

Abstract
Due to growing problems with drug resistance, there is an outstanding need for new, cost-effective drugs for the treatment of malaria. The 4-aminoquinolines have provided a number of useful antimalarials and Plasmodium falciparum, the causative organism for the most deadly form of human malaria, is generally slow to develop resistance to these drugs. Therefore, diverse screening libraries of quinolines continue to be useful for antimalarial drug discovery. We report herein the development of an efficient method for producing libraries of 4-aminoquinolines variant in the sidechain portion of the molecule. The effects of these substitutions were evaluated by screening this library for activity against P. falciparum revealing four potent compounds active in drug resistant strains.
Keywords: Parallel synthesis, parallel purification, antimalarial, 4-aminoquinoline
Introduction
Despite over a hundred years of drug development, malaria remains one of the most devastating infectious diseases in the world.1 The development of drug resistance to the potent and affordable drug chloroquine has left many developing countries without sustainable options for antimalarial chemotherapy.2 This burden has been heightened by a lack of industrial focus on malaria. Malaria is an infectious disease caused by parasitic protozoans of the genus Plasmodium. There are four species of Plasmodium that infect humans: falciparum, ovale, malaria, and vivax. Of these, Plasmodium falciparum is the most deadly and is responsible for the majority of the 300 – 500 million clinical cases each year that cause between 0.7 – 2.7 million deaths annually.3
Quinoline containing drugs, particularly 4-aminoquinolines, have a long and successful history as antimalarials4, 5. One of these, chloroquine, has been in worldwide use since the Second World War. However, resistance to chloroquine has become clinically significant in several areas of the world.6, 7 Prior work has shown that either shortening or lengthening the linker of the alkyl amine side chain in chloroquine leads to compounds that remain effective against drug-resistant strains of P. falciparum.8-10 Unfortunately many of these modifications have led to disadvantageous changes in the metabolism of these compounds, especially increased dealkylation of the distal amine. Dealkylation causes reduced lipid solubilities of the drugs and increased cross-resistance with chloroquine.11 Contrawise, modifications of the substitutions on the distal amine side chain to bulkier groups have been shown to increase in vivo efficacy and decrease cross-resistance with chloroquine, presumably by circumventing metabolic dealkylation.12 Bis-quinoline derivatives have also shown activity against chloroquine resistant strains of P. falciparum, but these compounds are known DNA intercalators and often cytotoxic.13 In consideration of these issues, we wished to explore the tolerance for various bulky substitutions on the distal amine group of shortened side-chain chloroquine analogs.
Our current aim is to synthesize a new series of chloroquine analogs with a shortened side chain and diverse functionalities on the alkyl amine side chain to develop a structure-activity relationship (SAR) for these modifications (Figure 1). Synthetic methods that allow access to substituted amine groups are of high interest for medicinal chemistry since alkyl amine side chains are found in many drugs. We have developed a robust synthetic method that allows the introduction of two independent points of diversity to the amine of the side chain by sequential indirect reductive aminations.
Figure 1.
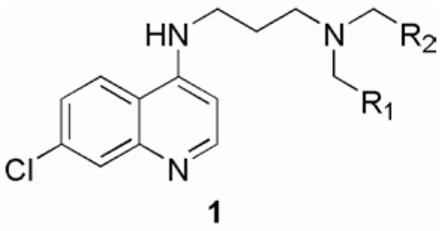
Structure of 7-chloro-4-aminoquinoline library 1.
Results and Discussion
Library Design
As described above, shortening the sidechain of chloroquine and replacing the alkyl substituents on the distal amine with bulky groups both independently increase stability and efficacy against chloroquine resistant strains of P. falciparum.8, 9, 12 Therefore, we fixed both the 7-chloroquinoline nucleus found in chloroquine and the shorter propyl amine side chain, while substituting the distal basic group to give a virtual library 1 (Figure 1). Retrosynthetically, we envisaged this library as arising from the double reductive amination of a pendant amine. A set of potential substitutions of the terminal amine group were chosen based on the commercial availability of the aldehyde reagents and used to populate a virtual library of all the theoretically possible products giving roughly 300,000 potential compounds. This virtual library was filtered to remove any “non-lead-like” candidates with non-organic atom types; reactive substructures; or compounds not obeying Lipinski's “Rule of Five”. Many of the compounds were relatively large by drug-likeness standards, therefore they were ranked by molecular weight. This procedure reduced the targeted library to roughly 850 members that were deconvoluted to the originating 97 aldehydes. When executed using protocols built in the Pipeline Pilot operating system, this filtering procedure required less than 10 minutes of computational time. A total of 25% of the aldehydes that passed through the screening filter as originators of this virtual library were chosen by hand based upon chemistry compatibility, structural diversity and inclusion of drug-like substructures (Figure 2). These 24 aldehydes were picked to sample the tolerance for diversity at this location and were combined with a second diversity element that represented either a small alkyl chain (propyl) or a bulky aromatic chain (benzyl). We report herein the synthesis and initial testing of the proofing library for this study.
Figure 2.
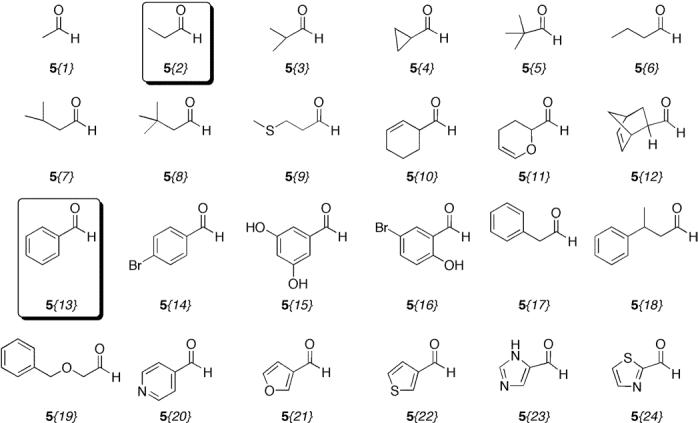
Structures of aldehyde diversity element 5{1-24}. Boxed elements represent the base structure used for R2. All elements were used for R1.
Synthesis
Our synthesis strategy was developed to quickly generate a series of high-purity compounds for biological screening. The recent recognition that many time consuming false positive “hits” from high throughput screening campaigns originate in residual impurities from synthesis has led to the increasing adoption of high throughput purification for production of screening libraries in industrial settings.14-16 Therefore, we chose to employ the combination of parallel synthetic and purification techniques. Since different reactions in the library synthesis give mixed purities, we elected to develop a general purification strategy so that we can purify any compounds of less than satisfactory purity. This first proofing library was made both to test the scope of the chemistry and to optimize handling procedures for subsequent larger libraries.
The proofing library for this approach was constructed as outlined in Scheme 1. The bulk common intermediate (3) was prepared using a modification of De's method in which 4,7-dichloroquinoline was allowed to react with neat diaminopropane to afford 3.17 In initial studies we consistently achieved lower yields then reported due to the formation of a coarse particulate precipitate during the wash step of the work-up. Analysis of this precipitate revealed it was made up predominately of the desired product, co-precipitated with a tarry byproduct. The precipitate was recovered by filtration and purified by continuous solid-liquid extraction in a Soxhlet extractor to leach the desired product from the side products. When both crops of the product were recombined, the procedure proceeded in an overall yield of 83%.
Scheme 1.
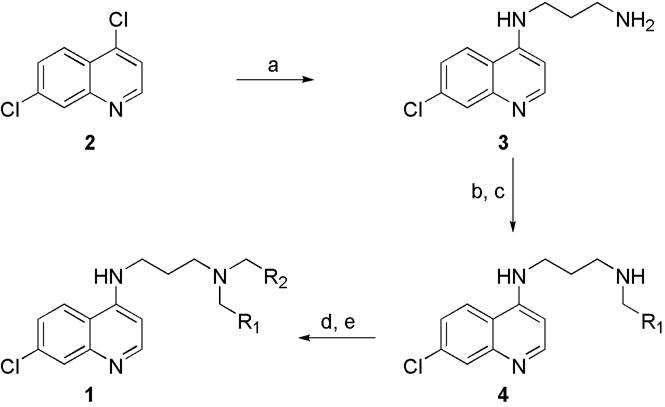
Synthesis of 4-amino-7-chloroquinoline library 1{1-24;2,13}. a) 1,3-diaminopropane (neat), reflux, 2 h, 83% yield; b) R1CHO (5{1-24}, 4 eq.), MeOH, rt, 24 h; c) NaBH4 (5 eq.), rt., 1 h; d) R2CHO (5{2,13}, 4 eq.), MeOH, rt, 24 h; e) NaBH4 (5 eq.), rt, 1 h.
The primary amine intermediate (3) was subjected to two sequential reductive aminations to introduce two points of chemical diversity. Reductive aminations have been commonly used in combinatorial chemistry due to the reliability of the reaction, commercial availability of aldehyde building blocks, and process simplicity.18 The challenge of doing two sequential reductive aminations lies in the fact that reductive aminations of primary amines usually afford a tertiary amine side product from double addition of the aldehyde under the reducing conditions due to the increased nucleophilicity of the secondary amine product. While this side product is minor and usually can be separated, our final products are also all tertiary amines and were difficult to separate from the side products using the common conditions imposed logistically by the library purification process.
In order to avoid the need to purify our secondary amine intermediate, we explored numerous reductive amination conditions to eliminate the formation of the tertiary amine side product. A survey of reducing conditions, in the presence or absence of various dehydrating reagents, revealed no conditions that produced only the monosubstituted product. Previous reports in the literature have circumvented this problem by carrying out a stepwise reductive amination procedure with a “preformed imine” and no excess aldehyde.18 This procedure did eliminate tertiary amine formation, but in many cases resulted in incomplete reaction progress leaving a mixture of the secondary amine and the primary amine starting material. The conditions that provided the best combination of yield of desired product and suppression of the undesired symmetrically substituted amine were: 1) formation of the imine with excess aldehyde and no dehydrating reagent in anhydrous methanol, followed by 2) concomitant reduction of the imine and the excess aldehyde using sodium borohydride. This procedure produced a primary alcohol side product, arising from reduction of the excess aldehyde, in the crude reaction mixture. However, this side product did not interfere with the subsequent reductive amination in our reaction sequence and was easily separated from our desired tertiary amine products by our final purification procedure. We have also explored the use of solid-phase extraction (SPE) methods using a strong-cation exchange (SCX) resin to remove the alcoholic byproducts. This optional prepurification method was generally successful in partially purifying our desired product, but we achieved comparable purities without this intermediate purification step (data not shown). Depending on purity standards for screening libraries, such a SPE technique could potentially eliminate the need for preparative HPLC purification. The use of excess aldehyde in the reductive amination also made the reaction setup more convenient since maintaining an exact stoichiometric ratio between the amine and aldehyde was no longer necessary. Also, methanol was found to be the optimal solvent choice since all of our reagents were sufficiently soluble in it and it has been shown to facilitate the most rapid formation of the imine.18 Thus in library production, intermediate 3 was split into 48 portions and treated in parallel with aldehyde diversity reagents 5{1-24} followed by the reducing reagent, to give the crude chloroquine derivatives 4{1-24,H}. The imine formation step was allowed to go for 24 hours since literature measurements of the kinetics of this reaction show reaction times varying from minutes to several hours, especially for sterically crowded aldehydes, imine formation can be quite slow.18 Finally, each of these intermediates was treated in parallel with aldehyde diversity reagents 513 to give the crude chloroquine derivatives 1{1-24;2,13}.
After the sequential amination procedure, the crude reaction mixtures were filtered to remove any remaining inorganic salts and the solutions were dried by evaporation in vacuuo. The crude mixtures were then directly purified by automated reversed-phase HPLC using UV triggered fractionation.16 The crude materials were dissolved into a loading buffer containing a small amount of the aqueous mobile phase in order to equilibrate the products to a constant pH. We explored several different loading conditions, including non-aqueous acid-workups, salt formation, and loading solvents. To optimize the chromatographic separation quality, we found that it was beneficial to pre-equilibrate the compounds to the high pH used for the separations. Other changes in loading conditions showed little or no effect on the purity and yields post-chromatography. Fraction collection was triggered by a threshold UV absorption at 254 nm. The resulting fractions were analyzed by flow injection electrospray mass spectrometry and fractions with the desired products were pooled together and dried by evaporation in vacuuo. Each purified product was then re-dissolved in a saturated methanolic solution of HCl to afford the HCl salts. Finally, solvent was removed to give the purified water-soluble forms of each compound. The final yield of each product was determined at this point by weighing.
All compounds were analyzed to determine purity and yields prior to biological testing. The overall yields were moderate with a 29% average yield, with losses primarily attributed to purification and handling processes. The overall yields could be improved by more careful and time-consuming handling and purification procedures, but our ultimate goal was to quickly and efficiently produce compounds of high purity in sufficient quantity for biological testing. Purity was assessed by analytical HPLC (C18 column) and monitored by a photodiode array (PDA) detector from 210 nm – 490 nm (Figure 3). The average purity for the compound library was 91% with only 4 of the 48 compounds failing to meet our purity standard of 80%. Of these, one 1{2,3} was determined to be of insufficient purity for assay.
Figure 3.
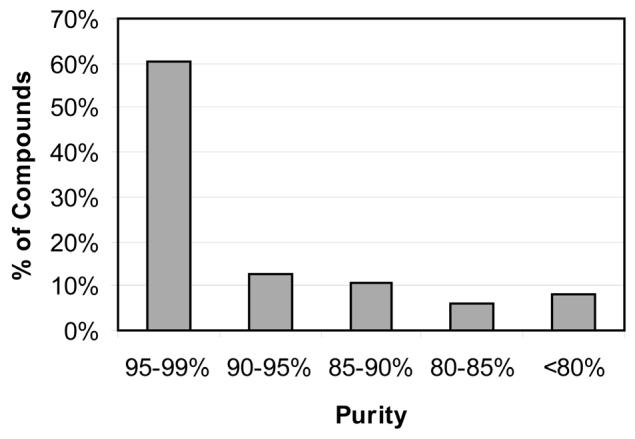
Histogram of purities of product library 1{2,13;1–24} after purification by reversed-phase HPLC. Purity was assessed by analytical HPLC analysis monitored at 220 nm on a reversed-phase C18 column (Column Engineering, ReliaSil C18 BDX, 5 μM).
The total amount of time spent creating this 48-member library, including purification and analysis, was about five days. Two days were spent carrying out the synthesis, four hours were spent doing the purification, and two days were spent doing fraction identification and sample drying. The actual number of man-hours spent working on the compounds is minimal since most of the steps are easily automated by commonly used robotics.
Screening and Biological Evaluation
All compounds were screened for antimalarial activity using in vitro cultures of Plasmodium falciparum strain 3D7 at two doses: 30 nM and 200 nM.19, 20 One compound 1{2,2} was lost during the screening process to a liquid handling error, therefore data are reported for 46 of the targeted 48 members of chemset 1. Chloroquine, which has a reported EC50 of about 20 nM,21 was used as a positive control. 4-amino-7-chloroquinoline, which is a structurally related inactive compound,22 was used as a negative control. The data is summarized in Figure 4 (tabulated screening data can be found in the supplementary materials). The members of the library exhibited a wide range of antimalarial activity, completely spanning that defined by the controls. When one alkyl group of the side chain was held constant as a propyl, a number of derivatives, including several with bulky aliphatic or aromatic rings, were at least as active as chloroquine. In contrast, when one alkyl group was held constant as the more sterically demanding benzyl, almost no derivatives had significant activity. Strikingly one compound, 1{13,23} exhibited activity with two bulky benzylic groups attached to the amine. The inactivity of the corresponding propyl derivative points to an unexpected synergy between the benzyl and imidazolylmethyl substituents and highlights the potential for cooperativity. A number of the compounds were active to within two-fold of the potency of chloroquine.
Figure 4.
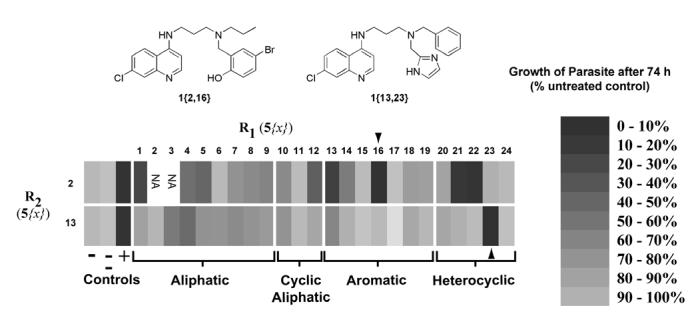
Relative activity of library 1{2,13;1–24} against Plasmodium falciparum 3D7 in cultured human erythrocytes. Substituents for R1 are arrayed on the X-axis, broken into groups representing the general structural class of substituents. Substituents for R2 are shown on the Y-axis. Grayscale shade represents the percent of parasite growth at 30 nm drug concentration relative to untreated cultures. “−“ refers to cultures treated with no drug. “− −“ refers to cultures treated with 30 nM negative control compound 4-amino-7-chloroquinoline. “+” refers to cultures treated with 30 nM Chloroquine. The structures of two representative active compounds are shown above the graphic.
The four most active compounds from our initial screen were assayed against a panel of three P. falciparum strains with varying degrees of drug resistance. The drug resistant strains we used were W2 and Dd2, which are both originally from Indochina. Both of these strains are resistant to all known quinoline drugs. The IC50s clearly show the superior activity of the new compounds compared to chloroquine against both the drug resistant strains. Interestingly, the compound 1{23,13} containing two bulky aromatic groups had substantially inferior activity in W2 and Dd2 when compared with the other three compounds containing a small propyl group, despite having comparable activity against the 3D7 strain.
Given the rather small number of compounds included in this study and the fact that they were designed to enforce maximal diversity rather than to probe congeneric series carefully to reveal detailed structure activity relationships, it is premature to comment generally upon the function of the alkyl groups decorating the amine. Two unexpected features do deserve comment. First, the only compound with two bulky groups that had strong activity included a third basic site on one of the alkyl groups. This leads one to believe that two bulky groups tends to suppress activity but that this trend can be overcome by biasing the equilibrium partition of the compound into the acidic food vacuole where the drugs act. Perhaps the most interesting feature is that all of the most active compounds against drug resistant strains contain a hydrogen bond acceptor on the alkyl substituent attached to the distal basic group. The basic group will be protonated at physiological pH allowing it to form an intramolecular hydrogen bond with the groups substituted on the amine. Both of these features will be included as design elements in subsequent studies.
Conclusions
The goal of this study was to develop a robust parallel synthesis / purification procedure for the production of moderate sized libraries (100's) of quinoline derivatives that will ultimately be coupled to similar approaches that allow variation of substituents on the A and B rings of the quinoline nucleus to afford large diverse screening libraries. The procedure reported herein is efficient, giving access to greater than 90% of the targeted compounds in greater than 80% purity using generalized procedures that have been adapted to the parallel format and automation. The method easily produces milligram amounts of material, sufficient for several hundred primary screens, with a cycle time of less than one week. This should be sufficiently robust to allow production of the desired screening libraries. Biological testing of the compounds produced in this proofing study revealed that a number of previously unaddressed substitutions at the distal base of chloroquine that afford active antimalarials. An exciting outcome of this study is that three of the compounds from the library that was designed to emphasize elements likely to convey activity against drug resistant strains of P. falciparum were indeed against multiple drug resistant strains. Additionally, this study revealed a previously unrecognized potential for synergistic advantage to the addition of two benzylic groups to the distal amine that is worthy of further study. The production of larger libraries of quinolines is underway and their synthesis and activity shall be reported in due course.
Experimental Methods
General
All reagents and starting materials were purchased from commercial sources and used without further purification; solvents were anhydrous HPLC grade. All parallel synthesis steps were carried out in polypropylene fritted FlexChem© 48-well reaction blocks. 1H-NMR spectra were recorded using a Varian 400 MHz spectrometer. Chemical shifts were measured in parts per million (δ) relative to tetramethylsilane (TMS) as the internal standard. Coupling constants (J values) were measured in hertz (Hz).
Virtual Library Filtering
All computations were carried out using the commercially available Pipeline Pilot software (SciTegic™, San Diego, CA) and were done in 6 minutes on a 1.2 GHz Pentium processor with 384 MB of RAM. The Lipinski filter was set to allow compounds with the following properties: Sum of N and O atoms ≤ 10, Molecular Weight ≤ 550, H-bond donors ≤ 5, and ALOGP ≤ 5. ALOGP was calculated using the Ghose/Crippen group-contribution estimate method.
N1-(7-Chloro-quinolin-4-yl)-propane-1,3-diamine (2)
A solution of 4,7-dichloroquinoline (25 g, 0.126 mol) in 1,3-diaminopropane (47 mL, 0.568 mol) was heated to reflux for 4 hours with stirring, and then allowed to cool to room temperature. The solution was diluted with methylene chloride (400 mL) and the resulting mixture was washed with sodium hydroxide (1 N, 400mL) and brine (400 mL), to give an aqueous layer, an organic layer, and a white course particulate precipitate (9.5 g). The organic layer was dried in vacuuo to give the product 2 as on off-white solid. The precipitate from the wash was filtered and extracted in a Soxhlet extractor with methylene chloride for 3 days. The methylene chloride was then removed in vacuuo to yield a second crop of 2 as an off-white solid (8.6 g). 1H-NMR analysis indicated the two solids were identical and pure, so they were combined and used without further purification (24.6 g, 0.104 mol, 83% yield); 1H NMR (CD3OD): δ 8.33 (d, J = 5.9 Hz, 1H), δ 8.05 (d, J = 8.8 Hz, 1H), δ 7.76 (d, J = 2.0 Hz, 1H), δ 7.36 (dd, J = 8.8, 2.0 Hz, 1H), δ 6.50 (d, J = 5.9, 1H), δ 4.90 (s, 2H), δ 3.40 (t, J = 6.8, 2H), δ 2.81 (t, J = 6.8, 2H), δ 1.90 (dt, J = 5.9, 5.9, 2H). 13C NMR (CD3OD): δ 152.9, 152.6, 149.9, 136.5, 127.8, 126.1, 124.5, 119.0, 99.9, 42.0, 40.5, 32.2. MS: ESI+ 236.3 [M + 1] Calc. Mass: 235.1.
General Procedure for Preparation of Chemset 1
N1-(7-Chloro-quinolin-4-yl)-propane-1,3-diamine (2) (1.44 g, 6.11 mmol) was dissolved in dry methanol (120 mL) to make a 50.9 mM stock solution. The stock solution was aliquoted to the wells of the 48-well reaction block (2.5 mL/well, 0.13 mmol/well). To each well was then added aldehydes 5 (4 equiv., 0.52 mmol). After clamping the block shut and rotating the block for 24 hours at room temperature, solid sodium borohydride (5 equiv.) was added to each well resulting in copious evolution of gas. CAUTION: Explosion hazard. The block was left open for 30 minutes to allow complete evolution of gas, then clamped shut and rotated for 2 hours to yield the secondary amine intermediate (3). The procedure was then repeated with a second aldehyde to yield the final the tertiary amine 1. The crude reaction mixtures were then collected into a 48-position deep well plate and solvent was removed on a GeneVac HT-4 (50 mbar, 3.5 hours, 35 °C).
General Procedure for Purification of Chemset 1
Crude library members were dissolved in 1.8 mL of a loading buffer (90% methanol, 10% ammonium acetate buffer, 20 mM, pH 6.8) then purified with a preparative YMC ODS-AQ reversed-phase column (20 mm χ 50 mm, particle size S-5) running a 10–100% gradient of ammonium acetate buffer (20 mM, pH 6.8)/ Methanol with a 25 mL/min flow rate on a Parallex Flex™ HPLC System. Chromatographs were monitored with a dual wavelength UV detector at 220 nm and 254 nm. Fraction collection was automatically triggered by UV absorption above 0.20 AU at 254 nm. Sample and fraction data was then transferred to the Waters OpenLynx operating software, which coordinated the injection, mass spectrometric analysis, and data processing for each fraction. A Gilson 215 liquid handler and a Gilson 208 injection module were used to inject samples into a Waters ZQ 4000 mass spectrometer rigged for flow injection with an electrospray probe and single quadrapole detector operating in positive ion mode. All fraction plates were then dried down using GeneVac Mega 980 solvent evaporator. Fractions that exhibited a peak in the mass spectra of the correct molecular weight were then dissolved in 1mL of freshly prepared saturated HCl in methanol and like fractions were combined. Combined fractions were then transferred to pre-weighed vials, solvent was removed on the GeneVac HT-4, and each vial was weighed to calculate final yields of the HCl salts of each compound.
Measurement of in Vitro Antimalarial Activity
The effects of compounds in library 1 on the growth of Plasmodium falciparum cultures in vitro were measured using flow cytometry.23 Synchronous cultures of ring stage parasites (500 μL, 0.8% parasitemia, 2% hematocrit) were grown in 24-well or 96-well tissue culture plates (Falcon) with 30 nm and 200 nm concentrations of experimental compounds. Cultures were grown in atmospherically regulated (6 % CO2, 5 %O2) incubators (Sanyo) at 37 °C for a total of 4 days. Each day additional media and drug were added to the cultures (300 μL on day 2, 700 μL on day 3, and 500 μL on day 4) to maintain nutrient-rich conditions necessary for rapid parasite growth. Aliquots of 100 μL were removed from each well at 74 hours post drug treatment and resuspended in 900 μL of 1 % paraformaldehyde, 1 nM YOYO-1 (Molecular Probes) in PBS. Each sample was then incubated for 16 h in the dark at 4 °C before being analyzed on a Becton-Dickenson LSR2 flow cytometer to measure the percent of parasitized red blood cells (RBCs). Percent of parasitized RBCs was directly read and subsequently growth inhibition values were calculated as the fraction of parasitized RBCs relative to cultures without drug. All screening was done in triplicate with two negative controls 1) no drug addition and 2) addition of non-active 4-amino-7-chloroquinoline compound) and a positive control, chloroquine. Growth inhibition was then measured using the same conditions as in the screen for the five most active compounds and chloroquine at 1, 5, 10, 15, 20, 30, 50, and 200 nm drug concentrations. Fifty percent inhibitory concentrations (IC50s) were estimated by analyzing the resulting log dose response curves via visual extrapolation.
Supplementary Material
SYNOPSIS TOC Due to growing problems with drug resistance, there is an outstanding need for new, cost-effective drugs for the treatment of malaria. The 4-aminoquinolines have provided a number of useful antimalarials and Plasmodium falciparum, the causative organism for the most deadly form of human malaria, is generally slow to develop resistance to these drugs. Therefore, diverse screening libraries of quinolines continue to be useful for antimalarial drug discovery. We report herein the development of an efficient method for the production of libraries of 4-aminoquinolines variant in the substitutions of the sidechain portion of the molecule and the results of screening this library for activity against P. falciparum.
Table 1.
IC50 values of the four most active compounds and CQ against the 3D7, W2 and Dd2 P. falciparum strains. Values are expressed as nM. These studies were run at a higher parasitemia than the screening studies in order to provide a more stringent test of activity thus leading to slightly elevated
| Strains |
|||
|---|---|---|---|
| Compound | 3D7 | W2 | Dd2 |
| CQ | 25 | >250 | >250 |
| 1{2,16} | 45 | 45 | 45 |
| 1{2, 21} | 50 | 70 | 45 |
| 1{2, 22} | 50 | 50 | 45 |
| 1{13, 23} | 50 | 130 | 150 |
ACKNOWLEDGMENT
NIH/NIAID AI53862-01 (J.L.D., R.K.G.), the Sandler Research Foundation (J.L.D., R.K.G.), the Burroughs Wellcome Quantitative Biology Fellowship (P.B.M.), and the National Science Foundation Graduate Fellowship Program (N.W.)
Footnotes
SUPPORTING INFORMATION AVAILABLE Tables of yield and purity data for all compounds, tabulated complete screening data, and 1H-NMR spectra for 20 library members. All data are available at http://malaria.ucsf.edu. Daughter plates of this library containing a total of 100 nmoles of each compound may be obtained, subject to availability, by contacting the corresponding author.
REFERENCES
- 1.Sachs J, Malaney P. Nature. 2002;415(6872):680–5. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 2.Winstanley PA, Breckenridge AM. Ann Trop Med Parasitol. 1987;81(5):619–27. doi: 10.1080/00034983.1987.11812163. [DOI] [PubMed] [Google Scholar]
- 3.Diseases, U. W. B. W. S. P. f. R. a. T. i. T . Tropical disease research : progress 1997-98 : fourteenth programme report : malaria / UNDP/WORLD BANK/WHO Special Programme for Research and Training in Tropical Diseases. World Health Organization; Geneva: 1999. TDR/PR14/MAL/99.1. [Google Scholar]
- 4.O'Neill PM, Bray PG, Hawley SR, Ward SA, Park BK. Pharmacol Ther. 1998;77(1):29–58. doi: 10.1016/s0163-7258(97)00084-3. [DOI] [PubMed] [Google Scholar]
- 5.Foley M, Tilley L. Pharmacol Ther. 1998;79(1):55–87. doi: 10.1016/s0163-7258(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 6.Krogstad DJ, Gluzman IY, Kyle DE, Oduola AM, Martin SK, Milhous WK, Schlesinger PH. Science. 1987;238(4831):1283–5. doi: 10.1126/science.3317830. [DOI] [PubMed] [Google Scholar]
- 7.Wellems TE, Walker-Jonah A, Panton LJ. Proc Natl Acad Sci U S A. 1991;88(8):3382–6. doi: 10.1073/pnas.88.8.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De D, Krogstad FM, Cogswell FB, Krogstad DJ. Am J Trop Med Hyg. 1996;55(6):579–83. doi: 10.4269/ajtmh.1996.55.579. [DOI] [PubMed] [Google Scholar]
- 9.Ridley RG, Hofheinz W, Matile H, Jaquet C, Dorn A, Masciadri R, Jolidon S, Richter WF, Guenzi A, Girometta MA, Urwyler H, Huber W, Thaithong S, Peters W. Antimicrob Agents Chemother. 1996;40(8):1846–54. doi: 10.1128/aac.40.8.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vennerstrom JL, Ellis WY, Ager AL, Jr., Andersen SL, Gerena L, Milhous WK. J Med Chem. 1992;35(11):2129–34. doi: 10.1021/jm00089a025. [DOI] [PubMed] [Google Scholar]
- 11.Bray PG, Hawley SR, Mungthin M, Ward SA. Mol Pharmacol. 1996;50(6):1559–66. [PubMed] [Google Scholar]
- 12.Stocks PA, Raynes KJ, Bray PG, Park BK, O'Neill PM, Ward SA. J Med Chem. 2002;45(23):4975–83. doi: 10.1021/jm0108707. [DOI] [PubMed] [Google Scholar]
- 13.Ryckebusch A, Deprez-Poulain R, Maes L, Debreu-Fontaine MA, Mouray E, Grellier P, Sergheraert C. J Med Chem. 2003;46(4):542–57. doi: 10.1021/jm020960r. [DOI] [PubMed] [Google Scholar]
- 14.Yan B, Fang L, Irving M, Zhang S, Boldi AM, Woolard F, Johnson CR, Kshirsagar T, Figliozzi GM, Krueger CA, Collins N. J Comb Chem. 2003;5(5):547–59. doi: 10.1021/cc030008f. [DOI] [PubMed] [Google Scholar]
- 15.Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C. J Comb Chem. 2003;5(3):322–9. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 16.Blom KF, Sparks R, Doughty J, Everlof JG, Haque T, Combs AP. Journal of Combinatorial Chemistry. 2003;5(5):670–683. [Google Scholar]
- 17.De D, Byers LD, Krogstad DJ. Journal of Heterocyclic Chemistry. 1997;34(1):315–320. [Google Scholar]
- 18.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. Journal of Organic Chemistry. 1996;61(11):3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 19.Saito-Ito A, Akai Y, He S, Kimura M, Kawabata M. Parasitol Int. 2001;50(4):249–57. doi: 10.1016/s1383-5769(01)00091-5. [DOI] [PubMed] [Google Scholar]
- 20.Bianco AE, Battye FL, Brown GV. Exp Parasitol. 1986;62(2):275–82. doi: 10.1016/0014-4894(86)90032-9. [DOI] [PubMed] [Google Scholar]
- 21.O'Neill PM, Hawley SR, Storr RC, Ward SA, Park BK. Bioorganic & Medicinal Chemistry Letters. 1996;6(4):391–2. [Google Scholar]
- 22.Egan TJ, Hunter R, Kaschula CH, Marques HM, Misplon A, Walden J. Journal of Medicinal Chemistry. 2000;43(2):283–291. doi: 10.1021/jm990437l. [DOI] [PubMed] [Google Scholar]
- 23.Barkan D, Ginsburg H, Golenser J. Int J Parasitol. 2000;30(5):649–53. doi: 10.1016/s0020-7519(00)00035-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.