

Abstract
The relative importance of CO2 and sympathetic stimulation in the regulation of cerebral and peripheral vasculatures has not been previously studied in humans. We investigated the effect of sympathetic activation, produced by isometric handgrip (HG) exercise, on cerebral and femoral vasculatures during periods of isocapnia and hypercapnia. In 14 healthy males (28.1 ± 3.7 (mean ± s.d.) years), we measured flow velocity (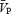 ; transcranial Doppler ultrasound) in the middle cerebral artery during euoxic isocapnia (ISO, +1 mmHg above rest) and two levels of euoxic hypercapnia (HC5, end-tidal PCO2, PET,CO2, = +5 mmHg above ISO; HC10, PET,CO2= +10 above ISO). Each PET,CO2 level was maintained for 10 min using the dynamic end-tidal forcing technique, during which increases in sympathetic activity were elicited by a 2-min HG at 30% of maximal voluntary contraction. Femoral blood flow (FBF; Doppler ultrasound), muscle sympathetic nerve activity (MSNA; microneurography) and mean arterial pressure (MAP; Portapres) were also measured. Hypercapnia increased
; transcranial Doppler ultrasound) in the middle cerebral artery during euoxic isocapnia (ISO, +1 mmHg above rest) and two levels of euoxic hypercapnia (HC5, end-tidal PCO2, PET,CO2, = +5 mmHg above ISO; HC10, PET,CO2= +10 above ISO). Each PET,CO2 level was maintained for 10 min using the dynamic end-tidal forcing technique, during which increases in sympathetic activity were elicited by a 2-min HG at 30% of maximal voluntary contraction. Femoral blood flow (FBF; Doppler ultrasound), muscle sympathetic nerve activity (MSNA; microneurography) and mean arterial pressure (MAP; Portapres) were also measured. Hypercapnia increased 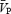 and FBF by 5.0 and 0.6% mmHg−1, respectively, and MSNA by 20–220%. Isometric HG increased MSNA by 50% and MAP by 20%, with no differences between ISO, HC5 and HC10. During the ISO HG there was an increase in cerebral vascular resistance (CVR; 20 ± 11%), while
and FBF by 5.0 and 0.6% mmHg−1, respectively, and MSNA by 20–220%. Isometric HG increased MSNA by 50% and MAP by 20%, with no differences between ISO, HC5 and HC10. During the ISO HG there was an increase in cerebral vascular resistance (CVR; 20 ± 11%), while 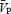 remained unchanged. During HC5 and HC10 HG,
remained unchanged. During HC5 and HC10 HG, 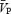 increased (13% and 14%, respectively), but CVR was unchanged. In contrast, HG-induced sympathetic stimulation increased femoral vascular resistance (FVR) during ISO, HC5 and HC10 (17–41%), while there was a general decrease in FBF below ISO. The HG-induced increases in MSNA were associated with increases in FVR in all conditions (r = 0.76–0.87), whereas increases in MSNA were associated with increases in CVR only during ISO (r = 0.91). In summary, in the absence of hypercapnia, HG exercise caused cerebral vasoconstriction, myogenically and/or neurally, which was reflected by increases in CVR and a maintained
increased (13% and 14%, respectively), but CVR was unchanged. In contrast, HG-induced sympathetic stimulation increased femoral vascular resistance (FVR) during ISO, HC5 and HC10 (17–41%), while there was a general decrease in FBF below ISO. The HG-induced increases in MSNA were associated with increases in FVR in all conditions (r = 0.76–0.87), whereas increases in MSNA were associated with increases in CVR only during ISO (r = 0.91). In summary, in the absence of hypercapnia, HG exercise caused cerebral vasoconstriction, myogenically and/or neurally, which was reflected by increases in CVR and a maintained 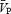 . In contrast, HG increased FVR during conditions of ISO, HC5 and HC10. Therefore, the cerebral circulation is more responsive to alterations in PCO2, and less responsive to sympathetic stimulation than the femoral circulation.
. In contrast, HG increased FVR during conditions of ISO, HC5 and HC10. Therefore, the cerebral circulation is more responsive to alterations in PCO2, and less responsive to sympathetic stimulation than the femoral circulation.
An increase in arterial PCO2 (Pa,CO2) has a potent vasodilatory effect on cerebral vessels, and to a lesser extent in the heart and limb vasculatures. It is also well established that increases in Pa,CO2 elevate sympathetic nerve activity (SNA). However, the role of increases in SNA during hypercapnia on the cerebral and limb circulations is unclear. Although studies in experimental animals have shown that the cerebral vessels are richly innervated with sympathetic fibres, the functional role for these fibres seems to be limited to modulation of the cerebrovascular response to more powerful local chemical influences (e.g. Pa,CO2; Edvinsson & Hamel, 2002).
Whether sympathetic outflow to cerebral vessels modulates cerebrovascular responses to CO2 in humans is a matter of controversy, with some investigators demonstrating modulation (Jordan et al. 1998, 2000) and others showing none (LeMarbre et al. 2003; Przybylowski et al. 2003). However, several of these previous studies have used baroreflex unloading to increase sympathetic outflow, and it is possible that this intervention, which is known to increase sympathetic outflow to many other vascular beds, may not affect the cerebral circulation.
Potential regional differences in the effect of sympathetic stimulation on vascular resistance during elevations in end-tidal (i.e. arterial) PCO2 (PET,CO2) have not been previously studied in humans. In order to investigate such differences, we used isometric handgrip exercise to induce sympathetic activation. Because hypercapnia attenuates the cerebral vasoconstriction elicited by increases in arterial pressure (i.e. autoregulation; (Brian, 1998), we hypothesized that handgrip-induced cerebral vasoconstriction, if it occurred, would also be attenuated during elevations in Pa,CO2. Because femoral vessels are not highly responsive to CO2, and because fatiguing handgrip exercise increases muscle sympathetic nerve activity (MSNA) and vascular resistance in the resting leg (Saito et al. 1988; Seals, 1989), we further hypothesized that handgrip-induced femoral vasoconstriction would be preserved during hypercapnia.
Methods
Subjects
Fourteen healthy male subjects (28.1 ± 3.7 (mean ± s.d.) years) participated in this study. All subjects received verbal and written instructions outlining the experimental procedure; written informed consent was obtained, studies conformed to the standards set by the Declaration of Helsinki, and the research study was approved by the Conjoint Health Research Ethics Board at the University of Calgary (Grant ID 17156). Participants were not taking any medication, all were non-smokers, and none had any history of cardiovascular, cerebrovascular, or respiratory disease.
Protocol
The experiments were conducted in our laboratory located at 1103 m above sea level (barometric pressure 659 ± 9 mmHg). Each subject was studied on two occasions. Subjects were requested not to eat or drink caffeine-containing beverages within 4 h before their scheduled testing sessions in the laboratory. During the initial visit, resting end-tidal gases and estimates of hypoxic and hypercapnic ventilatory responses were measured, and the subjects became familiarized with the apparatus and experimental testing procedures. The individual hypoxic and hypercapnic ventilatory sensitivities were used to calculate the estimated inspired gas concentrations that were required to maintain the desired end-tidal partial pressures during the hypercapnic challenge (described below). For the next visit, subjects reported to the laboratory and conducted two repetitions of the same protocol. Each protocol was separated by a 45 min rest period.
The subject's normal PET,CO2 and PET,O2 were measured prior to the experiment, while the subject was sitting quietly and comfortably for approximately 10 min. The respired gas was sampled continuously (20 ml min−1) via a fine catheter at the opening of one nostril by an adapted nasal O2 therapy kit, and analysed for PO2 and PCO2 by mass spectrometer (AMIS 2000, Innovision, Odense, Denmark). Values for PO2 and PCO2 were sampled by a computer every 10 ms. PET,O2 and PET,CO2 were identified and recorded for each breath using a computer and dedicated software (Chamber v1.00, University Laboratory of Physiology, Oxford, UK).
Isocapnic–hypercapnic protocol
The subject breathed normally through a mouthpiece with the nose occluded by a nose clip. Respiratory volumes were measured with a turbine device and volume transducer (VMM-400, Interface Associates, CA, USA). Respiratory flow direction and timing information were obtained with a pneumotachograph and differential pressure transducer (RSS100-HR, Hans Rudolf Inc., Kansas City, MO, USA). Accurate control of the end-tidal gases was achieved using the technique of dynamic end-tidal forcing and dedicated software (BreatheM v2.07, University Laboratory of Physiology, Oxford, UK). This technique of dynamic end-tidal forcing has been described in detail elsewhere (Robbins et al. 1982a, b; Ainslie & Poulin, 2004).
After an 8-min lead-in period of isocapnic euoxia (PET,CO2 held at 1.0 mmHg above predetermined resting value; PET,O2= 88 mmHg), subjects were instructed to perform a 2-min isometric handgrip at 30% of their predetermined maximal voluntary contraction. Subjects were instructed not to move or tense muscles other than those involved with the hand contractions, and each subject performed the handgrip exercise with their right arm. During each of the handgrip periods, subjects received visual feedback, via an oscilloscope, and verbal feedback from the experimenter to help them maintain the contraction at the desired level. A further 4 min of isocapnic euoxia followed the 2-min handgrip period. Over the next 20 min, two increases in PET,CO2 of +5.0 and +10.0 mmHg relative to isocapnia were maintained. Each PET,CO2 level was maintained for 10 min. During each PET,CO2 level, from minutes 4–6, subjects completed another 2-min isometric handgrip. Following the hypercapnic periods, one final 10-min period of isocapnic euoxia was administered with a handgrip during minutes 4–6. The experimental protocol is illustrated in Fig. 1.
Figure 1. Schematic of the experimental protocol illustrating the time-related alterations of end-tidal PCO2 (PET,CO2), end-tidal PO2 (PET,O2), ventilatory responses and periods of handgrip (shaded columns).

Data represents 15 s averages (mean ± s.d.; n = 14).
Measurement of middle cerebral blood flow velocity and Doppler power signal
Backscattered Doppler signals from the middle cerebral artery (MCA) were measured continuously during the protocol using a 2 MHz pulsed Doppler ultrasound system (TC22, SciMed, Bristol, England), as previously described (Poulin & Robbins, 1996). In this study, the peak blood velocity (VP) of the Doppler waveform, the velocity associated with the intensity-weighted mean frequency of the Doppler spectrum (VIWM), and the total power of the Doppler spectrum (P) were acquired every 10 ms and averaged over each heart beat (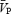 ,
, 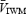 , and
, and 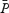 , respectively, as previously defined (Poulin et al. 1998). The
, respectively, as previously defined (Poulin et al. 1998). The 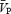 was used as the primary index of cerebral blood flow (Poulin & Robbins, 1996). In all subjects, we measured ipsilateral MCA flow (i.e. right MCA) to minimize the effects of neuronal activation on cerebral blood flow.
was used as the primary index of cerebral blood flow (Poulin & Robbins, 1996). In all subjects, we measured ipsilateral MCA flow (i.e. right MCA) to minimize the effects of neuronal activation on cerebral blood flow.
Estimations of cerebrovascular resistance
Previous studies using transcranial Doppler ultrasound (TCD) to assess changes in cerebral blood velocity in response to hypercapnia or exercise have typically measured the 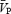 or the
or the 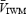 velocity of the Doppler signal. These indices are proportional to flow only if the cross-sectional area of the vessel remains constant (Poulin & Robbins, 1996). An alternative index of cerebral blood flow is
velocity of the Doppler signal. These indices are proportional to flow only if the cross-sectional area of the vessel remains constant (Poulin & Robbins, 1996). An alternative index of cerebral blood flow is 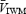 multiplied by the overall power of the signal
multiplied by the overall power of the signal 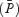 . Because the total power of the signal is proportional to the cross-sectional area, the proposed index
. Because the total power of the signal is proportional to the cross-sectional area, the proposed index 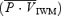 allows for any changes that may occur in the cross-sectional area of the MCA (Poulin & Robbins, 1996). Therefore, to account for any cross-sectional area changes of the MCA, we used two methods to estimate cerebrovascular resistance (CVR).
allows for any changes that may occur in the cross-sectional area of the MCA (Poulin & Robbins, 1996). Therefore, to account for any cross-sectional area changes of the MCA, we used two methods to estimate cerebrovascular resistance (CVR).
| (1) |
| (2) |
Measurement of heart rate and blood pressure
Mean arterial blood pressure (MAP) and heart rate (HR) were measured continuously using finger photoplethysmography (Portapress, TPD Biomedical Instrumentation, the Netherlands) and a three-lead ECG arrangement (Micromon 7142B monitor, Kontron Medical, France), respectively. The blood pressure and ECG signals, along with the transcranial Doppler waveforms, were updated every 10 ms by using a specialized data-acquisition package (BreatheM v 2.07, University Laboratory of Physiology, Oxford, UK).
Measurement of femoral blood flow
An ultrasound Doppler system (Sonos 1000, Hewlett-Packard, Andover, Massachusetts, USA) equipped with a linear phased array transducer probe, operating at an imaging frequency of 7.5 MHz and variable Doppler frequencies of 4.0–6.0 MHz, was used to measure two-dimensional (2D) femoral artery diameter and blood flow velocity at rest and during the intervention period. All measurements were performed below the inguinal ligament on the superficial femoral artery, approximately 3–5 cm distal to the bifurcation of the common femoral artery of the left leg. This position was chosen to prevent displacement of the probe during periods of heightened ventilation. Diameter measurements of the superficial femoral artery were obtained during the first 20 s of each minute under fixed perpendicular insonation at 7.5 MHz. Blood velocity measurements were obtained at a frequency of 4.0–6.0 MHz during the last 40 s of each minute, and were performed with the probe in the lowest insonation angle possible (45–60 deg). The sample volume was positioned in the centre of the artery and the ultrasound gate was adjusted to encompass the entire width of the vessel. Vessel diameter and blood velocity measurements were recorded onto a videocassette by means of an internal VCR (Panasonic, Matsushita Electrical Industrial Co., Japan), which was later used for off-line analysis.
Off-line analysis of femoral measurements
Superficial femoral artery diameter was determined from three longitudinal 2D frames, taken at end-diastole, during the initial 20 s of each minute. For each frame, three independent diameter measurements were made at different points along the vessel segment (Corretti et al. 2002). These diameter measurements were averaged and used to calculate the circular cross-sectional area (CSA) of the artery (A = πr2). The envelope trace of 9–12 consecutive waveforms, during the last 40 s of each minute, was used to determine the average velocity time integral (VTI). This value multiplied by the corresponding HR and CSA, gives femoral blood flow (FBF) through the following equation:
| (3) |
where FBF is measured in ml min−1; VTI in cm beat−1; CSA in cm2 and HR in beats min−1. Femoral vascular resistance (FVR) was calculated from:
| (4) |
Muscle sympathetic nerve activity recordings
Multi-unit, postganglionic sympathetic efferent nerve activity recordings were obtained in seven subjects from the peroneal nerve using the technique of Vallbo et al. (1979). Placement of the recording electrode within a muscle nerve fascicle was confirmed by (1) weak electrical stimulation through the electrode elicited involuntary muscle contraction but not paresthesias; (2) tapping or stretching the muscles or tendons supplied by the impaled fascicle elicited afferent discharge, whereas stroking the skin did not; and (3) the pulse-synchronous nature of the nerve activity. MSNA recording with signal-to-noise ratios of at least 3 : 1 were considered acceptable. Neural recordings that showed evidence of alpha-motoneurone activity or mechanoreceptor activity were excluded from the analysis.
Neurogram analysis
The sympathetic neurogram was integrated off-line with the electrocardiogram (ECG), blood pressure, and respiratory signals on a time series by an interactive computer program. Sympathetic bursts were identified by computer-assisted inspection of the mean voltage neurogram. For purposes of quantification, MSNA was expressed as burst frequency (bursts min−1), burst amplitude (arbitrary units), and total minute activity (burst frequency × mean burst amplitude). During the experimental protocol, MSNA was expressed as a percentage of the initial isocapnic baseline level.
Data and statistical analysis
The beat-by-beat values for 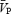 , MAP and HR, and the breath-by-breath values for ventilation and end-tidal gases, were initially averaged over 30-s intervals over each level of PET,CO2. Femoral blood flow, from each subject, was averaged over 60-s intervals. To calculate the percentage change from baseline, all data were averaged over the 5-min period of euoxic isocapnia immediately preceding any changes in PET,CO2 or handgrip exercise. The percentage changes in CVR1 and FVR (Fig. 3) are expressed as changes from the initial 5 min euoxic isocapnic baseline. Data were initially tested for normality of distribution using the Shapiro–Wilk test, before being analysed by repeated-measures analysis of variance, where appropriate. Post hoc tests (Tukey) were performed to isolate any significant differences. Following a simple main effect and interaction, Bonferroni-corrected paired-sample t tests were employed to make a posteriori comparisons at each handgrip period and PET,CO2 level of the between-subjects factor. Non-parametric equivalents included Wilcoxon matched pairs signed ranks and Kruskal–Wallis tests. Because there were no discernible differences between the two repetitions completed by each subject, data from the two protocols were combined to optimize the signal to noise ratio and for statistical analysis. The relationship between selected dependent variables was assessed using a Pearson product moment correlation (Fig. 4). Significance for all two-tailed tests was established at an alpha level of P < 0.05, and data are expressed as means ± 1 standard deviation (s.d.).
, MAP and HR, and the breath-by-breath values for ventilation and end-tidal gases, were initially averaged over 30-s intervals over each level of PET,CO2. Femoral blood flow, from each subject, was averaged over 60-s intervals. To calculate the percentage change from baseline, all data were averaged over the 5-min period of euoxic isocapnia immediately preceding any changes in PET,CO2 or handgrip exercise. The percentage changes in CVR1 and FVR (Fig. 3) are expressed as changes from the initial 5 min euoxic isocapnic baseline. Data were initially tested for normality of distribution using the Shapiro–Wilk test, before being analysed by repeated-measures analysis of variance, where appropriate. Post hoc tests (Tukey) were performed to isolate any significant differences. Following a simple main effect and interaction, Bonferroni-corrected paired-sample t tests were employed to make a posteriori comparisons at each handgrip period and PET,CO2 level of the between-subjects factor. Non-parametric equivalents included Wilcoxon matched pairs signed ranks and Kruskal–Wallis tests. Because there were no discernible differences between the two repetitions completed by each subject, data from the two protocols were combined to optimize the signal to noise ratio and for statistical analysis. The relationship between selected dependent variables was assessed using a Pearson product moment correlation (Fig. 4). Significance for all two-tailed tests was established at an alpha level of P < 0.05, and data are expressed as means ± 1 standard deviation (s.d.).
Figure 3. Percentage changes in middle cerebral artery peak blood flow velocity, femoral blood flow and cerebral and femoral vascular resistance during the isocapnic (ISO1 and ISO2) and hypercapnic (HC5, PET,CO2 5 mmHg above ISO1; HC10, PET,CO2 10 mmHg above ISO1) protocols.

Periods of handgrip are shown by the open circles and squares. *Significant within-condition difference (P < 0.05) between rest and handgrip. Cerebrovascular resistance =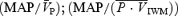 (n = 14) and femoral vascular resistance = MAP/FBF (n = 11). Data show the marked effect of hypercapnia alone in producing vasodilatation in the cerebral circulation (indicated by the decrease in CVR and increase in
(n = 14) and femoral vascular resistance = MAP/FBF (n = 11). Data show the marked effect of hypercapnia alone in producing vasodilatation in the cerebral circulation (indicated by the decrease in CVR and increase in 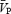 , •), whereas there is little change in FVR and FBF in the limb with hypercapnia (▪). In the femoral circulation, handgrip-induced muscle sympathetic nerve activity elicited a marked increase in FVR during isocapnic and hypercapnic conditions (□); in the cerebral circulation, the change in CVR is only evident in the isocapnic conditions (○). Arrows (→) denote significant impact of handgrip.
, •), whereas there is little change in FVR and FBF in the limb with hypercapnia (▪). In the femoral circulation, handgrip-induced muscle sympathetic nerve activity elicited a marked increase in FVR during isocapnic and hypercapnic conditions (□); in the cerebral circulation, the change in CVR is only evident in the isocapnic conditions (○). Arrows (→) denote significant impact of handgrip.
Figure 4. Relationship between handgrip-induced increases in muscle sympathetic nerve activity (MSNA) and cerebral vascular resistance (CVR, left panel) and femoral vascular resistance (FVR, right panel) during isocapnic and hypercapnic conditions.

Note the strong relationships between MSNA and CVR are lost during periods of hypercapnia.
Results
General observations
All 14 subjects completed the two trials, providing full data sets for the cardiorespiratory and cerebrovascular responses. MSNA data were successfully obtained from seven subjects in both tests. The unsuccessful MSNA recordings were due to alterations in nerve recording sites, loss of nerve signal, or failure to find an adequate MSNA signal. Successful FBF data were obtained in 11 subjects from both tests. The cardiorespiratory, cerebrovascular and MSNA data were not statistically different between the two trials. Therefore, data from the two tests were combined together. As illustrated in Fig. 1, the PET,CO2 and PET,O2 levels were well controlled at the desired levels throughout the experimental protocol. The general effect of both hypercapnia and handgrip-induced sympathetic activation on selected cardiorespiratory and cerebrovascular responses is outlined in Fig. 2.
Figure 2. Representative recordings from one subject (ID, 0039) during conditions of isocapnia (A), 30 s before handgrip and during the last 30 s of handgrip (shaded column); and (B) during conditions of hypercapnia (PET,CO2 10 mmHg above resting baseline value), 30 s before handgrip and during the last 30 s of handgrip (shaded column).

Note that during the isocapnic conditions, 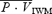 is maintained despite elevations in MAP, indicating strong cerebral autoregulation. Conversely, during the hypercapnic conditions, increases in MAP are reflected in parallel increases in
is maintained despite elevations in MAP, indicating strong cerebral autoregulation. Conversely, during the hypercapnic conditions, increases in MAP are reflected in parallel increases in 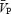 , suggesting a loss of cerebral autoregulation.
, suggesting a loss of cerebral autoregulation.
Cardiorespiratory responses: effect of static handgrip exercise
During the periods of handgrip-induced sympathetic activation, there were significant increases in MSNA, MAP, heart rate, and minute ventilation during the isocapnic and hypercapnic conditions (P < 0.05; Table 1; Fig. 2). On average, isometric handgrip increased muscle sympathetic nerve activity by ∼30–50% (P < 0.01), with no differences between conditions of isocapnia and hypercapnia. Mean arterial blood pressure increased significantly (P < 0.05) during all the periods of handgrip, with no differences between the isocapnic (ISO1) and the hypercapnic (HC) conditions (ISO1= 17.1 ± 9.7 mmHg; HC5, PET,CO2+5.0 = 19.5 ± 8.7 mmHg; HC10, PET,CO2+10.0 = 18.4 ± 10.1 mmHg). Similar increases in MAP were observed during the final isocapnic (ISO2) condition. Likewise, minute ventilation was increased significantly above baseline during each period of handgrip (ISO1= 4.3 ± 7.1 l min−1; HC5 = 8.7 ± 13.1 l min−1; HC10 = 9.4 ± 14.3 l min−1). A comparable increase from baseline in minute ventilation (7.2 ± 8.5 l min−1; P < 0.05) was apparent during the final handgrip in the ISO2 condition.
Table 1.
Selected responses during isocapnia, hypercapnia and periods of sympathetic nerve activation
| Condition |
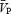 (cm s−1) (cm s−1) |
Power (V) | CVR1 (mmHg (cm s−1)−1) | CVR2 (mmHg %−1) | HR (beats min−1) | MAP (mmHg) | MSNAa (%) | FBFb (ml min−1) | FVRc (mmHg (ml min−1)−1) |
|---|---|---|---|---|---|---|---|---|---|
| ISO1 | 60.2 ± 8.9 | 2.8 ± 0.4 | 1.49 ± 0.23 | 0.90 ± 0.14 | 60.4 ± 11.7 | 89.7 ± 9.8 | 100 | 534.4 ± 227.7 | 0.17 ± 0.09 |
| ISO1 HG | 63.6 ± 11.2 | 2.6 ± 0.7 | 1.80 ± 0.32* | 1.52 ± 1.9* | 64.4 ± 19.2* | 114.7 ± 13.8* | 147.2 ± 41.9* | 478.0 ± 229.1* | 0.24 ± 0.11* |
| HC5 | 72.7 ± 11.3* | 2.7 ± 0.4 | 1.28 ± 0.20 | 0.82 ± 0.15 | 65.0 ± 13.2* | 93.3 ± 11.0 | 120.3 ± 32.1* | 526.4 ± 233.3 | 0.18 ± 0.08 |
| HC5 HG | 81.9 ± 11.6*† | 2.6 ± 0.4 | 1.37 ± 0.32 | 1.10 ± 0.30 | 72.8 ± 14.4*† | 111.9 ± 16.3*† | 176.4 ± 40.2*† | 506.9 ± 219.1 | 0.22 ± 0.10† |
| HC10 | 87.8 ± 15.0* | 2.6 ± 0.4 | 1.13 ± 0.24* | 0.79 ± 0.16* | 74.4 ± 15.6* | 99.6 ± 12.1* | 165.0 ± 83.0* | 567.4 ± 263.5 | 0.18 ± 0.09 |
| HC10 HG | 100.4 ± 13.9*‡ | 2.5 ± 0.6 | 1.20 ± 0.20* | 0.99 ± 0.41 | 84.7 ± 15.8*‡ | 120.3 ± 14.7*‡ | 189.8 ± 111.9*‡ | 568.0 ± 242.0 | 0.21 ± 0.10b |
| ISO2 | 57.2 ± 10.5 | 2.7 ± 0.5 | 1.61 ± 0.33* | 1.15 ± 0.41* | 66.0 ± 14.1* | 92.3 ± 19.7 | 117.8 ± 49.3 | 564.3 ± 277.8 | 0.16 ± 0.09 |
| ISO2 HG | 65.4 ± 11.3 | 2.5 ± 0.7 | 1.90 ± 0.31*§ | 1.68 ± 1.56*§ | 76.5 ± 16.8 *c | 124.0 ± 19.5*§ | 167.0 ± 81.3§ | 532.4 ± 260.4 | 0.23 ± 0.12c |
Significantly different from isocapnic baseline (P < 0.05);
significantly different from hypercapnia +5 mmHg (P < 0.05);
significantly different from hypercapnia +10 mmHg (P < 0.05);
significantly different from final isocapnic level (P < 0.05). 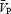 ,
, 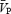 .
.
MSNA, data expressed as percentage change from initial isocapnic baseline;
femoral blood flow;
femoral vascular resistance, MAP/femoral blood flow.
Cerebrovascular and femoral vascular responses: effect of static handgrip exercise
The total power of the Doppler signal remained stable during the periods of handgrip (Table 1), indicating that the cross-sectional area of the MCA remained relatively unchanged. Application of handgrip-induced sympathetic activation produced an increase in both indices of CVR (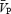 during both the ISO1 and ISO2 conditions (P < 0.05; Table 1). Consistency between
during both the ISO1 and ISO2 conditions (P < 0.05; Table 1). Consistency between 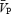 and
and 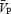 suggest that
suggest that 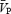 is an appropriate estimation of cerebral blood flow under the experimental conditions of the present study. During the isocapnic HG there was a 20% increase in calculated CVR1 (Table 1), while
is an appropriate estimation of cerebral blood flow under the experimental conditions of the present study. During the isocapnic HG there was a 20% increase in calculated CVR1 (Table 1), while 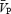 remained unchanged. Conversely, during the hypercapnic conditions,
remained unchanged. Conversely, during the hypercapnic conditions, 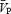 increased significantly by 13% and 14% during HC5 and HC10, respectively (Table 1), whereas CVR1 remained unchanged. There were no significant differences in CVR1 during HG-induced sympathetic activation at either HC5 or HC10. In contrast, HG-induced sympathetic stimulation produced comparable increases in FVR during the final minute of HG during each of the ISO and HC conditions (17–41%; Table 1; Fig. 3). During HG in ISO1, there was a decrease in FBF below baseline (P < 0.05). Likewise, during HG in HC5 and ISO2 there was a similar trend for a decrease in FBF (P= 0.09 and 0.07, respectively), whereas FBF was maintained during HG in HC10.
increased significantly by 13% and 14% during HC5 and HC10, respectively (Table 1), whereas CVR1 remained unchanged. There were no significant differences in CVR1 during HG-induced sympathetic activation at either HC5 or HC10. In contrast, HG-induced sympathetic stimulation produced comparable increases in FVR during the final minute of HG during each of the ISO and HC conditions (17–41%; Table 1; Fig. 3). During HG in ISO1, there was a decrease in FBF below baseline (P < 0.05). Likewise, during HG in HC5 and ISO2 there was a similar trend for a decrease in FBF (P= 0.09 and 0.07, respectively), whereas FBF was maintained during HG in HC10.
Cerebrovascular, cardiorespiratory and femoral responses to hypercapnia
On average, hypercapnia produced increases in minute ventilation and 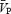 of approximately 5.7 l min−1 mmHg−1 and 2.7 cm s−1 mmHg−1, respectively. In ISO2, the baseline minute ventilation remained significantly elevated when compared with ISO1 (12.2 ± 5.6 versus 23.9 ± 7.0 l min−1; P < 0.01). There were no significant differences in
of approximately 5.7 l min−1 mmHg−1 and 2.7 cm s−1 mmHg−1, respectively. In ISO2, the baseline minute ventilation remained significantly elevated when compared with ISO1 (12.2 ± 5.6 versus 23.9 ± 7.0 l min−1; P < 0.01). There were no significant differences in 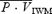 between the ISO1 and ISO2 conditions. Compared to ISO1, there were significant decreases in CVR during the HC10 level (P < 0.05; Table 1). Conversely, during the ISO2 test, the baseline CVR was elevated when compared to ISO1 (P < 0.05; Table 1; Fig. 2). The hypercapnic stimulus produced significant increases in heart rate, MAP and MSNA, when compared to ISO1 (Table 1). During ISO2, while heart rate remained elevated, there were no differences in MAP or MSNA, when compared to ISO1.
between the ISO1 and ISO2 conditions. Compared to ISO1, there were significant decreases in CVR during the HC10 level (P < 0.05; Table 1). Conversely, during the ISO2 test, the baseline CVR was elevated when compared to ISO1 (P < 0.05; Table 1; Fig. 2). The hypercapnic stimulus produced significant increases in heart rate, MAP and MSNA, when compared to ISO1 (Table 1). During ISO2, while heart rate remained elevated, there were no differences in MAP or MSNA, when compared to ISO1.
Hypercapnia did not produce any significant changes in FBF. However, during HC10, there was a trend for an increase in FBF (534 ± 228 ml min−1versus 568 ± 242 ml min−1; P= 0.07; Table 1) when compared with ISO1. As illustrated in Fig. 3, the conditions of hypercapnia produced unremarkable changes in FVR.
Relationships between changes in cerebral vascular resistance, femoral vascular resistance and muscle sympathetic nerve activity
Moderately strong, significant correlations were evident between the handgrip-induced increases in MSNA and CVR during ISO1 (r = 0.91; P < 0.01) and ISO2 (r = 0.78; P < 0.05). However, such relationships were not evident during HC5 (r = –0.43, NS) or HC10 (r = 0.32, not significant) above resting (Fig. 4). In contrast, moderately strong, significant correlations were evident between MSNA and FVR in all isocapnic and hypercapnic conditions (ISO1, r = 0.76; P < 0.05: HC5, r = 0.87; P < 0.05: HC10.0, r = 0.80; < 0.05: ISO2, r = 0.67; P < 0.05). These relationships are illustrated in Fig. 4.
Discussion
Major findings
Under isocapnic conditions, static handgrip exercise elicited an increase in CVR with unchanged 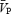 . However, under hypercapnic conditions CVR did not increase, and
. However, under hypercapnic conditions CVR did not increase, and 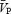 rose, tracking the handgrip-induced rises in arterial pressure. In contrast to the cerebral circulation, HG exercise produced significant increases in FVR in each of the isocapnic and hypercapnic conditions. Hypercapnia alone (before initiation of the HG exercise) produced small changes in FBF (0.6% mmHg−1) that were unremarkable compared to those in the cerebral circulation (5.0% mmHg−1). Collectively, our findings indicate that the cerebral circulation is more responsive to alterations in PCO2, and less responsive to sympathetic stimulation than the femoral circulation. The following discussion considers the evidence, methodological assumptions and the relevance underlying the findings of this study.
rose, tracking the handgrip-induced rises in arterial pressure. In contrast to the cerebral circulation, HG exercise produced significant increases in FVR in each of the isocapnic and hypercapnic conditions. Hypercapnia alone (before initiation of the HG exercise) produced small changes in FBF (0.6% mmHg−1) that were unremarkable compared to those in the cerebral circulation (5.0% mmHg−1). Collectively, our findings indicate that the cerebral circulation is more responsive to alterations in PCO2, and less responsive to sympathetic stimulation than the femoral circulation. The following discussion considers the evidence, methodological assumptions and the relevance underlying the findings of this study.
Influence of autonomic nerves on cerebral blood flow
The large cerebral arteries are innervated by adrenergic fibres originating from the ipsilateral superior cervical ganglion (Itakura et al. 1977). Smaller arteries or arterioles have much less adrenergic innervation, and are innervated mainly by secondary systems arising from the locus ceruleus (Nielsen, 1967; Itakura et al. 1977). Because of differences in distribution of α and β receptors, β receptor-mediated vasodilatation occurs mainly in small cerebral vessels, while α receptor-mediated vasoconstriction preferentially affects large cerebral arteries (Fitch et al. 1975). Thus, at least in experimental animals, sympathetic stimulation causes vasoconstriction in large vessels. However, as long as arterial pressure remains in the autoregulatory range, there is little change in cerebral blood flow because of a concomitant decrease in pial vessel resistance (Baumbach & Heistad, 1983), caused by either autoregulatory or β-adrenergic vasodilatation (Sohn, 1998). In the present study, the maintainance of 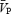 in the presence of handgrip-induced sympathetic stimulation and arterial pressure increase during isocapnia is broadly comparable to previous results in the highly controlled rabbit and cat models (Baumbach & Heistad, 1983).
in the presence of handgrip-induced sympathetic stimulation and arterial pressure increase during isocapnia is broadly comparable to previous results in the highly controlled rabbit and cat models (Baumbach & Heistad, 1983).
Cerebral vasoconstriction versus myogenic response
During HG performed under isocapnic conditions, we observed an increase in CVR along with unchanged 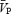 . However, from the present experiments, we cannot be certain whether this cerebral vasoconstriction was caused by sympathetic activation per se, or was a myogenic response elicited by the HG-induced rise in arterial pressure. Regardless of the mechanism, such increases in CVR served to maintain a constant cerebral blood velocity in the face of a large pressure response during isocapnic handgrip. In contrast, under hypercapnic conditions,
. However, from the present experiments, we cannot be certain whether this cerebral vasoconstriction was caused by sympathetic activation per se, or was a myogenic response elicited by the HG-induced rise in arterial pressure. Regardless of the mechanism, such increases in CVR served to maintain a constant cerebral blood velocity in the face of a large pressure response during isocapnic handgrip. In contrast, under hypercapnic conditions, 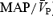 tracked the handgrip-induced rise in arterial pressure, indicating a failure of neural and/or autoregulatory mechanisms. While we acknowledge that correlations cannot establish causal relationships, we did observe strong correlations between increases in MSNA and CVR during isocapnic conditions (Fig. 4), which provide some support for the notion of sympathetically mediated vasoconstriction. The mechanism(s) underlying this finding requires further investigation.
tracked the handgrip-induced rise in arterial pressure, indicating a failure of neural and/or autoregulatory mechanisms. While we acknowledge that correlations cannot establish causal relationships, we did observe strong correlations between increases in MSNA and CVR during isocapnic conditions (Fig. 4), which provide some support for the notion of sympathetically mediated vasoconstriction. The mechanism(s) underlying this finding requires further investigation.
Handgrip-induced femoral vasoconstriction
Our findings, which are consistent with previous reports that leg vascular resistance and MSNA are highly correlated during fatiguing exercise (Saito et al. 1988; Seals, 1989), support the concept that sympathetic engagement is a major, direct vasoconstrictor mechanism in the lower limb. Although pressure-dependent (i.e. myogenic) mechanisms may also play a role (Shoemaker et al. 2000), we would not expect FBF to be reduced below the baseline level, as it was in this study, if only myogenic mechanisms were operative.
Differential responses between the cerebral and femoral vasculatures
In the present study, sympathetic engagement during conditions of hypercapnia caused vasoconstriction in the femoral, but not the cerebral vasculature. Why should blood vessels in the brain and leg respond differently when hypercapnia is superimposed on sympathetic activation? Several explanations for these differential sensitivities can be proposed. First, it has long been appreciated that the vasodilatory effect of hypercapnia is much more profound in the cerebral vessels than the leg vessels (Lennox & Gibbs, 1932). Indeed, in the present study, the CO2 reactivity in the cerebral circulation was a ∼5% increase in 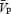 for every 1 mmHg increase in PET,CO2, whereas in the femoral circulation it was ∼8-fold lower. Hypercapnic cerebral vasodilatation is a local effect with complex and perhaps redundant mechanisms (nitric oxide synthase, cyclooxygenase, P-450 oxygenase pathways have all been implicated (Iadecola & Zhang, 1994; Pelligrino et al. 1999; Niwa et al. 2001). It is not clear why CO2 is a less potent vasodilator in the limb circulation. Second, the vasoconstrictor responsiveness of the cerebral and limb vessels is likely to be influenced by anatomical differences in the two vascular beds. There is a higher distribution of α-adrenoreceptors in the peripheral and dependent vascular beds than in the cerebral circulation, and the presence of the endothelial blood–brain barrier limits access of many circulating vasoconstrictors to vascular smooth muscle in the cerebral vessels (Faraci & Heistad, 1998). For example, recent studies which have examined the effects of intravenous infusion of various sympathomimetic drugs (ephedrine, dobutamine, dopexamine (Moppett et al. 2004)) and noradrenaline (Kimmerly et al. 2003), reported no effect of the drugs on cerebrovascular control in healthy humans.
for every 1 mmHg increase in PET,CO2, whereas in the femoral circulation it was ∼8-fold lower. Hypercapnic cerebral vasodilatation is a local effect with complex and perhaps redundant mechanisms (nitric oxide synthase, cyclooxygenase, P-450 oxygenase pathways have all been implicated (Iadecola & Zhang, 1994; Pelligrino et al. 1999; Niwa et al. 2001). It is not clear why CO2 is a less potent vasodilator in the limb circulation. Second, the vasoconstrictor responsiveness of the cerebral and limb vessels is likely to be influenced by anatomical differences in the two vascular beds. There is a higher distribution of α-adrenoreceptors in the peripheral and dependent vascular beds than in the cerebral circulation, and the presence of the endothelial blood–brain barrier limits access of many circulating vasoconstrictors to vascular smooth muscle in the cerebral vessels (Faraci & Heistad, 1998). For example, recent studies which have examined the effects of intravenous infusion of various sympathomimetic drugs (ephedrine, dobutamine, dopexamine (Moppett et al. 2004)) and noradrenaline (Kimmerly et al. 2003), reported no effect of the drugs on cerebrovascular control in healthy humans.
Comparison with previous studies: the cerebrovascular ‘escape’ phenomenon
Results from experimental animals, during sustained sympathetic activation have shown that a ‘vasomotor escape’ phenomenon occurs, suggesting that neural control of the cerebral circulation may be more effective under dynamic than steady-state conditions (Sercombe et al. 1979; Baumbach & Heistad, 1983). The results from the present study, under 2 min of HG-induced sympathetic activation, are not consistent with such a ‘vasomotor escape’ phenomenon. However, the unphysiological frequencies (1–50 Hz) and patterns of stimulation used in animal studies (Sercombe et al. 1979; Baumbach & Heistad, 1983; Lacroix et al. 1988) make comparisons with human studies unrealistic.
Methodological considerations
Transcranial Doppler ultrasound
We used Doppler ultrasound to measure flow velocity in the middle cerebral artery. While this is not a direct measurement of cerebral blood flow, we believe that velocity is a reasonable estimate of flow in our experiments because: (1) the diameter of the middle cerebral artery has been shown to vary by less than 4% during changes in arterial pressure and changes in CO2 tension (Giller et al. 1993; Serrador et al. 2000), and (2) velocity and flow through the middle cerebral artery are highly correlated (Kirkham et al. 1986). Our estimates of cerebrovascular CO2 reactivity (2.7 cm s−1 mmHg−1 (∼5% mmHg−1) for hypercapnia) are consistent with previous reports which have utilized a range of different methodological techniques to assess cerebrovascular CO2 reactivity (Kety & Schmidt, 1948; Rostrup et al. 1994; Ide et al. 2003). As evident in the present study, the total power of the Doppler signal remained stable during the periods of handgrip, indicating that the cross-sectional area of the MCA remained relatively unchanged. Furthermore, our contribution of a MCA flow index (i.e. 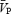 ) does help to interpret the results, in that this index corroborates the velocity measurements, as an index of flow, reported in this study. This is consistent with our previous work (Poulin & Robbins, 1996; Ainslie & Poulin, 2004) during periods of hypercapnia and hypoxia, which showed no change in the Doppler power signal. Finally, despite the limitations associated with transcranial Doppler ultrasound, this is the only method with sufficient temporal resolution to address the aims of this study.
) does help to interpret the results, in that this index corroborates the velocity measurements, as an index of flow, reported in this study. This is consistent with our previous work (Poulin & Robbins, 1996; Ainslie & Poulin, 2004) during periods of hypercapnia and hypoxia, which showed no change in the Doppler power signal. Finally, despite the limitations associated with transcranial Doppler ultrasound, this is the only method with sufficient temporal resolution to address the aims of this study.
Cerebrovascular resistance
Vascular resistance is estimated as the ratio of the pressure drop to flow across the vascular bed. In the case of CVR, calculation is complicated by unknown values of intracranial and venous pressures. However, since our subjects were positioned in a semisupine position, it is likely that the major determinant of cerebral perfusion pressure was MAP. Recent comparisons of different approaches to determining CVR have supported the use of the MAP : cerebral blood flow ratio (Schondorf et al. 2001). From the present study, however, it cannot be determined whether the cerebrovascular responses in the distribution of the MCA reflect global cerebral effects; e.g. discreet regions of the brain may respond differently to changes in sympathetic stimulation (Sercombe et al. 1975; Roatta et al. 2003). The novel approach in the present study is the use of a Doppler flow index in order to account for any changes in the cross-sectional area of the MCA.
Control of end-tidal gases
The combined techniques of dynamic end-tidal forcing, Doppler ultrasound and microneurography provided means to overcome many of the technical shortcomings apparent in previous studies. In the majority of these earlier studies, which have considered the role of sympathetic activation in the cerebral circulation of humans, it was not possible to achieve accurate control over the end-tidal gases because of limitations in the techniques used to administer the changes of PET,CO2 and PET,O2. A number of these studies did not control for PET,CO2 levels (Grubb et al. 1991; Levine et al. 1994; Bondar et al. 1995; Zhang et al. 1998; Jordan et al. 1998; Sohn, 1998; Giller et al. 2000; Zhang et al. 2002), raising the possibility that the reported decrease in CBF was due, at least in large part, to the influence of hypocapnia rather than any influence from the autonomic nervous system. The studies that did attempt to control the end-tidal gases, involved manual switches between different fixed inspired gas mixtures (Jordan et al. 2000; Jordan et al. 2002). As such, several minutes are required before the desired end-tidal levels are reached. The dynamic end-tidal forcing system uses prediction and feedback correction (via the end-tidal gas values) to adjust the inspired gas composition on a breath-by-breath basis to hold the subject's PET,CO2 and PET,O2 levels constant despite changes in ventilation. It is well established that arterial PCO2 is the most potent regulator of the cerebral circulation (Brian, 1998). Since we observed modest but significant increases in ventilation (4–10 l min−1) during the periods of handgrip, it is clear that precise control of the PET,CO2 gas levels is critical in the interpretation and understanding of the influence of sympathetic activation on CBF regulation. Inadequate control of PET,CO2 during periods of sympathetic stimulation may well be one important factor contributing to the controversy surrounding the neural control of the cerebral circulation in humans. While this study did not include direct measurements of arterial PCO2 and PO2, end-tidal values of PCO2 and PO2 have been shown to be appropriate estimates of arterial values in similar conditions to those of the present study, in individuals free of respiratory disease (Robbins et al. 1990).
Summary and relevance
During isocapnia, HG caused an increase in CVR, while 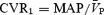 was unchanged; conversely, under hypercapnic conditions, CVR was unchanged and
was unchanged; conversely, under hypercapnic conditions, CVR was unchanged and 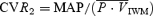 was elevated. In the femoral circulation, HG exercise significantly elevated FVR in both isocapnic and hypercapnic conditions. Hypercapnia (before initiation of the HG exercise) produced unremarkable changes in FBF (0.6% mmHg−1) compared to those in the cerebral circulation (5.0% mmHg−1).
was elevated. In the femoral circulation, HG exercise significantly elevated FVR in both isocapnic and hypercapnic conditions. Hypercapnia (before initiation of the HG exercise) produced unremarkable changes in FBF (0.6% mmHg−1) compared to those in the cerebral circulation (5.0% mmHg−1).
The fact that the circulatory adjustments of the cerebral vasculature are seemingly opposite to those of the femoral vasculature is of teleological relevance. If vessels in the cerebral and femoral vasculatures were both to dilate to the same extent in response to increased arterial PCO2, there would be a dramatic fall in systemic blood pressure. Sympathetic stimulation seems to play a major vasoconstrictor role in the femoral vasculature, but a less prominent role in the cerebral vasculature. The main functional relevance of sympathetically mediated cerebral vasoconstriction may be to protect the blood–brain barrier from overperfusion during periods of acute hypertension, when the limits of autoregulation are compromised (Mayhan et al. 1987). The high resting metabolic requirements of the brain, compared with that of the femoral vasculature, might be another reason why this circulatory arrangement is desirable, i.e. high CO2 sensitivity is a way for the brain to match metabolism with flow. On the other hand, sympathetic vasoconstriction is a way to redistribute cardiac output from non-essential to essential vascular beds under conditions of environmental change (posture, exercise, heat/cold). Since there is probably no condition where the cerebral circulation would be considered non-essential, it seems desirable for the brain to have limited capacity for vasoconstriction.
Acknowledgments
This project was supported by the Alberta Heritage Foundation for Medical Research (AHFMR), Heart and Stroke Foundation of Alberta, NWT, & Nunavut and the Canadian Institutes of Health Research (CIHR). PNA is supported by a Focus-on-Stroke postdoctoral fellowship (Heart and Stroke Foundation of Canada, the Canadian Stroke Network, CIHR, and AstraZeneca Canada). MJP is a CIHR New Investigator and AHFMR Medical Scholar. We extend our gratitude to Professor PA Robbins for assistance in setting up the dynamic end-tidal forcing technique in Calgary, and to JS Vantanajal for skilled technical support.
References
- Ainslie PN, Poulin MJ. Ventilatory, cerebrovascular, and cardiovascular interactions in acute hypoxia: regulation by carbon dioxide. J Appl Physiol. 2004;97:149–159. doi: 10.1152/japplphysiol.01385.2003. [DOI] [PubMed] [Google Scholar]
- Baumbach GL, Heistad DD. Effects of sympathetic stimulation and changes in arterial pressure on segmental resistance of cerebral vessels in rabbits and cats. Circ Res. 1983;52:527–533. doi: 10.1161/01.res.52.5.527. [DOI] [PubMed] [Google Scholar]
- Bondar RL, Kassam MS, Stein F, Dunphy PT, Fortney S, Riedesel ML. Simultaneous cerebrovascular and cardiovascular responses during presyncope. Stroke. 1995;26:1794–1800. doi: 10.1161/01.str.26.10.1794. [DOI] [PubMed] [Google Scholar]
- Brian JE. Carbon dioxide and the cerebral circulation. Anesthesiology. 1998;88:1365–1386. doi: 10.1097/00000542-199805000-00029. [DOI] [PubMed] [Google Scholar]
- Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–265. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Hamel E. Perivascular nerves in brain vessels. In: Edvinsson L, Krause DN, editors. Cerebral Blood Flow and Metabolism. 2nd. Philadelphia: Lippincott. Williams & Wilkins; 2002. pp. 43–67. [Google Scholar]
- Faraci FM, Heistad DD. Regulation of cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- Fitch W, MacKenzie ET, Harper AM. Effects of decreasing arterial blood pressure on cerebral blood flow in the baboon. Influence of the sympathetic nervous system. Circ Res. 1975;37:550–557. doi: 10.1161/01.res.37.5.550. [DOI] [PubMed] [Google Scholar]
- Giller CA, Bowman G, Dyer H, Mootz L, Krippner W. Cerebral arterial diameters during changes in blood pressure and carbon dioxide during craniotomy. Neurosurgery. 1993;32:737–742. [PubMed] [Google Scholar]
- Giller CAGI, Iler AM, Cooper CR, Hatab MR. Evaluation of the cerebral hemodynamic response to rhythmic handgrip. J Appl Physiol. 2000;88:2205–2213. doi: 10.1152/jappl.2000.88.6.2205. [DOI] [PubMed] [Google Scholar]
- Grubb BP, Gerard G, Roush K, Temesy-Armos P, Montford P, Elliott L, Hahn H, Brewster P. Cerebral vasoconstriction during head-upright tilt-induced vasovagal syncope. A paradoxic and unexpected response. Circulation. 1991;84:1157–1164. doi: 10.1161/01.cir.84.3.1157. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F. Nitric oxide- dependent and independent components of cerebrovasodilation elicited by hypercapnia. Am J Physiol. 1994;266:R546–R552. doi: 10.1152/ajpregu.1994.266.2.R546. [DOI] [PubMed] [Google Scholar]
- Ide K, Eliasziw M, Poulin MJ. The relationship between middle cerebral artery blood velocity and end-tidal PCO2 in the hypocapnic-hypercapnic range in humans. J Appl Physiol. 2003;95:129–137. doi: 10.1152/japplphysiol.01186.2002. [DOI] [PubMed] [Google Scholar]
- Itakura T, Yamamoto K, Tohyama M, Shimizu N. Central dual innervation of arterioles and capillaries in the brain. Stroke. 1977;8:360–365. doi: 10.1161/01.str.8.3.360. [DOI] [PubMed] [Google Scholar]
- Jordan J, Shannon JR, Black BK, Paranjape SY, Barwise J, Robertson D. Raised cerebrovascular resistance in idiopathic orthostatic intolerance – Evidence for sympathetic vasoconstriction. Hypertension. 1998;32:699–704. doi: 10.1161/01.hyp.32.4.699. [DOI] [PubMed] [Google Scholar]
- Jordan J, Shannon JR, Diedrich A, Black B, Costa F, Robertson D, Biaggioni I. Interaction of carbon dioxide and sympathetic nervous system activity in the regulation of cerebral perfusion in humans. Hypertension. 2000;36:383–388. doi: 10.1161/01.hyp.36.3.383. [DOI] [PubMed] [Google Scholar]
- Jordan J, Shannon JR, Diedrich A, Black BK, Robertson D. Increased sympathetic activation in idiopathic orthostatic intolerance – Role of systemic adrenoreceptor sensitivity. Hypertension. 2002;39:173–178. doi: 10.1161/hy1201.097202. [DOI] [PubMed] [Google Scholar]
- Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest. 1948;27:484–492. doi: 10.1172/JCI101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmerly DS, Tutungi E, Wilson TD, Serrador JM, Gelb AW, Hughson RL, Shoemaker JK. Circulating norepinephrine and cerebrovascular control in conscious humans. Clin Physiol Funct Imaging. 2003;23:314–319. doi: 10.1046/j.1475-0961.2003.00507.x. [DOI] [PubMed] [Google Scholar]
- Kirkham FJ, Padayachee TS, Parsons S, Seargeant LS, House FR, Gosling RG. Transcranial measurement of blood velocities in the basal cerebral arteries using pulsed Doppler ultrasound: velocity as an index of flow. Ultrasound Med Biol. 1986;12:15–21. doi: 10.1016/0301-5629(86)90139-0. [DOI] [PubMed] [Google Scholar]
- Lacroix JS, Stjarne P, Anggard A, Lundberg JM. Sympathetic vascular control of the pig nasal mucosa (1): Increased resistance and capacitance vessel responses upon stimulation with irregular bursts compared to continuous impulses. Acta Physiol Scand. 1988;132:83–90. doi: 10.1111/j.1748-1716.1988.tb08301.x. [DOI] [PubMed] [Google Scholar]
- LeMarbre G, Stauber S, Khayat RN, Puleo DS, Skatrud JB, Morgan BJ. Baroreflex-induced sympathetic activation does not alter cerebrovascular CO2 responsiveness in humans. J Physiol. 2003;551:609–616. doi: 10.1113/jphysiol.2003.046987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennox WG, Gibbs EL. The blood flow in the brain and leg of man, and the changes induced by alteration of blood gases. J Clin Invest. 1932;11:1155–1177. doi: 10.1172/JCI100470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine BD, Giller CA, Lane LD, Buckey JC, Blomqvist CG. Cerebral versus systemic hemodynamics during graded orthostatic stress in humans. Circulation. 1994;90:298–306. doi: 10.1161/01.cir.90.1.298. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Werber AH, Heistad DD. Protection of cerebral vessels by sympathetic nerves and vascular hyperthrophy. Circulation. 1987;75:I107–I112. [PubMed] [Google Scholar]
- Moppett IK, Wild MJ, Sherman RW, Latter JA, Miller K, Mahajan RP. Effects of ephedrine, dobutamine and dopexamine on cerebral haemodynamics: transcranial Doppler studies in healthy volunteers. Br J Anaesth. 2004;92:39–44. doi: 10.1093/bja/aeh014. [DOI] [PubMed] [Google Scholar]
- Nielsen KC. Adrenergic innervation of pial arteries related to the circle of Willis in the cat. Brain Res. 1967;6:770–772. doi: 10.1016/0006-8993(67)90134-5. [DOI] [PubMed] [Google Scholar]
- Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001;88:600–608. doi: 10.1161/01.res.88.6.600. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Santizo RA, Wang Q. Miconazole represses CO2-induced pial arteriolar dilation only under selected circumstances. Am J Physiol. 1999;277:H1484–H1490. doi: 10.1152/ajpheart.1999.277.4.H1484. [DOI] [PubMed] [Google Scholar]
- Poulin MJ, Liang P-J, Robbins PA. Fast and slow components of the cerebral blood flow response to step decreases in end-tidal PCO2 in humans. J Appl Physiol. 1998;85:388–397. doi: 10.1152/jappl.1998.85.2.388. [DOI] [PubMed] [Google Scholar]
- Poulin MJ, Robbins PA. Indexes of flow and cross-sectional area of the middle cerebral artery using Doppler ultrasound during hypoxia and hypercapnia in humans. Stroke. 1996;27:2244–2250. doi: 10.1161/01.str.27.12.2244. [DOI] [PubMed] [Google Scholar]
- Przybylowski T, Bangash MF, Reichmuth K, Morgan BJ, Skatrud JB, Dempsey JA. Mechanisms of the cerebrovascular response to apnoea in humans. J Physiol. 2003;548:323–332. doi: 10.1113/jphysiol.2002.029678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roatta S, Canova D, Bosone D, Micieli G, Passatore M. Noradrenergic constriction of cerebral arteries as detected by transcranial Doppler (TCD) in the rabbit. Ultrasound Med Biol. 2003;29:1397–1404. doi: 10.1016/s0301-5629(03)00977-3. [DOI] [PubMed] [Google Scholar]
- Robbins PA, Conway J, Cunningham DA, Khamnei S, Paterson DJ. A comparison of indirect methods for continuous estimation of arterial PCO2 in men. J Appl Physiol. 1990;68:1727–1731. doi: 10.1152/jappl.1990.68.4.1727. [DOI] [PubMed] [Google Scholar]
- Robbins PA, Swanson GD, Howson MG. A prediction correction scheme for forcing alveolar gases along certain time courses. J Appl Physiol. 1982a;52:1353–1357. doi: 10.1152/jappl.1982.52.5.1353. [DOI] [PubMed] [Google Scholar]
- Robbins PA, Swanson GD, Micco AJ, Schubert WP. A fast gas-mixing system for breath-to-breath respiratory control studies. J Appl Physiol. 1982b;52:1358–1362. doi: 10.1152/jappl.1982.52.5.1358. [DOI] [PubMed] [Google Scholar]
- Rostrup E, Larsson HBW, Toft PB, Thomsen C, Ring P, Sondergaard L, Henriksen O. Functional MRI of CO2 induced increase in cerebral perfusion. NMR Biomed. 1994;7:29–34. doi: 10.1002/nbm.1940070106. [DOI] [PubMed] [Google Scholar]
- Saito M, Mano T, Iwase S, Koga K, Abe H, Yamazaki Y. Responses in muscle sympathetic activity to acute hypoxia in humans. J Appl Physiol. 1988;65:1548–1552. doi: 10.1152/jappl.1988.65.4.1548. [DOI] [PubMed] [Google Scholar]
- Schondorf R, Stein R, Roberts R, Benoit J, Cupples W. Dynamic cerebral autoregulation is preserved in neurally mediated syncope. J Appl Physiol. 2001;91:2493–2502. doi: 10.1152/jappl.2001.91.6.2493. [DOI] [PubMed] [Google Scholar]
- Seals DR. Sympathetic neural discharge and vascular resistance during exercise in humans. J Appl Physiol. 1989;66:2472–2478. doi: 10.1152/jappl.1989.66.5.2472. [DOI] [PubMed] [Google Scholar]
- Sercombe R, Aubineau P, Edvinsson L, Mamo H, Owman CH, Pinard E, Seylaz J. Neurogenic influence on local cerebral blood flow. Effect of catecholamines or sympathetic stimulation as correlated with sympathetic innervation. Neurology. 1975;25:954–963. doi: 10.1212/wnl.25.10.954. [DOI] [PubMed] [Google Scholar]
- Sercombe R, Lacombe P, Aubineau P, Mamo H, Pinard E, Reynier-Rebuffel AM, Seylaz J. Is there an active mechanism limiting the influence of the sympathetic system on the cerebral vascular bed? Evidence for vasomotor escape from sympathetic stimulation in the rabbit. Brain Res. 1979;164:81–102. doi: 10.1016/0006-8993(79)90008-8. [DOI] [PubMed] [Google Scholar]
- Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke. 2000;31:1672–1678. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
- Shoemaker JK, Herr MD, Sinoway LI. Dissociation of muscle sympathetic nerve activity and leg vascular resistance in humans. Am J Physiol Heart Circ Physiol. 2000;279:H1215–H1219. doi: 10.1152/ajpheart.2000.279.3.H1215. [DOI] [PubMed] [Google Scholar]
- Sohn YH. Cerebral hemodynamic changes induced by sympathetic stimulation tests. Yonsei Med J. 1998;39:322–327. doi: 10.3349/ymj.1998.39.4.322. [DOI] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE, Torebjork HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD. Autonomic neural control of dynamic cerebral autoregulation in humans. Circulation. 2002;106:1814–1820. doi: 10.1161/01.cir.0000031798.07790.fe. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zuckerman JH, Levine BD. Deterioration of cerebral autoregulation during orthostatic stress: insights from the frequency domain. J Appl Physiol. 1998;85:1113–1122. doi: 10.1152/jappl.1998.85.3.1113. [DOI] [PubMed] [Google Scholar]