

Leishmania species cause a spectrum of human diseases in tropical and subtropical regions of the world. We have sequenced the 36 chromosomes of the 32.8-megabase haploid genome of Leishmania major (Friedlin strain) and predict 911 RNA genes, 39 pseudogenes, and 8272 protein-coding genes, of which 36% can be ascribed a putative function. These include genes involved in host-pathogen interactions, such as proteolytic enzymes, and extensive machinery for synthesis of complex surface glycoconjugates. The organization of protein-coding genes into long, strand-specific, polycistronic clusters and lack of general transcription factors in the L. major, Trypanosoma brucei, and Trypanosoma cruzi (Tritryp) genomes suggest that the mechanisms regulating RNA polymerase II–directed transcription are distinct from those operating in other eukaryotes, although the trypanosomatids appear capable of chromatin remodeling. Abundant RNA-binding proteins are encoded in the Tritryp genomes, consistent with active posttranscriptional regulation of gene expression.
Infection with pathogenic Leishmania results in a spectrum of human diseases, termed the leishmaniases, with an annual incidence of 2 million cases in 88 countries (1). Leishmania parasites are transmitted by sand flies as proliferative promastigotes, which differentiate into nondividing metacyclic forms before inoculation into the vertebrate host and phagocytosis by macrophages. The metacyclics subsequently differentiate into amastigotes, which proliferate in the phagolysosome, leading to macrophage lysis and serial infection of other macrophages (2). The outcome of infection is determined by the infecting species, host genetic factors, and the immune response.
Old World Leishmania (L. donovani and L. major groups) have 36 chromosome pairs (0.28 to 2.8 Mb) (3), whereas New World species have 34 or 35, with chromosomes 8+29 and 20+36 fused in the L. mexicana group and 20+34 in the L. braziliensis group (4). Gene order and sequence are highly conserved among the ∼30 Leishmania species (5). The genome sequence of L. major MHOM/IL/81/Friedlin was determined on a chromosome-by-chromosome basis. Here we present the structure and content of the L. major genome, with an emphasis on fundamental molecular processes such as chromatin remodeling, transcription, RNA processing, translation, posttranslational modification, and protein turnover. We also discuss the synthesis of complex surface glycoconjugates that are characteristic of Leishmania species and essential at the host-parasite interface. Discussion of cytoskeleton, metabolism, and transport can be found in the accompanying description of the Trypanosoma brucei genome (6), while signaling pathways, DNA repair, recombination, and replication are discussed in the Trypanosoma cruzi article (7).
Genome structure and content
The 32,816,678–base pairs (bp) in the current assembly (version 5.2) were obtained by shotgun sequencing large-insert clones and purified chromosomal DNA. A single contiguous sequence was generated for each of the 36 chromosomes (Table 1, Plate 4, and table S1), although the “right” end of chromosome 8 lacks a small amount of subtelomeric sequence and telomeric hexamer repeats. The accuracy of sequence assemblies was assessed by comparison to an optical map (8), with only a few discrepancies occurring in regions of repetitive sequence. Although the genome is partially aneuploid (9) and there are three large-scale allelic differences (table S1), there are very few (<0.1%) sequence polymorphisms, contrasting notably with the genomes of T. brucei (6) and T. cruzi (7).
Table 1.
Summary of the L. major genome.
| Parameter | Number |
|---|---|
| The genome | |
| Size (bp) | 32,816,678 |
| G+C content (%) | 59.7 |
| Chromosomes | 36 |
| Sequence contigs | 36 |
| Percent coding | 47.9 |
| Protein-coding genes | |
| Genes | 8272 |
| Pseudogenes | 39 |
| Mean CDS length (bp) | 1901 |
| Median CDS length (bp) | 1407 |
| G+C content (%) | 62.5 |
| Gene density (genes per Mb) | 252 |
| Intergenic regions* | |
| Mean length (bp) | 2045 |
| G+C content (%) | 57.3 |
| RNA genes | |
| tRNA | 83 |
| rRNA† | 63 |
| slRNA† | 63 |
| snRNA | 6 |
| snoRNA | 695 |
| srpRNA | 1 |
Region between protein-coding CDS.
The exact number cannot be determined because of misassembly.
Analysis of the L. major sequence using several algorithms (10) predicts 911 RNA genes, 39 pseudogenes, and 8272 protein-coding genes (Table 1), of which 3083 cluster into 662 putative families of related genes (table S2). Most of the smaller (<10 members) gene families appear to have arisen from tandem gene duplication, whereas most members of larger (>10 members) families have multiple loci containing single genes and/or tandem arrays (Table 2); many of the latter contain Leishmania-specific genes.
Table 2.
L. major Friedlin protein-coding gene families.
| Family size* | Gene product(s) | L. major–specific | Organization† | Chromosome(s) |
|---|---|---|---|---|
| 491 | Hypothetical proteins (several annotations) | Some | D | Multiple |
| 189 | Kinesins/hypothetical proteins | Some | T+D | Multiple |
| 60 | Protein kinases (several groups) | Some | T+D | Multiple |
| 46 | Amastins | Most | T+TI+D | 8, 31, 34, 36 |
| 32 | Protein kinases (CMGC group) | One | D | Multiple |
| 32 | PSA-2 (GP46) | All | T+D | 12, 21, 31, 35 |
| 29 | RNA helicases/eIF-4a | None | T+D | Multiple |
| 27 | ATPase/serine peptidases | None | D | Multiple |
| 29 | Hypothetical proteins (kinesin-like) | One | D | Multiple |
| 25 | Protein phosphatases | None | T+D | Multiple |
| 25 | Tuzins | Some | TI+D | 8, 34, 36 |
| 24 | Protein kinases (STE group) | Some | D | Multiple |
| 23 | Amino acid permeases | Some | T+D | Multiple |
| 19 | HSP83 | None | T+D | 29, 33 |
| 18 | DNA helicases | Some | D | Multiple |
| 18 | β-tubulins | None | T+D | 8, 21, 33 |
| 17 | Hypothetical proteins (LACK) | One | D | Multiple |
| 17 | Hypothetical proteins | Some | T+D | 11, 13, 21, 29, 31, 36 |
| 15 | Calpain-like cysteine peptidases | Some | T+D | 4, 20, 25, 31, 36 |
| 14 | HSP70 and related proteins | None | T+D | 1, 18, 26, 28, 30, 35 |
| 14 | Phosphoglycan β 1,3 galactosyltransferases | Some | T+D | 2, 7, 14, 21, 25, 31, 35, 36 |
| 14 | Dynein heavy chain | One | D | Multiple |
| 14 | RNA helicases | None | D | Multiple |
| 14 | α,γ,ε-tubulins | Bone | T+D | 13, 21, 25 |
| 13 | Hypothetical proteins (PIPK-like protein) | One | D | Multiple |
| 13 | Pteridine transporters | Some | T+D | 4, 6, 10, 19, 35 |
| 13 | Microtubule-associated proteins | All | T | 9 |
| 13 | ABC transporters/P-glycoproteins | Some | T+D | 23, 25, 26, 31, 32, 33, 34 |
| 12 | Protein kinases (NEK group) | One | D | Multiple |
| 12 | DNAJ chaperones | Some | D | Multiple |
| 12 | Hypothetical proteins | None | T+D | 12 |
| 11 | Long-chain fatty acid CoA ligases | Some | T+D | 1, 13, 19, 28 |
| 11 | Protein kinases (DYRK and CLK families) | One | D | Multiple |
| 11 | Translation elongation factors | None | T+D | 11, 17, 18, 34, 35 |
| 10 | Cyclophilins | None | D | Multiple |
| 10 | ABC transporters | Some | T+D | 2, 11, 15, 27, 29 |
| 10 | ATPases | None | T+D | 4, 7, 17, 18, 33, 35 |
| 10 | Clan CA, family C1 cysteine peptidases | Some | T+D | 8, 19, 29 |
| 10 | Hypothetical proteins (possible peptidases) | None | T+D | 15, 16, 27 |
Families correspond to Tribe-MCL clusters (table S2) obtained using inflation value 4, and BlastP cut-off of e−15. Under these conditions, more divergent, but nonetheless functionally related proteins, may not get classified into a given gene family.
Tandem (T), tandem interspersed (TI), distributed (D).
L. major telomeres are heterogeneous in structure and quite distinct from those in T. brucei and T. cruzi (11). The extremity of each L. major chromosome contains the tripartite “repeated-repeat” structure previously reported (12). Six telomeres contain 0.7 to 25 kb of the Leishmania subtelomeric repeat (LST-R) sequences (9) between the TAS region and the first gene. Five additional groups of telomeres share varying amounts of sequence, including two cases (chromosome 17 and chromosome 10) in which the ends of a single chromosome contain the same subtelomeric sequence.
Most L. major genes have orthologs in the T. brucei and T. cruzi genomes (11); however, 910 L. major genes have no orthologs in the other two Tritryp genomes, 74 orthologous groups contain only L. major and T. brucei genes, and 482 orthologous groups contain genes from only L. major and T. cruzi. These “Leishmania-restricted” genes are randomly distributed in the genome with respect to relative chromosomal position or distance from the telomere, although some are adjacent to strand-switch regions (see below) and synteny break points (11). Although some Leishmania-restricted genes are responsible for key metabolic differences between Leishmania and T. brucei and T. cruzi (e.g., certain peptidases, transporters, and glycoconjugate biosynthesis components), most (68%) have unknown functions. Of particular interest, however, are two closely related genes (LmjF33.1740 and LmjF33.1750), whose predicted proteins both contain a macrophage migration inhibition factor (MIF) domain and show 30 to 40% identity to MIF homologs from several other organisms (including other Leishmania species). The L. major MIFs are predicted to retain the tautomerase activity found in other species, but lack the thiol oxidoreductase activity found in higher eukaryotes. Phylogenetic analysis places the Leishmania MIF genes into a bacterial clade (fig. S1). Human MIF has been shown to activate macrophages to kill Leishmania parasites via a T helper cell 1 (TH1)–type pathway (13); the MIF homolog in the filarial worm, Brugia malayi, directs macrophages to a TH2 pathway (14). Thus, it is tempting to speculate that Leishmania MIF may similarly modulate the host macrophage response and promote parasite survival.
RNA genes
RNAs participate directly in several cellular processes, including DNA replication (lagging-strand primers and telomerase RNA), splicing [spliced leader RNA (slRNA) and small nuclear RNAs (snRNAs)], RNA processing and modification [small nucleolar RNAs (snoRNAs) and ribonuclease P (RNaseP)], translation [ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs)], translation regulation (microRNA and antisense RNA), and protein translocation across membranes [signal recognition particle (SRP) or 7slRNA]. Although all three genomes encode very similar repertoires of RNA genes, their organization differs between L. major and the two Trypanosoma species. For example, the 28S, 18S, and 5.8S rRNA genes occur as a single large tandem array in L. major, but in dispersed loci in several different chromosomes in T. brucei and T. cruzi. The 5S rRNA genes are found at 11 different loci on several chromosomes in L. major, but in a single tandem array in T. brucei and T. cruzi.
All three Tritryp genomes encode tRNAs with 45 of the 61 possible anticodons, but the number of genes and their locations (in numerous loci of up to five genes) differ between species (table S3). Eight of the 16 unrepresented anticodons can be covered by third-position wobble, whereas the others require modification of cognate anticodons. Six snRNAs (U1 to U6) for RNA splicing were found in the three genomes, generally associated with tRNA clusters. SnoRNAs function in 2′-O-methylation (C/D snoRNAs) and pseudouridylation (H/ACA snoRNAs) of rRNA, slRNA, and snRNA by formation of guide RNA-target duplexes. In the Tritryp genomes, the snoRNAs are encoded by several hundred genes organized in clusters of tandem repeats of one to five different genes in several loci on a number of different chromosomes (table S3); most occur on the same strand as the adjacent protein-coding genes. All three trypanosomatid genomes have a single gene encoding 7slRNA, located in a 5S rRNA/tRNA cluster in L. major, and a tRNA/snRNA cluster in T. brucei and T. cruzi. It has been suggested that a tRNA-like molecule found in the trypanosomatid SRP complex (15) provides (in trans) the translation elongation arrest function normally associated with the 7SL RNA Alu domain, which is absent in the Tritryp molecule.
Chromatin remodeling
Trypanosomatids contain multiple copies of the four core (H2A, H2B, H3, and H4) and linker (H1) histone genes, which package chromosomal DNA into nucleosomes in eukaryotes and regulate access by the RNA polymerase transcription complexes. Most of these genes are clustered in discrete single tandem arrays in T. brucei and T. cruzi, but each of the gene types occurs in two or more separate loci in L. major; dispersed single-copy variants are found in all three genomes (table S4). The H2A variant is a homolog of the highly conserved H2A.Z, which protects “active” chromatin from silencing in yeast (16). The H2B, H3, and H4 variants appear to be novel, but may have roles in gene silencing, gene expression, DNA repair, and centromere function. Centromeres have been reported in T. cruzi (17), but no homologs were found in the Tritryps for CenH3, which is required for kinetochore assembly during mitosis. The Tritryp genomes encode a number of enzymes involved in histone modification (table S4) that may influence transcription, replication, repair, and recombination. These include two families of acetyltransferases, at least three families of methyltransferases, and all three known classes of histone deacetylases, at least two of which are essential in T. brucei (18). Two of the acetyltransferases have putative methyl-lysine–binding chromodomains, implying an association with chromatin methylation. The genomes also encode at least four putative acetyl-lysine–binding bromodomain proteins, one (in L. major and T. cruzi) with a chromatin-associated CW-type zinc finger. The Tritryp parasites thus possess a range of chromatin-remodeling activities typical of eukaryotes, although there are some notable differences.
Transcription
Little is known about the mechanism of transcription initiation in trypanosomatids, and only a few promoters have been functionally analyzed (19). The chromosomes are characterized by their unique arrangement of directional gene clusters (DGCs), previously described in L. major (20, 21) and T. brucei (22, 23). The full extent of this organization is now evident. The L. major genome is organized into 133 clusters of tens to hundreds of protein-coding genes, with unrelated predicted functions, on the same DNA strand (Plate 4). The clusters can span up to 1259 kb and are separated by 0.9- to 14-kb divergent or convergent strand-switch regions, which show an unusual base composition (24). Experimental evidence suggests that polycistronic transcription by RNA polymerase II (RNAP II) initiates bidirectionally within the divergent strand-switch regions (21, 25, 26) and terminates within the convergent strand-switch regions, which often contain tRNA, rRNA, and/or snRNA genes (26). Several long DGCs contain intervening tRNA or snRNA genes (which are transcribed by RNAP III), suggesting that they may represent more than one polycistron. At most chromosome ends (55 of 72 for L. major), transcription proceeds toward the telomere, and in 12 cases, the DGC closest to the telomere is very short (one to three genes).
Eukaryotic RNAP I, II, and III contain 14, 12, and 17 protein subunits, respectively; in yeast, five shared and six conserved polypeptides are present in all three polymerases, with all other components specific to each complex (27, 28). Tritryps have all the shared and conserved subunits except for ABC10α and A43, but many homologs for RNAP-specific subunits are absent (table S5 and Fig. 1). The Tritryp genomes contain two or three different genes encoding both ABC27 and ABC23, suggesting that these subunits may not be shared by the different RNAP complexes as they are in yeast. Tritryp RPB1 lacks the heptad repeats found in the higher eukaryotic C-terminal domain (29). Thus, the Tritryp RNAP I, II, and III components differ appreciably from those in other eukaryotes.
Fig. 1.
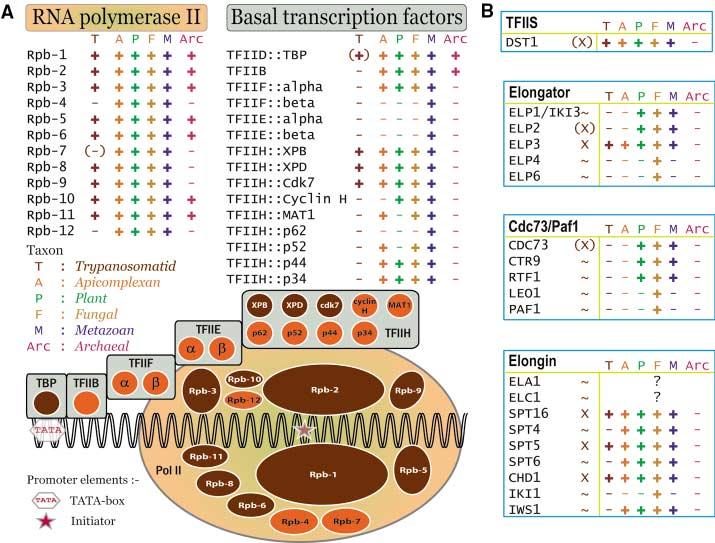
Trypanosomatid RNA polymerase subunits and transcription factors. (A) The TRIBE-MCL (table S2) protein families containing subunits of human RNA polymerase II (Rpb-1 to -12) and basal transcription factors (TBP and TFII-B, -E, -F, -H) are shown as ovals and circles respectively. Families containing Tritryp sequences are colored sepia and indicated by “+” or “(+)” (when the Tritryp gene is not a direct ortholog), whereas families lacking Tritryp sequences are indicated by “−” or “(−)” (when a Tritryp ortholog was detected only by BlastP analysis). Orthologs present in other taxa are indicated by “+”. The genomes queried for each taxon are detailed in (10). (B) Subunits of the yeast TFIIS, Elongator, Cdc73/Paf1, and Elongin complexes are displayed in blue boxes, with the first column of the row indicating whether Tritryp sequences with high “X”, weak “(X)”, or no “∼” similarity were detected. The remaining columns of the row are marked as in (A); “?” indicates that the S. cerevisiae sequence is not present in the TAP reference set.
Few potential homologs of RNAP II basal transcription factors found in other eukaryotes could be identified (table S6 and Fig. 1); the three subunits of TFIIH present also function in DNA repair and cell cycle control (30). TRF4 (which is related to TATA-box binding protein component of TFIID and TFIIIB) is essential for transcription by all three RNAPs (31). Potential homologs of BTF3b, BRF, La antigen, SNAP50, and a trypanosomatid-specific SL RNA promoter-binding factor were identified, along with four genes encoding proteins similar to TFIIS (Fig. 1B), and a SNF2-like DNA heli-case (table S6). A systematic search for Pfam domains relevant to gene expression revealed substantially fewer potential transcriptional regulators in the Tritryps than in most other eukaryotes (Fig. 2). By contrast, the Tritryp genomes contain a disproportionately higher number of proteins with CCCH-type zinc finger domains, which are found in RNA-binding proteins (see below). These findings, along with the polycistronic gene organization and paucity of RNAP II initiation sites, are consistent with posttranscriptional control mechanisms being the primary determinants of Tritryp gene expression (19).
Fig. 2.

Protein domains associated with regulation of gene expression in trypanosomatids. The Pfam accession numbers for HMMs that match Tritryp predicted proteins are shown at the left, with the next column indicating the total number of sequences matched in the Tritryp genomes. The matches in individual genomes (normalized by genome size) are shown by the bar graphs, expressed per 10,000 genes. Abbreviations: L. major (Lm); T. brucei (Tb); T. cruzi (Tc); P. falciparum (Pf); Cryptosporidium parvum (Cp); S. pombe (Sp); S. cerevisiae (Sc); Magnaporthe grisea (Mg); N. crassa (Nc); A. thaliana (At); C. elegans (Ce); D. melanogaster (Dm); Mus musculus (Mm); Rattus norvegicus (Rn); H. sapiens (Hs). The two numbers immediately to the right of the bar graphs show the maximum and minimum matches observed for all genomes, and the next column shows the mean ± SD for the 10 free-living eukaryotes. The Pfam description of the HMM is shown at the right, with the normalized and actual (in square brackets) number of matches in the L. major, T. brucei, and T. cruzi genomes shown beneath.
RNA processing
Trypanosomatid mRNA processing is distinctive: In addition to the trans-splicing of a spliced-leader RNA to the 5′ end of almost all mRNAs, the site of polyadenylation is determined by trans-splicing of the downstream mRNA, rather than by an AAUAAA and downstream G/U-rich tract (32). Only four cases of cis-splicing could be identified (table S7). Two have been previously described (33, 34); the two novel examples are both hypothetical proteins that are predicted to be capable of RNA binding. Both cis- and trans-splicing appear to be catalyzed by the Tritryp spliceosome (35). All snRNAs (table S4) and most spliceosomal proteins (table S8) were identified, but not all snRNP particle protein components were found. It was possible to identify many putative Tritryp splicing regulatory proteins, including those containing domains for 3′ splice site and branch point recognition factors, as well as several heterogeneous nuclear RNP (hnRNP) and sarcoplasmic reticulum (SR) proteins implicated in splicing and alternative splicing. Thus, it appears that regulation of splicing may have arisen early in eukaryotic evolution.
There are two dissimilar Tritryp poly(A) polymerases, which may have distinct functional roles. Homologs of the cleavage and polyadenylation specificity factor (CPF/CPSF) complexes, conserved between yeast and mammals, are also found in the Tritryps (table S9). However, no homologs of the CstF complex are evident, except for a possible CstF50 homolog. In mammalian cells, CstF50 interacts with the C-terminal domain of RNAP II and provides a link between polyadenylation and transcription initiation/termination. The absence of an RNAP II C-terminal domain may reflect the polycistronic transcription and resultant uncoupling of transcription termination and polyadenylation in the Tritryps.
Degradation of mRNAs is important in regulating trypanosomatid gene expression, and appears to resemble the situation in mammals, in which the exosome plays a dominant role. Tritryps have homologs of the deadenylation complexes, in addition to two poly(A) binding proteins. Although homologs of decapping proteins themselves were not found, a helicase involved in the process, and the exonucleases required to degrade decapped mRNAs, were (table S10). The six pleckstrin homology (PH)-domain exonucleases and three S1-domain proteins of the exosome “core” are conserved in these organisms and are essential in T. brucei (36). However, exosome-associated proteins that confer RNA processing and degradation specificity have not been found. The existence of nonsense-mediated mRNA decay in trypanosomatids is uncertain, because most of the genes required in yeast have not been identified in the Tritryp genome.
The paucity of Tritryp genes for transcriptional regulation (see above) implies a reliance on posttranscriptional control of gene expression (19) and is consistent with the presence of numerous genes for proteins with RNA-binding motifs. The total number of RNA recognition motifs (RRMs) is similar in yeast and Tritryps proteins (e.g., 103 in T. brucei versus 70 in Schizosaccharomyces pombe), but Tritryps have more small proteins with single RRMs (table S11), which may reflect unique Tritryp functions or cooperation between proteins. The Tritryp genomes encode ∼40 proteins with canonical CCCH-type zinc finger RNA-binding domains (Fig. 2 and table S11C), compared to only seven in Saccharomyces cerevisiae and 12 in S. pombe. Nearly all ∼40 Tritryp proteins have only a single CCCH domain, whereas two domains are typically required for RNA binding in other systems. We have identified a novel CCCH domain variant (Cx10Cx5CxH) that is occasionally found in association with a Cx8Cx5Cx3H finger. The roles of many of these proteins, beyond RNA binding, remain to be determined.
Translation and co-/posttranslational modification
Most major components of the translation machinery are found in the Tritryps (table S12), with similar copy numbers (one to seven) as observed in other lower eukaryotes. However, there appears to be paralogous expansion of the eIF-4A gene, with 15 copies showing 30 to 57% amino acid identity to that from S. cerevisiae, and ∼100 with <30% identity. Most contain adenosine 5′-triphosphate (ATP)–dependent DEAD-box RNA helicase domains, implying nucleic acid binding, perhaps for transcriptional or translational processes. There are also numerous copies of eEF-1α, which complexes with guanosine 5′-triphosphate (GTP) and aminoacyl-tRNAs for ribosomal A site binding during translation, but also functions in processes such as actin binding/bundling in cytokinesis in Tetrahymena (37). Functionally, L. major eIF-2B is predicted to also have mannose-1-phosphate guanyltransferase activity (LmjF23.0110) (38), whereas the eEF-1B complex has trypanothione S-transferase and peroxidase activity (39). Thus, the expanded number of potential translation factors in the Tritryps suggests a high degree of specialization.
Protein modification within the Tritryps involves typical eukaryotic processes, including phosphorylation, glycosylation, and lipidation for stabilization and/or activation. Several major modifications have been well characterized and shown to be essential, namely, glycosylphosphatidylinositol (GPI)–anchor addition, acylation (including N-myristoylation and palmitoylation), and prenylation, all of which facilitate membrane attachment and/or protein-protein interactions. The Tritryp genomes include a substantial number of proteins containing motifs for putative N-myristoylation (table S13) or prenylation (table S14), suggesting that the enzymes that catalyze these modifications may be promising drug targets, given their large number of possible substrates.
Surface molecules
The surface of the Leishmania parasite is distinguished by the presence of a variety of novel glycoconjugates implicated in various aspects of the infectious cycle in the sand fly and mammalian host. These include lipophosphoglycan (LPG), glycoinositolphospholipids (GIPLs), and membrane proteophosphoglycan (PPG), as well as glycosylated GPI-anchored proteins (e.g., GP63, PSA-2/GP46). The secreted acid phosphatase and other PPGs also contain similar posttranslational modifications and vary in structure, expression, and function among Leishmania species, as well as between the Tritryps. Although genes of the ether phospholipid synthetic pathway have been identified (6), mapping the entire LPG synthetic pathway remains incomplete (table S15), because many of the currently identified genes appear to be novel (40).
LPG is assembled in the lumen of the Golgi and requires the provision of activated sugars by nucleotide sugar transporters. The LPG2 gene product transports GDP-mannose, as well as GDP-arabinose and GDP-fucose, for LPG and phosphoglycan (PG) synthesis; and several potential UDP-Gal transporters can be found in L. major. Other nucleotide sugar transporters are present, but their specificity is unknown. Genes for several glycosyltransferases with likely roles in LPG and PG synthesis have been identified, as have genes with roles in synthesis of the LPG glycan core, PG repeating units, and species-and stage-specific PG repeat unit modification. Many, but not all, of these genes appear to be L. major–specific, although T. brucei and T. cruzi both contain a number of genes encoding glycosyltransferases with different specificities (table S15). Seven genes (SCG1 to SCG7) encoding PG-galactosyltransferases and three copies of a gene (LPG1G) encoding a possible GIPL β-galactofuranosyltransferase have telomeric locations. Six SCG-related (SCGR) genes are found in a cluster on chromosome 2, where they are interspersed with the SCA1 and SCA2 genes, which mediate the arabinosyl capping of the Gal-[PG] repeat necessary for parasite midgut release during metacyclogenesis (41). The functional significance of these nonrandom genomic locations is not yet apparent.
Sphingolipids are essential membrane components in all eukaryotic cells, and their metabolites also function in intracellular signaling. The primary sphingolipid species in the Tritryps is inositol phosphorylceramide (IPC), a target for drug development in pathogenic fungi, because it is not made in mammals. Genes for most of the sphingolipid biosynthetic pathway are present in the Tritryps, with the important exception of IPC synthase (table S16). However, this pathway is not essential for intracellular survival, because L. major scavenges lipid precursors from the host and remodels them to generate parasite-specific IPCs (42, 43).
The zinc metallopeptidase GP63 (leishmanolysin, MSP, or PSP) is the major insect-stage surface protein of Leishmania. It facilitates resistance to complement-mediated lysis on host cell entry and is also implicated in receptor-mediated uptake of Leishmania (44). In L. major, there is a tandem array of GP63 genes on chromosome 10 (some of which encode proteins with predicted GPI-anchors), a single GP63 gene on chromosome 28, and a related gene on chromosome 31 (table S17). T. brucei and T. cruzi both contain a tandem cluster of GP63 genes orthologous to the L. major chromosome 10 locus, as well as five genes in two other loci in T. brucei, and >350 (including pseudogenes) GP63-like genes in T. cruzi. Amastin was first described as an abundant amastigote surface protein in T. cruzi, where it occurs in tandem clusters, alternating with another putative surface protein, tuzin (45). L. major has 57 amastin genes, most of which are also located in tandem clusters on several chromosomes (table 2), but tuzin genes are found in only three of the loci (chromosomes 8, 34, and 36). T. brucei has only single, separate, orthologs of amastin and tuzin. All L. major amastins have transmembrane domains; 38 have predicted signal peptides, and some also have predicted GPI-anchors. Expression analyses in L. major and L. infantum demonstrate that some amastins are lifecycle stage–specific, and that the expression profiles of the various orthologs are dissimilar between these two species (46). Another large gene family that encodes GPI-anchored glycoproteins, alternatively known as PSA-2 or GP46 (depending on the Leishmania species), is found on chromosome 12, with five divergent copies at other loci (Table 2). The PSA-2 proteins function in macrophage binding and show structural similarity (but not sequence identity) with the T. cruzi mucins (7); there are no closely related orthologs in T. brucei.
Proteolysis
As found in most eukaryotes, peptidases represent ∼2% of the protein-coding genes in the Tritryps, and some have already been identified as virulence factors, vaccine candidates, and drug targets. Peptidases are structurally and functionally diverse and are grouped according to intrinsic evolutionary relationships (47). Only two aspartic peptidase homologs were identified in the Tritryps: presenilin 1 and signal peptide peptidase that cleave type I and II membrane proteins, respectively (table S18). Tritryps lack the pepsin-like aspartic peptidases (e.g., plasmepsins) that abound in apicomplexan parasites, but have many papain family (including the abundant and well-studied CPB/cruzipain lysosomal enzymes and CPA, which is unique to L. major) and calpain cysteine peptidases, as well as ubiquitin C-terminal hydrolases. Trypanosomatids, which are the only known eukaryotes with both a proteasome (48) and a eubacterial HslVU complex (49), also have numerous ubiquitin-conjugating enzymes, indicative of an active nonlysosomal cytosolic protein degradation system. In addition, the presence of two ATG4 cysteine peptidases, their potential substrate ATG8, and other ATG genes suggests that autophagy operates in organelle and protein turnover. The Tritryps lack caspases but contain several metacaspases, consistent with a caspase-independent cell death mechanism (50).
No trypsin/chymotrypsin family serine peptidases were found in Tritryps. Other serine peptidase families are present, however, including a subtilisin-like serine peptidase with a signal peptide and thus likely involved in the processing of secreted proteins. Other serine peptidases identified include six putative prolyl oligopeptidase family proteins [which have been shown to be important for cell invasion in T. cruzi (51) and are potential drug targets], a type I signal peptidase, a 26S regulatory proteasome subunit, a nucleoporin homolog, and several orthologs of rhomboidlike intramembrane serine peptidases, which might have signaling functions. Although GP63 is the most abundant metallopeptidase family, especially in T. cruzi (see above), there are 14 other families of metallopeptidases. Seven metallopeptidases, belonging to three paralogous families, show evidence of lateral gene transfer from prokaryotes (6), as do a putative peptidase T and a serine peptidase.
No representatives of the almost 200 mammalian peptidase inhibitors (e.g. serpins and cystatins) were found. However, Tritryps encode inhibitors of cysteine peptidases (ICP) that mammals seem to lack; one of these is chagasin, a potent inhibitor of cruzipain and mammalian cathepsin-L. Recent data suggest that Leishmania ICP play an important role in host-parasite interaction (52), whereas T. cruzi chagasin has a role in modulating parasite differentiation and invasion of mammalian cells (53). Curiously, the Tritryps also encode inhibitors of serine peptidases (ISPs) that are similar to ecotins, which are normally found in only a few bacterial species, including Escherichia coli. Ecotins are inhibitors of trypsin/chymotrypsin-like serine peptidases, which are notably absent in the Tritryps. However, because these peptidases are abundant in both mammals and insects, ISPs very likely play an important role in host-parasite interactions.
Implications and concluding remarks
The Tritryp genome sequences provide insights not only into the unique aspects of the biology of these parasites, but also eukaryote evolution, given their early divergence. Key differences from other eukaryotes include the manner in which the genome is organized into polycistronic gene clusters, a simplified transcriptional machinery, and mRNA trans-splicing coupled with polyadenylation. Although trypanosomatids can dynamically modify histones, implying an early origin of chromatin regulatory pathways, they primarily rely on posttranscriptional mechanisms for regulating gene expression. The lack of transcriptional control mechanisms is further manifested in the use of gene duplication/amplification as a means of increasing expression levels. Gene duplication and divergence are also exploited for the generation of antigenic diversity, particularly in T. brucei and T. cruzi. Trypanosomatids exhibit extensive posttranslational protein modification, especially for surface and secreted proteins, and have substantial species-specific arrays of glycoconjugate biosynthetic enzymes. The Tritryp genomes have much in common but display important differences (11), reflecting different survival requirements and pathogenesis in the specific niches they exploit.
The availability of the entire genetic content of one Leishmania species provides the foundation for the identification and in-depth functional analysis of virulence factors, critical enzymes in key metabolic pathways, and potential vaccine candidates. All provide crucial information for the development of new therapies for the leishmaniases. Genome sequence comparisons to additional Leishmania strains and species, projects that are ongoing, may elucidate the contribution of parasite factors to tropism and disease pathology.
Supplementary Material
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/309/5733/436/DC1
References and Notes
- WHO/TDR (World Health Organization–The Special Programme for Research and Training in Tropical Diseases) Web site on leishmaniasis, www.who.int/tdr/diseases/leish .
- 2.Alexander J, Russell DG. Adv. Parasitol. 1992;31:175. doi: 10.1016/s0065-308x(08)60022-6. [DOI] [PubMed] [Google Scholar]
- 3.Wincker P, et al. Nucleic Acids Res. 1996;24:1688. doi: 10.1093/nar/24.9.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Britto C, et al. Gene. 1998;222:107. doi: 10.1016/s0378-1119(98)00472-7. [DOI] [PubMed] [Google Scholar]
- 5.Ravel C, et al. Nucleic Acids Res. 1999;27:2473. doi: 10.1093/nar/27.12.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berriman M, et al. Science. 2005;309:416. [Google Scholar]
- 7.El-Sayed NM, et al. Science. 2005;309:409. [Google Scholar]
- 8.Zhou S, et al. Mol. Biochem. Parasitol. 2004;138:97. doi: 10.1016/j.molbiopara.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Sunkin SM, Kiser P, Myler PJ, Stuart KD. Mol. Biochem. Parasitol. 2000;109:1. doi: 10.1016/s0166-6851(00)00215-2. [DOI] [PubMed] [Google Scholar]
- 10. Materials and methods are available as supporting material on Science Online.
- 11.El-Sayed NM, et al. Science. 2005;309:404. [Google Scholar]
- 12.Chiurillo MA, et al. Exp. Parasitol. 2000;94:248. doi: 10.1006/expr.2000.4499. [DOI] [PubMed] [Google Scholar]
- 13.Juttner S, et al. J. Immunol. 1998;161:2383. [PubMed] [Google Scholar]
- 14.Falcone FH, et al. J. Immunol. 2001;167:5348. doi: 10.4049/jimmunol.167.9.5348. [DOI] [PubMed] [Google Scholar]
- 15.Liu L, et al. J. Biol. Chem. 2003;278:18271. doi: 10.1074/jbc.M209215200. [DOI] [PubMed] [Google Scholar]
- 16.Van Leeuwen F, Gottschling DE. Cell. 2003;112:591. doi: 10.1016/s0092-8674(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 17.Obado SO, Taylor MC, Wilkinson SR, Bromley EV, Kelly JM. Genome Res. 2005;15:36. doi: 10.1101/gr.2895105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ingram AK, Horn D. Mol. Microbiol. 2002;45:89. doi: 10.1046/j.1365-2958.2002.03018.x. [DOI] [PubMed] [Google Scholar]
- 19.Clayton CE. EMBO J. 2002;21:1881. doi: 10.1093/emboj/21.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myler PJ, et al. Proc. Natl. Acad. Sci. U.S.A. 1999;96:2902. [Google Scholar]
- 21.Worthey E, et al. Nucleic Acids Res. 2003;31:4201. doi: 10.1093/nar/gkg469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall N, et al. Nucleic Acids Res. 2003;31:4864. doi: 10.1093/nar/gkg674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Sayed NM, et al. Nucleic Acids Res. 2003;31:4856. doi: 10.1093/nar/gkg673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tosato V, et al. Curr. Genet. 2001;40:186. doi: 10.1007/s002940100246. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Calvillo S, et al. Mol. Cell. 2003;11:1291. doi: 10.1016/s1097-2765(03)00143-6. [DOI] [PubMed] [Google Scholar]
- 26.Martinez-Calvillo S, Nguyen DT, Stuart KD, Myler PJ. Eukaryot. Cell. 2004;3:506. doi: 10.1128/EC.3.2.506-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geiduschek EP, Bartlett MS. Nat. Struct. Biol. 2000;7:437. doi: 10.1038/75820. [DOI] [PubMed] [Google Scholar]
- 28.Huang Y, Maraia RJ. Nucleic Acids Res. 2001;29:2675. doi: 10.1093/nar/29.13.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evers R, et al. Cell. 1989;56:585. doi: 10.1016/0092-8674(89)90581-3. [DOI] [PubMed] [Google Scholar]
- 30.Zurita M, Merino C. Trends Genet. 2003;19:578. doi: 10.1016/j.tig.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Ruan JP, Arhin GK, Ullu E, Tschudi C. Mol. Cell. Biol. 2004;24:9610. doi: 10.1128/MCB.24.21.9610-9618.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthews KR, Tschudi C, Ullu E. Genes Dev. 1994;8:491. doi: 10.1101/gad.8.4.491. [DOI] [PubMed] [Google Scholar]
- 33.Mair G, et al. RNA. 2000;6:163. doi: 10.1017/s135583820099229x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ullu E. personal communication. [Google Scholar]
- 35.Liang XH, Haritan A, Uliel S, Michaeli S. Eukaryot. Cell. 2003;2:830. doi: 10.1128/EC.2.5.830-840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Estevez AM, Kempf T, Clayton C. EMBO J. 2001;20:3831. doi: 10.1093/emboj/20.14.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Numata O, Kurasawa Y, Gonda K, Watanabe Y. J. Biochem. (Tokyo) 2000;127:51. doi: 10.1093/oxfordjournals.jbchem.a022583. [DOI] [PubMed] [Google Scholar]
- 38.Garami A, Ilg T. EMBO J. 2001;20:3657. doi: 10.1093/emboj/20.14.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vickers TJ, Wyllie SH, Fairlamb AH. J. Biol. Chem. 2004;279:49003. doi: 10.1074/jbc.M407958200. [DOI] [PubMed] [Google Scholar]
- 40.Dobson DE, et al. J. Biol. Chem. 2003;278:15523. doi: 10.1074/jbc.M301568200. [DOI] [PubMed] [Google Scholar]
- 41.Dobson DE, Scholtes LD, Myler PJ, Turco SJ, Beverley SM. in preparation. [Google Scholar]
- 42.Zhang K, et al. Mol. Microbiol. 2005;55:1566. doi: 10.1111/j.1365-2958.2005.04493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denny PW, Goulding D, Ferguson MA, Smith DF. Mol. Microbiol. 2004;52:313. doi: 10.1111/j.1365-2958.2003.03975.x. [DOI] [PubMed] [Google Scholar]
- 44.Yao C, Donelson JE, Wilson ME. Mol. Biochem. Parasitol. 2003;132:1. doi: 10.1016/s0166-6851(03)00211-1. [DOI] [PubMed] [Google Scholar]
- 45.Teixeira SM, Russell DG, Kirchhoff LV, Donelson JE. J. Biol. Chem. 1994;269:20509. [PubMed] [Google Scholar]
- 46.Rochette A, et al. Mol. Biochem. Parasitol. 2005;140:205. doi: 10.1016/j.molbiopara.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 47.Rawlings ND, Tolle DP, Barrett AJ. Nucleic Acids Res. 2004;32(Database issue):D160. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang CC, et al. J. Biol. Chem. 2003;278:15800. doi: 10.1074/jbc.M300195200. [DOI] [PubMed] [Google Scholar]
- 49.Couvreur B, et al. Mol. Biol. Evol. 2002;19:2110. doi: 10.1093/oxfordjournals.molbev.a004036. [DOI] [PubMed] [Google Scholar]
- 50.Zangger H, Mottram JC, Fasel N. Cell Death Differ. 2002;9:1126. doi: 10.1038/sj.cdd.4401071. [DOI] [PubMed] [Google Scholar]
- 51.Burleigh BA, Woolsey AM. Cell. Microbiol. 2002;4:701. doi: 10.1046/j.1462-5822.2002.00226.x. [DOI] [PubMed] [Google Scholar]
- 52.Besteiro S, Coombs GH, Mottram JC. Mol. Microbiol. 2004;54:1224. doi: 10.1111/j.1365-2958.2004.04355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos CC, et al. J. Cell Sci. 2005;118:901. doi: 10.1242/jcs.01677. [DOI] [PubMed] [Google Scholar]
- 54. We thank our colleagues in the Leishmania Genome Network (LGN) for their support and encouragement. We thank the other members of the Tritryp Sequencing Consortium for their help with comparative genome annotation; special thanks to J. Donelson and S. Melville, who together have played a key driving role in the coordination, discussion, and collation of these manuscripts. Funding for this project was provided by grants from WHO TDR (T23/181/1 ID:940509), Burroughs Wellcome Fund (BWF) (APP#0500), and National Institute of Allergy and Infectious Diseases (NIAID) (RO1 AI040599) to SBRI; Wellcome Trust (WT) (054394/Z/98/Z, 060491/Z/00/Z, and 063272/Z/00/Z) to the Wellcome Trust Sanger Institute; the European Union (BIO4-CT98-0079) to the EULEISH consortium; NIAID (RO1 AI060645) to A.C.F.; and a Fundação de Amparo à Pesquisa do Estado de São Paulo fellowship (01/13461-9) to J.C.R. WHO TDR, WT, and NIAID also provided funds for several LGN meetings. Accession numbers: EMBL: CT005244 to CT005272, AL389894 and AL139794; GenBank: CP000078 to CP000081, AE001274, and NC_004916. All data are available in GeneDB (http://www.genedb.org. ).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.