

Abstract
Identifying genetic variations predictive of important phenotypes, such as disease susceptibility, drug efficacy, and adverse events, remains a challenging task. There are individual polymorphisms that can be tested one at a time, but there is the more difficult problem of the identification of combinations of polymorphisms or even more complex interactions of genes with environmental factors. Diseases, drug responses or side effects can result from different mechanisms. Identification of subgroups of people where there is a common mechanism is a problem for diagnosis and prescribing of treatment. Recursive partitioning (RP) is a simple statistical tool for segmenting a population into non-overlapping groups where the response of interest, disease susceptibility, drug efficacy and adverse events are more homogeneous within the segments. We suggest that the use of RP is not only more technically feasible than other search methods but it is less susceptible to multiple-testing problems. The numbers of combinations of genegene and gene environment interactions is potentially astronomical and RP greatly reduces the effective search and inference space. Moreover, the certain reliance of RP on the presence of marginal effects is justifiable as was found by using analytical and numerical arguments. In the context of haplotype analysis, results suggest that the analysis of individual SNPs is likely to be successful even when susceptibilities are determined by haplotypes. Retrospective clinical studies where cases and controls are collected will be a common design. This report provides methods that can be used to adjust the RP analysis to reflect the population incidence of the response of interest. Confidence limits on the incidence of the response in the segmented subgroups are also discussed. RP is a straightforward way to create realistic subgroups, and prediction intervals for the within-subgroup disease incidence are easily obtained.
Introduction
Despite the remarkable success in mapping single genes affecting important human traits, such as disease predisposition, relatively little progress has been made in finding multiple interacting genes. Many traits, including disease risk, appear highly heritable; however, often only a small proportion of the phenotypic variation can be attributed to a single polymorphism. The implied involvement of many genes is through epistatic or heterogeneous mechanisms. Unfortunately, such combinations of genetic polymorphisms are very difficult to pin down. When large collections of polymorphisms (e.g., SNPs) are screened, multiple testing becomes an important issue. Association genome scans may require over 100,000 SNPs (Goldstein et al., 2003), so the multiple testing becomes a formidable problem even when testing single markers for association with the trait. The goal of identifying genuine relationships with the trait using combinations of markers is even more difficult and many seemingly strong associations are likely to turn out to be false positives. This is a consequence of increasing the prior probability of the hypothesis of no association, Pr(H0), as will be discussed later.
Yet another additional challenge is the characterization of the nature of the relationship, which can be complicated and nonlinear. Recursive partitioning (RP) is a simple data mining tool that can, nevertheless, adequately handle these problems (Young et al., this issue). RP has been previously applied to genetic problems as a tool for dealing with the detection of interaction and identification of homogeneous subgroups (Rao 1998; Zhang et al., 2000; Czika et al., 2001; Province et al., 2001; Shannon et al., 2001; Costello et al., 2003). However, RP can be criticized for over-reliance on the presence of “marginal effects”.
When RP makes the first sweep through predictor variables (e.g., a set of genetic markers) it tries to identify those variables that result in a significant association with the trait. The data are then divided into subgroups corresponding to selected predictor categories, and the association test is repeated in the subgroups using the remaining predictors. At this stage the interaction can be detected. Suppose the first RP split divides the sample into two subgroups with individuals carrying the genotype AA vs. Aa + aa; the test among individuals in the AA group can detect interaction of AA with another marker. This interaction can only be detected if the first split is “significant” (i.e. there is substantial “marginal” effect at the marker A). One purpose of this article is to illustrate that it is reasonable to assume that most interactions will induce such marginal effects at one or more of the markers involved in the interacting set.
From the clinical perspective, a most useful characteristic of a predictive set of genetic polymorphisms is its ability to provide estimates of the probability of an event (condition) Y, such as an adverse event or disease. For a binary trait (event present/absent, denoted by Y, N), we also describe estimates of probabilities of developing the condition Y given that a random individual is classified into a particular node of the RP tree according to its multilocus genetic profile. Along with these estimates, interval estimates will also be provided. These are available by using a hold-out sample (i.e., part of the sample set aside for the validation purposes).
In this paper, frequencies of haplotypes, which are defined by joint frequencies of SNPs on the same gamete, are used to illustrate the approach and the analysis. More generally, RP is concerned with describing interactions among predictors. In practice, genetic data are likely to be in the form of diploid genotypes and haplotypes, which may not be directly observed.
Reliance on marginal effects and the recursive partitioning justification
Searching for sets of predictive markers results in many putative models being taken into consideration. In the extreme case, all possible combinations are evaluated and a criterion, such as a measure of statistical association, is scored; the combination with the best score is taken. This combination may be examined in an independent follow-up study to verify that it is not a “false positive”. If there are k markers to start with, there are k(k − 1)/2 pairs, and, at most, L = 2k − 1 combinations, including individual markers, all possible marker pairs, triples, and so on. A considerable problem with this approach is that the probability of a false positive increases with the number of combinations. The probability that a p-value of p or smaller represents a false positive is related to the false discovery rate, FDR (Morton 1998, Storey, 2002). At a given fixed value of p,
| (1) |
where F(p) is the p-value cumulative distribution function under the alternative hypothesis, which reflects the power. For example, with the χ2 test, where Ψd,λ and Ψd,0 are the cumulative distribution functions of central and non-central χ2, respectively, and d denotes the degrees of freedom.
Supposing that statistical tests and their p-values are used to rank the hypotheses and that there is a single “true” association, HA, among L−1 null hypotheses, H0, where L is the number of combinations, then the probability that a randomly picked hypothesis represents a false discovery is
If we let ℒ(FDR) = FDR/(1 − FDR), then this is an increasing function of FDR. Using Pr(H0) = 1 − 1/L,
| (2) |
One interpretation of this formula is that the power gained by considering combinations must increase more sharply than the number of combinations, L, otherwise FDR is going to increase. If L is an exponential function of the number of markers, k, the condition that FDR should not increase is difficult to achieve. Practically, this means that when L is large, the chances that a claimed association turns out to be a false positive are high.
For example, assume the power to detect a single true combination at α = 0.05 is 80%. Our analysis depends on the definition of a “true combination” and for illustrative purposes we assume that there is only a single subset. One can use a different definition, whereby every subset that partially overlaps with the combination of interest does represent a true discovery, which will reduce L in half. If we start with k = 10 markers and examine all L = 2k−1 = 1023 combinations, then among p-values ≤ 0.05, 98% are going to be false positives, with the assumption of a single true combination.
It is worthy to note that simple multiple testing correction alone does not easily get around this problem: suppose the testing is at the Bonferroni-corrected α = 0.05/1023 and that the proportion of false positives among p-values as small as 0.05/1023 ≈ 4.9 × 10−5 is examined. The proportion of false positives still remains high; about 32%. The situation worsens with a larger k. For example, when k = 15 the proportion of false positives among significant p-values after applying the Bonferroni correction (1.5 × 10−6 or smaller) is 69%.
This problem is very hard to circumvent, because L quickly increases with the number of markers, but the sample size does not increase correspondingly (see Witte et al., 2000, for requirements on that). On the other hand, RP has the implicit assumption that at least one of the individual markers in the “true” associated set, as well as the smaller subsets (combinations) of markers in this set, should show some degree of association. As a consequence, the number of combinations remains linear as a function of the number of markers, keeping FDR relatively low (e.g., a tree with three splits will make approximately 3 × k comparisons).
How reasonable is the assumption that at least one of the individual effects will be present? One might think it is easy to come up with examples of marker interactions that preclude “marginal” effects. Consider the simplest possible scenario: a binary case (e.g., the presence/absence of a disease) and the control phenotype (Y, N) and two diallelic loci (two binary predictors), x1 = {0, 1} and x2 = {0, 1}. If one considers a situation where allele ”1” has to be present at both markers in order for the disease risk to be high, then this is a clear case of interaction; however, the “case” category is likely to be enriched with the alleles of type ”1” at both markers compared to controls. A more subtle and seemingly “no-marginal effects” scheme is the following: ”1,1” or ”0,0” combinations are both associated with high risk, whereas ”0,1” and ”1,0” have low risk. Similar situations have been considered as justification for methods that go after “pure interactions”. Jannot et al. 2003 considered such interactions for the case of genotypic interactions where the penetrance array is 3 × 3 for two loci. They evaluated an approach wherein all 2k−1 marker combinations were analyzed and p-values associated with the best combination were obtained by a permutation algorithm. Lin et al. 2004 took a similar approach using transmission disequilibrium tests. They state that because of the usage of the permutation approach to multiple tests “the gains from considering haplotypes in our exhaustive allelic method are not overshadowed by the penalty paid for doing far more tests”. These approaches assume that the accurate type-I error protection through permutations is balanced with the appropriate increase in power provided by considering the proper subset. Ritchie et al. 2001 proposed an algorithm that starts with a multi-dimensional table that allows for all possible interactions for a given subset of markers. The multi-dimensional table is represented by one of 2k−1 combinations of k markers. The high-risk cells are then pooled into one group and the low-risk cells into another group, and a cross-validation is used to assess robustness of the reduced model. A combinatorial partitioning method (CPM) proposed by Nelson et al. 2001 evaluates all partitions of a cells (multilocus genotypes) into b groups. The number of partitions is given by:
which is substantially larger than the number of marker subsets. For example, with three diallelic markers there are 23−1 = 7 marker subsets that could be tested separately. However, assuming we start with the binary classification at each locus and a = 23 multilocus combinations, there are 127 ways to partition the genotypes into two groups, and 966 ways to partition them into three groups. Another combinatorial algorithm by Tahri-Daizadeh et al. 2003 considers increasingly complex models and evaluates proposed models on the basis of information criterion (Akaike, 1974). For quantitative traits, Culverhouse et al. 2004 proposed a modification of CPM that reduces this number of putative models by successfully merging cells with similar values of response. Such short-cut algorithms may consider a single model after the collapsing is finished. Significance levels are adjusted accordingly by a permutation algorithm. Nevertheless, the null distribution obtained via permutations essentially accounts for the number of looks through the data. This can be thought of as the adjustment by the ”effective” number of tests, Le ≤ L.
While methods optimized for discovery of pure interactions are important, even the most contrived penetrance configurations allow for marginal effects – the feature exploited by the RP. The advantage of the RP method is in drastically reducing the number of potential models, thus lowering Pr(H0) in (1) and, therefore, reducing the probability of a false discovery. Cordell and Clayton (2002) proposed a stepwise logistic regression procedure for evaluating the effects of the different polymorphisms within a gene. Curran (2003) compared such approach using the combination of forward and backward selection with the performance of RP. The research indicated a remarkable similarity between the two methods in terms of the predictive power and the effects and interactions entering the final model.
Returning to the “no-marginal effects” scheme (”1,1”/”0,0” vs. ”0,1”/”1,0”) it may seem that all alleles are going to be present at equal frequencies in both phenotype groups. However, this is generally not the case and it is possible to discover such haplotype-trait associations by looking at just a single marker. The single marker effect may be smaller than a multilocus effect; however the reduction in degrees of freedom or the number of tests may balance a reduction in the effect magnitude.
The four possible haplotypes formed by the two SNPs are X = {00, 01, 10, 11}, with frequencies P = {p00, …, p11}. Four haplotype penetrance values are:
With random pairing of haplotypes and multiplicativity, the population prevalence of Y is:
| (3) |
In terms of the two SNPs that constitute a haplotype, the values γij define two-locus interactions with respect to the binary phenotype. For example, a high value of Pr(Y | X = 11) relative to γ indicates that both x1 = 1 and x2 = 1 are required for this probability to be above the population average. Suppose we have allele information at one of the markers, x1. Then the association can be detected if the penetrance of one of the alleles, e.g. γ1·. = Pr(Y | x1 = 1) is different from γ. This marginal allele penetrance value is
| (4) |
using the allele probabilities among the case and the control groups:
In turn, these are calculated from the frequencies of haplotypes and the penetrance values as
Equation (4) can also be written as
| (5) |
When γ1· ≠ γ, the marginal effect of a SNP can be detected. Thus, in general, this effect depends not only on the haplotype penetrances but on the population frequencies of the haplotypes as well. Looking at the “no marginal effect” difference,
| (6) |
it can be seen that for a plausible “pure interaction” penetrance configuration, , the additional requirement on the population frequencies of haplotypes is such that the two pairs of frequencies should match:
| (7) |
As an example let the haplotype penetrance array {γ00, γ01, γ10, γ11} be Γ = {0.9, 0.2, 0.2, 0.9}, which may seem to imply that an SNP effect is unlikely, because of the ”orthogonal” structure of Γ. As often happens in reality, one of the four haplotypes can be of relatively high population frequency. In such a case, the marginal (SNP) effects can be quite pronounced, as can be seen from two following examples.
Example 1. Consider a situation when one of the low penetrance haplotype has a high frequency, P = {0.05, 0.05, 0.85, 0.05}. In this case, γ = 0.27 and γ1· = 0.55. The frequencies among the cases and the controls are Pr(x1 = 1 | Y) = 0.204 and Pr(x1 = 1 | N) = 0.062 corresponding to the loge of relative risk of 1.191.
Example 2. Now suppose that one of the high penetrance haplotypes has a high frequency, P = {0.05, 0.05, 0.05, 0.85}. The values become γ = 0.83 and γ1· = 0.55. The frequencies among the cases and the controls are Pr(x1 = 1 | Y) = 0.066 and Pr(x1 = 1 | N ) = 0.265 corresponding to the log relative risk of −1.39. Such value provides 99% power (at α = 0.05) with samples of 160 cases and 160 controls.
Allelic penetrance equation (5) can be easily generalized to multiple genetic loci. Let ”j” index all possible haplotypes that contain allele ”1” at the locus of interest. Then the marginal effect associated with allele ”1” is
| (8) |
where the denominator gives the frequency of ”1”. The condition of no marginal effect associated with allele ”1” is again γ1· = γ.
Example 3. As a three-locus illustration using RP, consider three diallelic loci, x1, x2, x3, with eight possible haplotype combinations, {000, 001, ..., 111}, with penetrances and population frequencies sampled from the Dirichlet(1/4, ..., 1/4) distribution, as given in Table 1.
Table 1.
Population parameters for the RP example
| Haplotype | Penetrance | Population frequency |
|---|---|---|
| 000 | 0.9 | 0.010668810 |
| 001 | 0.1 | 0.000005347 |
| 010 | 0.1 | 0.000000255 |
| 011 | 0.1 | 0.446030100 |
| 100 | 0.1 | 0.016740010 |
| 101 | 0.1 | 0.477861900 |
| 110 | 0.1 | 0.007107833 |
| 111 | 0.9 | 0.041585780 |
In this example there are two high risk haplotypes (000 and 111) carrying different alleles. By looking at the penetrance values alone, a technique that relies on the marginal effects associated with one of the three SNPs may appear unlikely to be successful. For simplicity, we considered a haploid population and sampled 250 case and 250 control haplotypes. A sample RP tree using HelixTree® is shown in Figure 1. HelixTree is a software system that uses recursive partitioning algorithms customized to the field of genetics (www.goldenhelix.com).
Figure 1.
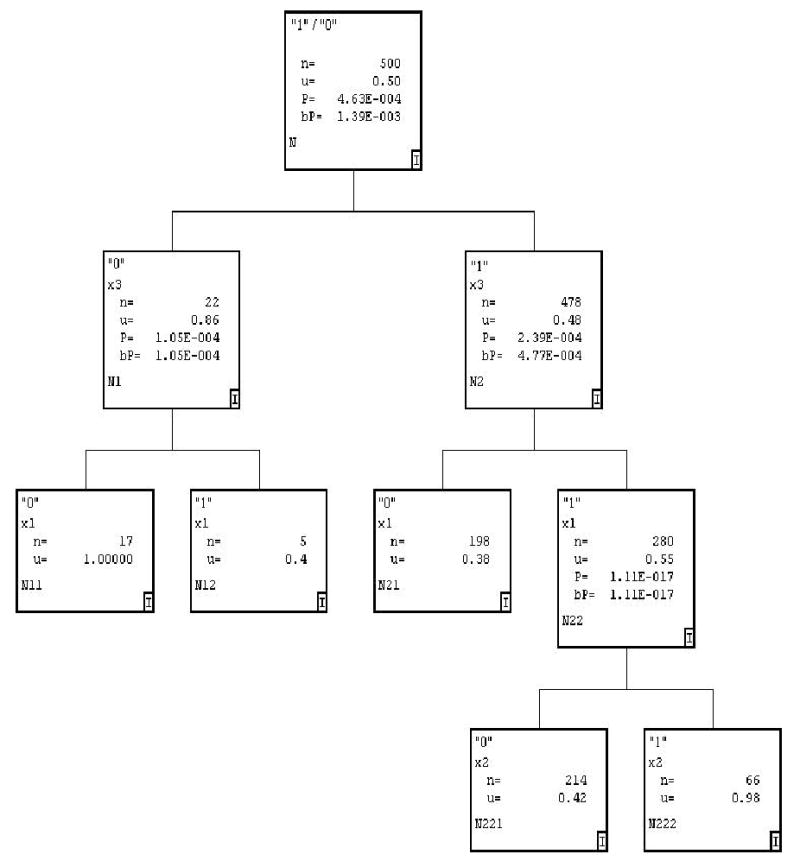
“No marginal effects” RP tree with 3 predictors; ’x1’, ’x2’, ’x3’ – predictor (SNP) labels; ’n’ – node-specific sample size; ’u’ – node-specific mean response value (proportion of cases in the node; ’P’ – association p-value; ’bP’ – Bonferroni-corrected association p-value (adjusted by the number of non-monomorphic SNPs at the current split)
The first split is on x3 with a p-value 4.6 ×10−4, signifying a substantial effect associated with this SNP. Allele ”0” at this SNP appears predictive of the cases, with the sample prevalence increasing from 0.5 in the root node to 0.86 at the first split. Following splits yield even smaller p-values. It appears that as the interaction is successfully uncovered, the p-values can still get progressively smaller even though the sample size is decreased. The right branch involving all three SNPs is significant with a p-value of 1.1 × 10−17, indicating that there is substantial interaction among SNPs, i.e. “haplotype effect”. Therefore, even in this odd situation, RP found one haplotype perfectly (x1 = x2 = x3 = 1). It also found two of the three alleles defining the second high penetrance haplotype (x1 = x2 = x3 = 0).
To examine the “no marginal effect” condition in more detail, the haplotype classes need to be reordered so that the first i = 1, ..., k of the total of m haplotypes contain the SNP of interest. For example, suppose we are looking at the effect of allele ”0” at the first SNP in Table 1. Then i = 1, ..., 3. The marginal penetrance of allele ”0” is
| (9) |
The marginal penetrance of allele ”1” (or all remaining alleles) is
| (10) |
and the population prevalence is
| (11) |
The condition of ”no marginal effect” can be expressed as
| (12) |
Consider some specific situations.
Obviously, the equality holds when all susceptibilities γi are the same.
-
Next, assume very high haplotype diversity. In the extreme, pi = 1/m for all i. In this case (12) simplifies toIf markers are diallelic, k = m/2, then (12) simplifies to
The ”pure interaction” penetrance considered above with the condition given in (7) corresponds to this case.
To examine the “orthogonal penetrance” case across various frequencies of haplotypes, we simulated the penetrance configuration similar to that given in Table 1 with haplotype population frequencies following the Dirichlet(1) distribution. In each of the 50,000 simulations we uniformly sampled penetrances of high risk haplotypes (000, 111) between 0.2 and 0.9. Penetrances of low risk haplotypes were set equal to each other and sampled from the interval (0, 0.1) at each simulation. Figure 2 shows the simulation results. Haplotype and SNP risk distribution refer to the risk of high susceptibility haplotype or SNP relative to the population prevalence that is specific to a particular simulation run. We denote this quantity by RRP: “risk of a genetic variant relative to the population prevalence”. The overall distribution of the population prevalence, as well as the prevalence among the carriers of a susceptibility SNP, is given in the second row. The 95% quantile of the population prevalence is 0.326. However, the last graph (SNP penetrance) is shifted to the right relative to the prevalence distribution, and 25% of its distribution is above the 95% quantile for the population prevalence, indicating the presence of effects associated with SNPs.
Many penetrance configurations, e.g. the case of a single highly penetrant haplotype, will induce marginal effects associated with SNPs. It is noteworthy to mention that even if the equality (12) holds for a particular SNP, the indices i = 1, ...k, i = k + 1, ...m would need to be rearranged each time a different SNP is considered, which is likely to result in the marginal effect at one of the SNPs. A situation we examined numerically is when there is one high frequency haplotype with the frequency p′ and the penetrance γ′, while other haplotypes are relatively rare. When all haplotypes except p′ are of the same frequency, the condition (12) becomes
Figure 2.
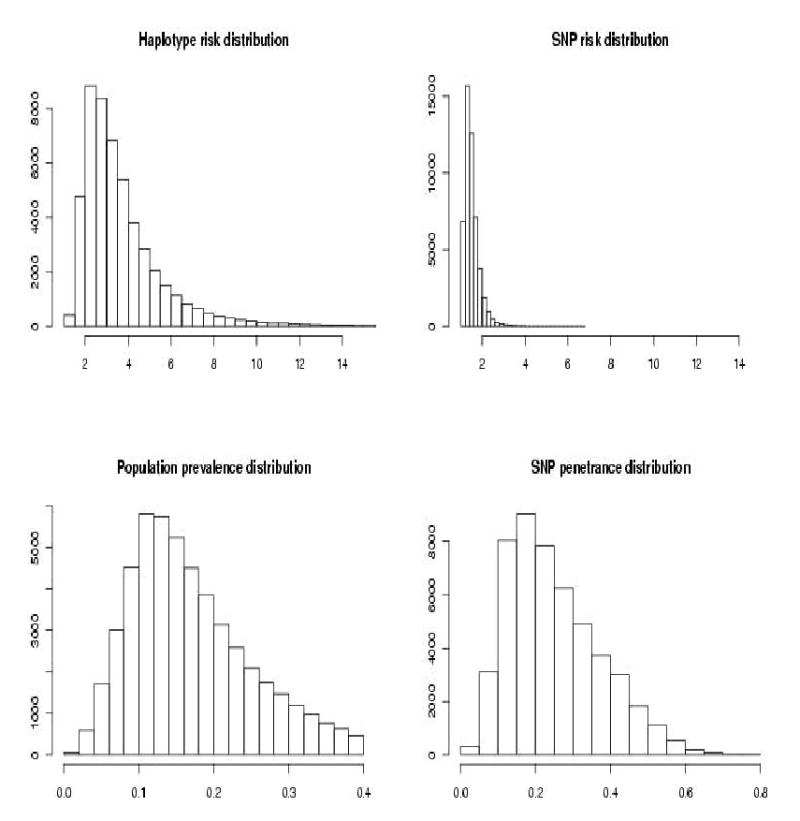
Results of simulations with two “orthogonal penetrance”, high risk (000, 111), random frequency haplotypes.
where and . The following results are based on 150,000 simulations.
The high susceptibility haplotype has the lowest frequency (Figure 3). Mean SNP relative risk (RRP, as defined above) was 1.903. Mean haplotype RRP was 5.843. The prevalence 95% quantile was 0.089. The proportion of SNP penetrance distribution above that value was 0.627.
The high susceptibility haplotype has an intermediate (random) but never the highest frequency (Figure 4). Mean SNP relative risk was 2.076. Mean haplotype RRP was 5.269. The prevalence 95% quantile was 0.097. The proportion of SNP penetrance distribution above that value was 0.695.
The high susceptibility haplotype has the highest frequency (Figure 5). Mean SNP relative risk was 1.243. Mean haplotype RRP was 1.516. The prevalence 95% quantile was 0.348. the proportion of SNP penetrance distribution above that value was 0.170.
Figure 3.
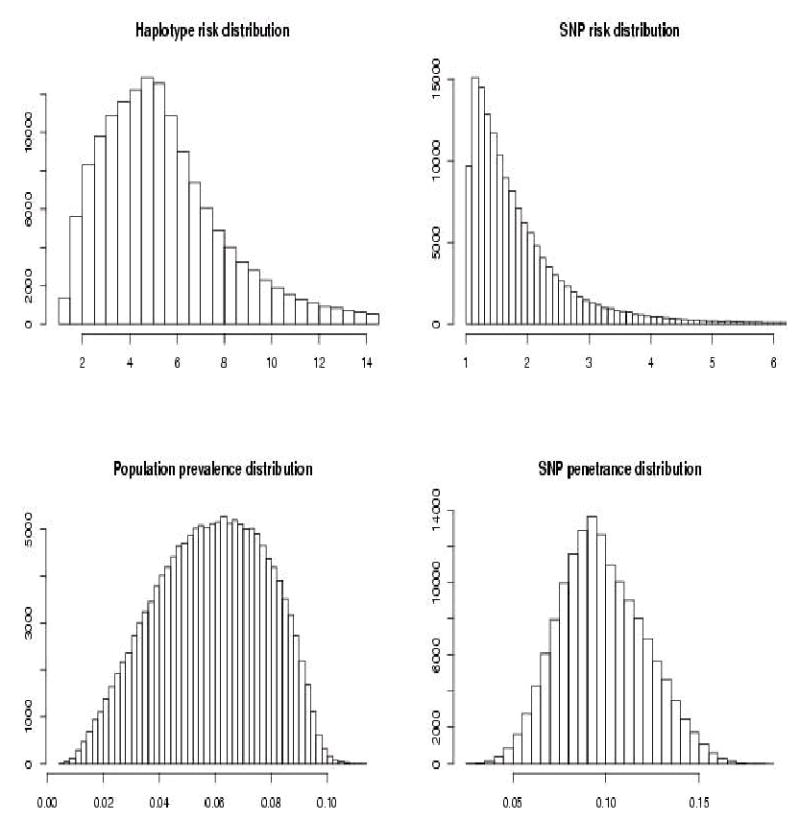
Results of simulations with a single high risk lowest frequency haplotype.
Figure 4.
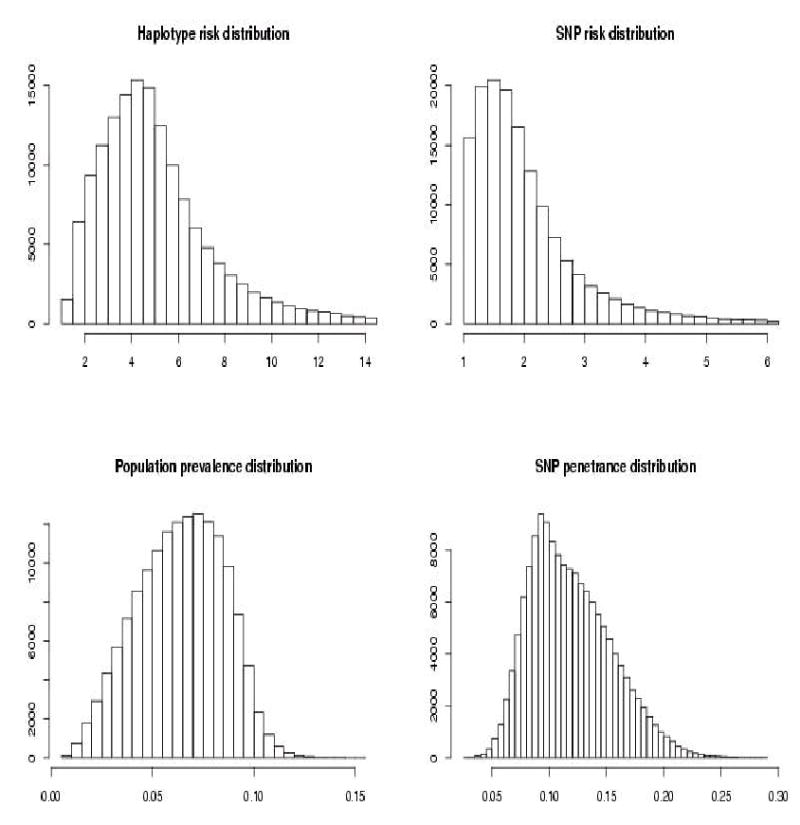
Results of simulations with a single high risk random frequency haplotype.
Figure 5.
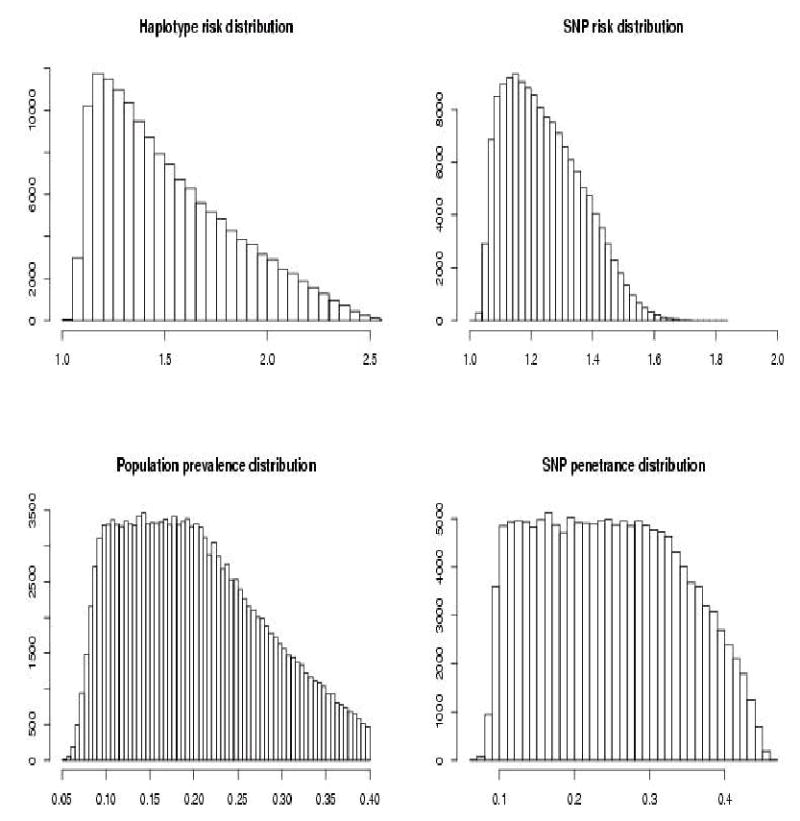
Results of simulations with a single high risk highest frequency haplotype.
Examples and simulation results presented in this section suggest that even in situations where the penetrance configuration is in favor of no marginal SNP effect, there needs to be a very specific set of population frequencies of haplotypes in order for the marginal effect to be absent. We considered highly interacting models so that the effect of a high-penetrance joint set of predictors (e.g., “haplotype”) is expected to be larger than that of a SNP. Nevertheless, one would need to balance the potential increase in the effect size with the number of looks through potential models. In this light, an analyst using the RP algorithm makes a very reasonable “bet” that marginal effects are going to be present among the markers that form the best predictive combination. The multiple testing implication of this sequence of order k searches is the reduction in the false discovery rate.
An interesting observation from Figures (3–5) is that the difference in magnitude between the haplotypic and SNP effects decreases as the frequency of the high-penetrance haplotype increases. It follows that “common” susceptibility haplotypes can be mapped by looking at individual SNP associations. The relative risk discrepancy is higher for rare susceptibility haplotypes. This case deserves further consideration. Low frequency of high-risk haplotypes implies problems with frequency estimation in the case of an unobserved haplotype phase, as well as a lack of statistical power for analysis using haplotypes as predictors. In this case, the relatively higher frequency of SNPs may still provide better power despite the smaller effect size.
Predictive values and interval estimation
Samples from clinical trials are likely to be cast in case-control form. To optimize power, observations may be collected in pairs – one case and one control – so the proportion of cases in the sample may be considerably different from the population prevalence, φ0 = Pr(Y). It is desirable and possible to estimate genetic susceptibility (penetrance) associated with a genetic marker M for the population. Suppose one of the genotypes (AA) is considered. Then φ1 = Pr(Y | AA) is obtained using the relation
| (13) |
where p = Pr(AA | Y), and q = Pr(AA | N). An estimate is obtained by plugging in estimates of p, q, using sample frequencies of AA in cases and controls,. If the genotype AA is predictive of Y, then is the positive predictive value (PPV) of AA.
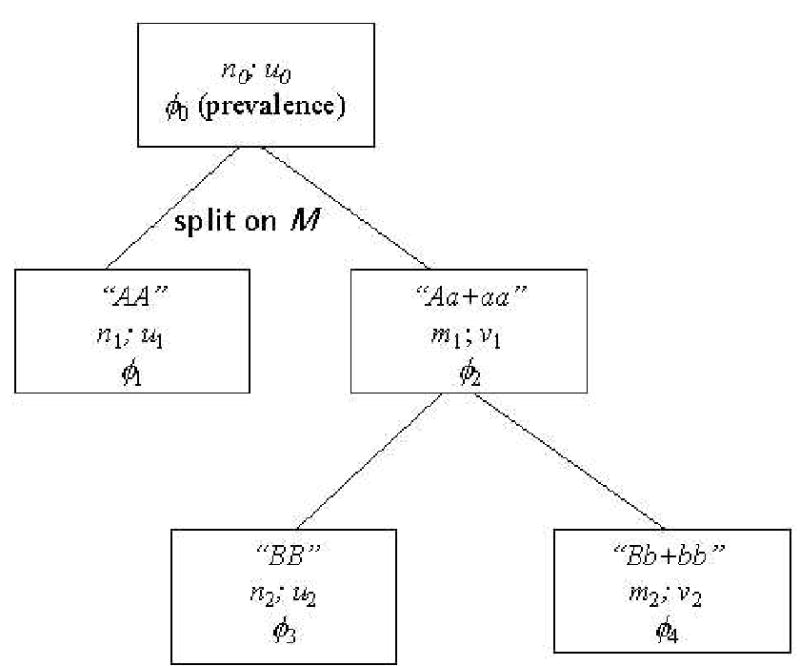
Quite usefully, the very first split of a RP tree allows a direct calculation of . Figure 6 shows a two-level tree where the first split is made based on the presence of a predictive marker M.
Figure 6.
Sample RP tree
n0, u0: sample size and the proportion of cases in the whole sample
ni, mi: node-specific numbers of subjects in the right and the left nodes
ui, vi: proportion of cases in the right and the left nodes
φi: node-specific positive predictive value
We can classify Figure 6 observations at the first split as follows:
| AA | : (Aa + aa) | |
|---|---|---|
| Cases (Y) | n1u1 | m1v1 |
| Controls (N) | n1(1 − u1) | m1(1 − v1) |
(For multi-way RP splits, the counts in the second ”not-AA”() column are obtained by adding up the counts from all nodes except ”AA”). The frequency estimates in cases and controls are
Now we can estimate susceptibilities at the first two ”AA” and ”Aa + aa” nodes as
| (14) |
| (15) |
Note that (PPV at the right node) is one minus the quantity referred to as the “negative predictive value”, NPV, (Pepe, 2003) at the left (”AA”) node.
Susceptibility estimation can proceed to subsequent RP splits using the same approach. Consider , which is the probability that an individual with the genotype ”Aa or aa” and ”BB” will develop the condition Y,
The estimate is
| (16) |
where
Note that equation (16) is similar to (14), except that it is based on the estimates conditional on the previous (”AA” vs. ”Aa + aa”) split. For example, in (16) is the estimated prevalence among individuals carrying ”Aa + aa” genotypes, whereas in (14) is the unconditional population prevalence. Thus, node-specific genetic susceptibility can be calculated recursively by using counts and proportions of cases at the node that give estimates, as well as the susceptibility at the previous split,.
A more direct estimate of is obtained by recursively substituting the definition of into , etc. up to . After simplification, this leads to the following general formula:
| (17) |
with products giving joint multilocus (defined by the RP branch above the node) frequencies in cases and controls. This expression assumes that the population prevalence, φ0, is known or well estimated from the external data. When interval statements are made about , the uncertainty in φ0 may become an issue. For clarity, we will assume here that φ0 is known, however it is possible to incorporate uncertainty in φ0 into the interval estimate of susceptibility (Zaykin et al., 2004).
The right hand side of (17) simplifies on the logit scale:
| (18) |
where are estimated frequency of the multilocus genetic profile defining the ith node in cases and controls respectively. Referring to Figure 1, node ”N12”, containing 5 observations with the proportion of cases 0.4, the frequency of the profile (x3 = 0, x1 = 1) in the cases is and the frequency of that profile in the controls is .
Define . Assuming φ0 is constant, the variance of the logit is
| (19) |
using first-order Taylor series approximation. Assuming binomial sampling,
| (20) |
Then the estimated variance in terms of the node counts is
| (21) |
The binomial assumption in (20) implies random sampling from the case and the control population as well as that the tree is not built in “exploratory” mode. Typically, a tree is built using either the most significant predictor for a split or is “guided” by the scientist. Both situations can result in biased phenotypic proportions in the nodes. In this exploratory mode, the RP splits are usually chosen so that the association test statistic value is one of the largest among potential split variables (genetic markers). The distribution of niui will follow the distribution of one of the first order statistics from the binomial distribution. Thus, the variance calculated by (21) may be underestimated. If there is a hold-out sample, it can be used to validate a particular RP tree built in the exploratory mode and will provide unbiased estimates. Using such a hold-out sample, it is possible to construct confidence intervals for estimates. The asymptotic normal-based confidence interval for the logit is calculated as:
| (22) |
Then the interval endpoints (l, u) are inverted as and , respectively to produce an approximate (1 − α)% interval estimate of φi.
Discussion
Methods for the detection of an association between traits and interacting genetic polymorphisms are being rapidly developed. Many approaches are considering important situations where haplotypes of consecutive markers can be defined and tested for association with the trait. This can incorporate different sampling designs, as well as the haplotype phase uncertainty (Schaid et al. 2002; Zaykin et al. 2002; Stram et. al. 2003; Epstein and Satten 2003; Lin 2004). These methods are most successful when a relatively small set of tightly linked markers can be selected a priori, such as a set of physically ordered markers within a gene that could, for example, tag a common transcript. It is a much more complex problem to identify associations caused by markers that are located in different genes or genomic regions. The problem is not only of searching through an inconceivably large number of marker combinations; even if an effective algorithm is designed that can identify the most predictive combination, the chances of it representing a false positive are unacceptably high. This issue is not solved by applying stringent significance levels because of the reduced power. When power is low, true positives are not guaranteed to rank among the most significant results and can appear among the bulk of the results after sorting by the significance or the association statistic value. In this paper we do not distinguish between the issues of identification of etiological polymorphisms and the detection of markers associated with causative variants through the population linkage disequilibrium. Nielsen and Weir (2001) expressed marker penetrance values via the actual penetrances at the susceptibility locus, allele frequencies, and link- age disequilibrium. Unless there is perfect correlation between the marker and the susceptibility locus, the induced effects are expected to be lower at the marker, reducing statistical power.
Although RP does not provide a perfect solution, its appealing feature is the remarkably reduced number of putative models. Given that the distribution of haplotype frequencies is often skewed and is unlikely to match the corresponding susceptibilities in the sense of satisfying the condition (12), we suggest that marginal effects at individual markers are likely to be expected. Moreover, the indices in the sums of that condition need to be rearranged for every different SNP that is considered, greatly increasing chances of marginal effects associated with at least one of the SNPs.
Practically, RP algorithm is computationally very efficient. The resulting partitioning tree provides clear biological and statistical interpretation. According to the multilocus profile, each individual can always be uniquely classified into one of the terminal RP nodes with readily defined population risks and corresponding interval estimates.
Highlights
Many data-mining methods share the unfortunate feature of making a multitude of ‘looks’ through the data. The more you look, the more likely a statistically promising result is a false positive.
Shotgun approaches that blindly evaluate all conceivable models increase the number of false associations so that true associations end up buried in the bulk of statistical noise; type I error control through permutations or otherwise does not resolve this issue. In other words, the false discovery rates associated with most significant results can be prohibitively high.
It is important to design and evaluate algorithms that minimize the number of comparisons. Recursive partitioning has the advantage over shotgun search approaches since it minimizes the number of comparisons, which is justified by the likelihood of marginal effects (e.g., effects associated with SNPs or their small subsets).
As the molecular technology improves, we predict that it will soon be feasible to directly discover genetic polymorphisms in study-specific, casecontrol samples. This will be done without relying on common variation that mostly represents ’wild-type’ polymorphisms, characteristic of population controls.
Large parts of candidate genes or genomic regions will be sequenced using samples enriched with individuals sharing the phenotype of interest. The potential for uncovering disease- and drug-related variation should improve. At the same time, the number of polymorphisms and the number of their putative explanatory combinations will further increase. Advances in statistical methodology are important for finding models that decrease the odds of finding false associations.
Acknowledgments
Mike Mosteller, Liling Warren, Clive Bowman and two reviewers provided valuable comments.
References
- Akaike H. New look at statistical-model identification. IEEE Transactions on automatic control. 1974;19:716–723. [Google Scholar]
- Cordell HJ, Clayton DG. A unified stepwise regression procedure for evaluating the relative effects of polymorphisms within a gene using case/control or family data: application to HLA in type 1 diabetes. Am J Hum Genet. 2002;70:124–141. doi: 10.1086/338007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello TJ, Swartz MD, Sabripour M, Gu X, Sharma R, Etzel CJ. Use of tree-based models to identify subgroups and increase power to detect linkage to cardiovascular disease traits. BMC Genet 4 Suppl. 2003;1:S66. doi: 10.1186/1471-2156-4-S1-S66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culverhouse R, Klein T, Shannon W. Detecting epistatic interactions contributing to quantitative traits. Genet Epidemiol. 2004;27:141–152. doi: 10.1002/gepi.20006. [DOI] [PubMed] [Google Scholar]
- Curran MD 2003 Statistical Modeling for Genetics: Pharmacogenetics, Molecular Evolution and Complex Traits. Dissertation. University of North Carolina at Chapel Hill.
- Czika WA, Weir BS, Edwards SR, Thompson RW, Nielsen DM, Brocklebank JC, Zinkus C, Martin ER, Hobler KE. Applying data mining techniques to the mapping of complex disease genes. Genet Epidemiol 21 Suppl. 2001;1:S435–440. doi: 10.1002/gepi.2001.21.s1.s435. [DOI] [PubMed] [Google Scholar]
- Epstein MP, Satten GA. Inference on haplotype effects in case-control studies using unphased genotype data. Am J Hum Genet. 2003;73:1316–1329. doi: 10.1086/380204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB, Ahmadi KR, Weale ME, Wood NW. Genome scans and candidate gene approaches in the study of common diseases and variable drug responses. Trends in Genetics. 2003;19:615–622. doi: 10.1016/j.tig.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Jannot AS, Essioux L, Reese MG, Clerget-Darpoux F. Improved use of SNP information to detect the role of genes. Genet Epidemiol. 2003;25:158–167. doi: 10.1002/gepi.10256. [DOI] [PubMed] [Google Scholar]
- Lin DY. Haplotype-based association analysis in cohort studies of un-related individuals. Genet Epidemiol. 2004;26:255–264. doi: 10.1002/gepi.10317. [DOI] [PubMed] [Google Scholar]
- Lin S, Chakravarti A, Cutler DJ. Exhaustive allelic transmission disequilibrium tests as a new approach to genome-wide association studies. Nat Genet. 2004;36:1181–1188. doi: 10.1038/ng1457. [DOI] [PubMed] [Google Scholar]
- Morton NE. Significance levels in complex inheritance. Am J Hum Genet. 1998;62:690–697. doi: 10.1086/301741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Kardia SL, Ferrell RE, Sing CF. A combinatorial partitioning method to identify multilocus genotypic partitions that predict quantitative trait variation. Genome Res. 2001;11:458–470. doi: 10.1101/gr.172901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DM, Weir BS. Association studies under general disease models. Theor Popul Biol. 2001;60:253–263. doi: 10.1006/tpbi.2001.1539. [DOI] [PubMed] [Google Scholar]
- Pepe MS 2003 The statistical evaluation of medical tests for classification and prediction Oxford University Press.
- Province MA, Shannon WD, Rao DC. Classification methods for confronting heterogeneity. Adv Genet. 2001;42:273–86. doi: 10.1016/s0065-2660(01)42028-1. [DOI] [PubMed] [Google Scholar]
- Rao DC. CAT scans, PET scans, and genomic scans. Genet Epidemiol. 1998;15:1–18. doi: 10.1002/(SICI)1098-2272(1998)15:1<1::AID-GEPI1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF, Moore JH. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet. 2001;69:138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon WD, Province MA, Rao DC. Tree-based recursive partitioning methods for subdividing sibpairs into relatively more homogeneous subgroups. Genet Epidemiol. 2001;20:293–306. doi: 10.1002/gepi.1. [DOI] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society, Series B. 2002;64:479–498. [Google Scholar]
- Stram DO, Leigh Pearce C, Bretsky P, Freedman M, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, Thomas DC. Modeling and E-M estimation of haplotype-specific relative risks from genotype data for a case-control study of unrelated individuals. Hum Hered. 2003;55:179–190. doi: 10.1159/000073202. [DOI] [PubMed] [Google Scholar]
- Tahri-Daizadeh N, Tregouet DA, Nicaud V, Manuel N, Cambien F, Tiret L. Automated detection of informative combined effects in genetic association studies of complex traits. Genome Res. 2003;13:1952–1960. doi: 10.1101/gr.1254203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte JS, Elston RC, Cardon LR. On the relative sample size required for multiple comparisons. Stat Med. 2000;19:369–372. doi: 10.1002/(sici)1097-0258(20000215)19:3<369::aid-sim335>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Young SS, Ge N. Large recursive partitioning analysis of complex disease pharmacogenetic studies. I. Motivation and overview. Pharmacogenomics. 2005;6:65–75. doi: 10.1517/14622416.6.1.65. [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered. 2002;53:79–91. doi: 10.1159/000057986. [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Meng Z, Ghosh SK. Interval estimation of genetic susceptibility for retrospective case-control studies. BMC Genetics 2004. 2004;5:9. doi: 10.1186/1471-2156-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bonney G. Use of classification trees for association studies. Genet Epidemiol. 2000;19:323–332. doi: 10.1002/1098-2272(200012)19:4<323::AID-GEPI4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]