

Abstract
Although shape-memory metal alloys have wide use in medicine and other areas, improved properties, particularly easy shaping, high shape stability, and adjustable transition temperature, are realizable only by polymer systems. In this paper, a polymer system of shape-memory polymer networks based on oligo(ɛ-caprolactone) dimethacrylate as crosslinker and n-butyl acrylate as comonomer was introduced. The influence of two structural parameters, the molecular weight of oligo(ɛ-caprolactone) dimethacrylate and the weight content of n-butyl acrylate, on macroscopic properties of polymer networks such as thermal and mechanical properties has been investigated. Tensile tests above and below melting temperature showed a decrease in the elastic modulus with increasing comonomer weight content. The crystallization behavior of the new materials has been investigated, and key parameters for the programming procedure of the temporary shape have been evaluated. Shape-memory properties have been quantified by thermocyclic experiments. All samples reached uniform deformation properties with recovery rates above 99% after 3 cycles. Whereas strain recovery increased with increasing n-butyl acrylate content, strain fixity decreased, reflecting the decreasing degree of crystallinity of the material.
Metallic alloys with shape-memory properties like NiTi alloys are already used in different areas, e.g., biomedical applications (1).§ However, their mechanical properties can be adjusted only within a limited range, and they are not degradable.
Here, we report an AB-polymer network showing shape-memory properties. The advantage of this polymer system is its versatility. Several macroscopic properties such as transition temperature and mechanical properties can be varied in a wide range by only small changes in their chemical structure and composition.
Shape-memory polymers are characterized by two features, triggering segments having a thermal transition Ttrans within the temperature range of interest and crosslinks determining the permanent shape. Depending on the kind of crosslinks, shape-memory polymers can be thermoplastic elastomers or thermosets. By using this technology, shape-memory polymers potentially can be tailored for specific applications.
For this polymer system, oligo(ɛ-caprolactone) has been selected as a crystallizable triggering segment that can fix a temporary shape by physical crosslinks. These segments were incorporated covalently into the thermosets. Therefore, oligo(ɛ-caprolactone) diols were functionalized with polymerizable methacrylate endgroups. The molecular weight of this macromonomeric crosslinker represents a first parameter on the molecular level to adjust crystallinity and mechanical properties of the polymer networks. For that purpose, especially for temperatures below the transition temperature, an additional monomer is introduced representing a second parameter. As comonomer, n-butyl acrylate, has been selected. Because of the low glass transition temperature of poly(n-butyl acrylate) [Tg = −55°C (2)], a softening effect can be expected for the networks. The polymers of the selected compounds poly(ɛ-caprolactone) and poly(n-butyl acrylate) are biocompatible (3–6).
Hydrophilic crosslinked polymers have been investigated widely as water-swollen hydrogels, including those having shape-memory properties (7–12). However, they are limited in their mechanical performance and stability. By the use of hydrophobic components, the swellability of the networks in aqueous media can be reduced significantly. In contrast to the crosslinking process for hydrogels containing acrylate moieties that usually is driven by photoinitiators like 2-methoxy-2-phenyl acetophenon (13), the polymer system introduced here having methacrylate endgroups has been crosslinked by photopolymerization without an initiator.
Materials and Methods
Chemicals.
All solvents (1,2-dichloroethane, chloroform, hexane, tetrahydrofurane, ethyl acetate, methanol, diethyl ether, dichloromethane) were of pro analysis or HPLC quality. Tetrahydrofurane was dried with calcium hydride and distilled before use. n-Butyl acrylate <99% (Sigma–Aldrich) was stored at 4°C, and the inhibitor (4-methoxyphenol) was removed with an anion exchange column before use. Oligo(ɛ-caprolactone) diols with number average molecular weights Mn of 2,000 and 10,000 g·mol−1 were purchased from Sigma–Aldrich. Other oligo(ɛ-caprolactone) diols were synthesized according to the standard ring opening polymerization procedure (14) and characterized by multidetector gel permeation chromatography. The synthesis of oligo(ɛ-caprolactone) dimethacrylates (PCLDMA) was performed according to the method described in literature (15). Triethylamine pro analysis (p.a.) (Fluka) was distilled and stored over molecular sieves (pore size 0.4 nm). Methacryloyl chloride p.a. (Sigma–Aldrich) was stored at 4°C and used without further purification.
Determination of Molecular Weight.
The setup for multidetector gel permeation chromatography consists of a HP1100 HPLC pump (Hewlett–Packard), two 300-mm × 7.5-mm PL-gel mixed-D columns (Polymer Laboratories, Amherst, MA), a combination of a differential viscosimetry/light-scattering detector (T60, Viscotek, Houston, TX), and an Optilab differential diffractometer (Wyatt Technology, Santa Barbara, CA). Solvent is chloroform at a flow rate of 0.7 ml·min−1 at room temperature and a pressure of 58 bar.
Determination of Degree of Methacrylation.
The degree of methacrylation was determined by using 1H NMR spectroscopy. The 1H NMR spectra of the oligo(ɛ-caprolactone) dimethacrylates were recorded on a Bruker 400 MHz NMR-spectrometer in D6-DMSO or deuterochloroform (CDCl3).
Synthesis of Polymer Networks.
A mixture of oligo(ɛ-caprolactone) dimethacrylate and the proper amount of n-butyl acrylate was heated to 10°C above the melting temperature (Tm) and filled into a mold formed by two glass plates (25 mm × 75 mm) and a Teflon spacer of 0.60 mm thickness. To achieve a good homogeneity, the mold was stored at Tm for another hour. Photo curing was performed with a 100 W mercury arc lamp (Ultracure 100ss plus, Efos, Mississauga, ON, Canada) on a heated plate at Tm and lasted for 15 min, unless stated differently. The distance between lamp head and sample was 5.0 cm. After cooling down to room temperature, the weight of the isolated film was determined (miso). The sample was swollen and extracted with a 100-fold excess of dichloromethane overnight, washed carefully, and weighed (msw). After drying at room temperature under reduced pressure, the sample was weighed again (md).
Gelation Degree and Degree of Swelling.
The gelation degree G was calculated from the isolated weight miso of the sample and the dry weight md after extraction:
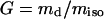 |
The swelling degree Q of the films in dichloromethane was calculated by (16):
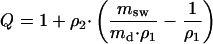 |
where msw is the weight of the sample in the swollen state, md is the weight of the dry extracted sample, and ρ1 and ρ2 are the specific densities of the swelling medium and the polymer, respectively. ρ2 was determined by weighing a sample piece of precisely known length, width, and thickness.
Differential Scanning Calorimetry (DSC).
DSC experiments were performed on a Perkin–Elmer TA7. Different programs were performed, all consisting of cooling down the samples from the melt at Th = 70°C at a cooling rate βc to a temperature Tl, keeping it at Tl for a time tl, and warming it up again at the heating rate βh = 10°C⋅min−1 to Th = 70°C.
For standard thermal analysis, βc was 10°C⋅min−1, Tl is −20°C, and tl was 2 min. The influence of the cooling rate on the melting properties was investigated by varying βc between −2°C⋅min−1 and −50°C⋅min−1; Tl is −70°C, and tl is 0. The influence of the crystallization temperature Tcr on the melting temperature Tm was determined by a series of experiments, with βc being 500°C⋅min−1 and Tl = Tcr varying from −20°C to 38°C. Tl was held constant until no further change in crystallization enthalpy was observed.
Mechanical and Thermomechanical Experiments.
Tensile tests at room temperature were carried out on an Instron 3100 tensile tester (Instron, Canton, MA). Experiments at extended temperature and thermocyclic experiments were performed on a Zwick 1410 tensile tester with thermo chamber (Zwick, Ulm, Germany) and temperature controller (Eurotherm Regles GmbH, Limburg, Germany). The strain rate was 10 mm·min−1 in all experiments. The thermocyclic program consisted of (i) heating up the sample to Th, extending it to ɛm, cooling it down to Tl in the extended state, and (ii) unloading it to 0% extension at Tl. Th and Tl were held for at least 10 min before loading or unloading the sample. Cycles i and ii were repeated five times each.
Results and Discussion
We have synthesized AB-polymer networks with shape-memory properties. Two oligo(ɛ-caprolactone) dimethacrylates with different molecular weights of 2,000 (PCLDMA2000) and 10,000 (PCLDMA10000) have been copolymerized as crosslinkers with n-butyl acrylate in different molar ratios by application of UV light. The degree of functionalization of the oligo(ɛ-caprolactone) diols with methacrylate endgroups determined by 1H NMR spectroscopy was between 85 and 93%. The crosslink density of the synthesized AB networks was varied by using different contents of n-butyl acrylate from 11 to 90 wt % in the case of PCLDMA2000 and from 20 to 71 wt % in the case of PCLDMA10000. Data of the synthesized networks are in Table 1.
Table 1.
Copolymerisates of PCLDMA2000 or PCLDMA10000 and n-butyl acrylate
| Sample identification* | PCLDMAm, mg |
n-butyl acrylate
|
Curing temperature T, °C | Gelation degree G, % | Degree of swelling Q, % | |
|---|---|---|---|---|---|---|
| V, μl | rB† | |||||
| C2 | 300.0 | — | — | 50 | 94 | 410 |
| C2B(11) | 267.9 | 36 | 1 | 50 | 94 | 490 |
| C2B(38) | 187.5 | 148 | 5 | 45 | 92 | 570 |
| C2B(54) | 136.8 | 183 | 10 | 40 | 96 | 660 |
| C2B(65) | 106.5 | 216 | 15 | 40 | 95 | 650 |
| C2B(75) | 76.2 | 250 | 25 | 30 | 92 | 680 |
| C2B(90) | 31.5 | 300 | 75 | 23 | 97 | 730 |
| C10 | 300.0 | — | — | 60 | 90 | 760 |
| C10B(20) | 239.4 | 68 | 10 | 55 | 97 | 800 |
| C10B(39) | 183.7 | 130 | 25 | 50 | 93 | 800 |
| C10B(50) | 149.1 | 169 | 40 | 50 | 90 | 840 |
| C10B(60) | 119.2 | 202 | 60 | 45 | 97 | 880 |
| C10B(71) | 85.0 | 241 | 100 | 40 | 93 | 1020 |
| ‡ | 57.7 | 271 | 166 | 35 | n.d. | n.d. |
The two-digit number in brackets of the sample identification gives the weight percentage wB of n-butyl acrylate in the monomer mixture.
rB: molar ratio of comonomers; rB = nn-butyl acrylate/nPCLDMA10000.
No stable film is obtained.
For all materials, the gelation degree G was between 90 and 97%. The degree of swelling Q in dichloromethane increased from about 400 to 1,000%, with decreasing content and increasing chain length of crosslinker, indicating a decreasing crosslink density. This result was in good agreement with the theoretical prediction for the swelling behavior of polymer networks made by Flory (17), as expected because of a higher contribution of entropy elasticity.
Polymer networks have been characterized thermally by differential scanning calorimetry and mechanically with tensile experiments. Shape-memory properties have been investigated by cyclic thermomechanical tests.
Thermal Properties of Polymer Networks.
The thermal characterization of polymer networks was performed with differential scanning calorimetry. In a standard procedure, melting temperature and heat of fusion were determined for thermosets with different comonomer ratios. To evaluate essential parameters for the thermocyclic tests, additional experiments concerning crystallization behavior were performed for selected materials.
The incorporation of n-butyl acrylate into the polymer network influences the thermal properties of the formed AB networks. However, this effect is stronger in the case of the PCLDMA2000 series than the PCLDMA10000 series. For thermosets formed from PCLDMA2000 only, the material with the lowest content of n-butyl acrylate, C2B(11) [network of oligo(ɛ-caprolactone) dimethacrylate with Mn = 2,000 and 11 wt % n-butyl acrylate], showed a melting peak at 25°C with a relatively low heat of fusion ΔHm of 12.4 J⋅g−1 compared with the C2 homonetwork having a melting peak at 32°C and a heat of fusion of 35.5 J⋅g−1. The other samples were completely amorphous. The thermal properties of thermosets formed by PCLDMA10000 and n-butyl acrylate are shown in Fig. 1. The melting temperature of the thermosets decreased by up to 5–46°C in the presence of 71 wt % n-butyl acrylate. The partial heat of fusion ΔHmC also decreased strongly because of introduction of noncrystallizable segments of n-butyl acrylate. None of the investigated materials showed a glass transition between −20 and 70°C.
Figure 1.

Melting temperature Tm (■) and partial heat of fusion ΔHmC (○) of thermosets prepared from PCLDMA10000 and n-butyl acrylate as a function of comonomer content of n-butyl acrylate wB in weight percent.
Apart from the standard procedure, the crystallization behavior of the thermosets was influenced by varying cooling rate βc and lower limit temperature Tl to observe the consequences on melting properties. Conclusions could be drawn from the relation between the variables of the crystallization process and the resulting crystallinity as well as for parameters for the programming process of the temporary shape. The heat of crystallization is essentially independent from the cooling rate for all examined materials (Fig. 2), indicating the same tendency for crystallinity.
Figure 2.

Crystallization enthalpy ΔHcr as a function of cooling rate βc for thermosets based on oligo(ɛ-caprolactone)-dimethacrylates or oligo(ɛ-caprolactone)-dimethacrylate/n-butyl acrylate. ▴, C2, Mn of macromonomer: 2,000 g⋅mol−1; ■, C3, Mn of macromonomer: 3,500 g⋅mol−1; ●, C6, Mn of macromonomer: 6,500 g⋅mol−1; ⧫, C10, Mn of macromonomer: 10,000 g⋅mol−1; ▵, C10B(39), wB = 39 wt %, Mn of macromonomer: 10,000 g⋅mol−1; ○, C10B(50), wB = 50 wt %, Mn of macromonomer: 10,000 g⋅mol−1; □, C10B(71), wB = 71 wt %, Mn of macromonomer: 10,000 g⋅mol−1.
The dependence of the melting temperature Tm of selected networks on the crystallization process has been investigated in isothermal crystallization experiments at different crystallization temperatures Tcr. For crosslinked PCLDMA10000 (C10), Tm did not vary with Tcr for crystallization temperatures up to 25°C and increased up to 5°C in a good linear fit for Tcr above 25°C (Fig. 3). Similar correlations are shown for crosslinked PCLDMA3500 (C3) and crosslinked PCLDMA10000 with 39 wt % n-butyl acrylate [C10B(39)].
Figure 3.

Hoffmann–Weeks plot of isothermal crystallization processes. ▴, C3, Mn of macromonomer: 3,500 g⋅mol−1; ■, C10, Mn of macromonomer: 10,000 g⋅mol−1; ●, C10B(39), wB = 39 wt %, Mn of macromonomer: 10,000 g⋅mol−1; —, Tm = Tcr.
On the basis of these data, a Hoffman–Weeks analysis (18) has been performed by extrapolating the straight line formed by plotting Tm against Tcr to a line that represents Tm = Tcr, the intersection led to the equilibrium melting temperature, Tm∞. For C10, this method led to a Tm∞ of 64°C, a value also given for high molecular poly(ɛ-caprolactone) (19). However, the same method used on C3 and C10B(39) in both cases gave a Tm∞ of 55°C, showing that a lower molecular weight of oligo(ɛ-caprolactone) segments or the presence of an amorphous phase of poly(n-butyl acrylate), respectively, led to a large change in the crystallization behavior of the oligo(ɛ-caprolactone) phase. These results emphasize the importance of crystallization behavior on the programming process of the temporary shape of a memory polymer network. Although the cooling rate is not critical for this procedure, the crystallization temperature (the lower limit temperature Tl in the thermocyclic tests described below) directly influences Tm as triggering temperature.
Mechanical Properties.
The materials have been mechanically characterized by tensile tests above and below Tm. The results of the tensile tests at room temperature for all thermosets are shown in Table 2.
Table 2.
Tensile properties of copolymerisates of n-butyl acrylate and PCLDMA2000 or PCLDMA10000 at room temperature
| Sample identification | E, MPa | ɛm, % | σm, MPa | ɛR, % | σR, MPa |
|---|---|---|---|---|---|
| C2 | 35 ± 3 | 21 ± 1 | 4.7 ± 0.1 | 20.6 ± 0.3 | 4.7 ± 0.1 |
| C2B(11) | 5.1 ± 0.2 | 13 ± 1 | 0.6 ± 0.2 | 12.8 ± 1.3 | 0.6 ± 0.2 |
| C2B(38) | 1.6 ± 0.1 | 28 ± 3 | 0.4 ± 0.1 | 29 ± 3 | 0.4 ± 0.1 |
| C2B(54) | 1.2 ± 0.1 | 41 ± 5 | 0.4 ± 0.1 | 44 ± 6 | 0.4 ± 0.1 |
| C2B(65) | 1.1 ± 0.1 | 32 ± 3 | 0.3 ± 0.1 | 34 ± 3 | 0.3 ± 0.1 |
| C2B(75) | 0.9 ± 0.1 | 71 ± 8 | 0.3 ± 0.1 | 74 ± 8 | 0.3 ± 0.1 |
| C2B(90) | 0.5 ± 0.1 | 129 ± 24 | 0.3 ± 0.1 | 130 ± 23 | 0.3 ± 0.1 |
| C10 | 71 ± 2 | 290 ± 30 | 16.2 ± 0.5 | 290 ± 30 | 15.7 ± 0.9 |
| C10B(20) | 58 ± 8 | 421 ± 21 | 14.6 ± 1.2 | 423 ± 23 | 14.1 ± 1.2 |
| C10B(39) | 49 ± 6 | 553 ± 28 | 10.3 ± 0.3 | 555 ± 30 | 10.3 ± 0.3 |
| C10B(50) | 30 ± 1 | 422 ± 11 | 9.2 ± 0.3 | 422 ± 11 | 9.2 ± 0.2 |
| C10B(60) | 7 ± 3 | 433 ± 30 | 8.0 ± 0.4 | 435 ± 42 | 8.0 ± 0.4 |
| C10B(71) | 7.8 ± 0.2 | 406 ± 63 | 4.3 ± 0.4 | 406 ± 63 | 4.3 ± 0.4 |
E is the elastic modulus (Young's modulus), σm is the maximum stress, ɛm the elongation at σm, ɛR is the elongation, and σR is the stress at break.
Increasing content of n-butyl acrylate influences the mechanical properties of the PCLDMA2000 series and PCLDMA10000 series in the same way. The elastic modulus E as well as tensile strength σm and stress at break σR decreases up to about one order of magnitude. Overall, these values are one order of magnitude larger for the PCLDMA10000 series, including ɛm (elongation at σm) and ɛR (elongation at break). The correlation of E, σm, and content of n-butyl acrylate is illustrated in Fig. 4 for PCLDMA2000 thermosets at room temperature. None of these thermosets showed a yield point. The results of the tensile tests at 70°C for PCLDMA10000 networks are presented in Table 3.
Figure 4.

Tensile properties of copolymerisates of PCLDMA2000 and n-butyl acrylate as a function of comonomer content wB at room temperature. ■, Young's modulus E; ○, elongation at break ɛR.
Table 3.
Tensile properties of copolymerisates of n-butyl acrylate and PCLDMA10000 at 70°C
| Sample identification | E, MPa | σm, MPa | ɛR, % |
|---|---|---|---|
| C10 | 0.70 ± 0.09 | 0.79 ± 0.10 | 210 ± 7 |
| C10B(20) | 1.01 ± 0.05 | 1.06 ± 0.17 | 187 ± 3 |
| C10B(39) | 0.84 ± 0.03 | 0.87 ± 0.08 | 222 ± 28 |
| C10B(50) | 0.80 ± 0.11 | 0.85 ± 0.12 | 229 ± 24 |
| C10B(60) | 0.60 ± 0.02 | 1.12 ± 0.01 | 382 ± 8 |
| C10B(71) | 0.43 ± 0.03 | 0.62 ± 0.11 | 354 ± 13 |
E is the elastic modulus (Young's modulus), σm is the maximum stress, and ɛR is the elongation at break.
Fig. 5 shows stress–strain plots for PCLDMA10000 thermosets at room temperature and 70°C. Both plots demonstrate the elastic properties of the materials above and below the transition temperature, whereas σ exhibited one order of magnitude lower values at 70°C. Only the materials with a n-butyl acrylate content up to 20 wt % showed a yield point at room temperature. The elastic modulus E decreased with increasing weight content of n-butyl acrylate at room temperature (Fig. 6), whereas at 70°C no correlation could be observed.
Figure 5.

Stress–strain plots for copolymerisates of PCLDMA10000 and n-butyl acrylate of different comonomer content wB: (a) at room temperature; (b) at 70°C. —-, C10, wB = 0 wt %; ⋅⋅⋅⋅, C10B(20), wB = 20 wt %; -⋅-⋅, C10B(39), wB = 39 wt %; ⋅⋅-⋅⋅, C10B(50), wB = 50 wt %; - - -, C10B(71), wB = 71 wt %.
Figure 6.

Young's modulus E of copolymerisates of PCLDMA10000 and n-butyl acrylate as a function of comonomer content wB: ■, at room temperature; ○, at 70°C.
Shape-Memory Properties.
An example for the macroscopic shape-memory effect of an AB-polymer network is shown in Fig. 7. The permanent shape of the presented sample is a rod, the temporary shape, a spiral. The transition from the temporary to the permanent shape occurred within 20 s at 70°C.
Figure 7.

Series of photographs showing the macroscopic shape-memory effect of AB-polymer networks. Permanent shape is a rod, temporary shape is a spiral. The pictures show the transition from temporary to permanent shape at 70°C.
Fig. 8 illustrates the molecular mechanism of the shape-memory effect. Shaping the material above the transition temperature and cooling thereafter leads to the crystallization of the oligo(ɛ-caprolactone) segments (red and blue colored). The temporary shape is fixed by physical crosslinks. Heating the material above the transition temperature causes the crystallites to melt again, and the material returns to its original shape. For both procedures, shape programming and shape recovery, respectively, elasticity of the material is required. By incorporation of oligo(n-butyl acrylate) segments (gray colored) the polymer network can be softened.
Figure 8.

Molecular mechanism of the shape-memory effect of AB-polymer networks containing crystallizable switching segments. Ttrans is the temperature of the shape transition [oligo(n-butyl acrylate) segments: gray, oligo(ɛ-caprolactone) segments: blue/red].
The shape-memory effect has been quantified for AB-polymer networks containing PCLDMA10000 by thermocyclic experiments. A tensile tester equipped with a thermo chamber was used that allowed running complementary strain and thermal programs at the same time.
The cyclic experiments consisted of heating the sample above the melting point to 70°C (Th), expanding it to 200% (ɛm), cooling it down in the expanded state to 0°C (Tl) (causing it to crystallize), and drawing back the extension to 0%. This step leads to a bending of the sample because of the fixation of the extended state by crystallization at Tl. Afterwards, the system was warmed up again to Th, causing the crystallites to melt and therefore the physical crosslinks to detach. The sample returned to a shape close to the original one. This cycle was repeated another four times with the same sample, leading to stress–strain plots that are more and more similar. A typical stress–strain diagram for a thermocyclic test is shown in Fig. 9.
Figure 9.

Thermocyclic tensile experiment of C10B(50), Mn of macromonomer: 10,000 g⋅mol−1, wB = 50 wt % with Th = 70°C, Tl = 0°C, ɛm = 200%. —, cycle number n = 1; - - -, n = 2; ⋅⋅⋅⋅, n = 3; -⋅-⋅, n = 4; -⋅⋅-⋅⋅, n = 5.
Thermomechanical experiments of thermosets of PCLDMA10000 and n-butyl acrylate generally showed a hysteresis loop, as expected. However, the stress at ɛm decreased in the cooling process for thermosets with a high content of n-butyl acrylate.
The experiment allowed the determination of the strain fixity rate Rf, as well as the strain recovery rate Rr. The strain fixity rate quantifies the fixability of the temporary form. The strain recovery rate describes to what extent the permanent shape is recovered. The strain recovery rate Rr was calculated from ɛm and the extension at the tension-free states ɛP(N-1) and ɛP(N) while expanding the sample in two following cycles N-1 and N after ref. 20:
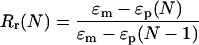 |
The total strain recovery rate Rr,tot is defined as the strain recovery after N cycles, compared with the original shape (therefore ɛp(N − 1) = 0), and is typically given for the last cycle, n = 5:
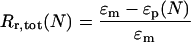 |
The strain fixity rate Rf is the quotient of the extension in the tension-free state ɛu(N), while drawing back the extension, and ɛm (20):
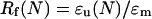 |
To compare the fixability of different materials or of the same material in different experiments, the average strain fixity rate Rf of all five cycles was taken into account.
All materials reached uniform deformation properties with strain recoveries Rr above 99% after 3 cycles, and the materials got slightly softer after the first deformation. Total strain recovery rates Rr,tot were between 93 and 98%, increasing with increasing content of n-butyl acrylate, and average strain fixity rates Rf about 95% (Fig. 10). Strain fixity was relatively low (82%) only for the thermoset with the highest content of n-butyl acrylate. This could be related to the lower crystallinity.
Figure 10.

Thermomechanical properties of copolymerisates of PCLDMA10000 and n-butyl acrylate as a function of comonomer content wB at Th = 70°C, Tl = 0°C, ɛm = 200%. □, total strain recovery rate Rr,tot; ●, average strain fixity rate R̄f.
Conclusions
The polymer system consisting of AB-polymer networks based on oligo(ɛ-caprolactone) dimethacrylates and n-butyl acrylate showed excellent shape-memory properties. Both structural parameters, the molecular weight of oligo(ɛ-caprolactone) dimethacrylate and the comonomer content of n-butyl acrylate, strongly influenced the macroscopic properties of the networks. In contrast to shape-memory alloys, the mechanical characteristics of the polymer system can be altered easily in a wide range. The versatility of these shape-memory AB-polymer networks potentially could enable the realization of numerous new applications in different areas, especially in the biomedical sector, because of the biocompatibility of the components.
Acknowledgments
Andreas Lendlein is grateful to Fonds der Chemischen Industrie for a Liebig fellowship. Financial support has been partly provided by Bundesministerium für Bildung und Forschung, BioFuture Award no. 0311867.
Abbreviations
- PCLDMA
oligo(ɛ-caprolactone) dimethacrylate
- PCLDMA10000
oligo(ɛ-caprolactone) dimethacrylate with Mn = 10,000
- C10 network of PCLDMA10000.
Footnotes
Besselink, B., Medical Device Technology Conference, March 2–3, 1999, London, U.K.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.031571398.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.031571398
References
- 1.Duerig T W, Pelton A R, Stöckel D. Biomed Mater Eng. 1996;6:255–266. [PubMed] [Google Scholar]
- 2.Roff W J, Scott J R, Pacitti J. Handbook of Common Polymers. London: Butterworth; 1971. [Google Scholar]
- 3.Shalaby S W, Johnson R A. In: Biomedical Polymers. Shalaby S W, editor. New York: Hansa; 1994. pp. 1–34. [Google Scholar]
- 4.Laurencin C T, Sobrasua I E M, Langer R S. In: Biomedical Applications of Synthetic Biodegradable Polymers. Hollinger J O, editor. Boca Raton: CRC; 1995. pp. 59–101. [Google Scholar]
- 5.Kohn J, Langer R. In: Biomaterials Science. Ratner B D, Hoffmann A S, Schoen F J, Lemons J E, editors. San Diego: Academic; 1996. pp. 64–73. [Google Scholar]
- 6.Lendlein A, Neuenschwader P, Suter U W. Macromol Chem Phys. 1998;199:2785–2796. [Google Scholar]
- 7.Hu Z, Zhang X, Li Y. Science. 1995;269:525–527. doi: 10.1126/science.269.5223.525. [DOI] [PubMed] [Google Scholar]
- 8.Osada Y, Matsuda A. Nature (London) 1995;376:219. doi: 10.1038/376219a0. [DOI] [PubMed] [Google Scholar]
- 9.He X, Oishi Y, Takahara A, Kajiyma T. Polym J. 1996;28:452–457. [Google Scholar]
- 10.Li Y, Hu Z, Chen Y. J Appl Polym Sci. 1997;63:9. , 1173–1178. [Google Scholar]
- 11.Sawhney A, Pathak C P, Hubbell J A. Macromolecules. 1993;26:581–587. [Google Scholar]
- 12.Sawhney A, Pathak C P, Rensburg J J, Dunn R C, Hubbell J A. J Biomed Mater Res. 1994;28:831–838. doi: 10.1002/jbm.820280710. [DOI] [PubMed] [Google Scholar]
- 13.Nathan A, Bolikal D, Vyavahare N, Zalipsky S, Kohn J. Macromolecules. 1992;25:4476–4484. [Google Scholar]
- 14.Lendlein A, Neuenschwader P, Suter U W. Macromol Chem Phys. 2000;201:1067–1076. [Google Scholar]
- 15.Aoyagi T, Miyata F, Nagase Y. J Contolled Release. 1994;32:87–96. [Google Scholar]
- 16.Malucelli G, Gozzelino G, Bongiovanni R, Priola A. Polymer. 1996;37:2565–2571. [Google Scholar]
- 17.Flory P J. Principles of Polymer Chemistry. Ithaca, NY: Cornell Univ. Press; 1953. [Google Scholar]
- 18.Hoffman J D, Weeks J J. J Res Natl Bur Stand Sect A. 1962;66:13. [Google Scholar]
- 19.Mandelkern L, Alamo R G. In: Physical Properties of Polymers Handbook. Mark J E, editor. New York: AIP; 1996. [Google Scholar]
- 20.Kim B K, Lee S Y, Xu M. Polymer. 1996;37:5781–5793. [Google Scholar]