

Abstract
High-sensitivity glycoprotein analyses are of particular interest in modern biomedical and clinical research, as well as in the development of recombinant protein products. The evolution of new hyphenated methodologies in high-sensitivity glycoprotein analysis is highlighted in this thematic review. These methodologies include, in particular, capillary LC/MALDI/TOF/TOF MS in conjunction with online permethylation platform, and silica-based lectin microcolumns interfaced to MS. The potential of these methodologies in glycomic and glycoproteomic analysis is demonstrated for model glycoproteins as well as total glycomes and glycoproteomes derived from biological samples. Additionally, the applications of CE-MS, CEC, and nanoLC with graphitized carbon in the areas of glycomics and glycoproteomics are described.
Keywords: Glycomics, Glycoproteomics, LC/MALDI/TOF/TOF MS, LC/MS, Lectin, Oligosaccharides, Online permethylation, Review, Tandem mass spectrometry
1 Introduction
1.1 Glycoscience and contemporary glycobiology
Biological sciences have recently undergone enormous expansion, generating many new fields. Their main directions notwithstanding, all rapidly developing fields in life sciences seem to feature one common trend: from empirical and descriptive toward more exact, molecular, and dynamic. New measurement technologies now further lend exciting opportunities for understanding quantitatively the molecular aspects of processes in living organisms and combining these extensive molecular data into “systems biology” knowledge [1–6]. Among different “-omics” (genomics, transcriptomics, proteomics, metabolomics, etc.), all closely related to new developments in analytical methodologies and instrumentation, the fields of glycoproteomics and functional glycomics are assuming an increasingly important role with the ever-increasing emphasis to understand eukaryotic cellular systems.
From a molecular scientist’s point of view, glycoscience encompasses a vast range of directions dealing with glycoconjugates or “sugar-like” molecules: from molecular/architectural aspects of large, natural glycomolecules (glycogen, starch, pectins, etc.) to biocompatible materials derived from the world’s most abundant polysaccharides (cellulose and chitin), to the glycoconjugates which play crucial roles in cellular recognition and signaling (glycoproteins, glycolipids, and proteoglycans). While there are common aspects of the analytical approaches to address this vast array of glycoconjugate structural and quantitation problems, this thematic review deals specifically with glycoprotein analyses at high sensitivity. High-sensitivity glycoprotein analyses are of particular interest in modern biomedical and clinical research, as well as in the quality control of recombinant protein products.
Different separation aspects of glycoconjugate analysis have been discussed previously [7–10], while the main structural directions of this field have been reviewed in 2002 [11]. Consequently, the thematic review presented here provides a brief account of activities since that time.
The term “glycobiology” emerged during the late 1980s to describe the field which is making a connection between the rapidly developing knowledge on the glycoconjugate structures and the functional aspects and roles of glycoconjugates in cellular development and function. Glycosylation has since been recognized as functionally the most important and perhaps most abundant posttranslational modification of proteins. It is estimated that more than 50% of all mammalian proteins are glycosylated. This fact alone represents a formidable analytical challenge that exceeds far the tasks of mainstream proteomics. Simultaneously, it has been increasingly evident that many structural selectivity determinants of biological recognition are unique glycan structures, the structural hierarchies of different glycans at the sites of glycosylation and, to some degree, the local peptide “landscape” at the sites of modification. However, the well-known propensity of glycan structures to form numerous isomers adds substantially to the difficulties of analytical glycobiology tasks.
Improved analytical methodologies in glycoproteomics and functional glycomics are clearly a key to the future of glycobiology research and its needed proliferation to clinical applications. The importance of glycosylation in health and sickness has been increasingly evident, for nearly all major human diseases have their glycoprotein attributes represented in the constantly growing number of “biomarkers”. This includes the most serious illnesses, such as various types of cancer, cardiovascular disease, and inflammation and immunological disorders [12, 13]. Additionally, a detailed knowledge of glycosylated structures could aid research efforts in developmental biology, plant sciences, agricultural research, etc.
2 Chromatographic approaches to glycoconjugate analysis
The difficulties of structural analysis in glycobiology mainly originate from (a) structural complexity of glycoconjugates featuring various forms of branching and other types of isomerism; (b) microheterogeneities at the sites of glycosylation; (c) extensive occurrence of glycosylation in eukaryotic proteins; (d) distribution of glycosylated proteins in different parts of living cells; and (e) considerably wide dynamic ranges in which different glycoproteins are encountered in biological mixtures (e.g., human blood). To address these issues at different degrees of complexity, analytical strategies have gradually evolved together with the development of specific reagents for glycoanalysis, such as fluorescence-tagging chemicals, exo- and endoglycosidase enzymes, and lectins. Since the introduction of ESI and MALDI into biomolecular MS, the MS-based structural determinations have become the norm in modern glycoanalysis. Some of the instrumental advances and approaches are clearly shared with the rapidly developing field of proteomics.
While the MS-based technologies combined with bioinformatic tools are most crucial to the final structural identifications of particular glycoconjugates, analytical separations are extremely important in securing optimum amounts of materials to be analyzed through MS. These important approaches range from the currently common uses of lectin chromatography isolation which allows the enrichment of trace amounts of glycoproteins from biological samples [14] as illustrated in Fig. 1; or 2-DE in conjunction with glycoproteins specific stains which allows global monitoring of glycosylation changes [15] as depicted in Fig. 2; to the sophisticated uses of multidimensional microcolumn chromatography in the online systems coupled to different mass analyzers. Modern glycoanalysis has clearly become a multimethodological task [11].
Figure 1.
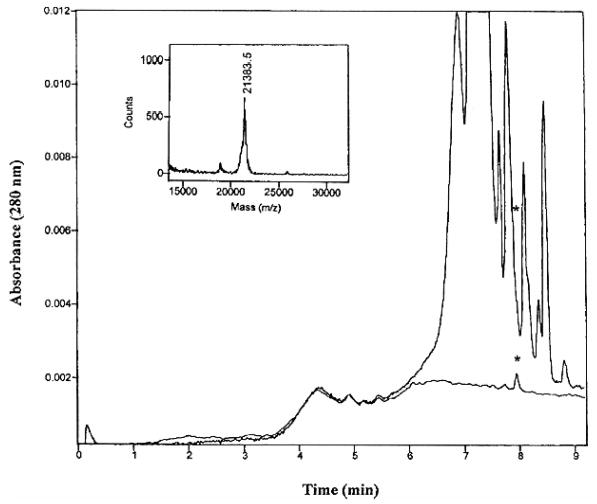
Anion-exchange chromatogram of the isolated MUP components (upper trace) and the Con A bound fraction (lower trace); inset is the mass spectrum of the isolated glycoprotein indicated with asterisk. Reproduced from [14], with permission.
Figure 2.

Detection of glycosylated and nonglycosylated proteins using Pro-Q Emerald 300 dye and SYPRO Ruby protein stain. (a) A 2-D glycoprotein map of human frontal cortex brain homogenates reveals approximately 50 glycosylated proteins. (b) Same gel, poststained with SYPRO Ruby protein stain, showing nearly 150 of the expressed proteins. Location of the Candy Cane molecular weight marker (kDa) is indicated along the y-axis of the gels. Reproduced from [15], with permission.
The strategies in glycoprotein analysis will differ, depending on the amount of available sample. Microgram to milligram quantities of isolated complex glycoproteins, or a recombinant product, are now easily amenable to complete chemical characterization, although controlled cleavages by proteases or glycanases could still result in fairly complex mixtures of peptides, glycopeptides, and glycans. Somewhat more challenging are the cases where glycoproteins have been chromatographically separated from a mixture of proteins through HPLC or isolated as gel spots in electrophoresis or blots. They still could be analytically demanding if the glycoproteins of interest are large biomolecules with low carbohydrate content (e.g., antibodies) or proteins glycosylated heavily at different sites. Yet more methodologically involved are the tasks where numerous glycosylated proteins are encountered in a complex biological medium, such as human physiological fluids or tissue extracts, at a very wide dynamic concentration range. These cases often present intricate analytical challenges. They represent an important direction in biomedical field, i.e., a typical search for biomarkers of different physiological conditions or disease. As discussed in some detail below, these types of studies will undoubtedly require selective preconcentration of analytes together with the most sensitive and informative tools of analysis.
The separations of intact glycoproteins from complex biological mixtures, apparently still a nontrivial task, may take advantage of new separation media and procedures that are becoming available for biomolecules. Microcolumn separations of intact proteins in different separation modes (size exclusion, ion exchange, hydrophobic interaction, RP, etc.) can potentially be applied toward the benefits of top-down proteomics. Additionally, the orthogonal nature of chromatographic and electrophoretic principles can be exploited in this regard.
It is most often necessary to fragment glycoproteins into the structurally amenable segments: peptides and glycopeptides and oligosaccharide entities, which then must typically be separated prior to MS/MS investigations. The samples of interest may be processed after the isolation of glycoprotein, or prior to that. In some high-sensitivity and online analyses of complex biological materials, the use of immobilized protease and glycosidase reactors [16, 17] is likely to grow in importance. While the separation procedures for protease-generated glycopeptides (a task of glycoproteomics in deriving the maximum information about the sites of glycosylation) closely parallel the methodologies of mainstream proteomics, the separation of glycans (in functional-glycomic studies) requires a different emphasis toward the final MS determinations of oligosaccharide structures.
The extraordinary complexity of biomolecular mixtures demands separation tools with a high resolving power. First, and perhaps most importantly, multidimensional separations have been emphasized in proteomic analyses [18–23] for several years. The overall separation efficiencies of chromatographic columns can be significantly enhanced through the combination of very small particles with increased column length, leading to the use of ultrahigh pressures in microcolumn LC [24–27]. Finally, the properties of monolithic capillary columns, operating in either the electromigration mode (CEC) [28–34] or a pressure-driven mode [35, 36] for the separations of both peptides and oligosaccharides, provide an additional route to solving the problems of peak overlap in proteomic and glycomic studies.
2.1 Capillary LC/MALDI/TOF/TOF MS: A platform for functional-glycomic investigations
A suitable general platform for functional glycomics must, first of all, consider the best possible structural technique that is capable of distinguishing different forms of isomerism at high sensitivity. During the recent past, distinctly different approaches and modern MS technologies were applied to satisfy this requirement. The use of sequential fragmentation in MS/MS (for example, MSn in an IT or quadrupole IT) was found to be effective in distinguishing isobaric structures among methylated branched oligosaccharides [37–43], albeit at the expense of a need for considerable sample quantity and limited compatibility with a prior LC separation. The effectiveness of a particular ionization technique, i.e., the use of ESI versus MALDI, must also be considered in view of the relatively inefficient ionization of oligosaccharides when compared to peptides. While MALDI forms exclusively the sodium adduct ions of oligosaccharides in the positive-ion mode, multiply charged states are formed in ESI that cause a loss in sensitivity as a result of splitting the signal of a single analyte.
For the branched structures and differently linked residues in the oligosaccharides from typical glycoproteins, it is not sufficient to generate secondary fragments due to the cleavages at the sites of glycosidic bonds; sufficient energies must be available to the analyte sugar molecules to form the more informative cross-ring fragments. As the low-energy CID in the IT analyzers [32, 34, 44] or the PSD in MALDI/TOF MS instruments [45] is generally insufficient to accomplish this fragmentation task, we have chosen a MALDI/TOF/TOF MS with high-energy CID capabilities to furnish extensive fragmentation of model neutral [46–50] and acidic [46, 51] glycans. An alternative approach pursued elsewhere for extensive fragmentation of glycans involves Fourier transform ion cyclotron resonance MS used in conjunction with sustained off-resonance irradiation (SORI) [52], CID [52, 53], IR multiphoton dissociation (IRMPD) [54–56], or electron capture dissociation (ECD) [54, 55]. In our MALDI/TOF/TOF MS experiments, we were able to distinguish a subtle form of isomerism in the mannose-rich glycans [47] (Fig. 3) from the ratios of diagnostic cross-ring fragments as well as different linkages of sialic acid residues and/or their attachment to a specific antenna within a branched structure [51]. Much of this structurally/functionally important information had previously been available only through NMR spectrometry which generally requires highly concentrated samples. Using new bioinformatic approaches [57] to MALDI/TOF/TOF MS fragmentation data, there is a distinct possibility to distinguish the finest structural details of pure glycans.
Figure 3.
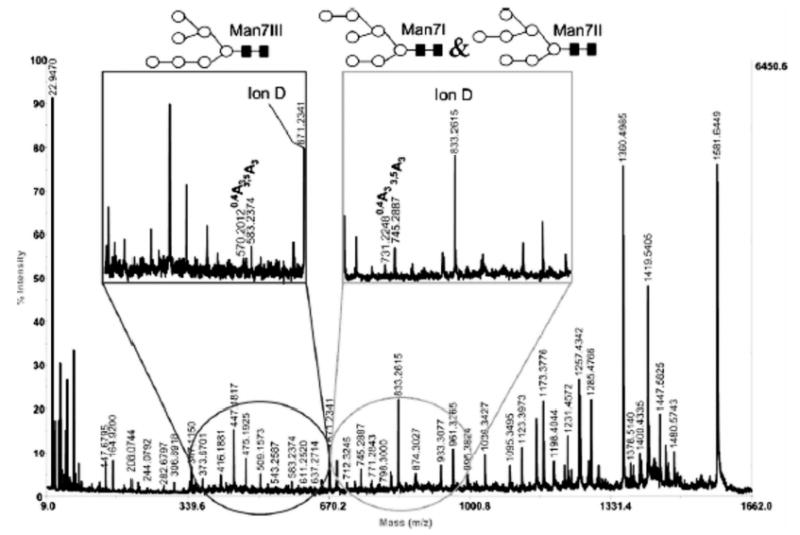
MS/MS recording of GlcNAc2Man7. Inset represents a zoomed-in section of the spectrum. Symbols: ▪, N-acetylglucosamine; ○, mannose. Reproduced from [47], with permission.
In choosing the MALDI/TOF/TOF MS approach for structural elucidation of glycans, it was natural to evaluate the merits of permethylation as an alternative for analyzing native glycans. While sample derivatization always introduces an additional procedural step, the advantages of permethylation for MS [58, 59] are substantial: stabilization of sialylated structures; enhanced sensitivity; and improvements in the MS/MS interpretation capabilities. Serendipitously, the oligosaccharides are made sufficiently hydrophobic through quantitative methylation to allow their separation from complex mixtures by means of capillary RP LC using “MS-friendly” mobile phases [60]. Permethylation also facilitates a simultaneous analysis of neutral and sialylated oligosaccharide solutes as well as offering predictable fragmentation.
Oligosaccharide permethylation has been employed prior to MS and MS/MS analyses of oligosaccharides and other glycoconjugates using two procedures that have been deemed suitable. The first, originally described by Hakomori [61], utilizes the anion DMSO− (commonly referred to as the dimsyl anion) to remove protons from the sample prior to their replacement with a methyl group. However, this methylation procedure tends to be nonquantitative, which is perhaps acceptable for MS studies of an isolated glycan, but not for LC-MS where just one solute may produce multiple chromatographic peaks. The second procedure, which is now more commonly accepted, was introduced in 1984 [58] and modified more recently [59]. It is based on the addition of methyl iodide to carbohydrates, which are dissolved in DMSO solution containing powdered sodium hydroxide and traces of water. The popularity of the modified procedure originates from its speed, experimental simplicity, and effectiveness for both O- and N-glycans and “cleaner” products. Unfortunately, when exploring this procedure at microscale, we found difficulties in recovering the small amounts of methylated glycans, which are presumably due to oxidative degradation and peeling reactions induced by the high pH and heat generated during the liquid-liquid extraction step. These difficulties led us to develop an online permethylation procedure using a sodium hydroxide-packed capillary as a microreactor [62] that can easily be incorporated into a valve-operated analytical system. This approach has been successfully utilized for the permethylation of glycans from model proteins as well as total glycomes derived from biological samples (Fig. 4) [62].
Figure 4.
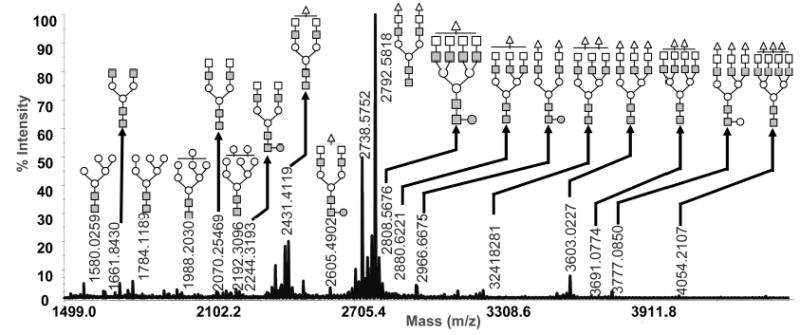
MALDI/TOF/TOF MS glycomic profile of permethylated N- and O-glycans derived from human blood serum. Symbols: ■, N-acetylglucosamine; ○, mannose; □, galactose; ●, fucose; ▵, N-acetylneuraminic acid.
Any successful glycomic/analytical platform will be dependent on an effective release of glycans from the polypeptide backbone of a glycoprotein or glycopeptides. When considering a permethylation as the next treatment step, a glycan cleavage procedure must be compatible with permethylation conditions. In our current approach, we prefer the recently developed microscale β-elimination [63] or glycanase-released glycans which are derivatized through a reductively stabilized Schiff base prior to permethylation. In fact, this step may be preferable for short-chain oligosaccharides, which are often obscured by the interferences associated with noise originating from the MALDI matrix clusters.
Serendipitously, the hydrophobicity of permethylated glycans renders them amenable to RP-LC separation which is popular, convenient, and easy-to-interface to ESI or MALDI/MS. Recently, in conjunction with the online permethylation, a system was developed, incorporating derivatization, C18 trapping of permethylated glycans, and C18 nanoLC separation of the glycans derived from model glycoproteins and biological samples. The online permethylation and LC/MALDI MS analysis of glycans derived from a mixture of glycoproteins are depicted in Fig. 5. RP separation allowed the resolving of structural isomers, the presence of which was confirmed by MALDI/TOF/TOF MS.
Figure 5.
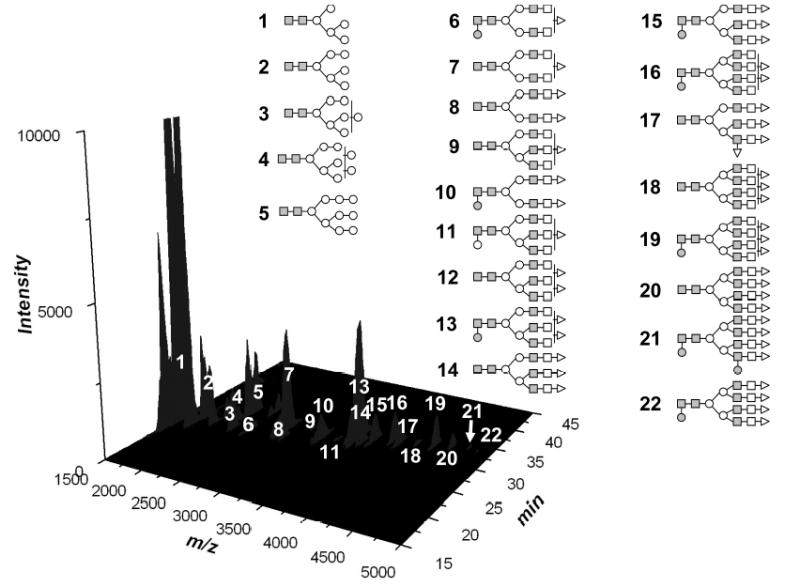
LC/MALDI/TOF/TOF MS of online permethylated glycans derived from a mixture of glycoproteins. Symbols: ■, N-acetylglucosamine; ○, mannose; □, galactose; ●, fucose; ▵, N-acetylneuraminic acid.
2.2 Interfacing other chromatographic techniques to MS
2.2.1 NanoLC with hydrophilic columns
Hydrophilic-interaction chromatography (HILIC), which was first coined by Alpert [64], has long been popular in carbohydrate analysis. Although LC of carbohydrates on unmodified silica was reported [65], it is more effective to use silica modified through chemical bonding of suitable polar functional groups such as amine [66–69] or amide [70–73].
Commonly, the utility of the HILIC approach for the analysis of oligosaccharides involves fluorescence labeling of glycans prior to LC separation [11]. However, this approach has been mainly based on the use of analytical columns employing very high flow rates, which adversely affect sensitivity. Recently, this problem has been rectified through the use of capillary amide columns at a flow rate of 300 nL/min, interfaced to IT-MS [74]. The use of nanoLC format permitted the detection of oligosaccharide mixtures at low-femtomole sensitivity. The approach was validated using N-glycans derived from keyhole limpet hemocyanine and horseradish peroxidase [74].
2.2.2 NanoLC with graphitized carbon
Although hydrophilic-phase chromatography using amine- and amide-bonded stationary phases have traditionally been considered most effective for oligosaccharide separations [11], graphitized carbon columns (GCC) are recently gaining popularity because of their ease of use, high capacity, high efficiency, and, more recently, their commercial availability as microcolumns (SGE, Ringwood, Australia). Carbohydrate retention in GCC is mainly based on adsorption, while some hydrophobic interactions are also expected. The unique selectivity of GCC and its unmatched ability to resolve isomeric and closely related compounds are brought about by its homogenous adsorptive nature.
The hydrophobicity of GCC is greater than what is observed with RP materials. Thus, a greater percentage of organic solvent in the mobile phase is required to assist the separation of compounds as hydrophilic as carbohydrates. Another major advantage of GCC is their ruggedness, which apparently allows repeated use without a loss of performance or reproducibility. Moreover, these columns are unaffected by strongly acidic or alkaline conditions and can be used throughout the entire pH range with a wide range of solvents. Thus far, these columns have been utilized in conjunction with MS for the analysis of glycans derived from different samples, including egg jelly surrounding Xenopus laevis eggs [53, 75, 76], human bronchial epithelial cell cultures [77], membrane proteins from premature aging Huchinson–Gilford progeria syndrome fibroblasts [78], human tear fluid [79], and human plasma [80]. Moreover, microliter flow rates were employed in all these applications, allowing only the detection of structures present in abundance. This situation was substantially improved recently as a result of the introduction of nanocolumns, which are utilized at much lower flow rates.
The use of graphitized carbon nanoLC/MS has allowed N-and O-glycans analysis at a substantially higher sensitivity, thus allowing the analysis to be performed on low-abundance glycoproteins. Three orders of magnitude increase in sensitivity was observed as a result of using nanoflow LC/MS instead of microflow LC/MS (Fig. 6) [81]. As a result, negative-ion MS in conjunction with graphitized carbon nanoLC/MS allowed the simultaneous detection of femtomole amounts of both acidic and neutral oligosaccharides. Although baseline resolution of several structural isomers is common in GCC, many of these structures were not detected using the larger column format at microflow rates. Moreover, N- and O-glycans derived from glycoprotein mixtures were isolated from mucosal surfaces and ovarian cancer cells [81].
Figure 6.
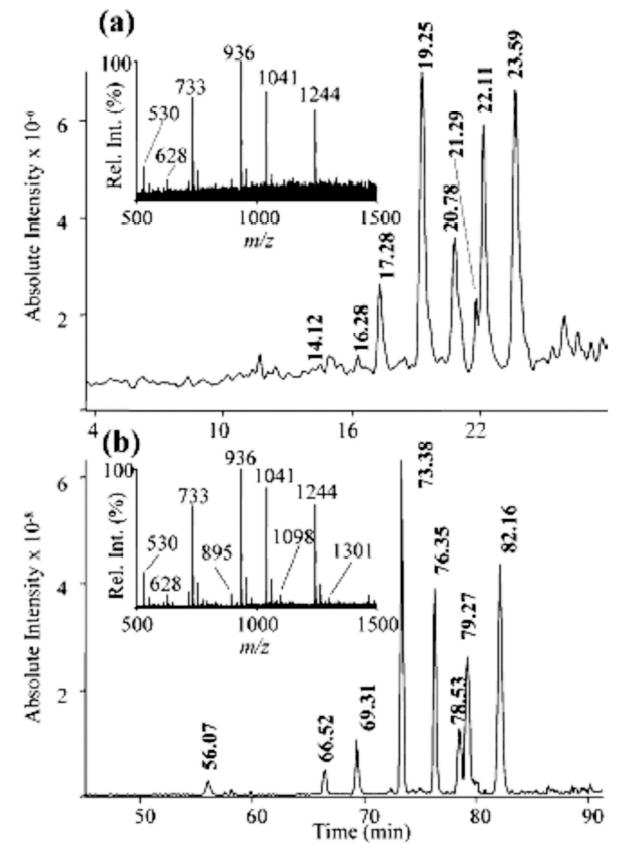
Comparison of negative ion (a) micro LC/MS versus (b) nanoLC/MS analysis of neutral O-linked oligosaccharides (5.5 ng) using graphitized carbon chromatography (base peak chromatograms). Combined MS1 mass spectra of the region where oligosaccharides were eluted are shown as insets. Reproduced from [81], with permission.
2.2.3 Anion-exchange chromatography-MS
Anion-exchange chromatography of glycans with pulsed-amperometric detection has become one of the standard methodologies in glycobiology because of its simplicity and quantitative reliability in the picomole range. While mass spectrometers are capable of detecting glycans at a level that is many orders of magnitude lower than amperometric detectors, interfacing this chromatographic technique to MS is not convenient due to the incompatibility of the mobile phases with MS. Several attempts [82–86] were made to address this problem, but neither approach was completely successful.
3 CE
For different applications in glycan analysis, there are other distinct separation options. While glycan profiles released from different glycoproteins (e.g., recombinant glycoprotein products) or even unfractionated glycoprotein mixtures can be displayed through MALDI/MS, fluorescently tagged mixtures separated by CZE and detected by LIF are often viewed as somewhat complementary to MALDI/MS, because the CZE approach is often capable of resolving certain isomers which are otherwise indistinguishable by MS [87]. As an example, the CE-LIF trace (Fig. 7a) of the fluorescently labeled standards (core-fucosylated biantennary, asialylated, and mono- and di-sialylated glycans) clearly illustrates the ability of this technique to separate structural isomers. Monosialylated structures which differ only in the attachment of a terminal sialic acid residue to 1–6 or 1–3 antenna were partially resolved in this CE-LIF analysis. The CE-LIF analysis of N-glycans derived from an mAb revealed the presence of seven major components in addition to many minor components (Fig. 7b) [87]. In this figure, the resolving power of CZE is also evident from the ability to baseline-resolve monofucosylated biantennary glycan structures lacking one terminal galactose residue.
Figure 7.
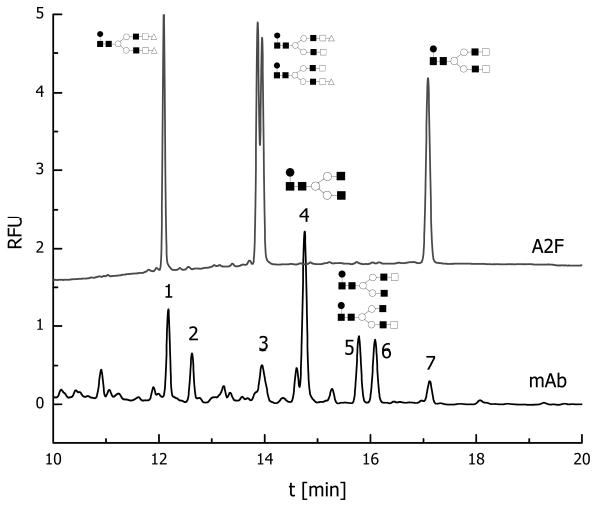
CE profile of APTS-labeled glycans derived from mAb. Upper trace represents standard core-fucosylated biantennary/disialylated, monosialylated, and asialylated glycans. Conditions: column, polyacrylamide-coated 50/365 mm ID/OD; length, 50.5 cm total, 40.5 cm effective length; temperature, 257°C; injection pressure, 0.5 psi for 5.0 s; voltage, 15 kV anodic EOF; λex 488 nm, λem 520 nm. Symbols: ▪, N-acetylglucosamine; ○, mannose; □, galactose; ▵, N-acetylneuraminic acid. Reproduced from [87], with permission.
The popularity of CZE-MS interfacing has substantially increased over the past several years due to improvements in sensitivity and reproducibility. This approach takes full advantage of the extreme selectivity and sensitivity of a mass spectrometer. In coupling CZE to MS, high mass accuracy and MS/MS operations were achieved. The application of CE-MS and MS/MS to glycoscreening in biomedical field has recently been highlighted [88]. More recently, CZE interfaced to a Q-Trap mass spectrometer was featured in the study of N-glycans from cellobiohydrolase I, with special interest in phosphorylated residues [89]. The mass spectrum was a modified triple quadrupole where the Q3 region can be operated as a conventional quadrupole mass filter or as a linear IT with axial ion ejection [90]. A simultaneous analysis of uncharged and charged glycans was achieved through fluorescence labeling with 8-aminopyrene-1,3,6-trisulfonate (APTS) which imparts negative charges to the labeled structures. Generally, APTS labeling of the oligosaccharides permits high-resolution CE, better ionization in the negative ion mode, a simultaneous detection of uncharged and charged oligosaccharides, and generation of predictable MS/MS spectra. A differentiation of phosphorylated glycan isomers was achieved through this system, while MS/MS data furnished additional structural information [89].
4 CEC-MS
Other approaches to separating native glycans using MS-friendly mobile phases lead to the use of columns featuring hydrophilic interactions. Among these efforts, CEC monolithic columns were recently shown [30, 32–34] where a high resolving power associated with this approach is capable of dealing with highly complex pools of glycans (Fig. 8). This is clearly illustrated for the case of bile salt-stimulated lipase from human breast milk, which is a relatively large glycoprotein consisting of 722 amino acid residues [91] with numerous O-glycosylation sites near the C-terminus. The CEC/IT-MS profile of chemically cleaved N- and O-glycans using hydrophilic monolithic columns is depicted in Fig. 8. The high selectivity of these columns assisted a partial or complete resolution of several structural isomers, as suggested by the detection of several m/z values at different retention times. These CEC columns were shown to be very effective in conjunction with ESI-MS, using both the IT [32, 34] and FT MS [33] mass analyzers, and MALDI-MS using a microdeposition device [92].
Figure 8.
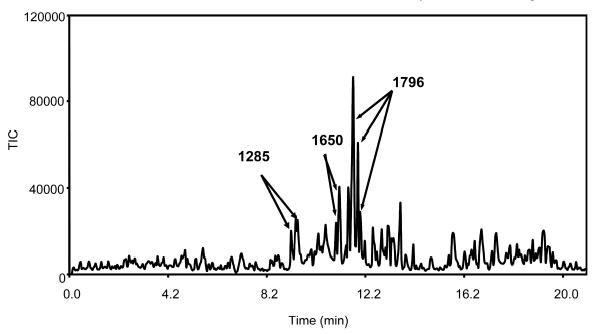
Mass electrochromatogram of a complex fraction of the O-linked glycans chemically released from human bile salt-stimulated lipase. Conditions: amino column 28 cm, field strength 500 V/cm, mobile phase ACN/water/ammonium formate buffer (240 mM, pH 3.0, 55:44:1 v/v/v), injection 1 kV, 10 s. Reproduced from [32], with permission.
5 Analytical uses of lectin microcolumns and a new platform for glycoproteomics
From numerous plant lectins described in the scientific literature [93–95], only a few have been widely used in glycoprotein isolation work. Lectins comprise a group of proteins with unique affinities toward carbohydrate structures. They have long been used in biomolecular isolation, carbohydrate chemistry, and histochemistry, and (more recently) as the mimicking agents in intercellular interaction studies. For a number of years, lectins have been utilized in various glycoprotein isolation efforts [93, 94, 96–98], albeit few addressed the quantitative aspects of this approach. Lectin affinity chromatography has now become a more widely applicable technique for purification of trace glycoproteins [95]. Lectins with high specificity toward oligosaccharides have been immobilized to agarose and other separation matrices. Some of these materials have been commercially available to isolate glycoproteins on the basis of their different glycan structures [93, 94, 96–98]. Due to the method’s high specificity, it is often advantageous to use lectin affinity chromatography in the early stages of glycoprotein isolation. While glycoproteins can be separated as based on their different glycan moieties [93, 94, 96] through lectin specificity, most cases of lectin affinity chromatography use either wheat germ agglutinin or Canavalia ensiformis (Con A), which have broad specificity [14, 98], to isolate the entire pools of glycoproteins rather than specific structural types. The uses of lectins in contemporary glycoproteomics and glycomics are likely to grow due to the frequent needs for fractionation and preconcentration for high-sensitivity MS determination [11].
One of the aims of our laboratory has been to develop lectin-based chemistries for high-sensitivity glycomic/glycoproteomic structural analysis. This necessitates departure from the most commonly used technique involving a small column containing 1–5 mL of agarose-based material [93, 94, 96–98] whereby the sample is loaded on such sorbents by using gravity-flow mode or low-pressure pumps. A notable exception has been a nonanalytical frontal-chromatography study of interactions [99]. In preparative uses, samples are commonly suspended in a high-ionic strength loading buffer, which is also used to wash the column prior to elution. Such composition of a loading buffer is necessary to eliminate nonspecific binding. Finally, glycoproteins or glycopeptides are eluted by displacement from the column with an elution buffer which has the composition of the loading buffer plus a haptene saccharide. However, this procedure is not compatible with the online pressurized systems that are intended to minimize sample handling, contamination, and sample losses. To promote the analytical uses of immobilized lectins as a part of pressurized chromatographic systems, we have synthesized silica-based materials and evaluated their performance in glycoproteomic analysis [100]. More recently, the silica-based lectin microcolumns have been employed in proteomic studies on complex biological materials such as blood serum [100] and soluble extracts of rat brain [101].
The lectin microcolumns utilized in our laboratory typically comprise small capillaries (5 cm × 500 μm, ID) packed with macroporous silica, featuring binding capacities higher than those offered by the commercially available (agarose-based) lectin media. A wide range of lectin materials have been successfully prepared including Con A, specific for mannose and glucose residues, Sambucus nigra (SNA), specific for α-(2–6)-sialylated structures, Phaseolus vulgaris (PHA-L), partially specific for carbohydrate containing Gal-β-(1–4)GlcNAc-β-(1,2)man sequence, and Ulex europaeus (UEA-I), specific for α-l-fucosylated moieties. Because of their high-pressure compatibility, these columns can readily be used in a valve-based analytical system [100].
Using additional system components (analytical columns and mass analyzers) together with the lectin microcolumns, we can employ them in a different analytical capacity: (a) separating exhaustively glycosylated and nonglycosylated molecules (glycoproteins or glycopeptides from other nonglycosylated molecules) on a serial arrangement of lectin microcolumns; (b) retaining fairly selective mixture components featuring unusual glycoconjugates in their structures through a particular lectin affinity; (c) using lectin microcolumns as a means of fractionating complex mammalian proteomes; and (d) through a removal of nonglycosylated peptides from a protease-digested mixture, increasing sensitivity of a site-of-glycosylation determination. An example for the latter use is depicted in Fig. 9, in which glycopeptides of bovine fetuin were isolated from the rest of tryptic peptides using online silica-based SNA lectin trapping column. Accordingly, the fetuin-derived glycopeptides associated with N 156, N 176, and N 99 amino acid moieties were not observed in the “unbound fractions” (Fig. 9a, 9c, and 9e, respectively), in comparison with “bound fractions” (Fig. 9b, 9d, and 9f, respectively). In addition, the isolation of glycopeptides from coeluting peptides enhanced glycopeptide ionization as a result of the elimination of competitive ionization. This isolation allowed a 5–20-fold increase in sensitivity.
Figure 9.
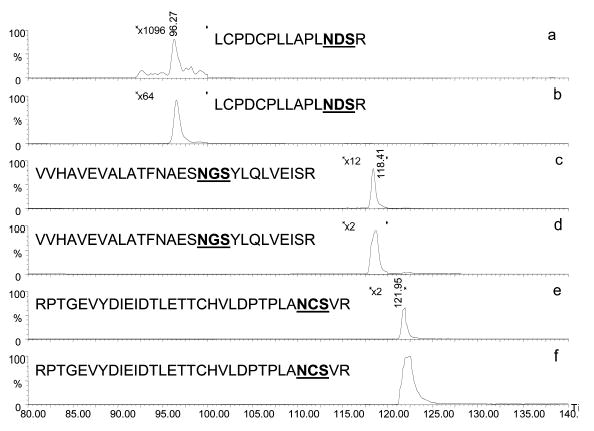
Extracted ion chromatograms of fetuin glycopeptides analyzed by online SNA-lectin trapping and LC/MS. Lectin unbound fractions (a, c, and e), and lectin bound fractions (b, d, and f). Reproduced from [100], with permission.
In a more recent study, the use of serial silica-based columns substantially enhanced a proteomic coverage of blood serum. A 5-μL aliquot of human blood serum was applied to three silica-based lectin columns (Con A, SNA, and PHA-L) which were serially connected. Glycoproteins were isolated from the mixture as a result of lectin enrichment, thus fractionating the sample into glycoprotein and protein fractions. These fractions were tryptically digested and analyzed by LC/MS/MS. The results were also compared to those attained without the lectin step (Fig. 10). Glycoprotein enrichment achieved by the serial lectin chromatography tripled the number of proteins observed.
Figure 10.
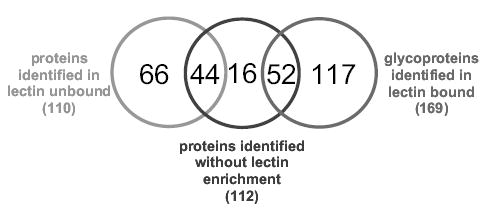
Comparison of the number of unique and common proteins identified with/without serial lectin enrichment approach.
Currently, the silica-based lectin columns, used both individually and serially, are utilized, as summarized in the flow chart depicted in Fig. 11. This analytical platform is intended to provide an exhaustive proteomic and glycoproteomic analysis of any complex sample. The analysis consists of two parts, which aim collectively at offering a highly sensitive analysis for as little as 5 μL of biological samples (e.g., blood serum). The approach is based on passing blood serum over a series of lectin columns with a wide range of specificity, thus ensuring effective trapping of nearly all glycoproteins. Proteins which are not trapped to lectin columns are then separated through a Zorbax C8 Poroshell column (Agilent, Wilmington, DE), which is superficially porous. This column allows fast and reliable HPLC separation of proteins. The separated proteins are fractionated prior to LC/MS/MS analysis. This furnishes proteomic information. Next, the trapped glycoproteins are eluted from the lectin columns, trapped on a C4 trapping cartridge, separated on Zorbax C8 Poroshell column, and fractionated again into a 96-well plate. Fractionated glycoproteins are again proteolytically digested and either analyzed by LC/MS/MS or loaded on another set of lectin traps prior to a different LC/MS/MS analysis to determine the site of glycosylation and its heterogeneity. This approach can potentially supply a comprehensive proteomic/glycoproteomic analysis, providing a wealth of structural information from a very small quantity of biological samples.
Figure 11.
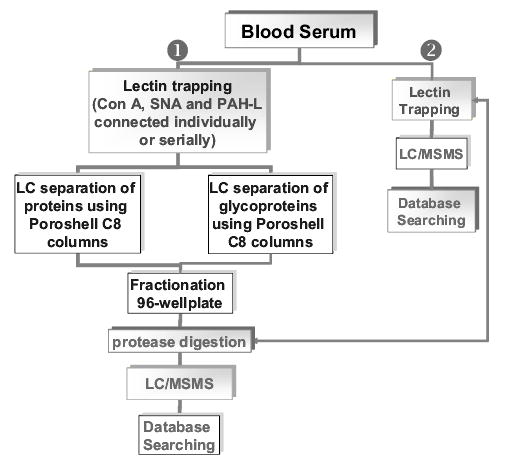
Analytical approach utilizing silica-based lectin columns for the fractionation and analysis of blood serum samples.
6 Conclusions
Modern glycoanalysis has clearly become a multimethodological task because of the inherent complexity of glycan structures originating from the microheterogeneity at the sites of glycosylation and the propensity of glycan structures to form numerous branches and isomers. These issues have been addressed very recently through the development of several analytical methodologies, which are mainly based on hyphenation of different techniques. The practical aspects of these methodologies are demonstrated and discussed in this thematic review. The application of RP-LC interfaced to MALDI/TOF/TOF MS in conjunction with online permethylation is demonstrated for the analysis of glycans derived from glycoproteins as well as from biological samples (e.g., blood serum). The use of different lectin materials used individually or serially to provide comprehensive glycoproteomic information is also highlighted. The review also discusses the application of nanoLC with graphitized carbon, CE-MS, and CEC-MS to glycans derived from various glycoproteins and different samples.
Acknowledgments
This work was supported by Grant No. (GM24349) from the National Institute of General Medical Sciences, U.S. Department of Health and Human Services and also supported by a grant from the Indiana 21st Century Research and Technology Fund.
References
- 1.Auffary C, Imbeaud S, Roux-Rouquie M, Hood L. Compt Rend Biol. 2003;326:879–892. doi: 10.1016/j.crvi.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 2.Ideker T. Nat Biotechnol. 2003;22:473–475. doi: 10.1038/nbt0404-473. [DOI] [PubMed] [Google Scholar]
- 3.Kell DB, Oliver SG. Bioessays. 2004;26:99–105. doi: 10.1002/bies.10385. [DOI] [PubMed] [Google Scholar]
- 4.Kitano H. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 5.Stevens CF. Curr Biol. 2004;14:R51–R52. doi: 10.1016/j.cub.2003.12.040. [DOI] [PubMed] [Google Scholar]
- 6.van der Greef J, Stroobant P, van der Heijden R. Curr Opin Chem Biol. 2004;8:559–565. doi: 10.1016/j.cbpa.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 7.El Rassi, Z. (Ed.), Carbohydrate Analysis by Modern Chromatography and Electrophoresis, Elsevier Science, Amsterdam, Netherland 2002.
- 8.Novotny MV. Methods Enzymol. 1996;270:101–133. doi: 10.1016/s0076-6879(96)70007-1. [DOI] [PubMed] [Google Scholar]
- 9.Novotny MV. J Chromatogr B. 1997;689:55–70. doi: 10.1016/s0378-4347(96)00398-2. [DOI] [PubMed] [Google Scholar]
- 10.Stefansson, M., Novotny, M. V., in: Townsend, R. (Ed.), Techniques in Glycobiology, Marcel Dekker, New York 1998.
- 11.Mechref Y, Novotny MV. Chem Rev. 2002;102:321–370. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]
- 12.Dennis JW, Granovsky M, Warren CE. Bioassays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 13.Lowe JB, Marth JD. Ann Rev Biochem. 2003;72:643–691. doi: 10.1146/annurev.biochem.72.121801.161809. [DOI] [PubMed] [Google Scholar]
- 14.Mechref Y, Zidek L, Ma W, Novotny MV. Glycobiology. 2000;10:1–5. doi: 10.1093/glycob/10.3.231. [DOI] [PubMed] [Google Scholar]
- 15.Kanninen K, Goldsteins G, Auriola S, Alafuzoff I, Koistinaho J. Neurosci Lett. 2004;367:235–240. doi: 10.1016/j.neulet.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Palm AK, Novotny MV. Rapid Commun Mass Spectrom. 2004;18:1374–1382. doi: 10.1002/rcm.1500. [DOI] [PubMed] [Google Scholar]
- 17.Palm AK, Novotny MV. Rapid Commun Mass Spectrom. 2005;19:1730–1738. doi: 10.1002/rcm.1979. [DOI] [PubMed] [Google Scholar]
- 18.Anderegg RJ, Wagner DS, Blackburn RK, Opiteck GJ, Jorgenson JW. J Protein Chem. 1997;16:523–526. doi: 10.1023/a:1026373830301. [DOI] [PubMed] [Google Scholar]
- 19.Evans CR, Jorgenson JW. Anal Bioanal Chem. 2004;378:1952–1961. doi: 10.1007/s00216-004-2516-2. [DOI] [PubMed] [Google Scholar]
- 20.Gygi SP, Rist B, Griffin TJ, Eng J, Aebersold R. J Proteome Res. 2002;1:47–54. doi: 10.1021/pr015509n. [DOI] [PubMed] [Google Scholar]
- 21.Janini GM, Conrads TP, Veenstra TD, Issaq HJ. J Chromatogr B. 2003;787:43–51. doi: 10.1016/s1570-0232(02)00616-5. [DOI] [PubMed] [Google Scholar]
- 22.Washburn MP, Wolters D, Yates JRI. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 23.Wolters DA, Washburn MP, Yates JRI. Anal Chem. 2001;73:5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 24.MacNair JE, Lewis KC, Jorgenson JW. Anal Chem. 1997;69:983–989. doi: 10.1021/ac961094r. [DOI] [PubMed] [Google Scholar]
- 25.MacNair JE, Patel KD, Jorgenson JW. Anal Chem. 1999;71:700–708. doi: 10.1021/ac9807013. [DOI] [PubMed] [Google Scholar]
- 26.Wu N, Collins DC, Lippert JA, Xiang Y, Lee ML. J Microcol Sep. 2000;12:462–469. [Google Scholar]
- 27.Xiang Y, Yan B, McNeff CV, Carr PW, Lee ML. J Chromatogr A. 2003;1002:71–78. doi: 10.1016/s0021-9673(03)00733-7. [DOI] [PubMed] [Google Scholar]
- 28.Allen D, El Rassi Z. J Chromatogr A. 2004;1029:239–247. doi: 10.1016/j.chroma.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 29.Huber CG, Choudhary G, Horvath C. Anal Chem. 1997;69:4429–4436. doi: 10.1021/ac970393t. [DOI] [PubMed] [Google Scholar]
- 30.Palm A, Novotny MV. Anal Chem. 1997;69:4499–4507. [Google Scholar]
- 31.Peters EC, Petro M, Svec F, Frechet JMS. Anal Chem. 1997;69:3646–3649. doi: 10.1021/ac970377w. [DOI] [PubMed] [Google Scholar]
- 32.Que A, Novotny MV. Anal Bioanal Chem. 2003;375:599–608. doi: 10.1007/s00216-003-1766-8. [DOI] [PubMed] [Google Scholar]
- 33.Que AH, Mechref Y, Huang Y, Taraszka JA, Clemmer DE, Novotny MV. Anal Chem. 2003;75:1684–1690. doi: 10.1021/ac025985c. [DOI] [PubMed] [Google Scholar]
- 34.Que AH, Novotny MV. Anal Chem. 2002;74:5184–5191. doi: 10.1021/ac025781w. [DOI] [PubMed] [Google Scholar]
- 35.Ishizuka N, Minakuchi H, Nakanishi K, Soga N, Nagayama H, Hosoya K, Tanaka N. Anal Chem. 2000;72:1275–1280. doi: 10.1021/ac990942q. [DOI] [PubMed] [Google Scholar]
- 36.Motokawa M, Kobayashi H, Ishizuka N, Minakuchi H, Nakanishi K, Jinnai H, Hosoya K, et al. J Chromatogr A. 2002;961:53–63. doi: 10.1016/s0021-9673(02)00133-4. [DOI] [PubMed] [Google Scholar]
- 37.Muhlecker W, Gulati S, McQuillen DP, Ram S, Rice PA, Reinhold VN. Glycobiology. 1999;9:157–171. doi: 10.1093/glycob/9.2.157. [DOI] [PubMed] [Google Scholar]
- 38.Reinhold VN, Sheeley DM. Anal Biochem. 1998;259:28–33. doi: 10.1006/abio.1998.2619. [DOI] [PubMed] [Google Scholar]
- 39.Sheeley DM, Reinhold VN. Anal Chem. 1998;70:3053–3059. doi: 10.1021/ac9713058. [DOI] [PubMed] [Google Scholar]
- 40.Viseux N, de Hoffmann E, Domon B. Anal Chem. 1997;69:3193–3198. doi: 10.1021/ac961285u. [DOI] [PubMed] [Google Scholar]
- 41.Viseux N, de Hoffmann E, Domon B. Anal Chem. 1998;70:4951–4959. doi: 10.1021/ac980443+. [DOI] [PubMed] [Google Scholar]
- 42.Weiskopf AS, Vouros P, Harvey DJ. Rapid Commun Mass Spectrom. 1997;11:1493–1504. doi: 10.1002/(SICI)1097-0231(199709)11:14<1493::AID-RCM40>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 43.Weiskopf AS, Vouros P, Harvey DJ. Anal Chem. 1998;70:4441–4447. doi: 10.1021/ac980289r. [DOI] [PubMed] [Google Scholar]
- 44.Baker AG, Alexander A, Novotny MV. Anal Chem. 1999;71:2945–2950. doi: 10.1021/ac980930p. [DOI] [PubMed] [Google Scholar]
- 45.Mechref Y, Baker AG, Novotny MV. Carbohydr Res. 1998;313:145–155. doi: 10.1016/s0008-6215(98)00264-x. [DOI] [PubMed] [Google Scholar]
- 46.Lewandrowski U, Resemann A, Sickmann A. Anal Chem. 2005;77:3274–3283. doi: 10.1021/ac048399n. [DOI] [PubMed] [Google Scholar]
- 47.Mechref Y, Novotny MV, Krishnan C. Anal Chem. 2003;75:4895–4903. doi: 10.1021/ac0341968. [DOI] [PubMed] [Google Scholar]
- 48.Morelle W, Slomianny MC, Diemer H, Schaeffer C, van Dorsselaer A, Michalski JC. Rapid Commun Mass Spectrom. 2004;18:2637–2649. doi: 10.1002/rcm.1668. [DOI] [PubMed] [Google Scholar]
- 49.Spina E, Sturiale L, Romeo D, Impallomeni G, Garozzo D, Waidelich D, Glueckmann M. Rapid Commun Mass Spectrom. 2004;18:392–398. doi: 10.1002/rcm.1350. [DOI] [PubMed] [Google Scholar]
- 50.Stephens E, Maslen SL, Gree LG, Williams DH. Anal Chem. 2004;76:2343–2354. doi: 10.1021/ac030333p. [DOI] [PubMed] [Google Scholar]
- 51.Mechref Y, Pilsoo K, Novotny MV. Anal Chem. 2005 (submitted). [Google Scholar]
- 52.Solouki T, Reinhold BB, Costello CE, O’Malley M, Guan S, Marshall AG. Anal Chem. 1998;70:857–864. doi: 10.1021/ac970562+. [DOI] [PubMed] [Google Scholar]
- 53.Zhang J, Lindsay LL, Hedrick JL, Lebrilla CB. Anal Chem. 2004;76:5990–6001. doi: 10.1021/ac049666s. [DOI] [PubMed] [Google Scholar]
- 54.Hakansson K, Cooper HJ, Emmett MR, Costello CE, Marshall AG, Nilsson CL. Anal Chem. 2001;73:4530–4536. doi: 10.1021/ac0103470. [DOI] [PubMed] [Google Scholar]
- 55.Hakansson K, Chalmers MJ, Quinn JP, McFarland MA, Hendrickson CL, Marshall AG. Anal Chem. 2003;75:3256–3262. doi: 10.1021/ac030015q. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J, Schubothe K, Li B, Russell S, Lebrilla CB. Anal Chem. 2005;77:208–214. doi: 10.1021/ac0489824. [DOI] [PubMed] [Google Scholar]
- 57.Tang H, Mechref Y, Novotny MV. Bioinformatics. 2005;21:i431–i439. doi: 10.1093/bioinformatics/bti1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ciucanu I, Kerek F. Carbohydr Res. 1984;131:209–217. [Google Scholar]
- 59.Ciucanu I, Costello CE. J Am Chem Soc. 2003;125:16213–16219. doi: 10.1021/ja035660t. [DOI] [PubMed] [Google Scholar]
- 60.Delaney J, Vouros P. Rapid Commun Mass Spectrom. 2001;15:325–334. doi: 10.1002/rcm.230. [DOI] [PubMed] [Google Scholar]
- 61.Hakomori SI. J Biochem. 1964;55:205. [PubMed] [Google Scholar]
- 62.Kang, P., Mechref, Y., Novotny, M. V., Rapid Commun. Mass Spectrom., in press. [DOI] [PMC free article] [PubMed]
- 63.Huang Y, Konse T, Mechref Y, Novotny MV. Rapid Commun Mass Spectrom. 2002;16:1199–1204. doi: 10.1002/rcm.701. [DOI] [PubMed] [Google Scholar]
- 64.Alpert AJ. J Chromatogr. 1990;499:177–196. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 65.Churms SC. J Chromatogr A. 1996;720:75–91. [Google Scholar]
- 66.Birrell H, Charlwood J, Lynch I, North S, Camilleri P. Anal Chem. 1999;71:102–108. doi: 10.1021/ac9806759. [DOI] [PubMed] [Google Scholar]
- 67.Charlwood J, Birrell H, Organ A, Camilleri P. Rapid Commun Mass Spectrom. 1999;13:716–723. doi: 10.1002/(sici)1097-0231(19990430)13:8<716::aid-rcm547>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 68.Morelle W, Strecker G. J Chromatogr B. 1998;706:101–111. doi: 10.1016/s0378-4347(97)00442-8. [DOI] [PubMed] [Google Scholar]
- 69.Thomsson KA, Karlsson H, Hansson GC. Anal Chem. 2000;72:4543–4549. doi: 10.1021/ac000631b. [DOI] [PubMed] [Google Scholar]
- 70.Guile GR, Rudd PM, Wing DR, Prime SB, Dwek RA. Anal Biochem. 1996;240:210–226. doi: 10.1006/abio.1996.0351. [DOI] [PubMed] [Google Scholar]
- 71.Rudd PM, Mattu TS, Masure S, Bratt T, van den Steen PE, Wormald MR, Kuster B, et al. Biochemistry. 1999;38:13937–13950. doi: 10.1021/bi991162e. [DOI] [PubMed] [Google Scholar]
- 72.Rudd PM, Wormald MR, Harvey DJ, Devasahayam M, McAlister MSB, Brown MH, Davis SJ, Barclay AN, Dwek RA. Glycobiology. 1999;9:443–458. doi: 10.1093/glycob/9.5.443. [DOI] [PubMed] [Google Scholar]
- 73.Taverna M, Tran NT, Valentin C, Level O, Merry T, Kolbe HVJ, Ferrier D. J Biotechnol. 1999;68:37–48. doi: 10.1016/s0168-1656(98)00187-4. [DOI] [PubMed] [Google Scholar]
- 74.Wuhrer M, Koeleman CAM, Deelder AM, Hokke CH. Anal Chem. 2004;76:833–838. doi: 10.1021/ac034936c. [DOI] [PubMed] [Google Scholar]
- 75.Xie Y, Liu J, Zhang J, Hedrick JL, Lebrilla CB. Anal Chem. 2004;76:5186–5197. doi: 10.1021/ac0496953. [DOI] [PubMed] [Google Scholar]
- 76.Zhang J, Xie Y, Hedrick JL, Lebrilla CB. Anal Biochem. 2004;334:20–35. doi: 10.1016/j.ab.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 77.Holmen JM, Karlsson NG, Abdullah LH, Randell SH, Sheehan JK, Hansson GC, Davis CW. Am J Physiol. 2004;287:L824–L834. doi: 10.1152/ajplung.00108.2004. [DOI] [PubMed] [Google Scholar]
- 78.Robinson LJ, Karlsson GN, Weiss AS, Packer NH. J Proteome Res. 2003;2:556–557. doi: 10.1021/pr034035k. [DOI] [PubMed] [Google Scholar]
- 79.Schulz BL, Oxley D, Packer NH, Karlsson GN. Biochem J. 2002;366:511–520. doi: 10.1042/BJ20011876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilson NL, Schulz BL, Karlsson GN, Packer NH. J Proteome Res. 2002;1:521–529. doi: 10.1021/pr025538d. [DOI] [PubMed] [Google Scholar]
- 81.Karlsson GN, Wilson NL, Wirth HJ, Dawes P, Joshi H, Packer NH. Rapid Commun Mass Spectrom. 2004;18:2282–2292. doi: 10.1002/rcm.1626. [DOI] [PubMed] [Google Scholar]
- 82.Conboy JJ, Henion JD. Biol Mass Spectrom. 1992;21:397–407. doi: 10.1002/bms.1200210806. [DOI] [PubMed] [Google Scholar]
- 83.Conboy JJ, Henion JD, Martin MW, Zweigenbaum JA. Anal Chem. 1990;62:800–807. [Google Scholar]
- 84.Simpson RC, Fenselau CC, Hardy MR, Townsend RR, Lee YC, Cotter RJ. Anal Chem. 1990;62:248–252. doi: 10.1021/ac00202a005. [DOI] [PubMed] [Google Scholar]
- 85.Torto N, Hofte AJP, van Der Hoeven RAM, Tjaden UR, Gorton L, Marko-Varga G, Bruggink C, van Der Greef J. J Mass Spectrom. 1998;33:334–341. [Google Scholar]
- 86.van der Hoeven RAM, Hofte AJP, Tjaden UR, van der Greef J, Torto N, Gorton L, Marko-Varga G, Bruggink C. Rapid Commun. Mass Spectrom1998;12:69–74. [Google Scholar]
- 87.Mechref Y, Muzikar J, Novotny MV. Electrophoresis. 2005;26:2034–2046. doi: 10.1002/elps.200410345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alina Z, Peter-Katalinic J. Electrophoresis. 2004;25:1949–1963. doi: 10.1002/elps.200405825. [DOI] [PubMed] [Google Scholar]
- 89.Sandra K, Van Beeumen J, Stals I, Sandra P, Claeyssens M, Devreese B. Anal Chem. 2004;76:5878–5886. doi: 10.1021/ac0493976. [DOI] [PubMed] [Google Scholar]
- 90.Sandra K, Devreese B, Stals I, Claeyssens M, van Beeumen J. J Am Soc Mass Spectrom. 2004;15:413–423. doi: 10.1016/j.jasms.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 91.Mechref Y, Chen P, Novotny MV. Glycobiology. 1999;9:227–234. doi: 10.1093/glycob/9.3.227. [DOI] [PubMed] [Google Scholar]
- 92.Tegeler TJ, Mechref Y, Boraas K, Reilly JP, Novotny MV. Anal Chem. 2004;76:6698–6706. doi: 10.1021/ac049341b. [DOI] [PubMed] [Google Scholar]
- 93.Cummings RD. Methods Enzymol. 1994;230:66–86. doi: 10.1016/0076-6879(94)30008-9. [DOI] [PubMed] [Google Scholar]
- 94.Cummings RD. Guide Tech Glycobiol. 1994;230:66–86. doi: 10.1016/0076-6879(94)30008-9. [DOI] [PubMed] [Google Scholar]
- 95.Gravel, P., Golaz, O., The Protein Protocols Handbook, Humana Press, Totowa, NJ 1996.
- 96.Carlsson, S. R., Glycobiology: A Practical Approach, Oxford University Press, Oxford, UK 1993.
- 97.Taketa K. Electrophoresis. 1998;19:2595–2602. doi: 10.1002/elps.1150191506. [DOI] [PubMed] [Google Scholar]
- 98.Yamamoto, K., Tsuji, T., Osawa, T., Glycoanalysis Protocols, Humana Press, Totowa 1998.
- 99.Zhang B, Palcic MM, Mo H, Goldstein IJ, Hindsgaul O. Glycobiology. 2001;11:141–147. doi: 10.1093/glycob/11.2.141. [DOI] [PubMed] [Google Scholar]
- 100.Madera M, Mechref Y, Novotny MV. Anal Chem. 2005;77:4081–4090. doi: 10.1021/ac050222l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Witzmann FA, Arnold RJ, Bail F, Hrncirova P, Kimpel MW, Mechref Y, McBride WJ, et al. Proteomics. 2005;5:2177–2201. doi: 10.1002/pmic.200401102. [DOI] [PMC free article] [PubMed] [Google Scholar]