.7 cells over-expressing CAPRI (filled column) or RAW264.7hCD2 control cells (empty column). IgG opsonized SRBCs were mixed with macrophages at 10:1 and incubated at 37°C for 15 min before counting. Data are mean+SD of triplicate determinants. Data are representative of 2 independent experiments.
We considered two possible roles of CAPRI in FcγR-mediated phagocytosis. The first possibility is that, similar to Vav, CAPRI functions as a GEF for Cdc42 and Rac1. Because CAPRI exhibits Ras-GAP activity only when transfected into CHO cells but not in its purified in vitro form18, we determined whether CAPRI functions as a GEF for Cdc42 and Rac1 by over-expressing it in RAW264.7 macrophage cell line. Using a retrovirus vector expressing hCD2 as a marker, polyclonal RAW264.7 cells expressing CAPRI were FACS sorted and the amounts of Cdc42-GTP and Rac1-GTP were determined in an affinity precipitation assay (Fig. 5b). As expected, Ras-GTP was decreased in RAW264.7 cells over-expressing CAPRI (data not shown). However, over-expression of CAPRI in RAW264.7 cells did not result in an obvious increase in the amount of Cdc42-GTP and Rac1-GTP before or after FcγR-stimulation (Fig. 5b). Furthermore, overexpression of CAPRI in RAW264.7 macrophage cells did not enhance their FcγR-mediated phagocytosis (Fig. 5c). These results argue against the notion that CAPRI has an intrinsic catalytic activity as a GEF for Cdc42 and Rac1.
An alternative possibility is that CAPRI functions as an adaptor for localized recruitment of Cdc42 and Rac1 to the sites of particle attachment during FcγR-mediated phagocytosis. To determine whether CAPRI interacts with Cdc42 and Rac1 in macrophages, we performed immunoprecipitation assays. Total cell lysates from wild-type peritoneal macrophages were precipitated with antibodies to Cdc42 or Rac1 before or after FcγR crosslinking with 2.4G2 mAb. The precipitates were blotted with anti-mouse CAPRI polyclonal antibody. CAPRI was readily precipitated by anti-Cdc42 or anti-Rac1 with or without FcγR crosslinking (Fig. 6a). To further confirm the interaction between CAPRI and Cdc42 or Rac1, we performed co-immunoprecipitation assays using CHO cells ectopically expressing human CAPRI18. Human CAPRI was also coimmunoprecipitated with Rac1 or Cdc42 in CHO cells (Fig. 6b). Mutation of hCAPRI GRD domain (Arg473 to Ser) that inactivates its GAP activity18 did not affect the interaction between hCAPRI and these two Rho GTPases (Fig. 6b). Furthermore, expression of either wild-type hCAPRI or the hCAPRI GRD domain mutants did not modulate the cellular levels of Cdc42-GTP or Rac1-GTP in CHO cells (Fig. 6c). Taken together, these results demonstrate that CAPRI interacts with Cdc42 and Rac1 constitutively, but it does not appear to modulate Cdc42-GTP or Rac1-GTP amounts when overexpressed.
Figure 6.

CAPRI interacts with Cdc42 and Rac1. (a) Co-immunoprecipitation of CAPRI with Rac1 and Cdc42 in primary macrophages. Total cell lysates of peritoneal macrophages from wild-type mice stimulated through FcγR by 2.4G2 were precipitated with anti-Rac1 and anti-Cdc42 and blotted with anti-CAPRI. Protein G agarose beads without antibodies served as a negative control. (b) Co-immunoprecipitation of human CAPRI with Cdc42 and Rac1 in CHO cells. Total cell lysates of parental CHO cells (lane 1), CHO cells stably expressing wild-type (lane 2 and 4) and mutant hCAPRI (lane 3) were precipitated with anti-Rac1 and -Cdc42 and blotted with anti-hCAPRI. Isotype-matched antibodies were used as negative controls. (c) Amount of Cdc42-GTP and Rac1-GTP in CHO cells over-expressing wild-type and mutant CAPRIs. Cell lysates from parental CHO cells (lane 1), CHO cells expressing wild-type (lane 2) or mutant CAPRI (lane 3 and 4) were assayed for Cdc42-GTP and Rac1-GTP with GST-PAK-1 PBD. Lane 3 and 4 showed results from two individual cell clones expressing the same hCAPRI GRD mutant. 10 μl of the cell lysates used for GTP assays were blotted for quantity control. (d) Co-localization of CAPRI and Rac1. Wildtype macrophages infected with retroviruses expressing myc-tagged mCAPRI were incubated with Ig opsonized SRBCs, fixed, stained with Alexa Fluor 488 labeled anti-myc, TRITC labeled anti-Rac1, and DAPI and analyzed under a confocal microscopy. Anti-Rac-1+ (red) but anti-myc− (green) macrophages were not infected by retroviruses and served as negative controls for anti-myc staining.
The facts that CAPRI constitutively interacts with Cdc42 and Rac1 and is translocated to phagocytic cups during FcγR-mediated phagocytosis suggest that CAPRI may co-localize with these GTPases in the membranes around phagocytic cups. We examined the localization of CAPRI and Rac1 in primary macrophages during FcγR-mediated phagocytosis using confocal microscopy. CAPRI co-localized with Rac1 not only on the membrane edges around phagocytic cups but also intracellularly (Fig. 6d). This data is consistent with the constitutive interaction between CAPRI and Rac1.
We next determined the structural basis underlying the interaction between CAPRI and Cdc42 or Rac1. To this end, we generated myc-tagged wild-type and domain-deleted CAPRI plasmids (Fig. 7a) and transiently expressed these constructs in HEK-293 cells (Fig. 7b). Co-immunoprecipitation assays revealed that CAPRI mutant 2 and 3, containing amino acid 1-555 and 240-802 of CAPRI respectively, retained the capacity to interact with Rac1 and Cdc42 (Fig. 7b). In contrast, CAPRI mutant 1 containing amino acid 1-240 did not interact with either Rac1 or Cdc42 (Fig. 7b). These results suggest that the interaction between CAPRI and Rac1 or Cdc42 is mediated through its GRD domain (Fig. 7a).
Figure 7.

Interaction of CAPRI deletion mutants with Cdc42 and Rac1 and restoration of CAPRI-deficiency by active Cdc42 and Rac1. (a) Schematic of CAPRI protein domains and 3 deletion mutants. Numbers in parentheses are the amino acids contained in each domain or mutant. The filled box at the C-terminus of each protein represents a myc tag. GRD: GAP-related domain. PH/Btk: pleckstrin homology domain and Bruton's tyrosine kinase motif. (b) Co-immunoprecipitation of CAPRI mutants with Rac1 or Cdc42. Cell lysates from HEK-293 cells transiently transfected with mutant CAPRI construct Mut1 (lane 1), Mut2 (lane 2), Mut3 (lane 3), and wild-type CAPRI (lane 4) were precipitated with anti-Rac1 or anti–Cdc42 and blotted with anti-myc (middle and right panel). Construct expression in the same cell lysates were examined by blotting a fraction of the lysates (left panel). The location of IgH and IgL is indicated. (c) Effect of active Cdc42 and Rac1 expression on FcγR-mediated phagocytosis by wild-type (+/+) and CARPI-deficient (−/−) macrophages. BM-derived macrophages were infected with retroviruses expressing active Cdc42GFP or Rac1GFP as indicated and examined for FcγR-mediated phagocytosis under a fluorescence microscopy. The phagocytosis index of GFP- wild-type macrophages (indicated as None) was calculated as 100%. Data are mean+SD from triplicate determinants.
The defective FcγR-mediated phagocytosis in CAPRI-deficient macrophages may also result from impairment in other signaling pathways in addition to Cdc42 and Rac1 activation. To test this hypothesis, we examined whether expression of activated Cdc42 or Rac1 in CAPRI-deficient macrophages restored the defect. We expressed constitutively active Cdc42 or Rac1 tagged with enhanced green fluorescence protein (EGFP) in wild-type and CAPRI-deficient macrophages by retrovirus-mediated gene delivery and examined their phagocytosis capability. Expression of active Cdc42 or Rac1 not only restored the defective FcγR-mediated phagocytosis of CARPI-deficient macrophages but also enhanced FcγR-mediated phagocytosis of both wild-type and CAPRI-deficient macrophages by 50-70% (Fig. 7c). These results suggest that the decreased activation of Cdc42 and Rac1 in CAPRI-deficient macrophages primarily accounts for the defective FcγR-mediated phagocytosis.
CAPRI in Ras-MAPK signaling. Given that CAPRI exhibits Ras-GAP activity when overexpressed in cell lines18, we determined whether CAPRI functions as a negative regulator in Ras-MAPK signaling pathway in macrophages after FcγR stimulation. Elicited macrophages from CAPRI-deficient and control mice were stimulated with mAb 2.4G2 and examined for ERK phosphorylation. CAPRI-deficient macrophages displayed a strong increase in the phosphorylation of ERK proteins 5 minutes after FcγR stimulation (Fig. 8a). We then examined the total protein phosphorylation in CAPRI-deficient macrophages after FcγR stimulation. A slight increase in phosphorylated proteins with molecular weights at the range of 50-80 kD in CAPRI-deficient macrophages before FcγR stimulation was observed (Fig. 8b). However, we did not find obvious difference in the global pattern of protein phosphorylation in mutant and control macrophages after FcγR crosslinking (Fig. 8b).
Figure 8.
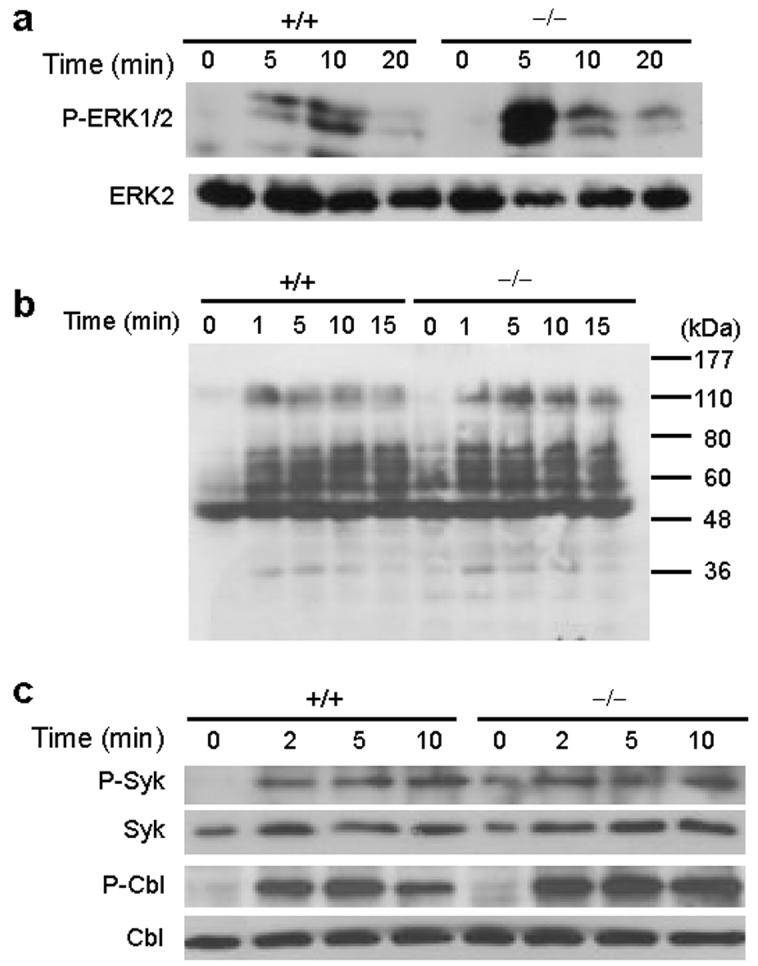
Effect of CAPRI deficiency on activation of ERK, Syk and Cbl. (a) Activation of ERK protein in CAPRI-deficient (−/−) and wild-type (+/+) macrophages. Equal number of elicited macrophages were incubated with anti-FcγRII/III mAb 2.4G2, crosslinked with anti-rat IgG for the indicated times. Total cell lysates were blotted with anti-phospho ERK1/2 or ERK2. (b) Total protein phosphorylation in CAPRI-deficient (−/−) and wild-type (+/+) macrophages. Macrophages were treated as in (a) and lysed for blotting with anti-phospho-tyrosine mAb 4G10. (c) Phosphorylation of Syk and Cbl in CAPRI-deficient macrophages. Cells were treated as in (a). Whole cell lysates were precipitated with anti-phospho-Syk and blotted with anti-Syk. Phospho-Cbl, total Syk and Cbl were determined by directly blotting the total cell lysates.
FcγR-mediated phagocytosis in macrophages is dependent on the Src family PTKs and Syk6-8. Our results demonstrate a critical role of CAPRI in FcγR-mediated phagocytosis and in the activation of Cdc42 and Rac1. To further investigate the possible link between CAPRI and Src PTKs/Syk activity, we examined the phosphorylation of Syk after FcγR crosslinking in CAPRI-deficient and control macrophages. Phosphorylation of Syk in CAPRI-deficient macrophages upon FcγR engagement was not impaired (Fig. 8c). Furthermore, phosphorylation of Cbl, a Src PTKs substrate, in CAPRI-deficient macrophages was comparable to that in control cells after FcγR stimulation (Fig. 8c). These data demonstrate that CAPRI-deficiency did not result in an impaired Src PTKs/Syk activity and suggest that CAPRI-mediated activation of Cdc42 and Rac1 may be in parallel to Src PTKs/Syk activity.
TLR signaling in CAPRI-deficient macrophages. Rac1 has been implicated in TLR2-mediated NF-κB activation24. Given the defective activation of Rac1 in CAPRI-deficient macrophages and the impaired innate immune response in CAPRI-deficient mice, we determined the role of CAPRI in TLR signaling. We stimulated elicited peritoneal macrophages from CAPRI-deficient and control mice with zymosan, peptidoglycan (PGN), lipoteichoic acid (LTA) and lipopolysaccharide (LPS) and measured tumor necrosis factor (TNF) and interleukin 6 (IL-6) production. Macrophages from CAPRI-deficient and control mice secreted similar amounts of TNF and IL-6 (Supplemental Fig. 2a). Furthermore, activation of the Ras-MAPK pathway in CAPRI-deficient and control macrophages was almost identical after stimulation with either heat-killed S. pneumoniae or S. aureus (Supplemental Fig. 2b and 2c). These results suggest that CAPRI does not play an active role in TLR induced signaling pathways.
Discussion
In this report we have demonstrated that CAPRI plays a critical role in FcγR-mediated macrophage phagocytosis and the innate immune response to bacterial infection. We show that CAPRI-deficient macrophages have defective activation of the Rho GTPases Cdc42 and Rac1. It is well established that activation of Cdc42 and Rac1 is required for FcR-mediated phagocytosis11-13. However, it was not clear what link(s) exists between FcγR and Cdc42 and Rac1 activities. Our data suggest that CAPRI functions as an adaptor protein that links FcγR to Cdc42 and Rac1 activities. It is proposed that FcγR clustering activates Syk and it will in turn activate a GEF for Rho GTPases2. Among the many GEFs for Rho GTPases, Vav activates Rac1, but not Cdc42, during FcγR-mediated phagocytosis17. However, the role of Vav in the activation of Rac1 is restricted to catalysis of GDP/GTP exchange on Rac rather than localized recruitment of Rac to the plasma membrane associated with aggregated FcγR, since inhibition of Vav exchange activity does not prevent Rac recruitment to the sites of particle attachment17. Thus, Rac1 recruitment depends on other unidentified molecules.
Four lines of evidence support the notion that CAPRI functions as an adaptor protein for the localized recruitment of both Cdc42 and Rac1 to the plasma membrane associated with clustered FcγR: CAPRI-deficient macrophages exhibit impaired FcγR-mediated but not complement receptor-mediated phagocytosis. This is accompanied by a defective activation of Cdc42 and Rac1 after FcγR crosslinking in both peritoneal and BM-derived macrophages. Both mouse and human CAPRI constitutively interact with Cdc42 and Rac1 in macrophages and cell lines. Our mapping study further suggests that the interaction is mediated through the GRD domain on CAPRI. CAPRI strongly accumulates in actin-rich phagocytic cups and the phagosomal membrane during FcγR-mediated phagocytosis and co-localizes with Rac1 in these areas. Finally, overexpression of mouse CAPRI in RAW264.7 macrophages and human CAPRI in CHO cells does not result in an increased Cdc42-GTP or Rac1-GTP, arguing against CAPRI serving as a GEF for Cdc42 and Rac1. Together with previous work on CAPRI18, our results suggest that upon FcγR clustering, CAPRI targets Cdc42 and Rac1 to the plasma membrane associated with aggregated FcγR. Activation of Cdc42 and Rac1 is then catalyzed by GEFs such as Vav.
The amount of Cdc42-GTP and Rac1-GTP in elicited peritoneal but not BM-derived macrophages from CAPRI-deficient mice without FcγR stimulation is lower than those in control cells. We consider that this may be related to the activation status of these macrophages. Thioglycollate-broth inflamed peritonum provide strong inflammatory stimuli to receptors other than FcRs on macrophages. These activated macrophages respond to chemoattractant signals and activate Cdc42 and Rac1 for their migration. Given the constitutive association of CAPRI with Cdc42 and Rac1 in macrophages, these data suggest that CAPRI may function in the activation of Cdc42 and Rac1 in other pathways in addition to FcγR-mediated signaling.
Defective activation of Cdc42 and Rac1 in CAPRI-deficient macrophages does not result from an impaired signaling events associated with Src PTKs/Syk activities. Previous studies have firmly established the role of Src family PTKs and Syk in FcγR-mediated phagocytosis6-8. Macrophages lacking three Src family PTKs hck, fgr and lyn exhibit impaired internalization of IgG-opsonized particles6. In addition, phosphorylation of Syk, Cbl and total cellular proteins was dramatically reduced or completely absent in macrophages from these triple knockout mice. Macrophages lacking Syk also exhibited similar defects to those lacking Src PTKs7,8, suggesting that Src PTKs/Syk pathway is essential for FcγR-mediated phagocytosis. However, the role of Src PTKs and Syk in actin assembly during FcγR-mediated phagocytosis is not conclusive7,25-27. The fact that macrophages from Syk-deficient mice have normal phagocytic cup formation but cannot complete the closure of the pocket generated by the advancing membrane edges7 suggests that Syk may mediate a signaling event for phagocytic cup closure during FcγR-mediated phagocytosis. Nevertheless, the normal phosphorylation of Syk, Cbl and total cellular substrate in CAPRI-deficient macrophages indicates that CAPRI is not required for these signaling processes. Moreover, the intact signaling of Src PTKs and Syk in CAPRI-deficient macrophages may account for the residual activation of Cdc42 and Rac1 during FcγR-mediated phagocytosis.
Our data suggest that CAPRI plays multiple roles in FcγR-induced signaling. In addition to acting as an adaptor for Cdc42 and Rac1, CAPRI also functions as a negative regulator of Ras-MAPK signaling pathway induced through the FcγR in macrophages. This is supported by the observation that FcγR-clustering of elicited peritoneal macrophages from CAPRI-deficient mice induces a stronger phosphorylation of ERK. Given the importance of Ras-MAPK pathway in TLR signaling28, however, it is a surprise to see that several ligands for TLR2 and TLR4 induce normal cytokine production and phosphorylation of ERK and JNK in CAPRI-deficient macrophages. These results suggest that CAPRI function is not essential in TLR-induced signaling pathway and other Ras-GAPs regulate TLR signaling in macrophages. In addition, the normal TLR2 signaling in CAPRI-deficient macrophages suggest that Rac1 activation by TLR2 signaling24 is independent of CAPRI.
Our data have clearly demonstrated an essential role of CAPRI in the innate immune response to bacterial infection. The inability to clear bacterial infection in CAPRI-deficient mice may reflect defective phagocytosis such as in S. pneumoniae infection. Surprisingly, although CAPRI-deficient macrophages do not exhibit obvious defects in the phagocytosis of H. influenzae and GBS, the in vivo clearance of these bacteria are still impaired. This is likely due to a dramatically reduced oxidative burst in CAPRI-deficient macrophages in vivo. Furthermore, FcR-mediated signaling is the main pathway for oxidative burst in macrophages1. Other phagocytic receptors on macrophages may compensate for the loss of FcR mediated internalization of H. influenzae and GBS but cannot substitute for the oxidative burst induced by FcR stimulation. Alternatively, CAPRI may participate in other pathways critical for clearance of these bacteria in vivo.
Methods
Generation of CAPRI-deficient mice.
Mouse Rasa4(CAPRI) cDNA was isolated by RT-PCR from the thymus of C57BL/6 mice based on sequences from a gene product obtained in a differential analysis. A BAC genomic clone containing the Rasa4 gene was purchased from Genome Systems (St. Louis, MI) and partially mapped by restriction digestion and sequencing. To generate a targeting construct of Rasa4 gene, a 1.8 kb HincII/XhoI fragment upstream of exon 5 of Rasa4 was cloned into the HincII/SalI sites on the pGK plasmid and a 7.2 kb NdeI/SmaI fragment downstream of exon 5 was cloned into the SmaI site. Successful targeting would replace a 1.4 kb of Rasa4 genomic DNA containing exon 5 encoding amino acids 101-143 of CAPRI protein with a neomycin-resistance gene cassette. A diptheria toxin gene was used as negative selection for non-homologously recombined clones. The targeting construct was transfected into TC1 embryonic stem cells derived from the 129 mouse strain29 by electroporation. ES cell clones with homologous recombination were screened by PCR and confirmed by Southern blot analysis. Three individual correctly targeted ES lines were used to generate chimeric mice. Heterozygous CAPRI-mutant mice after back-crossed to C57BL/6 mice for 7 generations were used to generate homozygous CAPRI-deficient mice and wild-type controls. All animals used in the experiments at 6-12 weeks of age were derived from the interbreeding of the above homozygous CAPRI-deficient mice and wild-type controls. Mice were housed in a specific pathogen free facility in Duke University Vivarium and used according to protocols approved by the Duke University Institutional Animal Care and Use Committee.
Bacterial infection of CAPRI-deficient mice.
S. typhimurium, S. pneumoniae, GBS and H. influenzae are clinical isolates provided by J.R. Wright at DUMC. S. aureus is from American Type Culture Collection (ATCC12598). Bacterial cultural plates including nutrient agar, Chocolate II Agar (with hemoglobin and Isovitalex) and Trypticase Soy Agar (with 5% sheep blood) plates were obtained from Becton Dickinson Microbiology Systems (Cockeysville, MD). Age-matched CAPRI-deficient and control mice were infected with S. typhimurium (1 × 105 CFU) by i.p. injection in 200 ml of PBS and observed for their survival for 3 days. Lung infection with bacteria in mice were performed as described30. Briefly, mice were anesthetized by i.p. injection of ketamine and xylazine and an anterior midline incision was made to expose the trachea. A 100 μl inoculum was dispersed into the lungs with a 30-gauge needle attached to a tuberculin syringe. All bacterial doses used for in vivo infection were determined by plating a fraction of the inoculum after the infection and were as follows: 1 × 108 CFU for S. pneumoniae survival assay, 1 × 105 CFU for S. pneumoniae bacterial clearance assay, 2 × 108 CFU for H. influenzae and 1.6 × 105 CFU for GBS in lung infections. Bacterial clearance in the lungs was determined at day 3 after the infection by homogenizing the lungs in 2 ml of sterile PBS and plating on appropriate agar plates for CFU count.
DNA blot and mRNA blot
Isolation of genomic DNA and RNA and blotting of DNA and mRNA were done as previously described31. 2 μg of mRNA from different tissues were analyzed in mRNA blot with a 1 kb cDNA fragment encoding part of CAPRI protein.
Macrophage phagocytosis
Peritoneal macrophages were collected from groups of 2-4 mice 3-4 days following injection of 1ml of 3% thioglycollate broth and isolated by adhering the cells to tissue plates at 37 °C for 1 h. BM-derived macrophages were generated from 6-day cultures of BM cells in DMEM containing 20% FBS and 30% L929 cell-conditioned medium30. The macrophage phagocytosis assay was carried out according to a standard protocol32. Briefly, resident peritoneal macrophages and bacteria opsonized with normal mouse serum were mixed in suspension at a ratio of 1:10 (cell:bacteria), rotated at 37 °C for 20-25 min, washed, stained with Diff-Quik, and counted for at least 200 macrophages per sample under oil-immersion microscopy. Phagocytic index= (% of macrophages containing at least one bacterium) × (mean number of bacteria per positive cell).
For examining FcγR mediated phagocytosis, peritoneal or BM-derived macrophages were cultured on glass coverslip overnight and incubated with SRBCs opsonized with rabbit anti-SRBC IgG (1 mg/ml) (Nordic Immunology, Tilburg, The Netherlands) at a ratio of 1:10. The incubation was carried out at 4 °C 15-30 min for binding and followed by 15-30 min at 37 °C for phagocytosis. Cell-surface bound SRBC were lysed with hypotonic buffer and the macrophages were fixed in 2.5% glutaraldehyde solution before counting.
Complement mediated phagocytosis was performed using FITC-labeled zymosan particles (Molecular Probes). Zymosan particles (5.4 × 107) were opsonized with 150 μl normal mouse serum at 37 °C for 30 min and mixed with macrophages at different ratios. The mixtures were then centrifuged and incubated at 4 °C for 30 min to allow zymosan binding. Unbound particles were washed with cold PBS. Pre-warmed medium was added to initiate phagocytosis. Macrophages were fixed with 3.7% paraformaldehyde after cell surface bound zymosan particles were quenched by trypan blue (2 mg/ml). Phagocytosed FITC-zymosan particles were counted under a fluorescence microscope.
Supplementary Material
Acknowledgements
This work was initiated in M.J. Bevan's laboratory at the University of Washington. We thank his continuous support. We thank W. Zhang, M. Krangel, J. Poe and H. Hartig for critical review of this manuscript. Supported by NIH grants AI54685, CA92123 and American Cancer Society Grant RSG-0125201 to Y.-W. H.
Footnotes
GenBank accession number.
The GenBank accession number for mouse CAPRI is AY591339.
Note: Supplementary information for additional methods and results is available on the Nature Immunology website.
Competing Interests Statement
The authors declare that they have no competing financial interests.
References
- 1.Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 2.Cox D, Greenberg S. Phagocytic signaling strategies: Fc(gamma)receptor-mediated phagocytosis as a model system. Semin. Immunol. 2001;13:339–345. doi: 10.1006/smim.2001.0330. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Garcia E, Rosales C. Signal transduction during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 2002;72:1092–1108. [PubMed] [Google Scholar]
- 4.Ravetch JV, Bolland S. IgG Fc receptors. Annu. Rev. Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 5.Daeron M. Fc receptor biology. Annu. Rev. Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 6.Fitzer-Attas CJ, et al. Fcgamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J. Exp. Med. 2000;191:669–682. doi: 10.1084/jem.191.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crowley MT, et al. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J. Exp. Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiefer F, et al. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell. Biol. 1998;18:4209–4220. doi: 10.1128/mcb.18.7.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr. Opin. Immunol. 2002;14:136–145. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- 10.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 11.Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- 12.Cox D, et al. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 1997;186:1487–1494. doi: 10.1084/jem.186.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massol P, Montcourrier P, Guillemot JC, Chavrier P. Fc receptor-mediated phagocytosis requires CDC42 and Rac1. Embo J. 1998;17:6219–6229. doi: 10.1093/emboj/17.21.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellano F, et al. Inducible recruitment of Cdc42 or WASP to a cell-surface receptor triggers actin polymerization and filopodium formation. Curr. Biol. 1999;9:351–360. doi: 10.1016/s0960-9822(99)80161-4. [DOI] [PubMed] [Google Scholar]
- 15.Castellano F, Montcourrier P, Chavrier P. Membrane recruitment of Rac1 triggers phagocytosis. J. Cell. Sci. 2000;113:2955–2961. doi: 10.1242/jcs.113.17.2955. [DOI] [PubMed] [Google Scholar]
- 16.Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 17.Patel JC, Hall A, Caron E. Vav regulates activation of Rac but not Cdc42 during FcgammaR-mediated phagocytosis. Mol. Biol. Cell. 2002;13:1215–1226. doi: 10.1091/mbc.02-01-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lockyer PJ, Kupzig S, Cullen PJ. CAPRI regulates Ca(2+)-dependent inactivation of the Ras-MAPK pathway. Curr. Biol. 2001;11:981–986. doi: 10.1016/s0960-9822(01)00261-5. [DOI] [PubMed] [Google Scholar]
- 19.Cullen PJ, et al. Identification of a specific Ins(1,3,4,5)P4-binding protein as a member of the GAP1 family. Nature. 1995;376:527–530. doi: 10.1038/376527a0. [DOI] [PubMed] [Google Scholar]
- 20.Maekawa M, et al. A novel mammalian Ras GTPase-activating protein which has phospholipid-binding and Btk homology regions. Mol. Cell. Biol. 1994;14:6879–6885. doi: 10.1128/mcb.14.10.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen M, Chu S, Brill S, Stotler C, Buckler A. Restricted tissue expression pattern of a novel human rasGAP-related gene and its murine ortholog. Gene. 1998;218:17–25. doi: 10.1016/s0378-1119(98)00394-1. [DOI] [PubMed] [Google Scholar]
- 22.Baba H, et al. GapIII, a new brain-enriched member of the GTPase-activating protein family. J. Neurosci. Res. 1995;41:846–858. doi: 10.1002/jnr.490410615. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto T, Matsui T, Nakafuku M, Iwamatsu A, Kaibuchi K. A novel GTPase-activating protein for R-Ras. J. Biol. Chem. 1995;270:30557–30561. doi: 10.1074/jbc.270.51.30557. [DOI] [PubMed] [Google Scholar]
- 24.Arbibe L, et al. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat. Immunol. 2000;1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 25.Cox D, Chang P, Kurosaki T, Greenberg S. Syk tyrosine kinase is required for immunoreceptor tyrosine activation motif-dependent actin assembly. J. Biol. Chem. 1996;271:16597–16602. doi: 10.1074/jbc.271.28.16597. [DOI] [PubMed] [Google Scholar]
- 26.Greenberg S, Chang P, Wang DC, Xavier R, Seed B. Clustered syk tyrosine kinase domains trigger phagocytosis. Proc. Natl. Acad. Sci. U. S. A. 1996;93:1103–1107. doi: 10.1073/pnas.93.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raeder EM, Mansfield PJ, Hinkovska-Galcheva V, Shayman JA, Boxer LA. Syk activation initiates downstream signaling events during human polymorphonuclear leukocyte phagocytosis. J. Immunol. 1999;163:6785–6793. [PubMed] [Google Scholar]
- 28.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 29.Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 30.He YW, et al. The extracellular matrix protein mindin is a pattern-recognition molecule for microbial pathogens. Nat. Immunol. 2004;5:88–97. doi: 10.1038/ni1021. [DOI] [PubMed] [Google Scholar]
- 31.Guo J, et al. Regulation of the TCRalpha repertoire by the survival window of CD4(+)CD8(+) thymocytes. Nat. Immunol. 2002;3:469–476. doi: 10.1038/ni791. [DOI] [PubMed] [Google Scholar]
- 32.Campbell PA, Canono BP, Drevets DA. Measurement of bacterial ingestion and killing by macrophages. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current Protocol in Immunology. John Wiley & Sons, Hoboken; New Jersey: 1996. pp. 14.6.1–14.6.13. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.