

Abstract
The 1679-nt single-stranded RNA genome of hepatitis delta virus (HDV) is circular in conformation. It is able to fold into an unbranched rodlike structure via intramolecular base-pairing. This RNA is replicated by host RNA polymerase II (Pol II). Such transcription is unique, because Pol II is known only for its ability to act on DNA templates. The present study addressed the ability of the HDV RNA replication to tolerate insertions of up to 1000 nt of non-HDV sequence either at an end of the rodlike RNA structure or at a site embedded within the rod. The insertions did not interfere with the ability of primary transcripts to be processed in vivo by ribozyme cleavage and ligation. The insertions greatly reduced the ability of genomes to replicate. However, when total RNA from such transfected cells was used to transfect new recipient cells, replicating HDV RNAs could be detected by Northern analyses. The size of the emerged RNAs was consistent with loss of the inserted sequences. RT-PCR, cloning, and sequencing showed that recovery involved removal of inserted sequences with or without small deletions of adjacent RNA sequences. Such restoration of the RNA genome is consistent with a model requiring intramolecular template-switching (RNA recombination) during RNA-directed transcription, in combination with biological selection for maintenance of the rodlike structure of the template.
Keywords: hepatitis delta virus, RNA-directed transcription, RNA polymerase II, template-switching, intramolecular RNA recombination
INTRODUCTION
Hepatitis delta virus (HDV) is a subviral agent, which uses hepatitis B (HBV) envelope proteins to assemble the 1.7-kb single-stranded circular RNA genome into viral particles (Rizzetto et al. 1980; Wang et al. 1986). The HDV genome replicates via RNA-directed RNA synthesis involving the production of an exactly complementary RNA, known as the antigenome (Chen et al. 1986). The genome and the antigenome contain unique and essential sequence domains that act as site-specific ribozymes (Kuo et al. 1988b; Macnaughton et al. 1993). HDV RNA replication is considered to involve a rolling-circle mechanism in which circular RNA templates are transcribed to produce greater than unit-length multimers of opposite polarity. These transcripts are processed by the ribozymes to release unit-length linear species. Some of these are ligated to produce circles that can be used as templates for further rounds of transcription and processing (Lai 2005; Taylor 2005). The antigenome contains the open reading frame for one small but essential protein, a 195–amino acid RNA-binding protein known as the delta antigen (δAg) (Chao et al. 1990; Lee et al. 1993). However, the δAg is translated from a third HDV RNA. This is a linear RNA of antigenomic polarity. Its size is less than unit-length, ∼0.8 kb. It has a 5′-cap and 3′-poly(A) tail and is considered to arise via an alternative processing of an antigenomic RNA transcript (Gudima et al. 2000; Nie et al. 2004).
Some of the most intriguing HDV questions center around how the HDV RNAs are transcribed. RNA replication is dependent upon one or more host RNA polymerases. At this time our interpretation (Taylor 2005; Chang et al. 2006a, b) is that the data favor that the major, if not sole, contributor is the RNA polymerase II (Pol II). This enzyme is otherwise known only for its ability to carry out transcription that is DNA-directed, typically leading to the production of 5′-capped and 3′-poly(A)-tailed mRNA species or, in some cases, to the production of certain small noncoding nuclear RNAs, such as U1–U5 (Dahlberg and Lund 1988).
Part of the essential nature of the δAg could be to help the host polymerase to transcribe an RNA template. Specifically, one interpretation of certain in vitro studies is that δAg can act as an inhibitor of a negative regulator of RNA transcription by Pol II from an RNA template (Yamaguchi et al. 2001). However, a major contributor to this redirected transcription is considered to be the structure of the genomic and antigenomic RNAs. These circular RNAs have the ability to fold, by intramolecular base-pairing involving ∼74% of all nucleotides, to produce the unbranched rodlike structure (Wang et al. 1986; Kuo et al. 1988a). There is, in addition, evidence that the δAg has not only RNA-binding ability but also the potential to make specific interactions with such rodlike HDV RNAs (Chao et al. 1991; Ryu et al. 1993).
Apparently there are significant constraints on the length and the sequence of the HDV genome. Examination of different natural isolates of HDV collected from around the world reveals that the size of these genomes is quite constant within a range of ∼30 nt (Radjef et al. 2004). In addition, several previous experimental studies have attempted to follow the consequences for replication of perturbations in the size and the sequence of the HDV RNA. For example, even after maintenance of the ribozyme domains and an attempt to conserve the rodlike folding, along with providing a separate source of δAg, such modified RNAs failed to replicate in transfected cells (Lazinski and Taylor 1994). In contrast, when relatively small changes, insertions, deletions, or even sequence replacements were made at either end of the rodlike structure, some of these RNAs were able to replicate (Netter et al. 1993, 1995a; Wu et al. 1997; Gudima et al. 1999). In another study, an 8-nt palindromic sequence was inserted at a series of sites around the genome. Some of these insertions were apparently tolerated; although in other situations, the inserted sequence was no longer detected after RNA replication was initiated (Wang et al. 1997).
This laboratory has previously demonstrated the ability of Pol II to carry out template-switching during HDV RNA–directed RNA transcription. We showed that such switching occurs when linear forms of HDV RNA are used to initiate replication. It is not that the linear RNA templates are first converted to a circle. Rather, there occurs during transcription intramolecular template-switching leading to the production of greater than unit-length primary transcripts. These, in turn, are processed to produce unit-length RNA circles that can act as templates for further replication (Chang and Taylor 2002; Gudima et al. 2004).
The present study was undertaken to further explore the mechanism and potential for template-switching during HDV RNA–directed RNA synthesis by Pol II. We mutated the HDV genomic RNA by inserting sequences of up to 1000 nt into the genome at either of two locations. One location was near an end of the rodlike RNA structure, and the other was embedded within the rod. We found that such insertions had no significant effect on the post-transcriptional processing and stabilization of RNA transcripts. However, those mutants with larger insertions seemed to lose the ability to achieve RNA-directed replication. However, after employing a procedure in which the total RNA from transfected cells was extracted and then used to transfect new recipient cells, we were able to detect replication of HDV RNA. Furthermore, these RNAs no longer contained the inserted non-HDV sequences. We present a model in which restored replication involved removal of the inserted sequences via intramolecular template-switching (that is, RNA recombination) on the original modified HDV RNA during an RNA-directed transcription event carried out by the host RNA Pol II.
RESULTS
Design of HDV RNA mutants
For the following studies, we initially considered it was necessary to design cDNA constructs that, when expressed in transfected cells, would produce mutated genomic RNAs that contained insertions of non-HDV sequences. Figure 1A is a representation of the unmodified expression construct, the nature of the RNA transcript, and the possible processing by the two genomic ribozymes, first to release unit-length linear RNA and followed by ligation to form circles.
FIGURE 1.

Generation in vivo of genomic HDV RNAs with insertion of non-HDV sequences. (A) An expression vector containing 1.2 copies of the HDV genome and two copies of the genomic ribozyme (shaded circles) was modified at either of two unique locations, the XbaI site at position 785/786 or the NheI site at position 430/431, using a published numbering system (Kuo et al. 1988a). If these DNA were transfected into cells, we would expect a primary transcript of genomic RNA, containing two ribozymes, to be processed by cleavage followed by ligation and produce unit-length RNA circles. (B) The folding predicted using M-fold (Zuker 2003) for the HDV genomic RNA at and around the XbaI site, which is near one end of the rodlike structure. (C) A similar folding at and around the NheI site, which is embedded within the rodlike structure.
We chose two sites for the insertion of non-HDV sequences. As indicated in Figure 1A, these corresponded to unique XbaI and NheI sites on an HDV cDNA. In terms of the predicted RNA folding, the XbaI site was very close to one end of the rod (Fig. 1B). In contrast, the NheI site was embedded in the rodlike folding (Fig. 1C). At the XbaI site, we inserted each of the sequences summarized in Table 1. These non-HDV insertions ranged from 1, 2, 3, 4, 10, and 12 nt up to 388 and 978 nt. We wanted to examine not only the size of insertion but also the influence of its structure. Therefore, we also made insertions of two known stem–loop elements. One was a GNRA tetraloop (Rudisser and Tinoco 2000); the other, a bacterial RNA polymerase terminator (Rosenberg and Court 1979). The tetraloop would exist only on the genomic RNA. In contrast, the terminator could only exist on the antigenome. At the NheI site, we only tested the 388- and 978-nt non-HDV sequences, but these were inserted in both orientations.
TABLE 1.
Sequence of non-HDV insertions introduced onto genome at XbaI site
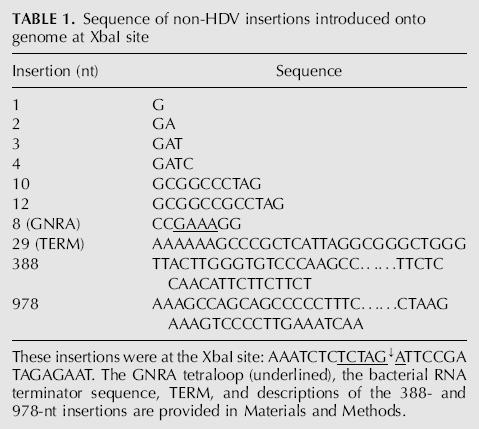
Post-transcriptional processing of nonreplicating RNA transcripts
Prior to addressing the replication competence of the modified HDV RNA constructs, it was necessary to test the assumption implicit in Figure 1: that the modified RNAs could first be transcribed and post-transcriptionally processed, by ribozyme cleavage and then ligation, to form RNA circles, which can be stabilized in vivo by the presence of the δAg (Lazinski and Taylor 1994; Moraleda et al. 1999). We studied this processing in the absence of possible replication. To suppress such replication, the expression plasmids were cotransfected into Huh7 cells along with a construct that expresses a form of δAg, known as large δAg, which under these conditions is known to inhibit RNA-directed RNA synthesis (Chao et al. 1990).
At 3 d after such cotransfections, the total RNAs were extracted, glyoxalated, and examined by Northern analysis using gels of 1.7% agarose for the presence of genomic HDV RNA species. Representative results are shown in Figure 2A. For all constructs, a major band was detected. For those with small inserts (lanes 2–5), the mobility of this band was indistinguishable from that of unit-length 1679-nt HDV RNA (lane 1). In contrast, for those with the larger insertions (lanes 6,7), the major bands had, as expected, a significantly reduced mobility because of the insertion of the extra 388 and 978 nt, respectively. For all constructs (not just those shown in lanes 2–7), the detected major band was of approximately the same intensity as that for the processed wild-type HDV RNA (lane 1). Our interpretation is that the insertions of non-HDV sequences did not produce a major defect in the transcription, processing, or accumulation of the processed DNA-directed RNA transcripts. However, at the same time, it was noted that in lanes 6 and 7, those corresponding to RNAs with the largest 388- and 978-nt insertions, there were also minor amounts of faster migrating species, some of which were at and about the migration for unit-length wild-type HDV RNAs.
FIGURE 2.

Northern analysis of processed genomic HDV RNA transcripts in the absence of replication. These RNAs were generated by transfection of Huh7 cells with DNA constructs (as described for Fig. 1) bearing various insertions at the XbaI site of non-HDV sequences as summarized in Table 1. (A) At day 3 after transfection, total RNA was extracted, glyoxalated, and subjected to Northern analyses using a gel of 1.7% agarose. Lane s refers to a gel-purified 1.1× genomic RNA standard. Lane u refers to untransfected cells. Lanes 1–7 refer to transfection of constructs with the inserts as indicated. (B) Samples 1, 6, and 7 were the same as in panel A but were examined here with more extensive electrophoresis and on a gel of 3% agarose. Based on previous experience, such conditions are sufficient to separate linear and circular forms of HDV RNA (Chen et al. 1986; Lazinski and Taylor 1994). (C) This shows two semilogarithmic plots of length of the modified proceed HDV RNAs vs. distance migrated. These plots are for the putative circular and linear forms shown in B.
To address both the nature of the major species and also those minor species, we performed Northern analyses using gels of 3% agarose, which have the ability to separate HDV RNA linear and circular conformations. Examples of such data are shown in Figure 2B. Consider first the migration of unmodified HDV RNAs in lane 1. Consistent with our previous studies (Chen et al. 1986; Lazinski and Taylor 1994), two species were observed. The faster species migrated as for a linear RNA of 1679 nt, while the slower species was as for a closed circular conformation of the same length. Lanes 6 and 7 show the species obtained when the RNAs have insertions of 388 and 978 nt, respectively. In each case there were two main bands, with the faster and slower being consistent with linear and circular conformations, respectively. Note that as the corresponding pairs of species became larger because of the introduced sequences, the distance between the bands increased. In fact, in Figure 2C, when represented in a semilogarithmic plot of the expected lengths versus the distance migrated, the data points for the species designated as linear and circles could each be fitted by a straight line.
Taken together, the data presented in Figure 2 supported our interpretation that for each construct we detected two major species, a linear and a circular conformation of the same length. We noted that in lanes 6 and 7 of Figure 2B there was an indication of a minor heterogeneous species migrating faster, but there were not any discrete-sized species that migrated as for 1679-nt linears or circles. Thus we interpreted that the heterogeneous species detected probably did not reflect any significant level of nucleolytic trimming out of the inserted sequences down to the size of the natural 1679-nt rodlike RNA structure. Instead, our interpretation was that they represented a variety of extents of endo- and exonucleolytic cleavage of ribozyme-processed HDV DNA–directed RNA transcripts.
Replication competence of mutated RNAs
The next question was to determine whether the various HDV RNAs with insertions were able to initiate genome replication. The same expression constructs were therefore transfected into 293-δAg cells, which contain an integrated copy of a cDNA for HDV mRNA under tetracycline (TET)on control (Chang et al. 2005a). Five days later, the total RNA was extracted and subjected to Northern analysis to detect antigenomic RNA. Typical results for genomes with insertions at the XbaI site are shown in Figure 3A.
FIGURE 3.

Northern analyses to detect the ability of the modified HDV RNAs to replicate in 293-δAg cells. (A) Cells were transfected with the same constructs expressing wild-type or mutated HDV as used in Figure 2A, and expression of δAg was induced by the addition of TET. At day 5, total cell RNA was extracted and examined by Northern analysis as in Figure 2A, except that the detection was for antigenomic HDV RNA. Indicated at the right side are the positions of unit-length HDV RNA (UL) and its less abundant dimer (2× UL). Also indicated is the DNA-directed mRNA produced from the cDNA integrated into the 293-δAg cells. (B) Aliquots of the RNAs shown in lanes 1–11 of A were DNase-treated and then used to transfect fresh 293-δAg cells. After 5 d, the total RNA was harvested, and the transfection process was repeated (retransfection) with fresh cells. After 5 d, the total RNAs were extracted and assayed by Northern analysis to detect genomic HDV RNA. In B, the mass of total RNA analyzed in lanes 1–5 was three times less than for lanes 6–11.
Within 2 d, TET induction leads to the production of >2 million copies per cell of the small form of δAg, which supports HDV genome replication (Chang et al. 2005a). As indicated in Figure 3A, this mRNA was readily detected. However, in terms of HDV genome replication, only those with insertions of 1–4 nt (lanes 2–5) were comparable to wild type (lane 1). For an insertion of 4 nt, the accumulation was already ∼30% relative to wild type (lane 5). For an insertion of 8 nt (the GNRA tetraloop), accumulation was just 6% (lane 8). For the insertions >10 nt, no more than a faint band at about the size of unit-length HDV RNA was detected. This same band was also detected in untransfected cells (lane u) and is considered to arise via cross-hybridization with 18S rRNA, which is present at ∼10 million copies per cell.
Thus, the HDV RNA accumulation detected following the transfection of genomes with inserts of ≥10 nt was, if anything, <4%, relative to wild type. However, it still remained possible that a small amount of replication was occurring for such modified genomes. To test this possibility, we adopted a strategy from studies with replicons of hepatitis C virus that demonstrated that dramatically increased amounts of replication per average cell could be achieved if, following an initial transfection, the total RNA was extracted and then used to transfect new cells (Zhu et al. 2003). Each of the total RNAs analyzed in Figure 3A was treated with DNase before transfection into fresh 293-δAg cells. Five days after TET induction, total RNAs from transfected cells were extracted and used in a second round of transfection. Finally, total RNAs were examined by Northern analysis for the presence of genomic RNA. As can be seen, all HDV constructs (lanes 1–11) gave significant genome replication (Fig. 3B). There were even detected minor amounts of a slower migrating band, known to be a dimer of the genome (Chen et al. 1986). (Minor amounts of a dimer that was antigenomic were also seen for lanes 1–22 of Fig. 3A.) As a negative control, after retransfection of RNA from cells that had not been previously transfected, no replication of HDV RNA was detected (Fig. 3B, lane u).
Even more interesting than the recovery of replicating RNAs was the finding that those HDV RNAs with initial insertions of 388 and 978 nt at the XbaI site had apparently reverted to a length more similar to that of the wild-type genome (see Fig. 3B, cf. lanes 10,11 and lanes 1–9). Similar results were also obtained when the initial insertions were at the NheI site rather than the XbaI site (data not shown). Specifically, large insertions at the NheI site interfered with the ability of RNA to replicate. However, after three retransfections the amount of replicating HDV RNA per average cell again increased to readily detectable levels. Also, according to Northern analysis, the replicating RNAs again were not distinguishable by size from the wild type.
Nucleotide sequence analysis of replicating RNA genomes
The following experiments show that the restoration involved changes at the original sites of insertions, and provide support for the interpretation of intramolecular template-switching in combination with a selective pressure to maintain the rodlike RNA structure.
We examined by RT-PCR, cloning, and nucleotide sequencing, the corresponding regions of those genomes that were now able to replicate. Table 2 summarizes the results for genomes that were initially subjected to insertions at the XbaI site. (Not shown in Table 2 are the findings for wild type and mutants with insertions of either one or two extra nucleotides, because even after second retransfection no sequence changes relative to the input were observed).
TABLE 2.
Nucleotide sequence changes detected on restored HDV genomes

For the recovered sequences following the insertion of 3 nt, it can be seen that the majority of the recovered clones (78%) maintained this insert. However, the remainder lost the insert along with either 9 or 10 nt of adjacent HDV RNA sequence.
For the genomes initially given a 4-nt insertion, only 25% maintained the insert, with the remainder of the genomes losing the insert together with 8 or 10 nt of HDV RNA sequence. A minor fraction could be considered as having maintained 3 nt of insert, along with changing the fourth.
Table 2 also shows the results when several RNAs with inserts of progressively increasing size were tested for their sequences following recovery. Some general comments can be made.
With only one exception, all the genomes that had inserts of >4 nt were found after recovery to have lost their inserted sequences. (The exception was for one sequence recovered following the 8-nt GNRA insertion. The insert was maintained, but it was associated with a deletion, nearby, of 13 nt of HDV RNA sequence).
In each case, the recovered genomes were heterogeneous in sequence at and around the insertion site.
In some cases the recovered genomes included species that were equivalent to reversion to the wild-type RNA sequence.
In other cases there was a spectrum of nucleotide deletions of HDV RNA sequences. These deletions were heterogeneous in terms of their 5′- and 3′-borders, as well as in terms of their length. Figure 4 shows the frequency of these as a function of size. While the sizes varied up to as much as 17 nt, it was striking that many of these deletions were at or about 10 nt in length. Furthermore, these multiple examples of 10-nt deletion were indistinguishable in terms of their 5′- and 3′-borders.
More rare were insertions of what might be nontemplated non-HDV RNA sequences. And, even for these “insertions,” it has to be allowed that at least some of them could also be explained as residues of the original inserted sequence.
In addition to the changes close to the original insertion site, we detected a number of single-nucleotide substitutions, often quite close to the original insertion site (as underlined in Table 2), but also at sites more distant (data not shown) that were predominantly of A to G or U to C. We interpreted these, based on previous experience with changes occurring during RNA replication, as having arisen as consequences of post-transcriptional RNA editing by ADAR (Casey and Gerin 1995; Netter et al. 1995b; Gudima et al. 2002; Chang et al. 2005b).
FIGURE 4.

The size distribution of nucleotide deletions relative to the wild type that were detected on the restored HDV genomes following initial insertions at the position of the XbaI site. The data for the restored genomes are summarized in Table 2. Indicated here are the relative frequencies for each net change relative to the wild-type sequence (excluding the removal of the inserted sequence).
The results so far showed that insertions of non-HDV RNA sequences could interfere with the efficiency of these RNAs to initiate replication and accumulation. Nevertheless, following application of a retransfection strategy, RNA replication could then be detected, and we would therefore infer that even after the primary transfection there must have been present low levels of replicating restored genomes. Furthermore, this restoration often occurred in a way such that the inserted foreign sequences were no longer present, even though restoration was not always perfect, in that it was often associated with deletions of adjacent HDV RNA sequences.
The above results were largely obtained for insertions at the XbaI site, located near to one end of the rodlike structure (Fig. 1). When we considered insertions at the NheI site, embedded within the rodlike folding, the results were both similar and strikingly different. They were similar in that the insertions interfered with the ability to initiate replication. They were similar in that after the retransfection strategy it was made clear that restoration had occurred and that the replicating genomes had lost the large non-HDV RNA insertions. However, the major difference, obtained from the nucleotide sequencing at and around this insertion site, was that all the restored genomes were identical to the unmodified wild-type sequence (data not shown).
As considered further in the Discussion, we interpret that for insertion sites either near an end of the rod (XbaI) or embedded within this folding (NheI), there was a need for intramolecular template-switching to achieve recovery. However, in addition, for insertions at the NheI site there was apparently zero tolerance for nucleotide sequence changes. It was as if only those RNAs that exactly maintained the wild-type sequence and rodlike folding were capable of replication.
Possible role of terminal redundancies of HDV sequence
As represented in Figure 1A, the DNA template used for insertion of non-HDV RNA sequences has a short terminal redundancy of HDV sequences. We were concerned that this might have contributed to the ability to achieve restoration. First, there was the formal possibility of a rare DNA–DNA recombination event on the expression vector. Second, even if the restoration was achieved from RNAs transcribed in vivo from the expression construct, the possibility existed that some form of homology-based RNA–RNA recombination might be involved. Such concerns were heightened for the large insertion at the NheI site, where we observed complete restoration to the wild-type sequence.
With these concerns in mind, we first reduced the redundancy of HDV sequences at the 5′-end from 355 to 167 nt (Fig. 1A). This means that there was no longer any redundancy at and around the NheI site. Such a DNA construct, with a 388-nt insert at the NheI site, was transfected into cells by using procedures as in Figure 3. Just as previously encountered, it was only after three retransfections of total cellular RNA that RNA replication was readily detected (data not shown). The mobility of this replicating RNA was consistent with the loss of the non-HDV RNA insertion. Furthermore, after one additional retransfection, we used RT-PCR cloning and sequencing, as in Table 2, to characterize the sequence of the restored genomes at and around the original site of insertion. We found that nine of nine clones were again of the same sequence as unmodified wild-type HDV RNA (data not shown). Therefore, under the experimental conditions used here, the redundancies of HDV sequence did not detectably contribute to the ability to achieve restoration to the wild-type sequence. In a similar set of experiments, we also reduced the sequence redundancy relative to the XbaI site, when the insertion at this site was of 978 nt. Again, with this construct we were able to achieve replication of restored genomes. Furthermore, when the sequences at the restored sites were examined, we observed both the deletion of the inserted sequence along with a variety of altered sequences, as in Table 2 (data not shown).
For all the above studies, an additional concern arose because the expression constructs were transfected into 293-δAg cells. These cells contain a single copy of cDNA for the δAg (Chang et al. 2005a). The precise region is from ScaI (position 1624) to XbaI (position 781) on the antigenomic sequence. Thus there was the remote possibility that these HDV cDNA sequences, even at the level of a single copy per cell, might have participated in some form of DNA–DNA recombination with the transfected expression constructs to produce restored replicating HDV genomes. To totally exclude this, and all other possibilities relating to DNA–DNA recombination, we performed the following confirmatory experiments in which cells were transfected not with expression constructs but with RNAs that had first been transcribed in vitro.
Genome restoration from RNA transcribed in vitro
In all of the studies presented above, the initial source of mutated RNA was an RNA transcribed in vivo from a DNA construct. Inside the cell this RNA was able to undergo specific post-transcriptional processing, to the point of accumulating unit-length circular RNA species (Fig. 2B). As an alternative approach, we also tested whether mutated RNA species transcribed in vitro would also be able to lead to restoration. We neither expected (Chang and Taylor 2002) nor observed (data not shown) that the linear HDV RNAs transcribed in vitro were processed in vivo to form a circular species. Nevertheless, we tested whether linear RNA that has been mutated by an insertion of non-HDV sequence was capable of initiating HDV genome replication.
The linear RNA was transcribed by using T7 RNA polymerase and an expression construct described in the previous section. We thus produced a greater than unit-length linear genomic RNA with a 388-nt insertion at the NheI site. This RNA was gel-purified and transfected into recipient 293-δAg cells. Four days later, total RNA was extracted and subjected to Northern analysis. We observed the accumulation of unit-length HDV RNAs, that is, apparently lacking the 388-nt insertion (data not shown). Furthermore, after one retransfection, we applied RT-PCR, cloning, and sequencing, and found that 10 out of 10 sequences were now wild type at the site of insertion (data not shown).
Our interpretation of these results is that genome restoration can also be achieved following transfection of RNA species that are linear in conformation. Thus, as discussed subsequently for Figure 5, we interpret that this particular restoration was achieved by two intramolecular switches during transcription from a linear RNA template. In addition, since these studies were initiated by transfection with RNA, the results exclude the possibility that some form of intermolecular DNA–DNA recombination, even at a very low frequency, was necessary for the ability to ultimately detect restored genomes.
FIGURE 5.

Comparison of the ability to achieve restoration following transfection of modified HDV RNAs. A shows the four genomic linear RNA species, P–S, that were transcribed in vitro and used to transfect 293-δAg cells. The 5′ and 3′ ends correspond to positions 716 and 715, respectively, and are located within the ribozyme domains (indicated by partial circles). Only species P contains a specific 4-nt change (X) that, as previously shown (Gudima et al. 2004), does not compromise replication ability and is used in Table 3 to distinguish and quantitate species P relative to species Q–S. Species Q–S contain insertions of non-HDV sequences: 388 nt at the NheI site and/or 978 nt at the XbaI site, as indicated. B shows the ethidium bromide staining of agarose gel analysis of the in vitro transcribed and gel-purified species P–S, prior to use in transfection. Note that species Q–S migrate slower than P, consistent with the presence of inserted non-HDV sequences. C shows Northern analyses of antigenomic RNA species detected in RNA extracted from 293-δAg cells at 6 d after transfection with species P–S, as indicated. At left (lane u) is a control of untransfected cells. Note that the detected RNAs are of approximately the same migration as unit-length HDV RNA. D shows a similar analysis when the RNAs from panel C were transfected into 293-δAg cells and after 6 d, examined by Northern. In C and D, antigenomic and genomic RNA was detected using a full-length uniformly 32P-labeled RNA probes, respectively.
As a follow-up to these results, a comparison was made of the ability to achieve restoration following insertion of non-HDV RNA sequences at one or both of two locations. As diagrammed in Figure 5A, species P is a control species of unit-length genomic RNA with no insertions. Species Q has an insertion of 388 nt at the NheI site, embedded within a region of the predicted rodlike folding. Species R has an insertion of 978 nt at the XbaI site, corresponding to one end of the rodlike folding. Finally, species S has the two insertions of Q and R. These four RNAs were transcribed in vitro and gel-purified, and the sizes were confirmed by gel electrophoresis with detection by ethidium bromide staining, as shown in Figure 5B. The four modified RNAs were then transfected into 293-δAg cells, and after 6 d the total cellular RNAs were extracted and examined by Northern analysis to detect HDV antigenomic RNA, with results as shown in Figure 5C. For the control species P, with no insertion, we detected significant HDV RNA replication. Species Q gave no detectable replication when considered relative to a sample of untransfected cells in lane u. However, species R and S did give a small amount of replication, and in addition, the detected RNAs migrated as if the insertions had been removed.
The total cellular RNAs from each of these initial transfections were then retransfected into new 293-δAg cells, and after 6 d the total RNA was extracted and examined by Northern analysis, with results as shown in Figure 5D. Now each of the modified species Q–S, as well as control species P, was producing readily detectable genome replication.
From the results in Figure 5, C and D, we can make several conclusions: (1) All of the replicating RNAs derived from transfections with Q–S were of approximately the same mobility as wild type, consistent with removal of the large inserts of non-HDV sequence(s); (2) such restoration was more readily observed after a retransfection; (3) of the three modified RNAs, R, with the insertion at one end of the rodlike structure, was more readily restored; and (4) even species S, with two insertions of non-HDV sequence, was able to lead to replicating genomes restored to wild-type size.
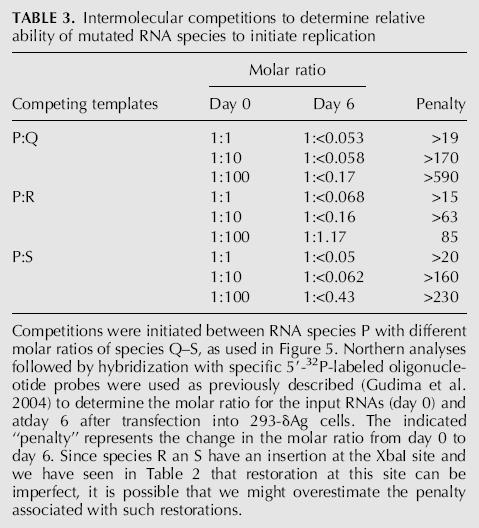
Species P is a unit-length HDV RNA sequence without inserted non-HDV sequences. Nevertheless, since it is of linear conformation, we assert that to achieve genome replication, there needs to be one template-switching event during transcription (Gudima et al. 2004). It follows then that species Q and R would require one additional template switch, and species S would require two. While the data in Figure 5, C and D, gave some idea of the replication penalties associated with such additional template switches, we made use of the following second approach. In previous studies we have successfully used intermolecular competition assays to measure the relative ability of two different HDV RNA species to initiate genome replication (Gudima et al. 2004). Therefore, the same approach was used to determine the penalty for RNA species Q, R, and S relative to species P.
For this purpose, species P was given a genetic marker (indicated in Fig. 5A by x), which has been shown to not interfere with replication ability but allows separate quantification by oligonucleotide hybridization of marked and unmarked RNAs. Three different molar ratios of species P and Q, P and R, and P and S were transfected into 293-δAg cells, and we determined how these ratios changed in the next 6 d. The results are summarized in Table 3. The change in the ratio is referred to as the penalty; that is, the reduction in the ability of the RNA relative to species P to achieve initiation of HDV RNA replication. From such intermolecular competitions, even when the input (day 0) amounts of Q, R, and S were 100 times more than that of P, we observed that at day 6, species P was the winner, with there being undetectable amounts of the competitor. The only exception was with species R, where accumulation was detected. We can therefore deduce that the penalty for R is ∼85-fold relative to P. For species Q and S, we can only deduce that the penalties are >590 and >230, respectively. In summary, these intermolecular competitions indicate that the penalties associated with the insertions of non-HDV sequences are significant (Table 3), and yet in the situations where the modified RNAs are transfected in the absence of a competitor, the replication of HDV RNA was in each case detected, and these RNAs had been restored to the wild-type length (Fig. 5C,D).
TABLE 3.
Intermolecular competitions to determine relative ability of mutated RNA species to initiate replication
DISCUSSION
This study focused on the consequences of insertion of relatively large sequences onto the HDV genomic RNA. As discussed below, our findings, some of which were particularly surprising, have implications for RNA-directed transcription by host cellular RNA Pol II and for the importance of the rodlike RNA structure ensuring the replication competence of the template. Our studies invoke what is referred to here as intramolecular template-switching, a process that is synonymous with what virologists often refer to as intramolecular RNA recombination (Olsthoorn et al. 2002; Alejska et al. 2005). In earlier studies, we have provided strong evidence that such template-switching can occur on HDV RNA templates (Chang and Taylor 2002; Gudima et al. 2004). However, what makes the HDV results unique is that it is the host RNA Pol II that performs such activity using RNA as a template. Objectively, it needs to be mentioned that two studies have interpreted that fragments of viral RNAs can somehow be restored by another mechanism not involving RNA transcription (Gmyl et al. 2003; Gallei et al. 2004). However, as explained elsewhere, we consider that a nonreplicative mechanism does not apply to our HDV studies (Gudima et al. 2005).
Consider first the observed phenomenon of genome restoration. Why did this process occur for each of the RNA templates that we mutated by insertion of non-HDV RNA sequences? One contributor would be enough copies of the mutated RNA in the initial cells. We are not suggesting that such abundance would facilitate the event. We mean only that with more potential templates we increase the opportunity for detecting a rare successful event that leads to genome restoration. Another contributor would be the provision of an abundance of the small δAg needed for replication. Specifically, we know that within each 293-δAg cell, at 1 d after TET induction, there were accumulated >2 million new copies of δAg (Chang et al. 2005a).
We propose that these two factors contribute to the ability to achieve some level of RNA polymerase binding and the initiation of RNA-directed transcription. Soon after this, when transcription reaches the site of an insertion on the template, these same two factors (the quantity of RNA templates and δAg) increase the chances that many and different intramolecular template-switching events could occur. Some of these switches could produce RNA transcripts in which the inserted sequences (and sometimes even flanking HDV RNA sequences) are not copied. If just one of these events leads to an RNA that can achieve genome replication, we will observe what we have referred to as restored genomes. We might expect that only a small fraction of all RNA-directed transcription events are successful in this way.
In Figure 6 we present a model with three possible pathways for genome restoration. Based on data present here and documented previously (Chang and Taylor 2002; Gudima et al. 2004, 2005), these pathways all depend upon intramolecular template-switching. (1) In the first, the RNA template with insertion is considered to fold into a rodlike structure, with or without help from the δAg, to produce an extrusion of the foreign sequence. This folding somehow facilitates the removal of the insert via template-switching (steps 1 and 2, followed by steps 5 and 6). (2) A second possibility is that prior to template-switching, extrusion of the inserted sequences makes them susceptible to some extent of endo- and then exonucleolytic cleavage by host ribonucleases. Such prior cleavage could thus demand that template-switching occur in order to rescue the nascent transcript and thence achieve genome replication (steps 1, 2, and 3, followed by steps 5 and 6). (3) The third explanation for the restoration phenomenon is different in that it refers to experiments in which we detected restoration following an initial transfection of cells with mutated RNA that had been transcribed not in vivo but in vitro. Since this RNA was linear in conformation, we favor the interpretation shown in step 4 followed by steps 5 and 6. That is, we consider this linear RNA was directly used as a template for transcription involving two template-switches. One of these switches was to remove the non-HDV RNA insert; the other, to remove the 5′- and 3′-sequences. This combination would lead to nascent RNA transcripts that can be processed to the wild-type unit-length species with the associated wild-type rodlike folding. In conclusion, it should be re-emphasized that in terms of the actual restoration at the site of insertion, all three pathways shown in Figure 6 have the same requirement for intramolecular template-switching during transcription as facilitated by attempted rodlike folding of the RNA template.
FIGURE 6.

A model of how restoration of replication competent HDV RNA species was achieved. As indicated, the input RNA is a greater than unit-length genomic RNA. It contains two copies of the genomic ribozyme (shaded circles) and an insertion of non-HDV sequence (shaded lines). Step 1 indicates the consequences of ribozyme cleavage, leading in step 2, to ligation to produce circular RNA, which may fold into the unbranched rodlike structure with extrusion of the non-HDV sequences. Implicit is the expectation that δAg might bind to and protect the HDV-specific structure. Step 3 indicates how these extruded non-HDV sequences might be the substrate for endo- and then exonucleolytic attack. Step 4 shows how in the absence of processing the nascent transcript might fold into the rodlike structure again with extrusion of the non-HDV sequences. Steps 5 and 6 represent how each of the three forms of RNA might act as templates for the transcription with intramolecular template-switching, leading to the processing of new restored anti-genomic RNAs that no longer contain the non-HDV sequences (broken line), and in turn can be transcribed to produce replicating genomic RNA.
We note that restoration was a rare event and, in many cases, required one or more retransfections of total cell RNA before we could detect restored genomes by Northern analysis. In the case of a large insertion at the NheI site, we determined that the penalty for such an insertion was at least 500-fold, relative to an RNA lacking this insertion (Fig. 5E).
The model, as shown in Figure 6, is drawn for insertion at the NheI site that is embedded within the rodlike folding. However, we also observed genome restoration when the inserts were located almost at one end of the rodlike folding, the XbaI site. This location difference might explain why the restoration of genomes with insertions at the NheI site were 100% perfect, in that the exact unmodified wild-type sequences was the only winner. In contrast, for the XbaI site, only some of the restored winners were wild type, while there was also a spectrum of others with deletions of adjacent HDV RNA sequences and sometimes also with short non-HDV insertions (Table 2). A common feature of both sets of results was that the winners restored overall rodlike folding that was similar or even identical to that of wild type. In this way we would explain the observation that such restoration at the XbaI site that was located at one end of the rod could be more tolerant of changes than for the NheI site that was embedded within the rod (Fig. 1B,C). We consider it hard to believe that the rodlike folding per se, in the initial template, did not facilitate the restorative template switch. Nevertheless, any molecule that was transcribed from this initial template and ultimately survived the subsequent biological pressures to be a winner maintained rodlike folding.
We can speculate as to what are or are not the biological pressures. At least in our experimental system, such pressures do not need to include the synthesis of new δAg, since this was produced via an integrated cDNA copy (Chang et al. 2005a). Nor is there a need for particle assembly and release. Furthermore, for our studies with Pol II DNA-directed transcripts of the modified HDV RNAs, we show here that the RNA transcripts have no major defect in their ability to be post-transcriptionally processed by ribozyme action, RNA ligation, and ultimate RNA stabilization (Fig. 2). Thus we would speculate that the major biological pressure, in addition to those also present for unmodified RNAs, is at the level of RNA-directed transcription, specifically in the ability to achieve a template-switching at the insertion site that removes the inserted sequence. Apparently this can be achieved for the insertions of 388 and 978 nt and not just for those as small as 4 and 3 nt.
In some cases the restored genomes included a series of different species distinguishable by the nucleotide sequence at the original insertion site. We wanted to know whether these RNAs were of comparable replication competence. For the case of one particular insertion (388 nt at the XbaI site), we therefore compared the sequences after one, two, or three consecutive rounds of retransfection. No significant difference was detected between these three sets of sequences in the relative abundance of the individual restored sequences (data not shown). Thus we conclude that the restored genomes were of uniform replication ability, both relative to each other and to the original wild-type sequence. Also, we considered there was no continued evolution of the restored sequences (at least, within the time period analyzed). This supports the model presented above, where restoration involved just one early template switch, one that occurred on the input modified RNA. That is, the subsequent retransfections only provided an opportunity to expand the number of cells replicating the restored genomes.
As can be noted, the procedure of retransfecting fresh cells with total RNA achieves an enhancement of genome spread to new cells that in some ways mimics the natural process of virus assembly and reinfection. A difference is that in the retransfection the recipient 293-δAg cells always provide multiple copies of the essential δAg.
This phenomenon of intramolecular template-switching by Pol II may be unique to HDV RNA templates, whether or not it is an integral part of normal HDV replication. Nevertheless, it opens the question of how this enzyme carries out such a process during RNA-directed transcription. There is still some controversy as to whether such switching can occur when the same enzyme carries out transcription that is DNA-directed (Kandel and Nudler 2002; Li and Zhang 2002). The observed ability on RNA templates may be related to the structure of the template and/or differences in the host and viral factors associated with such transcription. Of course, many animal viruses are able to carry out intramolecular RNA directed template-switching (Olsthoorn et al. 2002; Alejska et al. 2005). For some viruses, such as coronaviruses, it is even an essential part of each replication cycle (Sola et al. 2005).
In retrospect, it may not be too surprising that the host Pol II, just like the RNA polymerases of RNA viruses, is able to achieve intramolecular as well as intermolecular template-switching. The common feature is the opportunity provided by virus genome replication, to selectively amplify what would otherwise be rare events. The same may be said for the host DNA polymerases when they are used to support the replication of certain DNA viruses, such as SV40 and adeno-associated virus.
MATERIALS AND METHODS
Cloning, mutagenesis, and RNA transcription
Originally, constructs expressing mutated HDV genomes were based on vector pDL553, which expresses a 1.2× of wild-type HDV genomic RNA (Wu et al. 1997). As represented in Figure 1, non-HDV sequences were inserted at one of two unique locations: XbaI site (position 785/786) or NheI site (position 430/431), using the numbering of a published HDV RNA sequence (Kuo et al. 1988a). The insertions are summarized in Table 1. To introduce foreign sequences of size <30 nt at XbaI location, we used appropriate pairs of chemically synthesized oligonucleotides. These were annealed and inserted into pDL553 precut with restriction enzymes BstXI and XbaI. The insertions of 388 and 978 nt were excised by simultaneous XbaI and SpeI digestion from plasmid pA/BVDV/D9 containing cp7 D19c, a full-length replicon of bovine diarrhea virus (Meyers et al. 1996) and provided by Olaf Isken and Sven Behrens. These fragments were gel-purified and then inserted into the XbaI or NheI sites of pDL553. Both orientations of insert were made only at the NheI site. Alternatively, in order to eliminate any redundancies at and around the NheI site, the insert of 388 nt was also introduced into vector pDL542 (Lazinski and Taylor 1994), expressing StyI-StyI-XbaI greater than unit-length genomic RNA sequences. Also, to avoid redundancies at both insertion sites, we created a vector bearing a fragment (position 518–774) of the HDV genome, inserted into BamHI–XhoI sites of pSVL vector (Pharmacia) using PCR-based techniques. Based on this vector, the construct expressing greater than unit-length of RNA genome (position 518–518–774) with a 978-nt insert at XbaI location was created. For in vitro transcription of StyI-StyI-XbaI HDV RNA bearing 388-nt insert at NheI site, the appropriate mutated HDV sequences were introduced downstream of a T7 RNA polymerase promoter into pCDNA3 vector (Invitrogen). Resulting constructs were verified by nucleotide sequencing. In vitro RNA transcription and subsequent gel purification of RNA were performed by using previously described protocols (Gudima et al. 1997, 2004).
Cell lines and transfection
Huh7, a line of human hepatoma cells (Nakabayashi et al. 1982), was maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum without addition of antibiotics. The 293-δAg cell line was created from 293-TRex, a derivative of human embryonic kidney cells (Invitrogen) (Chang et al. 2005a). It contains a single copy of a cDNA for the small δAg, with expression under TET on control.
For transfection of both cell lines with cDNA or RNA, we used Lipofectamine 2000 reagent (Invitrogen).
For testing DNA-directed expression of mutated RNAs in absence of RNA replication we used transfection of Huh7 cells with the constructed expression vectors in combination with plasmid pDL445, expressing large δAg (Lazinski and Taylor 1993), and plasmid pSVTVA (Asselbergs and Grand 1993) in the mass ratio 1:1:0.1.
For testing the replication abilities of mutants, 293-δAg cells were transfected either with constructs, expressing RNAs with non-HDV insertions or with gel-purified in vitro transcribed modified HDV RNAs. Synthesis of δAg small was induced by adding tetracycline at 4 h, the time when the transfection mixture was removed.
Retransfection means the process in which total RNA was extracted from previously transfected 293-δAg or Huh7 cells and delivered to by Lipofectamine 2000 into fresh 293-δAg cells.
RNA preparation and Northern analysis
Unless indicated otherwise, cells were harvested at days 3, 4, or 5 after transfection, and total RNA was extracted by using Tri Reagent (Molecular Research Center). For Northern analyses, RNA was glyoxalated and then resolved on gels of 1.7 or 3% agarose. Genomic and antigenomic RNAs were detected using 32P-labeled riboprobes or 32P-5′-labeled oligonucleotides (Kuo et al. 1989; Chang and Taylor 2002; Gudima et al. 2004). Detection was via bio-imager (Fuji). Images were collected and quantitated by using Image Gauge V3.3 and prepared for publication by using Photoshop V7.0 and Canvas V9.0 software.
RT-PCR, cloning, and sequencing
Total RNA (0.2 μg) was treated with RNase-free RQ1 DNAse (Promega) prior to RT-PCR reactions. For PCR spanning the XbaI insertion site, we used primers 5′-728-ACATTCCGAGGGGACCGTC-746-3′ and 5′-865-ACCTGGGCATCCGAAGGAG-847-3′. Similarly, for around the NheI site, the primers were 5′-320-TCAGCGAACAAGAGGCGCTTCGA-342-3′ and 5′-507-GATGGGGACGATAAGTCGAGTTC-485-3′. The specific PCR products were cloned into pCR 2.1-Topo vector by using a TOPO TA cloning kit (Invitrogen). Plasmids were selected and subjected to automated nucleotide sequencing. Resulting sequences were analyzed relative to the HDV master sequence (Kuo et al. 1988a) using the program Sequencher 4.2.1 (Gene Codes).
ACKNOWLEDGMENTS
Nucleotide sequencing was carried out in the Fox Chase DNA Sequencing Facility by Anita Cywinski. Valuable constructive comments on the manuscript were provided by Glenn Rall, Richard Katz and William Mason. J.T. was supported by grants AI-26522 and CA-06927 from the NIH and by an appropriation from the Commonwealth of Pennsylvania. S.G. designed and performed all the experiments.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2328806.
REFERENCES
- Alejska M., Figlerowicz M., Malinowska N., Urbanowicz A. A universal BMV-based RNA recombination system: How to search for general rules in RNA recombination. Nucleic Acids Res. 2005;33 doi: 10.1093/nar/gni106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselbergs F.A.M., Grand P. A two-plasmid system for transient expression of cDNAs in primate cells. Anal. Biochem. 1993;209:327–331. doi: 10.1006/abio.1993.1128. [DOI] [PubMed] [Google Scholar]
- Casey J.L., Gerin J.L. Hepatitis D virus RNA editing: Specific modification of adenosine in the antigenomic RNA. J. Virol. 1995;69:7593–7600. doi: 10.1128/jvi.69.12.7593-7600.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Taylor J. In vivo RNA-directed transcription, with template-switching, by a mammalian RNA polymerase. EMBO J. 2002;21:157–164. doi: 10.1093/emboj/21.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Gudima S.O., Tarn C., Nie X., Taylor J.M. Development of a novel system to study hepatitis delta virus genome replication. J. Virol. 2005a;79:8182–8188. doi: 10.1128/JVI.79.13.8182-8188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Gudima S.O., Taylor J.M. Evolution of hepatitis delta virus RNA genome following long-term replication in cell culture. J. Virol. 2005b;79:13310–13316. doi: 10.1128/JVI.79.21.13310-13316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Nie X., Gudima S., Taylor J. Replication of the hepatitis delta virus genome. In: Hefferon K.L., editor. Recent advances in RNA virus replication. Transworld research network; Trivandrum, Kerala, India: 2006a. [Google Scholar]
- Chang J., Nie X., Gudima S.O., Taylor J.M. Action of inhibitors on the accumulation of processed hepatitis delta virus RNAs. J. Virol. 2006b;80:3205–3214. doi: 10.1128/JVI.80.7.3205-3214.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao M., Hsieh S.-Y., Taylor J. Role of two forms of the hepatitis delta virus antigen: Evidence for a mechanism of self-limiting genome replication. J. Virol. 1990;64:5066–5069. doi: 10.1128/jvi.64.10.5066-5069.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao M., Hsieh S.-Y., Taylor J. The antigen of hepatitis delta virus: examination of in vitro RNA-binding specificity. J. Virol. 1991;65:4057–4062. doi: 10.1128/jvi.65.8.4057-4062.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.-J., Kalpana G., Goldberg J., Mason W., Werner B., Gerin J., Taylor J. Structure and replication of the genome of hepatitis δ virus. Proc. Natl. Acad. Sci. 1986;83:8774–8778. doi: 10.1073/pnas.83.22.8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg J.E., Lund E. Small nuclear ribonucleoprotein particles. Springer; Berlin: 1988. [Google Scholar]
- Gallei A., Pankraz A., Thiel H.J., Becher P. RNA recombination in vivo in the absence of viral replication. J. Virol. 2004;78:6271–6281. doi: 10.1128/JVI.78.12.6271-6281.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmyl A.P., Korshenko S.A., Belousov E.V., Khitrina E.V., Agol V.I. Nonreplicative homologous RNA recombination: Promiscuous joining of RNA pieces? RNA. 2003;9:1221–1231. doi: 10.1261/rna.5111803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudima S.O., Kazantseva E.G., Kostyuk D.A., Shchaveleva I.L., Grishchenko O.I., Memelova L.V., Kochetkov S.N. Deoxyribonucleotide-containing RNAs: A novel class of templates for HIV-1 reverse transcriptase. Nucleic Acids Res. 1997;25:4614–4618. doi: 10.1093/nar/25.22.4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudima S., Dingle K., Wu T.-T., Moraleda G., Taylor J. Characterization of the 5′-ends for polyadenylated RNAs synthesized during the replication of hepatitis delta virus. J. Virol. 1999;73:6533–6539. doi: 10.1128/jvi.73.8.6533-6539.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudima S., Wu S.-Y., Chiang C.-M., Moraleda G., Taylor J. Origin of the hepatitis delta virus mRNA. J. Virol. 2000;74:7204–7210. doi: 10.1128/jvi.74.16.7204-7210.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudima S.O., Chang J., Moraleda G., Azvolinsky A., Taylor J. Parameters of human hepatitis delta virus replication: The quantity, quality, and intracellular distribution of viral proteins and RNA. J. Virol. 2002;76:3709–3719. doi: 10.1128/JVI.76.8.3709-3719.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudima S.O., Chang J., Taylor J.M. Features affecting the ability of hepatitis delta virus RNAs to initiate RNA-directed RNA synthesis. J. Virol. 2004;78:5737–5744. doi: 10.1128/JVI.78.11.5737-5744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudima S.O., Chang J., Taylor J.M. Reconstitution in cultured cells of replicating HDV RNA from pairs of less than full-length RNAs. . RNA. 2005;11:90–98. doi: 10.1261/rna.7164905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel E.S., Nudler E. Template switching by RNA polymerase II in vivo. Evidence and implications from a retroviral system. Mol. Cell. 2002;10:1495–1502. doi: 10.1016/s1097-2765(02)00777-3. [DOI] [PubMed] [Google Scholar]
- Kuo M.Y.-P., Goldberg J., Coates L., Mason W., Gerin J., Taylor J. Molecular cloning of hepatitis delta virus RNA from an infected woodchuck liver: Sequence, structure, and applications. J. Virol. 1988a;62:1855–1861. doi: 10.1128/jvi.62.6.1855-1861.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo M.Y.P., Sharmeen L., Dinter-Gottlieb G., Taylor J. Characterization of self-cleaving RNA sequences on the genome and antigenome of human hepatitis delta virus. J. Virol. 1988b;62:4439–4444. doi: 10.1128/jvi.62.12.4439-4444.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo M.Y.-P., Chao M., Taylor J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989;63:1945–1950. doi: 10.1128/jvi.63.5.1945-1950.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M. RNA replication without RNA-dependent RNA polymerase: Surprises from hepatitis delta virus. J. Virol. 2005;79:7951–7958. doi: 10.1128/JVI.79.13.7951-7958.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazinski D.W., Taylor J.M. Relating structure to function in the hepatitis delta virus antigen. J. Virol. 1993;67:2672–2680. doi: 10.1128/jvi.67.5.2672-2680.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazinski D.W., Taylor J.M. Expression of hepatitis delta virus RNA deletions: Cis and trans requirements for self-cleavage, ligation, and RNA packaging. J. Virol. 1994;68:2879–2888. doi: 10.1128/jvi.68.5.2879-2888.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-Z., Lin J.-H., McKnight K., Lai M.M.C. RNA-binding activity of hepatitis delta antigen involves two arginine-rich motifs and is required for hepatitis delta virus RNA replication. J. Virol. 1993;67:2221–2227. doi: 10.1128/jvi.67.4.2221-2227.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Zhang J. Intramolecular recombinations of Moloney murine leukemia virus occur during minus-strand DNA synthesis. J. Virol. 2002;76:9614–9623. doi: 10.1128/JVI.76.19.9614-9623.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnaughton T.B., Wang Y.-J., Lai M.M.C. Replication of hepatitis delta virus RNA: Effect of mutations of the autocatalytic cleavage sites. J. Virol. 1993;67:2228–2234. doi: 10.1128/jvi.67.4.2228-2234.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers G., Tautz N., Becher P., Thiel H.J., Kummerer B.M. Recovery of cytopathogenic and noncytopathogenic bovine viral diarrhea viruses from cDNA constructs. J. Virol. 1996;70:8606–8613. doi: 10.1128/jvi.70.12.8606-8613.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraleda G., Seeholzer S., Bichko V., Dunbrack R., Otto J., Taylor J. Unique properties of the large antigen of hepatitis delta virus. J. Virol. 1999;73:7147–7152. doi: 10.1128/jvi.73.9.7147-7152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi H., Taketa K., Miyano K., Yamane T., Sato J. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
- Netter H.J., Hsieh S.Y., Lazinski D., Taylor J. Modified HDV as a vector for the delivery of biologically-active RNAs. Prog. Clin. Biol. Res. 1993;382:373–376. [PubMed] [Google Scholar]
- Netter H.J., Lazinski D., Taylor J. Hepatitis delta virus. In: Vos J.-M.H., editor. Viruses in human gene therapy. Carolina Academic Press; Durham, NC: 1995a. pp. 33–52. [Google Scholar]
- Netter H.J., Wu T.-T., Bockol M., Cywinski A., Ryu W.-S., Tennant B.C., Taylor J.M. Nucleotide sequence stability of the genome of hepatitis delta virus. J. Virol. 1995b;69:1687–1692. doi: 10.1128/jvi.69.3.1687-1692.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X., Chang J., Taylor J.M. Alternative processing of hepatitis delta virus antigenomic RNA transcripts. J. Virol. 2004;78:4517–4524. doi: 10.1128/JVI.78.9.4517-4524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsthoorn R.C., Bruyere A., Dzianott A., Bujarski J.J. RNA recombination in brome mosaic virus: Effects of strand-specific stem-loop inserts. J. Virol. 2002;76:12654–12662. doi: 10.1128/JVI.76.24.12654-12662.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radjef N., Gordien E., Ivaniushina V., Gault E., Anais P., Drugan T., Trinchet J.C., Roulot D., Tamby M., Milinkovitch M.C.,, et al. Molecular phylogenetic analyses indicate a wide and ancient radiation of African hepatitis delta virus, suggesting a deltavirus genus of at least seven major clades. J. Virol. 2004;78:2537–2544. doi: 10.1128/JVI.78.5.2537-2544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzetto M., Hoyer B., Canese M.G., Shih J.W.K., Purcell R.H., Gerin J.L. δ agent: Association of δ antigen with hepatitis B surface antigen and RNA in serum of δ-infected chimpanzees. Proc. Natl. Acad. Sci. 1980;77:6124–6128. doi: 10.1073/pnas.77.10.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg M., Court D. Regulatory sequences involved in the promotion and termination of RNA transcription. Annu. Rev. Genet. 1979;13:319–353. doi: 10.1146/annurev.ge.13.120179.001535. [DOI] [PubMed] [Google Scholar]
- Rudisser S., Tinoco I., Jr. Solution structure of cobalt(III)hexammine complexed to the GAAA tetraloop, and metal-ion binding to G.A mismatches. J. Mol. Biol. 2000;295:1211–1223. doi: 10.1006/jmbi.1999.3421. [DOI] [PubMed] [Google Scholar]
- Ryu W.S., Netter H.J., Bayer M., Taylor J. Ribonucleoprotein complexes of hepatitis delta virus. J. Virol. 1993;67:3281–3287. doi: 10.1128/jvi.67.6.3281-3287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I., Moreno J.L., Zuniga S., Alonso S., Enjuanes L. Role of nucleotides immediately flanking the transcription-regulating sequence core in coronavirus subgenomic mRNA synthesis. J. Virol. 2005;79:2506–2516. doi: 10.1128/JVI.79.4.2506-2516.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J.M. Structure and replication of hepatitis delta virus RNA. In: Handa H., Yamaguchi Y., editors. Hepatitis delta virus. Landes Bioscience; Georgetown, TX: 2005. (in press) [Google Scholar]
- Wang K.-S., Choo Q.-L., Weiner A.J., Ou J.-H., Najarian C., Thayer R.M., Mullenbach G.T., Denniston K.J., Gerin J.L., Houghton M. Structure, sequence and expression of the hepatitis delta viral genome. Nature. 1986;323:508–513. doi: 10.1038/323508a0. [DOI] [PubMed] [Google Scholar]
- Wang H.-W., Wu H.-L., Chen D.-S., Chen P.-J. Identification of the functional regions required for hepatitis D virus replication and transcription by linker-scanning mutagenesis of viral genome. Virology. 1997;239:119–131. doi: 10.1006/viro.1997.8818. [DOI] [PubMed] [Google Scholar]
- Wu T.-T., Netter H.J., Lazinski D.W., Taylor J.M. Effects of nucleotide changes on the ability of hepatitis delta virus to transcribe, process and accumulate unit-length, circular RNA. J. Virol. 1997;71:5408–5414. doi: 10.1128/jvi.71.7.5408-5414.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y., Filipovska J., Yano K., Furuya A., Inukai N., Narita T., Wada T., Sugimoto S., Konarska M.M., Handa H. Stimulation of RNA polymerase II elongation by hepatitis delta antigen. Science. 2001;293:124–127. doi: 10.1126/science.1057925. [DOI] [PubMed] [Google Scholar]
- Zhu Q., Guo J.T., Seeger C. Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J. Virol. 2003;77:9204–9210. doi: 10.1128/JVI.77.17.9204-9210.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]