

Abstract
Genetically encoded fluorescent probes of neural activity represent new promising tools for systems neuroscience. Here, we present a comparative in vivo analysis of 10 different genetically encoded calcium indicators, as well as the pH-sensitive synapto-pHluorin. We analyzed their fluorescence changes in presynaptic boutons of the Drosophila larval neuromuscular junction. Robust neural activity did not result in any or noteworthy fluorescence changes when Flash-Pericam, Camgaroo-1, and Camgaroo-2 were expressed. However, calculated on the raw data, fractional fluorescence changes up to 18% were reported by synapto-pHluorin, Yellow Cameleon 2.0, 2.3, and 3.3, Inverse-Pericam, GCaMP1.3, GCaMP1.6, and the troponin C-based calcium sensor TN-L15. The response characteristics of all of these indicators differed considerably from each other, with GCaMP1.6 reporting high rates of neural activity with the largest and fastest fluorescence changes. However, GCaMP1.6 suffered from photobleaching, whereas the fluorescence signals of the double-chromophore indicators were in general smaller but more photostable and reproducible, with TN-L15 showing the fastest rise of the signals at lower activity rates. We show for GCaMP1.3 and YC3.3 that an expanded range of neural activity evoked fairly linear fluorescence changes and a corresponding linear increase in the signal-to-noise ratio (SNR). The expression level of the indicator biased the signal kinetics and SNR, whereas the signal amplitude was independent. The presented data will be useful for in vivo experiments with respect to the selection of an appropriate indicator, as well as for the correct interpretation of the optical signals.
Keywords: genetic indicators, GFP, neural activity, transgenic animals, optical imaging, calcium
Introduction
Studies in neurobiology usually rely on recordings of neural membrane potential by means of intracellular and extracellular electrodes. Despite its unmatched directness and temporal resolution, this method offers limited spatial resolution and introduces a bias toward larger neurons. These disadvantages were overcome with the advent of synthetic optical probes for neural activity reporting membrane potential (Cohen et al., 1978) or intracellular calcium (Grynkiewicz et al., 1985). When applied by intracellular injection or as AM-ester (or pressure ejection) (Stosiek et al., 2003), a spatially resolved recording of neuronal activity in single cells (Tank et al., 1988; Svoboda et al., 1997) or in multiple neurons simultaneously can be achieved. As a basic principle, the use of synthetic dyes is invasive, and they cannot be targeted to specific neuronal populations. Genetically encoded indicators (Miyawaki et al., 1997; Persechini et al., 1997; Romoser et al., 1997) offer a solution to this problem. Formed from amino acids, they can bypass the limitations of synthetic dye application by biosynthesis in situ. Different techniques (Brand and Perrimon, 1993; Basler and Struhl, 1994; Lee and Luo, 1999; Zemelman and Miesenboeck, 2001; Gossen and Bujard, 2002; Wong et al., 2002) allow to direct their expression to defined subsets of neurons and even to tag them to subcellular compartments (Miyawaki et al., 1997; Heim and Griesbeck, 2004) (G. Guerrero, G. Agarwal, R. W. Ball, C. S. Goodman, and E. Y. Isacoff, unpublished observation). Moreover, DNA-based probes have been developed for many cellular processes (Tsien and Miyawaki, 1998; Guerrero and Isacoff, 2001; Zhang et al., 2002; Miyawaki, 2003a,b) that could not be monitored previously. Engineered and improved with respect to pH and pCl sensitivity, dynamic range, spectral properties, maturation of the chromophore at 37°C, and subcellular targeting (Miyawaki et al., 1997, 1999; Baird et al., 1999; Griesbeck et al., 2001; Nagai et al., 2001, 2002, 2004; Mizuno et al., 2001; Nakai et al., 2001; Truong et al., 2001; Heim and Griesbeck, 2004), the genetically encoded calcium indicators (GECIs) and the pH-sensitive synapto-pHluorin (SpH) (Miesenboeck et al., 1998; Ng et al., 2002; Bozza et al., 2004) are particularly promising tools for the analysis of neural circuits in vivo. However, only a few of the growing multitude of indicators were applied to neurons of living animals in a limited number of preparations: Caenorhabditis elegans (Kerr et al., 2000; Shyn et al., 2003; Suzuki et al., 2003; Kimura et al., 2004), Drosophila (Fiala et al., 2002; Reiff et al., 2002; Liu et al., 2003; J.W. Wang et al., 2003; Yu et al., 2003; Y. Wang et al., 2004), and zebrafish (Higashijima et al., 2003). So far, direct comparison of the indicator properties was not possible because they were used in different animals and cell types. Many of the presently available indicators have not been used in vivo at all. Only recently, three different indicators were tested in mice and fish (Hasan et al., 2004) and rigorously analyzed in transfected mammalian brain slices (Pologruto et al., 2004). This emphasizes the need to analyze the fluorescence properties of the various indicators in vivo under reproducible and well controlled conditions. To tackle this problem, we engineered transgenic Drosophila flies and analyzed the performance of 10 GECIs plus synapto-pHluorin in presynaptic boutons of the larval neuromuscular junction (NMJ).
Materials and Methods
Flies and genetics. We used the Gal4/UAS-system (Brand and Perrimon, 1993) to direct the expression of indicators to cells of interest. Experimental animals were generated by crossing 10 virgin elavC155-Gal4 flies (generous gift from C. S. Goodman, University of California Berkeley, Berkeley, CA) to five UAS-indicator male flies. All animals were raised on standard corn medium supplemented with fresh yeast at 25°C.
The cDNA of nine different constructs was subcloned into the pUAST vector (Brand and Perrimon, 1993) and inserted into the Drosophila genome by P-element-mediated germ-line transfection (Spradling and Rubin, 1982). In addition, three fly lines, UAS-SpH, UAS-YC2.0, and one additional line, UAS-GCaMP1.3, were obtained from other laboratories (see below) and included in our analysis. The following indicator cDNAs were inserted into the pUAST vector (Brand and Perrimon, 1993). The cDNA of the Yellow Cameleon (YC) variants YC2.3, YC3.3, and Camgaroo-2 (Camg2) (Griesbeck et al., 2001), as well as troponeon-L15 (TN-L15) (Heim and Griesbeck, 2004) were inserted into the NotI site of pUAST; the cDNAs were kindly provided by Oliver Griesbeck [Max-Planck-Institute (MPI) of Neurobiology, Martinsried, Germany]. GCaMP-1.3 (Nakai et al., 2001) was cloned using 5′BglII/3′NotI. GCaMP-1.6 (Yunishi Nakai, unpublished observation) was amplified via PCR using primers containing additional 5′NotI and 3′XbaI sites. Both GCaMP-cDNAs were a generous gift from Yunishi Nakai (RIKEN Brain Science Institute, Wako City, Saitama, Japan). Camgaroo-1 (Camg1) (Baird et al., 1999) was kindly provided by Roger Y. Tsien (University of California, San Diego, La Jolla, CA) and inserted as NotI fragment. Flash-Pericam (FP) and Inverse-Pericam (IP) (Nagai et al., 2001) were generous gifts from Atsushi Miyawaki (RIKEN Brain Science Institute, Wako City, Saitama, Japan). Both cDNAs were amplified via PCR using primers with additional XhoI sites. All UAS-indicator constructs were verified by restriction analysis and sequencing before P-element-mediated germline transfection into white- flies (w-, “Bayreuth”; kindly provided by Christian Lehner, University of Bayreuth, Bayreuth, Germany).
For each indicator, 200-1000 embryos were injected with cDNA. Successful transformants were identified by eye color, and up to 50 independent fly stocks were raised per GECI. The effectiveness of Gal4-driven indicator expression was determined by crossing all new UAS-GECI lines to Gal4 flies (elavC155-Gal4) (Lin and Goodman, 1994) and subsequent antibody staining. Fly lines that gave rise to sufficiently strong expression were selected for the imaging experiments. In some cases, the exact P-element insertion location was mapped molecularly. UAS-YC2.0-2/6i flies (Reiff et al., 2002) containing the original “Yellow Cameleon” 2.0 cDNA (Miyawaki et al., 1997) were kindly provided by Christoph Schuster (University of Heidelberg, Heidelberg, Germany). UAS-SpH flies (Miesenboeck et al., 1998) and additional UAS-GCaMP-1.3 lines were a generous gift from Gero Miesenboeck (Yale University School of Medicine, New Haven, CT) and Yalin Wang (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY), respectively.
All six UAS-indicator fly lines that showed negative imaging results were analyzed to see whether the proper indicator sequence was inserted into the fly genome. Two independent fly lines were analyzed for Flash-Pericam, Camgaroo-1, and Camgaroo-2, respectively. Genomic DNA was extracted from the flies and purified using standard protocols (Dneasy; Qiagen, Hilden, Germany). The indicator cDNA, including ∼150 bp of the flanking pUAST sequences, was then amplified by PCR using forward (CAATCTGCAGTAAAGTGCAAG) and reverse (CTCTGTAGGTAGTTTGTCCAA) primers located to the left and right of the multiple cloning site of pUAST. The PCR products showed the right size, were purified and sequenced using the same set of primers and another set of primers located within the cDNA, and were elongated toward the 5′ and 3′ region of the cDNA, respectively. The obtained sequences of ∼700 bp in the start and stop region were aligned to the published indicator sequences, covered ∼90% of the entire indicator cDNA, and were identical to the published indicator sequences.
Antibody staining. Approximately 1000 filet preparations of F1 female late third-instar larvae were marked, stained, and analyzed under standardized conditions. Late third-instar larvae were dissected in HL3 (Stewart et al., 1994) and fixed for 5 min in ice-cold 4% paraformaldehyde, and 20-24 filets were marked and collected in a common tube in PBS containing 0.1% Triton X-100. After 5% normal goat serum treatment (S-2007; Sigma, St. Louis, MO) for 45 min, the filets were stained for 2 h using the anti-green fluorescent protein (GFP)-IgG conjugated to Alexa Fluor 488 (A-21311; Molecular Probes, Eugene, OR) at a dilution of 1:1000. The washed filets were equilibrated in glycerol 70%/PBS 30% and mounted for fluorescence microscopy.
Physiology and optical imaging. The larval dissection was done as described by Macleod et al. (2002). All experiments were performed in HL6 (Macleod et al., 2002) containing 7 mm glutamate and 1.5 mm calcium, unless otherwise stated. Glutamate effectively blocked postsynaptic muscle contractions at concentrations ≥5 mm without influencing presynaptic Ca dynamics (Macleod et al., 2004). In brief, late third-instar larvae were pinned to the bottom of a self-made recording chamber and opened up along the dorsal midline, and the trachea, gut, and fat body were removed. All segmental nerves were severed, the ventral brain was removed, and larval NMJs were recorded at muscle 6/7 in abdominal segment 2 or 3. Stimulation was done by placing the cut end of the segmental nerve in a suction electrode. Suprathreshold current pulses were applied (Iso-Stim 01-D; NPI Electronics, Tamm, Germany) time locked to CCD image acquisition (Cool Snap camera and MetaView software; Visitron Systems, Puchheim, Germany) and fluorescence excitation (Polychrome IV; T.I.L.L. Photonics, Martinsried, Germany). The temporal order of the applied stimulus paradigms (80, 40, 20, 10, 5, and 0 Hz) was changed randomly. Experiments were controlled by custom software written in Delphi (Borland, Scotts Valley, CA) using an analog-to-digital converter (DAS-1602/12; Computerboards, Middleboro, MA). A fixed-stage microscope (Axioskop; Zeiss, Oberkochen, Germany) with motorized focus drive (Luigs & Neumann, Ratingen, Germany) and a 60× water immersion objective (LumPlanFL; Olympus Optical, Tokyo, Japan) were used for wide-field epifluorescence detection. In all experiments, images were acquired at a rate of 10 Hz and 3 × 3 binning of the CCD, resulting in an ∼290 nm image plane per hyperpixel, also reflecting the approximate optical resolution limit of the optics. YC variants and TN-L15 were imaged by excitation at 430 nm using a 455 dichroic long pass (DCLP) and splitting the emission using a multispec viewer (Optical Insights, Tucson, AZ) with an additional 515 DCLP. SpH, GCaMP-1.3, GCaMP-1.6, Camg1, and Camg2 were imaged by excitation at 470 nm using a Q480LP and a HQ535/50 bandpass. IP was excited at 485 nm, and the emission was separated using a 490 DCLP and an HQ535/50 bandpass. All dichroics and filters were obtained from Chroma Technology (Rockingham, MA).
Data were analyzed using custom software programmed in IDL version 6 (RSI, Boulder, CO). Relative changes in fluorescence (ΔF/F) were calculated by subtracting a control image (Fcntrl) from each image Ft of the series and subsequently dividing the difference images by Fcntrl. The control image was obtained by averaging the images before stimulus onset. For background subtraction, a homogeneous region surrounding the individual boutons was selected. Its intensity was subtracted from the raw intensity of the bouton before calculating ΔF/F as described above. Double-exponential bleach correction was done by deleting the stimulation period before fitting. The resulting function perfectly matched the remaining fluorescence trace and was subsequently subtracted from the original fluorescence trace (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material).
For double-chromophore indicators (see Results), a ratio image sequence was calculated from the enhanced yellow fluorescent protein (EYFP) and the enhanced cyan fluorescent protein (ECFP) fluorescence images. The movie obtained in this way was processed as described above to yield relative changes in the fluorescence ratio (ΔR/R). Background subtraction was done by subtracting a background region from the raw bouton intensity in each of the two channels individually. Then, ΔR/R was calculated as described above.
Two-photon images were taken using a custom built two-photon microscope designed by W. Denk (MPI for Medical Research, Heidelberg, Germany). A mode-locked femtosecond Titan-Sapphire Laser (Tsunami; Spectra-Physics, Mountain View, CA) pumped by a diode-pumped solid-state laser (Millennia; Spectra-Physics) was used to excite YC3.3 at 860 nm. To create image stacks, a series of 256 × 256 images was taken at Δz = 1 μm. The CfNT software (MPI for Medical Research) was used to control the experiment.
Results
Analyzed GECIs and transgenic flies
Our main goal in this study was to characterize and compare different genetic indicators of neural activity in vivo. SpH and 10 different GECIs (Table 1) representing almost all currently known designs of genetic calcium indicators (Tsien and Miyawaki, 1998; Guerrero and Isacoff, 2001; Zhang et al., 2002; Miyawaki, 2003a,b; Rudolf et al., 2003; Griesbeck, 2004) were expressed in the nervous system of flies (see Materials and Methods). The analyzed GECIs were based on a single chromophore (sc), their circular permutated variants (cpsc), or double chromophores (dc). sc and cpsc indicators rely on calcium-dependent rearrangements of the GFP conformation, whereas dc indicators are based on calcium-dependent changes in the efficacy of fluorescence resonance energy transfer (FRET) (Habuchi et al., 2002; Jares-Erijman and Jovin, 2003) between two spectral GFP variants. In all indicators, the calcium-dependent conformational change is brought about by a calcium binding protein such as calmodulin (CaM) or troponin (Tpn). In dc indicators, this calcium binding region connects the fluorophores, in sc indicators, it is spliced into the fluorophore (Camgs), and, in cpsc indicators, the circularly permuted variant of wild-type GFP (GCaMPs) or YFP (pericams) is fused to M13 on the N terminus and to CaM on the C terminus of the chromophore. In detail, flies were equipped with the following cpsc probes: GCaMP-1.3 (Nakai et al., 2001), the new GCaMP1.6 (Nakai, unpublished observation), FP, and IP (Nagai et al., 2001). As sc probes, we chose Camg1 (Baird et al., 1999) and Camg2 (Griesbeck et al., 2001). Thus, we inserted low-affinity sc probes (Camg1 plus Camg2) lacking the CaM binding peptide M13 and high-affinity cpsc probes (GCaMPs and pericams) containing M13 in conjunction with various GFP or YFP variants (for Kd, see Table 1). These GECIs represent the entirety of known calcium probes (except the ratiometric pericam) that are based on a single chromophore (Table 1). Apart from IP, which decreases the emitted fluorescence during calcium binding, all other sc and cpsc indicators increase their fluorescence with increased calcium. All dc GECIs that we tested rely on FRET between ECFP and EYFP or citrine. Among them, we chose the improved cameleon variants YC2.3 and YC3.3 (Griesbeck et al., 2001). These indicators were compared with UAS-YC2.0-flies (original cameleon) (Miyawaki et al., 1997) from a former study (Reiff et al., 2002) and to flies carrying the “Troponeon” TN-L15 (Heim and Griesbeck, 2004). UAS-TN-L15 flies are the first transgenic animals with a calcium indicator whose calcium-binding moiety is derived from chicken Tpn C rather than CaM as in all other GECIs (detailed information is given in Table 1).
Table 1.
Design and in vitro and in vivo properties of genetic probes of neural activity
|
Probe |
Design |
Ca sensor |
Working principle |
In vitro pKaa |
In vitro Kd (μm) |
In vitro change (%) |
Change at 40 and 80 Hz (%) |
τ rise at 40 and 80 Hz (seconds)e |
τ decay at 40 and 80 Hz (seconds)e |
Visibility at restc |
|---|---|---|---|---|---|---|---|---|---|---|
| YC2.0 | ECFP/EYFP | CaM | FRET | 6.9 | 0.1 | 100 | 7.8 | 1.52 | 0.58/2.52 | High |
| (SST) | 11.0 | 11.6 | (0.53) | (0.47/3.39) | ||||||
| YC2.3 | ECFP/citrine | CaM | FRET | 5.7 | 0.1 | 100 | 5.5 | 0.72 | 0.53/3.48 | High |
| 4.3 | 9.4 | (0.35) | (0.53/2.37) | |||||||
| YC3.3 | ECFP/citrine | CaM | FRET | 5.7 | 1.5 | 100 | 5.9 | 1.07 | 0.62/3.02 | High |
| 9.6 | (0.42) | (0.51/2.84) | ||||||||
| TN-L15 | ECFP/citrine | Tpn Cd | FRET | 5.7 | 1.2 | 140 | 6.9 | 0.49 | 1.29 | High |
| 8.2 | (0.31) | (1.29) | ||||||||
| Camg1 | Split EYFP | CaM | Ca-induced | ∼7 | 7.0 | 700 | (—) | Very low | ||
| (SVQST) | pKa change | |||||||||
| Camg2 | Split citrine | CaM | Ca-induced | ∼7 | 5.3 | 700 | (—) | Moderate | ||
| pKa change | ||||||||||
| FP | cpEYFP | CaM | Ca-induced | ∼7 | 0.7 | 800 | None | |||
| pKa change | ||||||||||
| IP | cpEYFP | CaM | Ca-induced | ∼7 | 0.2 | down | −6.7b | 0.61 | 0.90 | High |
| pKa change | to 15 | −8.9b | (0.28) | (0.98) | ||||||
| GCaMP 1.3 | cpEGFP | CaM | Ca-induced | ∼7 | 0.235 | 450 | 8.2 | 0.84 | 0.46 | Moderate |
| pKa change | 16.1 | 0.31 | 0.48 | |||||||
| GCaMP 1.6 | cpEGFP | CaM | Ca-induced | ∼8.5 | 0.146 | 480 | 6.2 | 0.56 | 0.34 | High |
| pKa change | 17.8 | 0.16 | 0.35 | |||||||
| SpH | EGFP | pKa | ∼6 | 600 | 9.1 | 4.33 | f | Moderate | ||
|
|
|
|
|
|
|
|
16.5 |
2.49
|
f
|
|
Differences in the molecular design of the probes, their spectral properties, Kd, the calcium binding moiety, and pH sensitivity are summarized. The in vitro performance, given in the original literature, is compared with the results from our in vivo experiments. In general, the signal changes observed in vivo tended to be much smaller than the maximum change observed in vitro. Subtraction of the background increases the calculated amplitudes only by a factor of 3-5, without increasing the SNR (see Fig. 4). Only small signals were, in particular, observed for Camg1 plus Camg2 and FP; the latter did not exhibit any detectable fluorescence. The other indicators exhibited reliable fluorescence changes relative to baseline. Their amplitude and time constant for the rise and decay are shown. Reliable time constants in the absence of saturation of the indicator are in bold. Besides these functional aspects, the fluorescence of the indicators at resting calcium (rightmost column) is an important parameter for the identification of expressing neurons in live experiments, in particular at the single-cell and subcellular level. SST, Mutations S65G, S72A, T203Y; SVQST, S65G, V68L, Q69K, S72A, T203Y; cp, circularly permutated.
In dc probes, the relevant pKa of the longer wavelength chromophore.
Calculated after subtraction of experiments without stimulus (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Experimentally determined in presynaptic boutons ∼30 min after cutting the innervating axon.
Tpn C from chicken skeletal muscle.
Time constants were fit to a single exponential with R2 = 0.99. Only the decay of the cameleon indicators had to be fitted by a double-exponential function (τ1/τ2). The decay of TN-L15 signals followed a single-exponential time course.
SpH did not return to baseline by the end of the experiment.
Indicator expression was determined by crossing all new UAS-GECI lines to Gal4 flies (elavC155-Gal4, hereafter called “the Gal4-driver”) that drive expression in the entire nervous system (Lin and Goodman, 1994). The specificity of the GECI fluorescence was analyzed in F1 female animals by two-photon laser scanning microscopy (Denk et al., 1990). Figure 1A shows a section of a larval hemisegment in a YC3.3-expressing animal. GECI fluorescence was detected exclusively in the nervous system of the larvae. Because the fluorescence at rest is low in some of the sc and cpsc probes (see above and Table 1), the evaluation of the expression level of our new UAS-GECI lines was routinely done by screening with a fluorescent antibody against GFP (Alexa Fluor 488; for details, see Materials and Methods) (Fig. 1B-D). We found that the efficacy of Gal4-driven indicator expression varied strongly between individual lines (Fig. 1B-D), presumably depending on the chromosomal location of the P-element (Spradling and Rubin, 1983). Most fly lines containing probes based on cpsc fluorophores did not give rise to noteworthy expression. However, this approach allowed us to identify suitable UAS-GECI lines and to generate experimental animals with sufficiently high and different expression levels of each indicator. In total, the GECI expression level could be controlled in many ways: using lines with a single GECI copy but different expression efficacy (Fig. 1B-D), using lines with multiple GECI copies (see Figs. 3, 4, GCaMP1.3), or by rendering experimental flies heterozygous or homozygous for GECIs and/or the Gal4 source (Fig. 1E). Later, we will show how the expression level relates to the measured fluorescence changes (see Fig. 5).
Figure 1.

Specificity and expression level in newly generated UAS-GECI fly lines. A, A YC3.3-expressing larva is shown. Two-photon imaging revealed the specificity of elavC155-Gal4-driven UAS-GECI expression in the larval nervous system. Expression is specifically directed to axons and NMJs. B-D, Anti-GFP stainings at larval NMJs (see Materials and Methods) revealed varying expression levels in different fly lines (UAS-GCaMP1.6-29c, -48b, and -30i in B, C, and D, respectively, crossed to elavC155-Gal4). This protocol allowed us to routinely evaluate new UAS-GECI lines also for sc indicators for which fluorescence is disrupted at rest. E, Incident light (top) and a fluorescence light microscopy image (bottom) of the same adult flies: the level of GECI expression in experimental animals can also be “adjusted” by increasing the number of copies encoding Gal4 and/or the UAS-indicator cDNA. The leftmost animal is homozygous for Gal4 as well as for UAS-YC3.3, the rightmost animal is heterozygous for both, and the animal in the middle is a homozygous/heterozygous mixture. Scale bar, 20 μm.
Figure 3.

Comparison of presynaptic fluorescence changes of 10 different GECIs and SpH. Presynaptic boutons were imaged and analyzed as described (see Fig. 2 and Materials and Methods). Calcium influx and vesicle fusion were evoked by electrical stimulation (80, 40, and 0 Hz, from t = 1 to 3.2 s) of the nerve. The exhibited mean ΔF/F and ΔR/R of sc and dc indicators, respectively, was calculated from the unmodified raw data and plotted versus time. The number of analyzed animals, NMJs, and boutons is indicated in brackets. NMJs expressing GCaMP1.3 (A), GCaMP1.6 (B), IP (C), SpH (D), YC2.3 (E), YC3.3 (F), YC2.0 (G), and TN-L15 (H) exhibited reliable fluorescence changes. At 40 Hz (data not shown) and 80 Hz stimulation, no or only very small signals, respectively, were recorded from Camg1-expressing (I) and Camg2-expressing (J) boutons. Increasing the extracellular calcium concentration to 10 mm (J, light gray trace) resulted in a degradation of the fluorescence signal that we cannot explain. FP-expressing animals (n > 10) were imaged by focusing on the cleft between muscle 6/7 because the FP fluorescence could not be detected at rest. Stimulation did not result in a detectable increase in fluorescence. After the imaging experiment, we verified FP expression in these NMJs by GFP antibody staining (K). UAS-Camg1-, UAS-Camg2-, and UAS-FP-indicator flies were tested for proper indicator insertion (see Materials and Methods). A detailed description of the fluorescence changes is given in Results and Table 1.
Figure 4.
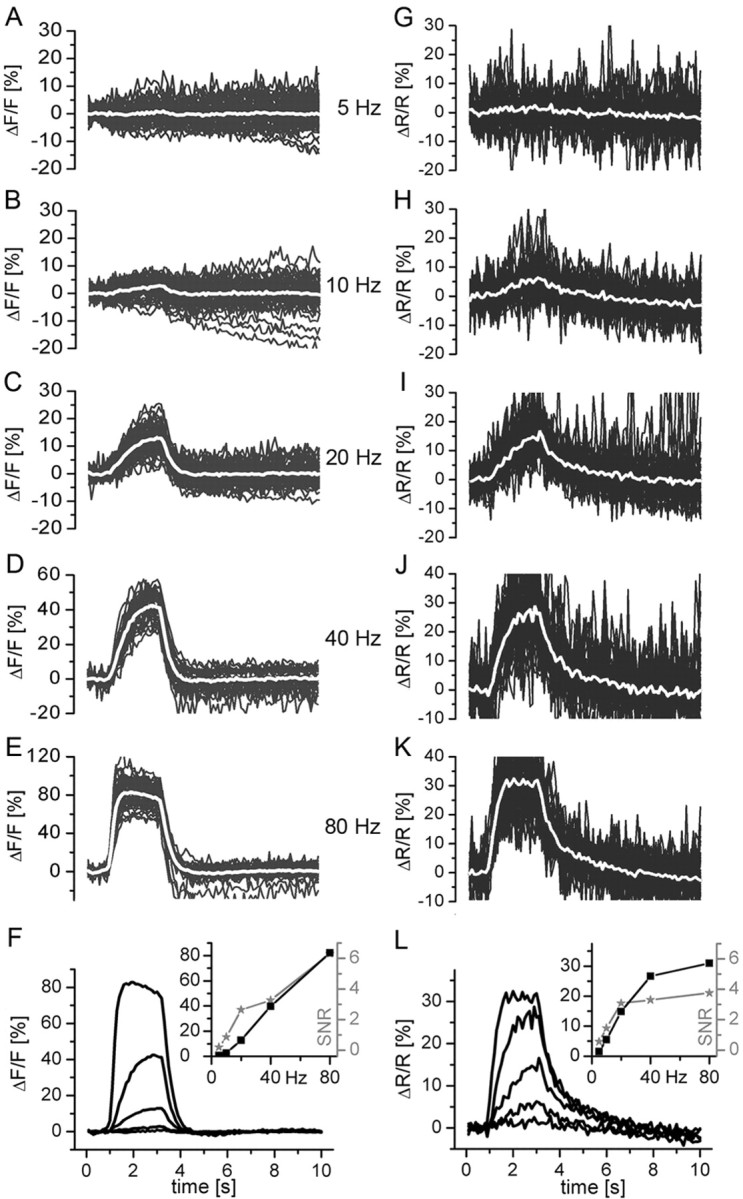
Linearity and SNR of the fluorescence signals. NMJs expressing either GCaMP1.3 (A-F) or YC3.3 (G-L) were stimulated at five different frequencies (5, 10, 20, 40, and 80 Hz). Signals from individual boutons are plotted in gray, and the corresponding average response is shown in white. Approximately 50 boutons were analyzed at each stimulus frequency. Signals of both indicators were corrected for the background fluorescence, and GCaMP1.3 signals were also corrected for the bleaching (see Materials and Methods) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In F and L, the mean fractional fluorescence changes are shown for the different stimulation frequencies. In the inset, the peak amplitudes (black squares and line) and SNR (gray asterisks and line) are given for the individual stimuli. For the calculation of the SNR, five time points surrounding the peak of the average response were taken per individual bouton and divided by the number of selected time points. The mean of these amplitudes was divided by the SD of the mean (SNR is mean amplitude/SD).
Figure 5.

The kinetics and SNR of the fluorescence change depend on the GECI expression level. Two different UAS-YC2.3 lines were analyzed that gave rise to different amounts of Gal4-driven indicator expression. Stimulation at 40 Hz evoked identical mean ΔR/R amplitudes (white traces) in weakly expressing animals (A) and strongly expressing animals (B). In A, line 5b is shown (6 animals, 71 boutons), and, in B, line 30l is shown (7 animals, 94 boutons). Individual boutons are plotted in gray. For the calculation of ΔR/R, the background fluorescence was subtracted in the individual channels (see Materials and Methods). C, Identical amplitudes but faster kinetics, in particular the rise of the fluorescence signals, were observed in the weakly expressing line (5b, τ = 594 ± 58 ms; 30l, τ = 909 ± 48 ms; single-exponential fit). Before subtraction of the background, similar results were found for the time constant of the decay. However, after subtraction of the background, this was less obvious. D, Quantification of the indicator expression level (judged by the citrine intensity after background subtraction), mean ΔR/R, and SNR (for SNR calculation, see Fig. 4) of the two lines. *p < 10-5 (unpaired t test). Details are given in Results. a.u., Arbitrary units.
Analysis of genetic indicators in presynaptic boutons
UAS-GECI stocks that gave rise to clearly detectable expression were crossed to the pan-neuronal Gal4-driver. At the level of wide-field epifluorescence microscopy, in conjunction with CCD imaging (see Materials and Methods), individual NMJs (Fig. 2A) at muscle 6/7 were readily identifiable. Thus, the specificity of the Gal4-driver allowed us to optically isolate structures of interest such as nerves, axons, and presynaptic terminals and to characterize the exhibited fluorescence changes in the different indicators on the level of individual boutons in vivo (Fig. 2A-C, YC3.3). Electrical stimulation of up to 80 Hz mimicked the natural activity of this synapse (Barclay et al., 2002). From the recorded raw image sequences, we calculated (see Materials and Methods) the fractional fluorescence change (ΔF/F) and the fractional change in FRET (ΔR/R). In Figure 2D, false-color images are shown that display changes in ΔR/R observed at the junction shown in Figure 2A (supplemental movie 1, available at www.jneurosci.org as supplemental material). Individual boutons were selected from the raw image, and their fluorescence changes were calculated (Fig. 2E). This was done for 5-20 boutons per movie of a single NMJ. During the stimulation period, dc indicators always showed negative fluorescence changes in the CFP channel and positive changes in the YFP or citrine channel (Fig. 2E, bottom graph), indicating that the observed fluorescence changes were not attributable to motion, bleaching, or other artifacts. The predominant number of the analyzed boutons was located in the central part of the NMJs and showed quite uniform fluorescence changes (Fig. 2E, bottom and top graphs, superimposed black traces), which is in accordance with a previous study (Reiff et al., 2002). We found ∼10% signal change when ΔR/R (Fig. 2E, top graph) was calculated compared with only ∼5% ΔF/F in the individual channels (Fig. 2E, bottom graph), and, at the same time, bleaching was reduced in the ΔR/R movies (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The background was free of fluorescence changes (signals from the pink background regions in Fig. 2A are represented by the pink traces in Fig. 2E) if no out-of-focus presynaptic structures were included.
Figure 2.
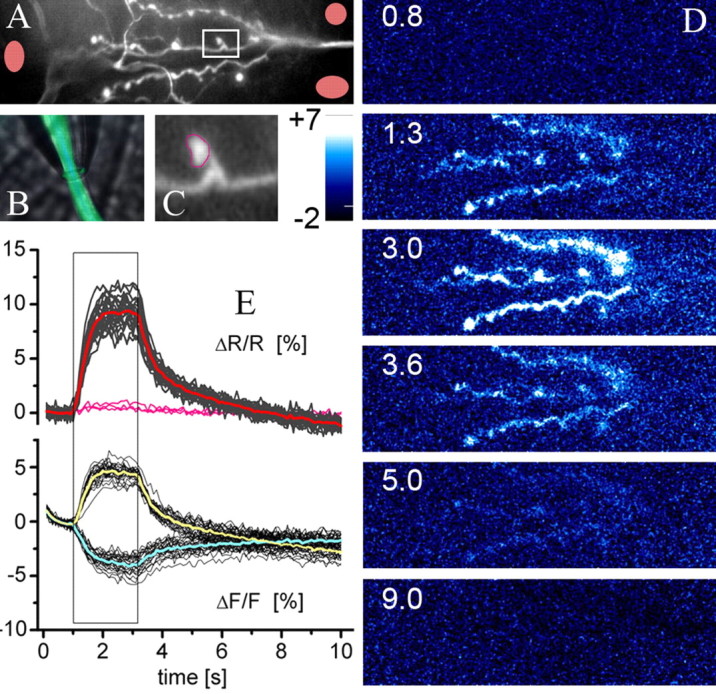
Analysis of indicator performance at the subcellular level in larval NMJs of transgenic Drosophila. A, The raw image of an NMJ at muscle 6/7 shows that boutons can be visually identified by the indicator fluorescence at rest. B, The segmental nerve that harbors the relevant axons was placed in a stimulation electrode. C, The close-up of the bouton (marked by the white box in A) shows that individual boutons can be reliably identified for off-line analysis of fluorescence changes. D, Spatially low-pass-filtered false-color images encoding ΔR/R (no subtraction of the background in D and E) during the time course of an experiment (80 Hz stimulation for 2.2 s; see black box in E). The numbers (in seconds) indicate the time points at which the images were calculated. E, Off-line analysis of the fluorescence changes in ∼20 individual presynaptic boutons during the same experiment. Thin black traces represent signals from individual boutons, and mean ΔF/F calculated for CFP and YFP is plotted in cyan and yellow, respectively. The mean ΔR/R is shown in red. Signals from background regions (pink areas in A) are represented by pink traces. The background (i.e., the muscle cells) does not show fluorescence changes.
In vivo comparison of 10 GECIs and SpH in presynaptic boutons
In presynaptic boutons of Drosophila and the crayfish NMJs, the fluorescence signal was shown previously to rapidly reach steady state, the amplitude of which depends linearly on the stimulus frequency (Tank et al., 1995; Macleod et al., 2002), matching the underlying steady-state calcium calculated in a single compartment model (Tank et al., 1995). If linearity is assumed for calcium binding to the GECIs, the fluorescence intensity should linearly increase with the stimulus frequency. Also, the time constant of the rise of the fluorescence signals should be identical for 40 and 80 Hz stimulation [but see GCaMP1.3 (Nakai et al., 2001)]. Deviations of this notion suggest saturation of the indicator and/or nonlinear calcium binding. In addition, other intrinsic properties such as bleaching and interference with the endogenous calcium buffers (i.e., the expression level of the indicator) might influence the exhibited fluorescence signals. In Figure 3, we present a compendium of 11 activity indicators analyzed in vivo. The indicators cover almost the complete history of GECI engineering and affinities for calcium, ranging from Kd of ∼0.1 to ∼ 10 μm when analyzed in vitro (Table 1). In-depth information for a selection of them is given in the following figures. The shown GECIs were used to record fluorescence changes linked to changes in residual presynaptic calcium. The fast nanodomain calcium, which directly controls triggered vesicle release, could not directly be measured. The presynaptic vesicle release itself was analyzed by including UAS-SpH flies in our study (superecliptic SpH) (Miesenboeck et al., 1998; Ng et al., 2002). Based on pH differences between the intravesicular and extracellular space, SpH makes it possible to directly measure presynaptic vesicle fusion, a key parameter for the analysis of neural activity. Using the described preparation and protocol, stimulation at either 40 Hz (Fig. 3A-H, gray traces) or 80 Hz (Fig 3A-H, black traces) produced robust fluorescence changes in SpH (Fig. 3D) and 7 of the 10 different GECIs. The responsive GECIs were GCaMP1.3, GCaMP1.6, IP, YC2.3, YC3.3, YC2.0, and TN-L15 (Fig. 3A-C,E-H). In Figure 3, the mean ± SEM ΔF/F or ΔR/R of large data samples (numbers indicated in the brackets) was calculated for sc, cpsc, and dc indicators, respectively, and plotted versus time. In addition, a control movie (Fig 3A-H, light gray traces) was taken in each experiment without any stimulation, resulting in a total of three stimulus conditions measured from each analyzed NMJ. The control movies allow a crude estimate of the corruption of the measured fluorescence changes by photobleaching, although bleaching is likely to be more pronounced in the more fluorescent state of the chromophore(s). This is suggested by the decrease of the GCaMP1.3 and particularly of the GCaMP1.6 fluorescence during the 80 Hz stimulation period (Fig. 3A,B) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Because of the very inhomogeneous fluorescence properties of the different indicators, none of the recorded fractional fluorescence changes in Figure 3 is corrected for photobleaching or background fluorescence. We rather show most original and unmodified data. Examples for the effect of background subtraction and bleach correction on the signal amplitudes are given in Figures 4 and 5. Table 1 summarizes the maximum relative fluorescence change or fluorescence ratio change, time constant of signal rise and decay, and visibility of the indicator at rest obtained from the data in Figure 3.
In most cases, two copies of UAS-GCaMP1.3 (on second and third chromosome) had to be used to result in clearly visible NMJs at rest (Table 1). GCaMP1.3 (Fig. 3A) showed a double-exponential bleaching whose fast component ended clearly before the onset of the action potential (AP) volley (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Looking at the relative change in the fluorescence during 80 Hz stimulation, when the fluorophore became highly fluorescent, the signal exhibited a peak response and did not clearly settle to a steady state. This was still observed after applying a double-exponential bleach correction (Fig. 4) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The missing steady state is in contrast to the reported time course of presynaptic calcium in this preparation measured with synthetic dyes (Macleod et al., 2002, 2004) during prolonged stimulation periods. At 40 Hz stimulation, no such transient peak is visible. A mild, long-lasting undershoot after stimulus offset was visible after both stimulation paradigms. This undershoot exceeded the bleaching in the control movies and indicates increased bleaching of the fluorophore in the more fluorescent state. The maximum ΔF/F of GCaMP1.3 increased twofold when the stimulation frequency was increased from 40 to 80 Hz (from 8 to 16%). A qualitatively similar result (i.e., a linear fluorescence increase over an expanded stimulus range) was also obtained after background subtraction and bleach correction (Fig. 4). In contrast, in the same activity regimen, an almost threefold increase was observed in GCaMP1.6 (from 6 to 17%). GCaMP1.6 (Fig. 3B) showed much higher fluorescence at rest (Table 1) when compared with GCaMP1.3, requiring only a single copy of cDNA for its detection. GCaMP1.6 showed stronger initial bleaching, exhibited a clear peak with no steady state after stimulation (at both 40 and 80 Hz), and showed a more pronounced undershoot after stimulation. This undershoot is not linearly linked to the fraction of indicator bound to calcium. At 80 Hz stimulation, the undershoot is only slightly more pronounced than at 40 Hz stimulation (Fig. 3B). These phenomena were similarly seen after background subtraction (data not shown). In summary, GCaMP1.6 was easier to detect at rest compared with GCaMP1.3 and showed faster kinetics (Table 1) of the fluorescence signal, offsetting the disadvantage of more pronounced bleaching. A narrow dynamic range would be expected for both indicators from their large Hill coefficient of n = 3.3 [GCaMP1.3 (Nakai et al., 2001)] and n = 3.8 [GCaMP1.6 (Nakai, personal communication)]. Moreover, we expected saturation of the GCaMPs in the experiments in Figure 3 because calcium changes of many hundred nanomoles up to the micromolar range can be expected in the Drosophila boutons under these conditions (Macleod at al., 2004), and Kd values of 235 and 146 nm have been reported for GCaMP1.3 (Nakai et al., 2001) and GCaMP1.6, respectively, when measured in vitro (Nakai, personal communication). This discrepancy cannot be explained by the interplay of several compensating factors (such as cooperative calcium binding, saturation of the probe, missing background subtraction, and bleaching) because a linear relationship of the GCaMP1.3 fluorescence to neural activity was observed (Fig. 4). These results rather suggest a shift in the Kd that is further supported by the work of Pologruto et al. (2004) who reported a Kd of 1.7 μm for GCaMP1.3 when expressed in hippocampal slices after virus transfection. The saturation of both GCaMPs that is at first glance suggested by the reduction of the time required for the rise of the fluorescence signals (from 840 to 310 ms and from 560 to 160 ms for GCaMP1.3 and GCaMP1.6, respectively) (Table 1) is most likely entirely mediated by the calcium-dependent kinetics of this indicator. In GCaMP1.3, the time for the rise is known to depend strongly on calcium, such that increased calcium leads to a much faster time to rise (Nakai et al., 2001).
In our study, the only other functional calcium indicator based on a single chromophore was IP (Fig. 3C). IP appeared a promising tool for experiments with single cell expression for its high fluorescence at rest and still reasonable big fluorescence change. However, it bleached quickly and was highly nonlinear. We found a decay of 20% ΔF/F within the first second after the onset of illumination, which corrupted signal detection substantially under these conditions. Because the initial time course matches perfectly (Fig. 3C, superimposed traces), it might be fair to subtract the control movies from the stimulus movies (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Then, and only in IP, presynaptic activity could be detected more clearly and was time locked to evoked neuronal activity. The maximum ΔF/F amplitude increased only slightly (from 6.7 to 8.9%) while the initial rate of rise was faster for 80 Hz stimulation, indicating that the evoked presynaptic calcium increase was sufficient to partially saturate IP. Clear saturation of IP is suggested by the reported in vitro Kd of 200 nm (Nagai et al., 2001). The found moderate saturation is in better accordance with the reported Kd of 900 nm in brain slices (Pologruto et al., 2004). Compared with the fast rise (τ = 600 ms), a rather slow decay (τ = 900 ms) of the fluorescence signal was observed at 40 Hz stimulation (Table 1).
In contrast to the cpsc indicators, the dc indicators YC2.3 and YC3.3 (Fig. 3E,F) showed a more stable fluorescence ratio (ΔR/R) before stimulus onset and only -2% signal change during the control movie. This suggests that the YC2.3 and YC3.3 signals were only moderately reduced by bleaching (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Also during 80 Hz stimulation, the signals increased to an approximately stable steady-state level, which is also visible after background subtraction (Fig. 4K). At 40 Hz, the plateau was just reached at the end of the stimulation period, and the rise of the YC2.3 and YC3.3 signals showed a τ of 700 ms and 1 s, respectively, which is clearly slower compared with TN-L15 (Fig. 3H). After stimulation, the signal decayed with a rather slow time course that was fitted by a double-exponential function (Table 1). The decrease of the time to rise and the sublinear increase in ΔR/R (from 5.5 to 9.4% in YC2.3 and from 5.9 to 9.6% in YC3.3) suggest beginning saturation. This can also be seen in Figure 4L, in which five different stimuli and subtraction of the background were applied to YC3.3.
In our experiments, these cameleons never reached the maximum value of 100% ratio change reported in vitro, and comparable results were obtained from YC2.0. In YC2.0, comparable changes in the time to rise and the fluorescence amplitude were found (Fig. 3G) at 40 and 80 Hz stimulation (7.8 and 11.6% ΔR/R, respectively). Initially published by Miyawaki et al (1997), this “original” cameleon performed well in our experiments as it did in a former study (Reiff et al., 2002). The YC2.0 signals follow the description of the other YC variants with only minor differences. The kinetics of the YC2.0 fluorescence change were slightly slower and the maximum signal changes were bigger compared with the cameleon.3 variants (Table 1). Both results correspond well to the high cameleon expression level in the YC2.0 experimental animals (for the influence of the indicator expression level, see Fig. 5). No pronounced deficits could be observed, although YC2.0 lacks the intended improvements (brighter fluorescence and pH stability) made in the acceptor of YC2.3 and YC3.3 (Miyawaki et al., 1999; Griesbeck et al., 2001). In fact, it is the fluorescence ratio at rest that was even more stable (Fig. 3G, light gray trace) for YC2.0 than for the others. Ratiometric imaging and good visibility at rest made these indicators easy to work with (Fig. 3E-G; Table 1). These aspects also apply to TN-L15.
The troponin C-based reporter TN-L15 showed the least sign of bleaching of all of the GECIs (Fig. 3H, light gray trace). Its fluorescence was stable before stimulus onset, and it then rose quickly during stimulation and rapidly reached an almost stable steady state at 80 Hz stimulation. Increasing the stimulation from 40 to 80 Hz increased the maximum ΔR/R only moderately from 6.9 to 8.2%, indicating that the indicator is close to saturation. At 40 Hz stimulation, the rise phase of the TN-L15 signal showed the fastest time constant of all indicators (τ = 490 ms), and the decay had a time constant of τ = 1.3 s. In contrast to the cameleons, the TN-L15 decay was well fit by a single exponential. This finding might be linked to the fact that binding of calcium to troponin C directly executes the rearrangement of the fluorophores with no interacting M13 linker compared with the cameleons.
Individual SpH-expressing boutons were identifiable even after long periods of rest, whereas axons were never visible (Table 1). Because the pH-sensitive SpH is nonfluorescent at pH <6 (Miesenboeck et al., 1998), a small pool must constitutively be present within the presynaptic cell membrane, most likely attributable to spontaneous vesicle fusion that happens at the Drosophila NMJ at a frequency of ∼1-3 Hz. Using SpH, neuronal activity could reliably be monitored (Fig. 3D), displaying kinetics that clearly differed from the calcium-dependent fluorescence changes in all of the GECIs. The SpH fluorescence increased to 9.1 and 16.5% ΔF/F, with a monoexponential time course displaying τ = 4.3 s and τ = 2.5 s at 40 and 80 Hz, respectively, with no plateau reached during the 2.2 s stimulation period. Recovery of SpH signals was incomplete by the end of the experiments, suggesting a slow retrieval of presynaptic membrane. Thus, referring to the slow rise and decay of the fluorescence signals, SpH produced the slowest temporal response of all indicators. Taken into account that SpH broadcasts changes in the net balance of vesicle release and vesicle recycling, a slower time course of the fluorescence change can be expected compared with the GECI signals.
Camg1 fluorescence was barely detectable (Table 1). In two animals only were we able to identify a fluorescent NMJ with a total of 22 boutons. At 40 Hz stimulation, no fluorescence change could be observed (data not shown). Only at 80 Hz stimulation a minimal fluorescence increase is suggested at the level of the plotted mean (Fig. 3I), whereas the signal-to-noise ratio (SNR) was too low to allow detection. Camg2 boutons were easier to find at rest, as expected by the higher basal fluorescence (Griesbeck et al., 2001). However, it did not perform better than Camg1 (Fig. 3J, black trace). This was unexpected because a high fluorescence change of up to sevenfold has been reported for both sc indicators in vitro (Baird et al., 1999; Griesbeck et al., 2001). Their low Kd (Table 1) cannot solely account for this result, because Camg2 signals could not be increased by raising extracellular calcium to 10 mm. Instead, we found a strong degradation of the signal that we cannot explain (Fig. 3J, light gray trace).
Only after antibody staining (anti-GFP-Alexa 488; see Materials and Methods) could FP be visualized at NMJs of experimental animals (Fig. 3K). No FP fluorescence at all could be detected in live animals (Table 1) and the filet preparations that were used for the imaging experiments. In these experiments, we focused on the cleft between muscle 6 and 7 and stimulated the nerve as usual. In all cases (n > 10), we failed to observe any increase in fluorescence (data not shown), although a suitable Kd for calcium of ∼700 nm and very large fluorescence changes (up to eightfold) have been reported in vitro (Nagai et al., 2001) (Table 1). Hasan et al. (2004) also reported negative results on FP when expressed in HeLa cells. However, our antibody stainings demonstrated that FP was expressed at the very same synapses.
In summary, GCaMP1.3 (Fig. 3A), all of the FRET-based GECIs (Fig. 3E-H), and SpH allowed the reliable detection of presynaptic fluorescence changes at the level of individual boutons in single trials (Figs. 2D,E, 6A-C, false-color images) (supplemental movies 1-3, available at www.jneurosci.org as supplemental material). TN-L15 exhibited the most stable and fastest rising signals at lower activity rates, whereas GCaMP1.6 produced the fastest signals at high rates of nerve activity. GCaMP1.6 and GCaMP1.3 signals were complicated by bleaching, as was the case for IP. Camg1 and Camg2 showed little or no response, and FP did not result in any detectable fluorescence in our hands. For the three “nonfunctional” indicators, we cannot absolutely rule out a systematic failure of nerve stimulation. However, this is extremely unlikely because such failures were never observed in other indicator animals, and “functional” indicators were successfully imaged between the individual Camg and FP experiments.
Figure 6.

Dynamic properties of GECIs: fluorescence changes in response to three consecutive AP volleys. The detection of a given signal critically depends on the indicator kinetics. We addressed how well sequential neuronal activity can be resolved from the exhibited fluorescence changes in GCaMP1.3-expressing (A), YC3.3-expressing (B), or SpH-expressing (C) boutons. Three consecutive AP volleys (80 Hz/1 s; horizontal black bars) were evoked, spaced by 2 s at rest and 7 s after the last stimulus. The fluorescence change is plotted as mean ΔF/F (A, C) or mean ΔR/R (B) (±SEM) for the following numbers of animals, NMJs, and boutons: A, 3, 3, and 36; B, 4, 4, and 61; and C, 3, 3, and 44, respectively. Images were taken at a rate of 10 Hz for 15 s. On the right, single boutons are shown in false-color code during an experiment at the time points indicated by the numbers (in seconds). Compared with the plotted traces, qualitatively similar changes can be reliably observed in individual boutons. This demonstrates the high signal-to-noise ratio of these indicators at the given stimulus frequency. Note that, in this graph, for the purpose of data display, the GCaMP1.3 fluorescence has been linearly corrected for bleaching.
GECI signals over a wide range of neural activity
We compared the fractional fluorescence changes in GCaMP1.3 (Fig. 4A-F) and YC3.3 (Fig. 4G-L) in response to neural activity at different rates (5, 10, 20, 40, and 80 Hz) and calculated the corresponding SNR. Fifty ± 2 boutons were analyzed per stimulus frequency in each genotype. The background was subtracted, and GCaMP1.3 signals were additionally bleach corrected (see Materials and Methods) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). GCaMP1.3 amplitudes increased fairly linear over the entire stimulus range (Fig. 4F, inset) (ΔF/F = 0.7, 2.7, 12.7, 39.8, and 82.3%), whereas YC3.3 amplitudes increased linearly only in the lower stimulus range and sublinearly in the upper range (ΔR/R = 1.5, 5.5, 14.9, 26.7, and 31.0%), indicating saturation of the probe. Because the response of YC3.3 did not fully reach steady state during the stimulation period, the respective graph (Fig. 4L, inset) might slightly underestimate the true saturation characteristics of this probe. In simulations (data not shown), this effect was proven to be not relevant for the interpretation of the data. For both indicators, the SNR depended on the action potential frequency in a way that followed the increase in the fluorescence amplitudes (Fig. 4F,L, insets), suggesting basically constant noise in the measurements. Thus, at high activity rates of 80 Hz, GCaMP1.3 showed a much higher SNR compared with YC3.3 (6.3 and 3.8, respectively), and, at 40 Hz, both indicators showed similar SNR (3.3 and 3.3, respectively). However, in the lower activity range, YC3.3 was slightly superior: at 5, 10, and 20 Hz, we calculated for GCaMP1.3 and YC3.3 an SNR of 0.2, 0.9, 2.6 and 0.5, 1.4, 3.1, respectively.
Kinetics and SNR of GECI signals depend on expression level
Fluorescence changes associated with changes in calcium depend on the concentration of the calcium reporter molecule itself, in a way that kinetics and amplitudes of the fluorescence change are or can be altered, respectively (Yasuda et al., 2004). To investigate this question, two lines of UAS-YC2.3 flies were analyzed that gave rise to a fourfold difference in the GECI expression level (Fig. 5A-D), as judged by quantifying the intensity of the citrine fluorescence in presynaptic boutons at rest. For precise quantification of the expression level, ΔR/R, and SNR of the signals, the background was subtracted locally. However, only the relative level of expression could be determined. The actual concentration at which GECIs were expressed in our experimental animals was unknown. For stimulation, we chose a nonsaturating frequency of 40 Hz. The exhibited mean (ΔR/R) is shown by the white traces for line 5b (Fig. 5A) and line 30l (Fig. 5B). The amplitudes were identical for both lines. The overlay of the mean signals (Fig. 5C) more clearly shows that the weak-expressing line 5b exhibited faster fluorescence signals in particular for the rise of the signals (τ = 594 ± 58 ms in 5b and 909 ± 48 ms in 30l). In Figure 5, A and B, signals from individual boutons are plotted in gray, and the higher SNR of the signals from the strong-expressing line in B is obvious. In Figure 5D, a summary of the results is shown: citrine intensity, 0.92 ± 0.45 and 3.80 ± 1.51 arbitrary units, p < 10-5; ΔR/R, 16.94 ± 0.78 and 16.68 ± 0.40%; SNR, 2.58 and 4.30 for YC2.3-5b (n = 71/6) and YC2.3-30l (n = 95/7), respectively (±SEM, n is boutons/animals).
Dynamic properties of GECIs
We investigated the signal dynamics of genetically encoded activity indicators in response to three consecutive AP volleys (Fig. 6A-C). In these experiments, signals from the background were not subtracted because this would not change the time course of the signals. This more dynamic stimulus pattern was used to evoke fluorescence changes in three clearly different activity indicators: GCaMP1.3, YC3.3, and SpH. Three consecutive AP volleys (80 Hz/1 s) were separated by 2 s without stimulation. Figure 6A shows that this was enough time for the GCaMP1.3 fluorescence to reach its maximum level or undershoot (τ decay = 480 ms) (Table 1) during the interstimulus periods and to reliably report each AP volley. There was basically no reduction in the ΔF/F amplitudes of the individual peaks. In contrast, the YC3.3 signals (Fig. 6B) showed a superposition of the newly evoked fluorescence changes on the decaying signal in response to the previous stimulation. However, there was no increment of the second and third maximum compared with the first as saturation (Fig. 4L) and bleaching of the probe took place. In contrast, a clear superposition of the fluorescence signals was found for the SpH signal (Fig. 6C), resulting almost in a staircase response to the three consecutive stimulus volleys. Because of sufficient SNR of these indicators at 80 Hz stimulation, the different signal kinetics could also be observed on the level of individual boutons. This is shown in the false-color images of a single bouton (Fig. 6, right) taken at the time (in seconds) indicated by the numbers in the image, as well as by the black dots on the x-axis in Figure 6C (supplemental movies 2, 3, available at www.jneurosci.org as supplemental material).
Reproducibility of the fluorescence changes
For most investigations of neuronal calcium dynamics, it is important to reliably image the same anatomical structure in a series of experiments. The reproducibility of the fluorescence changes evoked by 50 Hz stimulation was investigated by sequential imaging of NMJs in four runs. Between the runs, each NMJ was allowed to recover for 3 min. Animals expressing YC2.3 (Fig. 7A), TN-L15 (Fig. 7B), GCaMP1.3 (Fig. 7C), or IP (Fig. 7D) were chosen. In contrast to our previous experiments, a neutral-density 0.4 filter was used to reduce the excitation energy to just allow the visual identification of boutons. Under these conditions, the dc GECIs YC2.3 (Fig. 7A) and TN-L15 (Fig. 7B) exhibited a fully reproducible responses. In GCaMP1.3 (Fig. 7C), the fluorescence change decreased by ∼25%, from 8.1% ΔF/F in the first run to 6.1% ΔF/F in the fourth run. GCaMP1.3 also changed in the time course before stimulus onset (from t = 0 to t = 2 s; compare first run with second, third, and fourth). Because of random stimulus presentation and subsequent averaging of the signals, this effect was masked in Figures 3 and 4. The other circular permuted indicator IP (Fig. 7D) consistently showed an even larger change in the shape and time course of the fluorescence signal. Moreover, the strong signal degradation during the individual runs and the lack of reliable onset and decay of the fluorescence signal are clear disadvantages of this GECI when applied to living animals. These results are summarized in Figure 7E, in which we normalized the peak amplitudes of each run to the first run. The dc indicators fluctuated ∼100% accuracy, whereas both cpsc indicators deviated from the fluorescence change in the first run. We also quantified the raw fluorescence of individual boutons before stimulus onset in each run (Fig. 7F) and found that fluorescence at rest is fairly stable from run to run for the individual indicators. Thus, although bleaching is visible in the GCaMP1.3 and IP traces, fluorescence at rest fully recovers during the 3 min resting period. Because we used wide-field epifluorescence microscopy, bleaching also occurred in out-of-focus areas. Moreover, almost the entire NMJ was imaged. Thus, diffusion of indicator molecules is very unlikely to significantly contribute to the observed fluorescence recovery at rest. However, we cannot fully rule out this option. The changed time course and reduced fractional change after the first run rather suggests altered fluorescence properties than irreversible bleaching during the succession of experiments. Photoconversion and other unknown factors might account for the change in time course and the limited reproducibility of the cpsc indicator signals [i.e., a decrease in the GCaMP1.3 fractional fluorescence change (Fig. 6C,E) and an increase in the IP fractional fluorescence loss (Fig. 6D,E)]. dc indicators were clearly superior in this regard.
Figure 7.

Reproducibility of the fluorescence changes. Four sequential imaging experiments (runs) were done at each NMJ. Within each run, electrical stimuli were applied at a rate of 50 Hz for 2.2 s (stimulus interval, from 2 to 4.2 s). Between the individual runs, the preparation was allowed to recover for 3 min. The calculated fluorescence change is plotted versus time for the following indicators and number of animals, NMJs, and boutons: A, YC2.3, 2, 2, and 15; B, TN-L15, 3, 3, and 27; C, GCaMP1.3, 3, 3, 50; and D, IP, 2, 2, and 23, respectively. The perfect superposition of the individual runs in A and B demonstrates the high reproducibility of the exhibited fluorescence change in the dc indicators YC2.3 and TN-L15. E, The data in A-D are summarized by normalizing the peak amplitudes of each run to the first run. For the measurement of the peak fluorescence, the five nearby frames were averaged. For IP, this peak was subtracted from the signal at the onset of the stimulus. F, Quantification of the raw fluorescence intensity before stimulus onset as a measure of bleaching. The raw fluorescence of each run was normalized to the first run. We did not observe bleaching in any of the GECIs. In all experiments shown, a neutral-density 0.4 filter was used to attenuate the power of the epifluorescence excitation to a level that just allowed us to identify individual NMJs at rest.
Discussion
We present a comparative study of 11 genetic indicators of neural activity. A broad palette of eight reliable genetic indicators for the imaging of neural activity in intact animals was identified. Using Drosophila NMJ larval presynaptic terminals as model system, we performed well controlled and highly reproducible experiments to allow direct comparison of different sc, cpsc, and dc calcium indicators and SpH. As a basic principle, the recorded fluorescence signals differed substantially for the different indicators. Fluorescence changes were related to neural activity in a gradual way, but smaller than what has been observed on purified GECIs in vitro, very likely a shift in Kd of some indicators and more significant problems with photobleaching were observed. The kinetics and SNR of the fluorescence signals depended on the expression level of the indicator. The new sensors showed significant improvements: GCaMP1.6 showed increased fluorescence at rest and reported high rates of neural activity with the largest and fastest fluorescence changes. TN-L15 showed the fastest rise of all indicators at lower rates of nerve activity and had less complex signal kinetics compared with the CaM-based cameleons.
Nonfunctional or unreliable GECIs in vivo
Under our recording conditions, FP did not show any fluorescence in vivo. In vitro, similar results were described by Hasan et al. (2004), although Nagai et al. (2001) reported highly dynamic signals in vitro. Camg1 and Camg2 exhibited no or only small fluorescence changes, then with very low SNR. ΔF/F of 4% was previously reported in neurons of transgenic mice expressing Camg2 under tet control (Hasan et al., 2004). Yu et al. (2003) reported even larger Camg1 and Camg2 fluorescence changes in Drosophila mushroom bodies in vitro. In our experiments, the Camg2 fluorescence tended to drop unpredictably after stimulation (Fig. 3K), an effect also seen by Yu et al. (2003). An additional drawback was that Camg1-expressing NMJs were not visible because of low fluorescence at rest (Table 1) (Baird et al., 1999; Griesbeck et al., 2001; Yu et al., 2003). Hypothetically, our results might be attributable to the experimental procedures. In living animals, initial excitation of the chromophore is required for focusing on the structure of interest. This has been suggested to cause photoconversion of Camg2 to an almost completely calcium-insensitive form (Filippin et al., 2003). It is important to note that all nonfunctional indicator flies have been tested for proper indicator insertion. The indicator cDNA and flanking P-element sequences were amplified from the genomic fly DNA and sequenced (see Materials and Methods).
Reliable genetic indicators of neural activity in vivo
None of the indicators matched the requirements of optimal sensors of neural activity. Optimal sensors would provide a high signal-to-noise ratio at minimum stimulation and indicator concentration, brought about by bright fluorescence, large fluorescence change, and linear relationship to activity or calcium (Yasuda et al., 2004). Nevertheless, in agreement with previous publications, we achieved reliable optical recordings using SpH, GCaMP1.3, IP, and YC2.0 (Ng et al., 2002; Reiff et al., 2002; J.W. Wang et al., 2003; Bozza et al., 2004; Hasan et al., 2004; Suh et al., 2004; Y. Wang et al., 2004; Yu et al., 2004). In addition to these, we identified four other indicators, GCaMP1.6, YC2.3, YC3.3, and TN-L15, to reliably report neuronal activity in vivo.
GCaMP1.6 is a derivative of GCaMP1.3, carrying an additional 64 amino acids at its N terminus and two amino acid changes within the wild-type GFP backbone (Nakai, unpublished observation). These modifications strongly improved visibility at rest, rise and decay time constants, and the dynamics of the GCaMP1.6 signal, offsetting the limited photostability of this indicator. Limited photostability was also discovered in the fluorescence of the other cpsc indicators GCaMP1.3 and IP, causing lower reproducibility (Fig. 7C,D) and limited signal detection (Fig. 3C), respectively. However, GCaMP1.3 signals exhibited a nice linear relationship to neural activity (tested up to 80 Hz) and high SNR at high activity levels, and bleaching could be corrected with sufficient accuracy (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), making this indicator very useful.
Improved photostability was reported for YC2.3 and YC3.3, both of which carry amino acid changes within EYFP, rendering the longer wavelength component of cameleon brighter and more pH stable (citrine) (Griesbeck et al., 2001). In accordance with these results, accurate reproducibility of the signals was measured in YC2.3 and TN-L15 (Fig. 7), which also contain citrine. TN-L15 (Heim and Griesbeck, 2004) was developed by replacing the CaM-M13 by non-neuronal chicken troponin C. This new indicator shows great promise also in terms of SNR (our unpublished data) and the time resolution. TN-L15 exhibited a faster time constant for the rise (Table 1) compared with all other indicators when stimulated at 40 Hz. These key aspects confirm that TN-L15 might overcome the problems that have been attributed to the promiscuous behavior of CaM when applied to the intracellular space in vivo (Jurado et al., 1999; Benaim and Villalobo, 2002). These results are of particular importance because improved subcellular targeting has been demonstrated for TN-L15 (Heim and Griesbeck, 2004), a property that is retained in vivo (our unpublished data). In addition, the pKa of TN-L15 is ∼5.7, suggesting that mild pH changes, which can be induced by electrical activity (Wang et al., 1994), might not interfere with the function of this indicator. In contrast, the sc and cpsc indicators possess a pKa that is within the physiological range, and their fluorescence change is brought about by a change in the proto-nation of the chromophore during calcium binding (Baird et al., 1999; Griesbeck et al., 2001; Nagai et al., 2001; Nakai et al., 2001; Zhang et al., 2002). Thus, in summary, new dc indicators such as TN-L15 represent particularly promising calcium indicators.
Fluorescence signals and neural activity
NMJs were stimulated twice as long as the reported average length of motoneuron patterns during fictive larval locomotion (∼80 Hz/80 APs) (Cattaert and Birman, 2001; Barclay et al., 2002). When fast synthetic dyes (Macleod et al., 2002, 2004) were used, steady-state fluorescence signals with linear relationship to the stimulus frequency were observed in the same preparation. The described signals in Figure 3 and the linear relationship of GCaMP1.3 and YC3.3 signals to neural activity are in accordance with these results. Similar results were reported from presynaptic terminals at the crayfish NMJ (Tank et al., 1995) in which the underlying calcium dynamics was described by a single compartment model. In this model, calcium quickly reaches a plateau when influx and efflux/sequestration are in balance (Tank et al., 1995). A rather low endogenous buffer capacity and a fast monoexponential extrusion of calcium after stimulation were described. Thus, time to achieve steady state, amplitude, deviation, and time to return to baseline of the fluorescence can mostly be assigned to the intrinsic properties of the cell, the expression level of the indicator, and its dissociation constant. We showed for YC2.3 that the signal amplitudes were unchanged with increasing expression level (Fig. 5), implying that even at high expression the indicator concentration did not alter the capacity of the endogenous buffers, whereas the kinetics of the signal was slowed down (Fig. 5C), thus matching the expected results (Tank et al., 1995). Conversely, the SNR increased with the expression level, pointing out an important tradeoff between signal kinetics and SNR. Using rather low concentrations of Oregon-Green-BAPTA with a Kd of ∼500 nm, Macleod et al. (2002, 2004) showed that the decay in calcium after a single AP as well as after long stimulus trains was ∼60 ms in type Ib boutons of muscle 6/7 Drosophila NMJs. Thus, 60 ms is likely to correspond to the calcium extrusion rate, a value that we could not approach because the fastest time constant of the decay was 350 ms reported by GCaMP1.6. Also, single action potentials could not be resolved in our experiments, and even responses to low frequency AP trains were generally hard to detect (Fig. 4). Similar findings have been published recently in transfected cortical slices (Pologruto et al., 2004). In this context, it is interesting to see that, in our experiments, YC3.3 showed a better SNR at low activity rates when compared with GCaMP1.3 (Fig. 4). However, the generally rather low SNR made us chose robust stimuli to characterize the indicators in Figure 3. The 80 Hz stimuli appeared to saturate the high Kd dc indicators, whereas the low Kd GCaMP1.3 showed a linear relationship to neural activity over the entire stimulus range (for Kd values, see Table 1). Thus, dc indicators have a limited dynamic range in vivo, whereas GCaMP1.3 most likely has a substantial increase in its Kd in vivo. The latter has been reported for some synthetic dyes (Kurebayashi et al., 1993; Bassani et al., 1995; Baylor and Hollingworth, 2000) and recently for Camg2 and GCaMP1.3, which exhibited a 1.5-fold and 8-fold increase, respectively, in their Kd compared with in vitro (Pologruto et al., 2004). However, in the same study, a nonlinear relationship of the GCaMP1.3 fluorescence to neural activity has been shown, and similarly this has been suggested previously to underestimate the calcium rise in response to single APs and small bursts of APs in the Drosophila olfactory system (J. W. Wang et al., 2003; Y. Wang et al., 2004). Our data differed from these results because we found a linear behavior of both amplitudes and SNR for GCaMP1.3 (and YC3.3, which was saturated at 80 Hz).
The fluorescence change in SpH was different from all GECIs because its kinetics reflect synaptic vesicle cycling (Miesenboeck et al., 1998). Thus, SpH fluorescence during stimulation reflects the net result of vesicle fusion and competing endocytosis (Fernandez-Alfonso and Ryan, 2004). No sign of reaching a steady state was visible during the stimulation period, indicating that the ratio of endocytosis/exocytosis is rather small, leading to a surface accumulation of SpH. Moreover, the time course of SpH signals depended on the amount of net accumulation of synaptic membrane on the presynaptic cell surface (Nicholson-Tomishima and Ryan, 2004). This can be seen in Figure 6C, in which the signal rise slows down from one AP volley to the next, whereas the signal decay becomes faster. Small changes in the presynaptic release frequency will be effectively filtered and produce only small fluorescence changes (Ng et al., 2002). Such filtering properties might explain recent differences in results from electrophysiological recordings (Wilson et al., 2004) and optical imaging studies in the Drosophila olfactory system (SpH, Ng et al., 2002, Yu et al., 2004; GCamP1.3, J.W. Wang et al., 2003; Y. Wang et al., 2004) in which these genetic probes underreported the electrical response to low odorant concentrations. Thus, additional improvements in the design of indicators are required (Griesbeck, 2004; Miesenboeck, 2004). At present, the indicators reliably report higher-frequency bursts, which are central to natural activity in many systems (Rieke et al., 1997). New indicators would report calcium with better SNR and thus better resolve the underlying electrical events that happen on the timescale of milliseconds and are believed to be a prerequisite of information processing in neural circuits (Abeles, 1991; Shadlen and Newsome, 1994; de Ruyter van Steveninck et al., 1997; Borst and Theunissen, 1999; Ikegaya et al., 2004; Miesenboeck, 2004). Ratiometric indicators with increased maximum fluorescence change (Nagai et al., 2004) (Griesbeck, unpublished data) demonstrate that the field is heading toward such probes.
Footnotes
This work was supported by the Max-Planck-Society. We thank Winfried Denk for help with the two-photon laser microscope, Oliver Griesbeck, Atsushi Miyawaki, and Roger Tsien for providing DNA, Yalin Wang, C. Goodman, C. Schuster, C. Lehner, and Gero Miesenboeck for providing flies, and Juergen Haag for programming, help, and discussion. We thank Yong Choe for discussions and comments on this manuscript. In particular, we thank Wolfgang Essbauer and Christian Theile for excellent technical assistance.
Correspondence should be addressed to Dr. Dierk F. Reiff, Department of Systems and Computational Neuroscience, Max-Planck-Institute of Neurobiology, 82152 Martinsried, Germany. E-mail: reiff@neuro.mpg.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254766-13$15.00/0
D.F.R. and A.I. contributed equally to this work.
References
- Abeles M (1991) Corticonics. Cambridge, MA: Cambridge UP.
- Baird GS, Zacharias DA, Tsien RY (1999) Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci USA 96: 11241-11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay JW, Atwood HL, Robertson RM (2002) Impairment of central pattern generation in Drosophila cystein string protein mutants. J Comp Physiol [A] 188: 71-78. [DOI] [PubMed] [Google Scholar]
- Basler K, Struhl G (1994) Compartment boundaries and the control of Drosophila limb pattern by hedgehog protein. Nature 368: 208-214. [DOI] [PubMed] [Google Scholar]
- Bassani JW, Bassani RA, Bers DM (1995) Calibration of indo-1 and resting intracellular [Ca]i in intact rabbit cardiac myocytes. Biophys J 68: 1453-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor SM, Hollingworth S (2000) Measurement and interpretation of cytoplasmic [Ca2+] signals from calcium-indicator dyes. News Physiol Sci 15: 19-26. [PubMed] [Google Scholar]
- Benaim G, Villalobo A (2002) Phosphorylation of calmodulin. Functional implications. Eur J Biochem 269: 3619-3725. [DOI] [PubMed] [Google Scholar]
- Borst A, Theunissen FE (1999) Information theory and neural coding. Nat Neurosci 11: 947-957. [DOI] [PubMed] [Google Scholar]
- Bozza T, McGann JP, Mombaerts P, Wachowiak M (2004) In vivo imaging of neuronal activity by targeted expression of a genetically encoded probe in the mouse. Neuron 42: 9-21. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401-415. [DOI] [PubMed] [Google Scholar]
- Cattaert D, Birman S (2001) Blockade of the central generator of locomotor rhythm by noncompetitive NMDA receptor antagonists in Drosophila larvae. J Neurobiol 48: 58-73. [DOI] [PubMed] [Google Scholar]
- Cohen LB, Salzberg BM, Grinvald A (1978) Optical methods for monitoring neuron activity. Annu Rev Neurosci 1: 171-182. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW (1990) Two-photon laser scanning fluorescence microscopy. Science 248: 73-76. [DOI] [PubMed] [Google Scholar]
- de Ruyter van Steveninck RR, Lewen GD, Strong SP, Koberle R, Bialek W (1997) Reproducibility and variability in neural spike trains. Science 5307: 1805-1808. [DOI] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA (2004) The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron 41: 943-953. [DOI] [PubMed] [Google Scholar]
- Fiala A, Spall T, Diegelmann S, Eisermann B, Sachse S, Devaud J-M, Buchner E, Galizia CG (2002) Genetically expressed Cameleon in Drosophila melanogaster is used to visualize olfactory information in projection neurons. Curr Biol 12: 1877-1884. [DOI] [PubMed] [Google Scholar]
- Filippin L, Magalhaes PJ, Di Benedetto G, Pozzan T (2003) Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J Biol Chem 278: 39224-39234. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H (2002) Studying gene function in eukaryotes by conditional gene inactivation. Annu Rev Genet 36: 153-173. [DOI] [PubMed] [Google Scholar]
- Griesbeck O (2004) Fluorescent proteins as sensors for cellular function. Curr Opin Neurobiol 14: 636-641. [DOI] [PubMed] [Google Scholar]
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY (2001) Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem 276: 29188-29194. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440-3450. [PubMed] [Google Scholar]
- Guerrero G, Isacoff EY (2001) Genetically encoded optical sensors of neuronal activity and cellular function. Curr Opin Neurobiol 11: 601-607. [DOI] [PubMed] [Google Scholar]
- Habuchi S, Cotlet M, Hofkens J, Dirix G, Michiels J, Vanderleyden J, Subramaniam V, De Schryver FC (2002) Resonance energy transfer in a calcium concentration-dependent cameleon protein. J Biophys 8: 3499-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MT, Friedrich RW, Euler T, Larkum ME, Giese G, Both M, Duebel J, Waters J, Bujard H, Griesbeck O, Tsien RY, Nagai T, Miyawaki A, Denk W (2004) Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PLoS Biol 2: 763-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim N, Griesbeck O (2004) Genetically encoded indicators of cellular calcium dynamics based on Troponin C and green fluorescent protein. J Biol Chem 279: 14280-14286. [DOI] [PubMed] [Google Scholar]
- Higashijima S, Masino MA, Mandel G, Fetcho JR (2003) Imaging neuronal activity during zebrafish behavior with a genetically encoded calcium indicator. J Neurophysiol 6: 3986-3997. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Aaron G, Cossart R, Aronov D, Lampl I, Ferster D, Yuste R (2004) Synfire chains and cortical songs: temporal modules of cortical activity. Science 5670: 559-564. [DOI] [PubMed] [Google Scholar]
- Jares-Erijman E, Jovin TM (2003) FRET imaging. Nat Biotechnol 21: 1387-1395. [DOI] [PubMed] [Google Scholar]
- Jurado LA, Chockalingam PS, Jarrett HW (1999) Apocalmodulin. Physiol Rev 79: 661-682. [DOI] [PubMed] [Google Scholar]
- Kerr R, Lev-Ram V, Baird G, Vincent P, Tsien RY, Schafer WR (2000) Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans Neuron 26: 583-594. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Miyawaki A, Matsumoto K, Mori I (2004) The C. elegans thermosensory neuron AFD responds to warming. Curr Biol 14: 1291-1295. [DOI] [PubMed] [Google Scholar]
- Kurebayashi N, Harkins AB, Baylor SM (1993) Use of Fura red as an intracellular calcium indicator in frog skeletal muscle fibers. Biophys J 64: 1934-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L (1999) Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22: 451-461. [DOI] [PubMed] [Google Scholar]
- Lin DM, Goodman CS (1994) Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron 13: 507-523. [DOI] [PubMed] [Google Scholar]
- Liu L, Yermolaieva O, Johnson WA, Abboud FM, Welsh MJ (2003) Identification and function of thermosensory neurons in Drosophila larvae. Nat Neurosci 6: 267-273. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Hegstrom-Wojtowicz M, Charlton MP, Atwood HL (2002) Fast calcium signals in Drosophila motor neuron terminals. J Neurophysiol 88: 2659-2663. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Marin L, Charlton MP, Atwood HL (2004) Synaptic vesicles: test for a role in presynaptic calcium regulation. J Neurosci 24: 2496-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenboeck G (2004) Genetic methods for illuminating the function of neural circuits. Curr Opin Neurobiol 14: 395-402. [DOI] [PubMed] [Google Scholar]
- Miesenboeck G, De Angelis DA, Rothman JE (1998) Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394: 192-195. [DOI] [PubMed] [Google Scholar]
- Miyawaki A (2003a) Fluorescence imaging of physiological activity in complex systems using GFP-based probes. Curr Opin Neurobiol 13: 591-596. [DOI] [PubMed] [Google Scholar]
- Miyawaki A (2003b) Visualization of the spatial and temporal dynamics of intracellular signaling. Dev Cell 4: 295-305. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY (1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388: 882-887. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Griesbeck O, Heim R, Tsien RY (1999) Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci USA 96: 2135-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Sawano A, Eli P, Hama H, Miyawaki A (2001) Red fluorescent protein from Discosoma as a fusion tag and a partner for fluorescence resonance energy transfer. Biochemistry 40: 2502-2510. [DOI] [PubMed] [Google Scholar]
- Nagai T, Sawano A, Park ES, Miyawaki A (2001) Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci USA 98: 3197-3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20: 87-90. [DOI] [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A (2004) Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci USA 29: 10554-10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K (2001) A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat Biotechnol 19: 137-141. [DOI] [PubMed] [Google Scholar]
- Ng M, Roorda RD, Lima SQ, Zemelman BV, Morcillo P, Miesenbock G (2002) Transmission of olfactory information between three populations of neurons in the antennal lobe of the fly. Neuron 36: 463-474. [DOI] [PubMed] [Google Scholar]
- Nicholson-Tomishima K, Ryan TA (2004) Kinetic efficiency of endocytosis at mammalian CNS synapses requires synaptotagmin I. Proc Natl Acad Sci USA 101: 16648-16652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persechini A, Lynch JA, Romoser VA (1997) Novel fluorescent indicator proteins for monitoring free intracellular Ca2+ Cell Calcium 22: 209-216. [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Yasuda R, Svoboda K (2004) Monitoring neural activity and [Ca2+] with genetically encoded Ca2+ indicators. J Neurosci 24: 9572-9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff DF, Thiel PR, Schuster CM (2002) Differential regulation of active zone density during long-term strengthening of Drosophila neuromuscular junctions. J Neurosci 22: 9399-9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F, Warland D, de Ruyter van Steveninck RR, Bialek W (1997) Spikes—exploring the neural code. Cambridge, MA: MIT.
- Romoser VA, Hinkle PM, Persechini A (1997) Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J Biol Chem 272: 13270-13274. [DOI] [PubMed] [Google Scholar]
- Rudolf R, Mongillo M, Rizzuto R, Pozzan T (2003) Looking forward to seeing calcium. Nat Rev Mol Cell Biol 4: 579-586. [DOI] [PubMed] [Google Scholar]
- Shadlen MN, Newsome WT (1994) Noise, neural codes and cortical organization. Curr Opin Neurobiol 4: 569-579. [DOI] [PubMed] [Google Scholar]
- Shyn SI, Kerr R, Schafer WR (2003) Serotonin and Go modulate functional states of neurons and muscles controlling C. elegans egg-laying behavior. Curr Biol 21: 1910-1915. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM (1982) Transposition of cloned P-elements into Drosophila germ line chromosomes. Science 218: 341-347. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM (1983) The effect of chromosomal position on the expression of the Drosophila xanthine dehydrogenase gene. Cell 1: 47-57. [DOI] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF (1994) Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol [A] 175: 179-191. [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A (2003) In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci USA 12: 7319-7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh GS, Wong AM, Hergarden AC, Wang JW, Simon AF, Benzer S, Axel R, Anderson DJ (2004) A single population of olfactory sensory neurons mediates an innate avoidance behaviour in Drosophila Nature 431: 854-859. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Kerr R, Bianchi L, Frokjaer-Jensen C, Slone D, Xue J, Gerstbrein B, Driscoll M, Schafer WR (2003) In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron 39: 1005-1017. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW (1997) In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature 385: 161-165. [DOI] [PubMed] [Google Scholar]
- Tank DW, Sugimori M, Connor JA, Llinás RR (1988) Spatially resolved calcium dynamics of mammalian Purkinje cells in cerebellar slice. Science 242: 773-777. [DOI] [PubMed] [Google Scholar]
- Tank DW, Regehr WG, Delany KR (1995) A quantitative analysis of presynaptic calcium dynamics that contribute to short-term enhancement. J Neurosci 15: 7940-7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong K, Sawano A, Mizuno H, Hama H, Tong KI, Mal TK, Miyawaki A, Ikura M (2001) FRET-based in vivo Ca2+ imaging by a new calmodulin-GFP fusion molecule. Nat Struct Biol 8: 1069-1073. [DOI] [PubMed] [Google Scholar]
- Tsien RY, Miyawaki A (1998) Seeing the machinery of live cells. Science 280: 1954-1955. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Randall RD, Thayer SA (1994) Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J Neurophysiol 72: 2563-2569. [DOI] [PubMed] [Google Scholar]
- Wang JW, Wong AM, Flores J, Vosshall LB, Axel R (2003) Two-photon calcium imaging reveals an odor-evoked map of activity in the fly brain. Cell 112: 271-282. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guo HF, Pologruto TA, Hannan F, Hakker I, Svoboda K, Zhong Y (2004) Stereotyped odor-evoked activity in the mushroom body of Drosophila revealed by green fluorescent protein-based Ca2+ imaging. J Neurosci 24: 6507-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Turner GC, Laurant G (2004) Transformation of olfactory representation in the Drosophila antennal lobe. Science 5656: 366-370. [DOI] [PubMed] [Google Scholar]
- Wong AM, Wang JW, Axel R (2002) Spatial representation of the glomerular map in the Drosophila protocerebrum. Cell 109: 229-241. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K (2004) Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE 2004: pl5. [DOI] [PubMed] [Google Scholar]
- Yu D, Baird GS, Tsien RY, Davis RL (2003) Detection of calcium transients in Drosophila mushroom body neurons with Camgaroo reporters. J Neurosci 23: 64-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ponomarev A, Davis RL (2004) Altered representation of the spatial code for odors after olfactory classical conditioning; memory trace formation by synaptic recruitment. Neuron 42: 437-449. [DOI] [PubMed] [Google Scholar]
- Zemelman BV, Miesenboeck G (2001) Genetic schemes and schemata in neurophysiology. Curr Opin Neurobiol 11: 409-414. [DOI] [PubMed] [Google Scholar]
- Zhang J, Campbell RE, Ting AY, Tsien RY (2002) Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol 3: 907-918. [DOI] [PubMed] [Google Scholar]