

Abstract
The cardiac muscle ryanodine receptor (RyR2) functions as a calcium release channel in the heart. Up to 40 mutations in RyR2 have been linked to genetic forms of sudden cardiac death. These mutations are largely clustered in three regions of the sequence of the polypeptide: one near the N terminus, one in the central region, and the third in the C-terminal region. The central region includes 11 mutations, and an isoleucine-proline motif (positions 2427 and 2428) in the same region is predicted to contribute to the binding of FKBP12.6 protein. We have mapped the central mutation site in the three-dimensional structure of RyR2 by green fluorescent protein insertion, cryoelectron microscopy, and single-particle image processing. The central mutation site was mapped to a “bridge” of density that connects cytoplasmic domains 5 and 6, which have been suggested to undergo conformational changes during channel gating. Moreover, the location of this central mutation site is not close to that of the FKBP12.6-binding site mapped previously by cryoelectron microscopy.
Heart disease is the world’s leading cause of death, accounting for 38% of all deaths in the United States in 2002. Each year, ~900,000 people die from heart disease, and many millions more suffer from and eventually succumb to heart diseases such as coronary heart disease, stroke, heart failure, high blood pressure, cardiomyopathy, and arrhythmias (1). Sudden cardiac death is the sudden, abrupt loss of heart function; about half of all deaths from heart disease are unanticipated, occurring instantly or shortly after symptoms appear and, unfortunately, before emergency medical treatment can be administered. Sudden cardiac deaths account for 19% of deaths in children between 1 and 13 years of age, and 30% of those between 14 and 21 years (1). Many sudden cardiac death victims have no diagnosed heart disease, and post-mortem examinations reveal no morphologic abnormalities to explain the death (2). Recently, mutations of the cardiac muscle ryanodine receptor (RyR2; known as RyR2) have been associated with heart failure and sudden cardiac death (3–11). Molecular genetic analysis demonstrated that mutations in the cardiac muscle RyR gene may be associated with ~14% of these sudden cardiac death cases (11).
RyR2 functions as a calcium release channel in the heart, where it plays a crucial role in excitation-contraction coupling. Influx of Ca2+ through the cardiac L-type voltage-gated Ca2+ channel (dihydropyridine receptor (DHPR)) on the plasma membrane triggers RyR2 to release a large amount of Ca2+ from the sarcoplasmic reticulum into the cytoplasm. The resulting increase in cytoplasmic Ca2+ activates the myofilaments to generate cardiac muscle contraction (12). Aberrant release of calcium ions via RyR2 can lead to electrical instability, abnormal cell growth, and irregular heartbeat and can in turn lead to heart disease. To date, 40 naturally occurring mutations in RyR2 have been linked to genetic forms of sudden cardiac death (3–11). Of these, 34 mutations have been linked to catecholaminergic polymorphic ventricular tachycardia (CPVT) (3, 4, 7–11), and six have been linked to arrhythmogenic right ventricular dysplasia type 2 (ARVD2) (5, 6). CPVT is a genetic arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden cardiac death (3). ARVD2 is an autosomal dominant cardiomyopathy characterized by partial degeneration of the myocardium right ventricle, electrical instability, and sudden death, and it is most frequently found in juveniles and athletes (5).
The mechanism by which a single-point mutation among the ~5000 amino acids of RyR2 affects normal heart function and causes sudden cardiac death remains elusive. These mutations are largely clustered in three regions of the RyR2 primary sequence: one near the N terminus, one in the central region, and the third in the C-terminal region (see Fig. 1). Intriguingly, these three regions are homologous to the three malignant hyperthermia (and related central core disease) regions in the skeletal muscle RyR (known as RyR1): the N-terminal region comprises the first 614 amino acids of RyR1; the central region is located between amino acids 2163 and 2458; and the C-terminal region is between amino acids 4800 and 4900 (13).
FIGURE 1. The 40 mutations in RyR2 sequence implicated in sudden cardiac death.
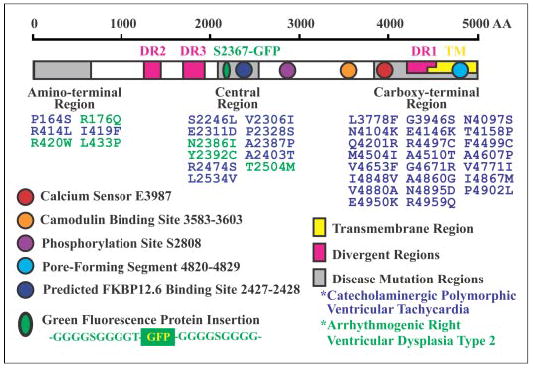
These mutations are largely clustered in three regions of the sequence (gray boxes). The ~500 amino acids (AA) beginning at the C terminus compose the transmembrane domain (TM; yellow box). The sequences of the three RyR isoforms differ primarily in the three divergent regions (DR; pink boxes). The proposed Ca2+ sensor, the calmodulin-binding site, a phosphorylation site, the proposed pore-forming segment, and a predicted FKBP12.6-binding site are indicated. GFP flanked by two glycine-rich spacers was inserted in the middle of the central mutation region after Ser-2367 (green oval).
For RyR2, the central region mutations include S2246L, V2306I, E2311D, P2328S, N2386I, A2387P, Y2392C, A2403T, R2474S, T2504M, and L2534V. An isoleucine-proline motif (positions 2427 and 2428) in the same region is predicted to be involved in binding FKBP12.6 (FK506-binding protein of 12.6 kDa), a protein thought to regulate RyR2 channel gating (14). Tiso et al. (15) have suggested that central region mutations in CPVT and ARVD2 influence the binding of FKBP12.6 to RyR2, which causes the closed state of mutant RyR2 to become destabilized so that unusually large amounts of Ca2+ are released from the sarcoplasmic reticulum into the cytoplasm, leading to faster heart beat and sudden death.
To explore the structural basis of central region mutations in sudden cardiac death, we identified the location of a central mutation site in the three-dimensional structure of RyR2. We inserted a green fluorescent protein (GFP) tag after Ser-2367, near the middle of the central region (RyR2S2367-GFP). RyR2S2367-GFP expressed in human embryonic kidney (HEK) 293 cells functions as a caffeine- and ryanodine-sensitive calcium release channel. Purified RyR2S2367-GFP was studied by cryoelectron microscopy (cryo-EM), and a three-dimensional structure was reconstructed by single-particle image processing. The location of the inserted GFP tag was determined by comparison with the reconstruction of wild-type (wt) RyR2 (RyR2wt). The inserted GFP tag (and consequently, Ser-2367) was mapped to the “bridge” between cytoplasmic domains 5 and 6. These domains have been suggested to undergo conformational changes during channel gating. Thus, our data suggest that disease-associated RyR2 mutations located in the central region alter the conformational changes that normally occur during channel gating. Moreover, the three-dimensional location of this central mutation site is not close to the FKBP12.6-binding site mapped previously, suggesting that the isoleucine-proline motif is unlikely to be the FKBP12.6-binding site.
EXPERIMENTAL PROCEDURES
cDNA Construction of RyR2S2367-GFP
The cloning and construction of the 15-kb cDNA encoding the mouse cardiac muscle RyR (known as RyR2) have been described previously (16). The DNA encoding GFP flanked by Gly-rich linkers and an AscI site (see Fig. 1) was obtained by PCR as described previously (17). The AscI site was introduced into the central mutation region of RyR2 after Ser-2367 in the sequence by overlap extension by PCR. The “outer” primers were as follows: forward, 5′-ACC AGT GGC TCC GGG CGC GCC CTT GAT ATA GAA GAG GAG-3′; and reverse, 5′-TTC TAT ATC AAG GGC GCG CCC GGA GCC ACT GGT TGG AGA-3′. The AscI fragment containing GFP and the linkers was then subcloned into full-length RyR2wt after Ser-2367. The sequences of all PCR fragments were verified by DNA sequencing.
Expression and Purification of RyR2S2367-GFP
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium as described previously (18). HEK293 cells grown on 100-mm tissue culture dishes for 18–20 h after subculture were transfected with 6–12 μg of RyRS2367-GFP cDNAs using Ca2+ phosphate precipitation (18). The transfected HEK293 cells were detergent-lysed in lysis buffer (25 mm Na-PIPES (pH 7.2), 140 mm NaCl, 5 mm EGTA, 2.5 mm dithiothreitol, 1% CHAPS, 0.5% egg lecithin, and protease inhibitor mixture (2 mm benzamidine, 4 μg/ml leupeptin, 2 μg/ml pepstatin A, 4 μg/ml aprotinin, and 0.75 mm phenylmethylsulfonyl fluoride)) and incubated on ice for 1 h. Insoluble material was removed by sedimentation, and sucrose (400 mm final concentration) and NaCl (400 mm final concentration) were added to the supernatant. Glutathione-Sepharose beads (100 μl) containing 400 μg of bound glutathione S-transferase (GST)-FKBP12.6 fusion protein were added to the cell lysate supernatant. The mixture was incubated at 4 °C with rotation for 2 days. The beads were collected by centrifugation and washed four times with lysis buffer containing 0.5% CHAPS and 0.25% egg lecithin. The RyR2 proteins were eluted from the beads by incubation with 100 mm glutathione in 25 mm Na-PIPES (pH 7.2), 400 mm NaCl, 400 mm sucrose, 1 mm EGTA, 0.2 mm CaCl2, 2.5 mM dithiothreitol, 0.4% CHAPS, 0.16% egg lecithin, 200 mm Tris/HEPES (pH 7.4), and protease inhibitor mixture for 15 min. The eluate was aliquoted, frozen in liquid nitrogen, and stored at −90 °C.
Ca2+ Release Measurement, [3H]Ryanodine Binding Assay, SDS-PAGE, and Immunoblotting
The free cytosolic Ca2+ concentration in the transfected HEK293 cells was measured with the fluorescent Ca2+ indicator dye fluo-3 as described previously (18). Equilibrium [3H]ryanodine binding to purified RyR2 proteins was carried out at various Ca2+ concentrations as described previously (19). The purified RyR2 proteins were denatured in Laemmli sample buffer and subjected to SDS-6% polyacrylamide mini-gel electrophoresis. Immunoblotting with anti-RyR antibody (clone 34C) or anti-GFP antibody (catalog no. PA1-980) (Affinity BioReagents, Golden, CO) was performed as described previously (20).
Cryo-EM and Image Processing
Purified RyR2S2367-GFP was diluted 5–10-fold with EM dilution buffer (20). Grids were prepared for cryo-EM according to standard methods. Micrographs were recorded using low-dose protocols on a Philips EM420 microscope, equipped with a low-dose kit and a Gatan 626 cryo-transfer holder with 100-kV operating voltage, in a defocus range between 2.0 and 2.5 μm and at a magnification of ×38,600× (±2%) as verified by a tobacco mosaic virus standard. Each exposure corresponded to an electron dose of ~10 e−/Å2. Micrographs were checked for drift, astigmatism, and presence of Thon rings by optical diffraction. Selected electron micrographs were digitized on a Zeiss imaging scanner (Z/I Imaging Corp., Huntsville, AL) with a step size of 14 μm. Images were processed using the SPIDER/WEB software package (21). Images of RyR2 proteins in the 4-fold symmetric orientation were aligned by cross-correlation methods, and two-dimensional averages were computed (22). Three-dimensional reconstructions were obtained from images of particles lying in all available orientations by the projection matching procedure (23). The final three-dimensional reconstructions of RyR2wt and RyR2S2367-GFP were computed from 5434 and 4105 particles, respectively, to ensure that the particle orientations covered the entire angular space and that each view would be represented by almost equal numbers of particles. Four-fold symmetry was enforced in both the two-dimensional averages and the three-dimensional reconstructions. The final resolutions were estimated to be 34 Å by Fourier shell correlation (with a cutoff of 0.5) for RyR2wt and 33 Å for RyR2S2367-GFP. The three-dimensional difference map was calculated by subtraction of the volume of RyR2wt from that of RyR2S2367-GFP.
RESULTS
Construction, Expression, Functional Characterization, and Purification of RyR2S2367-GFP
To gain insight into the structural basis of RyR2 malfunction caused by central region mutations, we mapped the location of a central mutation site in the three-dimensional structure of RyR2. A chimeric cDNA of RyR2 with GFP inserted in the middle of the central mutation region, after Ser-2367, was constructed (Fig. 1). cDNAs of modified RyR2 proteins with GFP insertions were expressed in HEK293 cells.
RyR2wt expressed in HEK293 cells has been shown to function as a Ca2+ release channel (17). We measured intracellular Ca2+ release of RyR2S2367-GFP using the fluorescent Ca2+ indicator dye fluo-3 acetoxymethyl ester. As shown in Fig. 2a, HEK293 cells transfected with RyR2S2367-GFP cDNA displayed multiple Ca 2+ release events in response to repeated stimulation by caffeine, an activator of RyR2. The addition of ryanodine led to a slow increase in fluorescence and diminished the response to multiple caffeine stimulations. This is most likely the result of modulation by ryanodine, which is known to bind to only the open state of the channel and to dramatically increase the channel open probability. As a consequence, the ryanodine-modified channel no longer responded to further caffeine stimulation. These results suggest that RyR2S2367-GFP is sensitive to caffeine and ryanodine modification. Similar Ca2+ release responses to caffeine and ryanodine have also been observed in HEK293 cells transfected with RyR2wt (17).
FIGURE 2. Biochemical characterization of RyR2S2367-GFP.
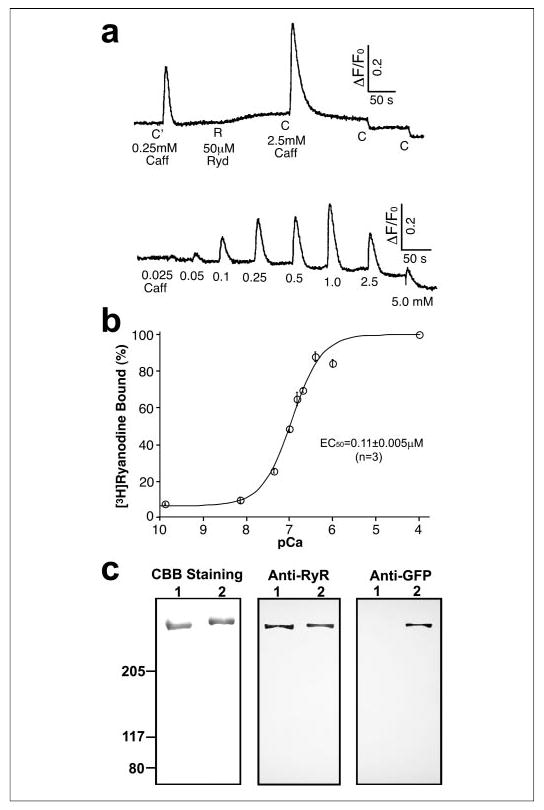
a, Ca2+ release activity detected following the addition of the RyR2 activator caffeine (Caff; C′ = 0.25 mm, C = 2.5 mm) or ryanodine (Ryd; R = 50 μM) to RyR2S2367-GFP cDNA-transfected HEK293 cells as measured by fluo-3 acetoxymethyl ester fluorescence intensity; b, [3H]ryanodine binding to the purified RyR2S2367-GFP proteins at various Ca 2+ concentrations; c, SDS-PAGE and Western blot of purified RyR2wt (lanes 1) and RyR2S2367-GFP (lanes 2). CBB, Coomassie Brilliant Blue.
The functional properties of the purified RyR2S2367-GFP proteins were further characterized by [3H]ryanodine binding studies. Fig. 2b shows [3H]ryanodine binding to purified RyR2S2367-GFP in the presence of a wide range of Ca2+ concentrations. Analysis of the Ca2+ dependence of [3H]ryanodine binding by the Hill equation yielded an EC50 of 0.11 ± 0.005 μm (mean ± S.E., n = 3). We also determined the specificity of [3H]ryanodine binding to the purified RyR2S2367-GFP proteins. Binding was carried out in the presence of 50 nm [3H]ryanodine in an attempt to saturate the binding and 100 μm Ca2+ to maximally activate the channel. Under these conditions, we obtained a binding specificity of 518 ± 51 pmol of [3H]ryanodine/mg of protein (n = 4), which is similar to those reported previously for the purified RyR2 and RyR1 proteins (24, 25). These results suggest that the insertion of GFP after Ser-2367 does not grossly alter the structural and functional properties of RyR2.
The expressed RyR2S2367-GFP protein was purified in a single step from detergent-solubilized cell lysates by affinity chromatography (glutathione-Sepharose) using GST-FKBP12.6 as the affinity ligand (17). Evidently, the insertion of GFP after Ser-2367 did not restrict the binding of GST-FKBP12.6 to RyR2. The purified protein was analyzed by SDS-PAGE and immunoblotting. A single high molecular mass band, which migrated at a slightly slower rate compared with RyR2wt (as expected, due to the addition of GFP), was detected in the purified sample of RyR2S2367-GFP (Fig. 2c). This band was recognized by both the anti-RyR and anti-GFP antibodies. On the other hand, the purified RyR2wt protein was recognized by the anti-RyR antibody, but not by the anti-GFP antibody.
Three-dimensional Localization of a Central Mutation Site
The purified RyR2wt and RyR2S2367-GFP samples were diluted to a suitable concentration, applied to carbon-coated EM grids, blotted, rapidly frozen, and imaged by electron microscopy. Fig. 3 shows a typical electron micrograph in which individual RyR2S2367-GFP channels are visible.
FIGURE 3. Cryo-EM of RyR2S2367-GFP.
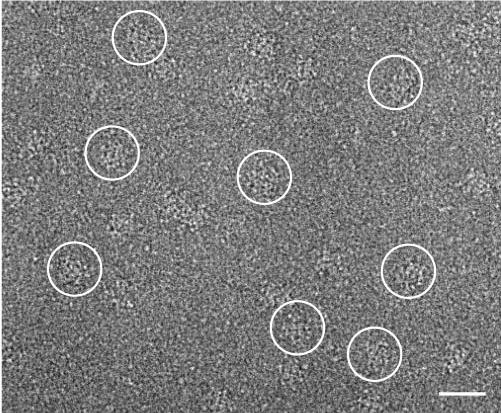
A portion of a cryoelectron micrograph of the purified RyR2S2367-GFP proteins embedded in a thin layer of vitreous ice is shown. The tetrameric structure of RyR2S2367-GFP is well preserved. Several individual RyR2S2367-GFP particles are circled. Scale bar = 600 Å.
Image processing was performed using the SPIDER/WEB software package (21). Fig. 4 (a and b) shows two-dimensional averages of RyR2S2367-GFP and RyR2wt, respectively, computed using images of selected particles that were lying with their 4-fold symmetry axes oriented perpendicular to the carbon support film; these images were aligned by cross-correlation methods (22). Subtraction of the two-dimensional average of RyR2wt from that of RyR2S2367-GFP provides a difference map that should resolve the projected location of the GFP insertion and perhaps also conformational differences between the two averaged projection structures. The brightest white areas shown in Fig. 4c correspond to the most significant positive densities, which represent protein mass present in RyR2S2367-GFP, but absent from RyR2wt. These regions are located near the center of the “clamp” structures that form the corners of the square-shaped receptor (26). From previous three-dimensional reconstructions, we know that the domains that lie in the vicinity of the inserted GFP tag are domains 5–8 (see the three-dimensional reconstruction in Fig. 5). Statistical analysis (Fig. 4d) of the difference between the two averaged images indicated that these regions show significant differences at a confidence level of >99.9% (27). These differences almost certainly correspond to the additional mass contributed by the GFP insertion in RyR2S2367-GFP. This interpretation is confirmed, and GFP is more precisely mapped in the three-dimensional reconstructions described below.
FIGURE 4. Two-dimensional averages of RyR2wt and RyR2S2367-GFP.

a, two-dimensional average of RyR2S2367-GFP (n = 251 particle images) in the “bottom” view. b, two-dimensional average of RyR2wt (n = 269 particle images) in the same view. c, difference map obtained by subtraction of b from a. The “top” and bottom views of RyR2 (both 4-fold symmetric) are the most frequent orientations assumed by RyR2 particles on EM grids; the bottom view depicted here represents the projection of the receptor as seen from the lumen side of the sarcoplasmic reticulum. The largest positive differences shown in c, corresponding to the additional masses due to the GFP insertions, are seen as bright white areas in the clamp domains (circled). d, map of statistically significant regions of difference obtained by the t test displayed at a >99.9% confidence level. The width of each frame is 544 Å.
FIGURE 5. Three-dimensional localization of GFP inserted at Ser-2367.

a, a surface representation of the three-dimensional reconstruction of RyR2S2367-GFP is shown in green. b, a difference map (RyR2S2367-GFP − RyR2wt) shown in green is superimposed on the three-dimensional reconstruction of RyR2wt (blue). Each of the green regions of the difference map corresponds to a GFP insertion in the central mutation region of a RyR2 subunit. The three-dimensional reconstructions in both panels are shown in three views: left, top view from the cytoplasmic surface that in situ would face the transverse tubule membrane; middle, bottom view that would face the sarcoplasmic reticulum lumen; right, side view. The side view shows that RyR2 has a small transmembrane assembly (TA) and a large cytoplasmic assembly as the main mass of the protein. The numerals 1–10 on the cytoplasmic assembly indicate the distinguishable domains according to previous nomenclature (26).
Three-dimensional reconstructions were obtained by a projection matching procedure (23). Fig. 5a shows surface representations of the three-dimensional reconstruction of RyR2S2367-GFP in three principal orientations. The reconstructed structure consists of two major components: a large cytoplasmic assembly composed of at least 10 distinct domains (labeled 1–10) (26) and a smaller transmembrane assembly (labeled TA). For the reconstruction of RyR2S2367-GFP, the final resolution was estimated to be 33 Å (see “Experimental Procedures”). Overall, the three-dimensional reconstruction of RyR2S2367-GFP is very similar to that of the RyR2wt (17), but close examination revealed some small differences. The most noticeable difference was found in the region between domains 5 and 6; specifically, the volume of the bridge linking domains 5 and 6 in RyR2S2367-GFP appears to be larger than the corresponding bridge volume in RyR2wt. To more precisely delineate the differences, we generated a three-dimensional difference map by subtracting the three-dimensional volume of RyR2wt from that of RyR2S2367-GFP. The difference regions are displayed in green and superimposed on the three-dimensional reconstruction of RyR2wt. This difference map clearly indicates one significant difference, located in each bridge connecting domains 5 and 6 in the cytoplasmic assembly. The GFP tag was inserted into each RyR2 monomer, and because RyR2 is a homotetramer composed of four identical monomers, the difference would be expected to repeat four times in the three-dimensional difference map.
We are confident that these differences are directly attributable to the excess mass contributed by the GFP insertion in RyR2S2367-GFP because they are the only significant differences that appear when the three-dimensional difference map is displayed at a density threshold nearly the same as that used to image RyR2S2367-GFP and RyR2wt. Furthermore, the calculated volume of each of the four difference features in Fig. 5b corresponds to a molecular mass of 28 kDa, assuming a protein density of 1.37 g/cm3; this value agrees well with the molecular mass of GFP (26). Other minor differences (located in both the clamp and handle domains in the cytoplasmic assembly as well as in the transmembrane assembly) can be regarded as less significant in terms of both volume and density and are unlikely to correspond to the inserted GFP tag. Reassuringly, the locations of the GFP sites in the three-dimensional reconstruction are consistent with the projected locations of the major differences seen in the two-dimensional analysis (Fig. 4). In summary, the central mutation region, as indicated by the inserted GFP tag, is located on the bridge between domains 5 and 6 in each of the clamp regions of RyR2.
DISCUSSION
The Central Mutation Region Is Not near the FKBP12.6-binding Site
FKBP12 (FK506-binding protein of 12 kDa) binds tightly to the skeletal muscle RyR (known as RyR1) and modulates RyR1 channel gating by stabilizing the closed state and by promoting transitions between the closed and open states (eliminating subconductance states) (28). A highly homologous protein (FKBP12.6) binds to the cardiac muscle RyR (known as RyR2) and stabilizes RyR2 in a similar manner (29, 30). An isoleucine-proline motif (positions 2427 and 2428) in the RyR2 sequence has been shown to be the binding site for FKBP12.6, and a similar motif is also found in the RyR1 sequence (Val-Pro at positions 2461 and 2462) (14). Ser-2808 has been identified as a site of phosphorylation in RyR2 (31). Marx et al. (14) have shown that hyperphosphorylation of RyR2 by protein kinase A at this site causes dissociation of the FKBP12.6 subunit from RyR2 and that such dissociation affects RyR2 functioning, thereby contributing to the ventricular arrhythmias that often occur during heart failure. Similarly, the mutations in RyR2 that cause CPVT and ARVD2 are thought to weaken the affinity of RyR2 for FKBP12.6 (30).
The FKBP12-binding site was previously mapped to a region at or near the junction of domains 3 and 9 in the three-dimensional structure of RyR1 (32), as indicated by magenta dots in Fig. 6a. Recently, the FKBP12.6-binding site has been mapped to a similar region in the three-dimensional structure of RyR2. It is important to know that the three-dimensional location of Ser-2367, as indicated by the localization of the inserted GFP tag, is not close to the FKBP12.6-binding sites. The distance between the central mutation location and the FKBP12.6-binding sites is >80 Å. Thus, it appears unlikely that the mutations in the central region disrupt the binding of FKBP12.6 through disruption of the structure of RyR2 in the immediate vicinity of the FKBP12.6-binding site. If the FKBP12.6/RyR2 interaction is weakened by such mutations, this weakening may occur by means of an allosteric effect acting over tens of angstroms.
FIGURE 6. Relationship of Ser-2367 to other landmarks on the RyR three-dimensional architecture.
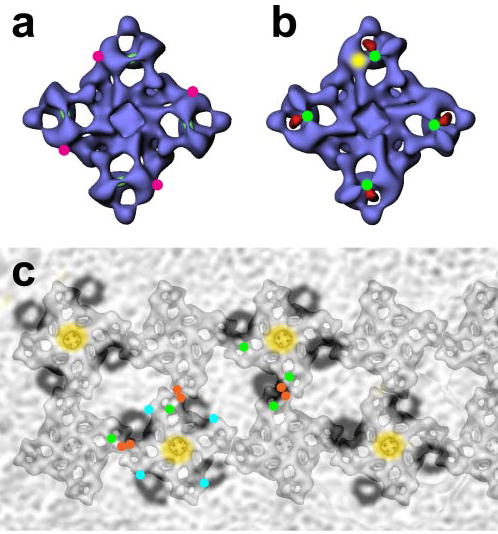
a, Ser-2367 is not close to the FKBP12.6-binding site in the three-dimensional structure of RyR. The binding sites of FKBP12 and FKBP12.6 have been mapped to the same location on the three-dimensional structures of RyR1 and RyR2 (magenta dots) (32). b, Ser-2367 lies adjacent to the N terminus and the N-terminal region mutation hot spot. The GST tag fused to the N terminus of RyR3 was mapped to the center of the clamp region (red) (23), and a region containing much of the N-terminal mutation region was mapped to domain 5 (yellow in one clamp structure) according to the work of Serysheva et al. (41). c, Ser-2367 is located in the clamp region, which is proposed to contact the DHPR. DHPR tetrad arrays are shown as semitransparent dark rings, which are superimposed on the RyR1 ordered arrays (from Paolini et al. (50)); the centers of the tetrads are shown in yellow. Green, red, and cyan dots represent the central mutation region and divergent regions 2 and 3, respectively.
Our results contribute to the growing body of evidences that disfavor the proposal that RyR2 Ile-2427 and Pro-2428 contribute to the binding site for FKBP12.6 (33–37). The site of the GFP insertion studied here is only a distance of 59 residues from the Ile-Pro motif in the primary sequence, but the rather large distance between this site and the actual FKBP12.6-binding site as found by three-dimensional cryo-EM (~80 Å) makes it unlikely (although not impossible) that Ile-2427 and Pro-2428 contribute directly to the binding site for FKBP12.6. Other recent studies have also raised doubts about the role of these residues in the FKBP12.6/RyR2 interaction. A C-terminal truncation analysis demonstrated that a region between positions 1815 and 1855, near divergent region 3, is essential for FKBP12.6 interaction (33). Consistent with this finding, we used cryo-EM to map a GFP insertion at Thr-1874 (in the middle of divergent region 3) to domain 9, which is adjacent to the FKBP12.6-binding site (33). A CPVT-linked RyR2 mutant in the central region (S2246L) also displays normal RyR2-FKBP12.6 binding (34). Moreover, in contrast to a previous report (14), recent studies have found that protein kinase A phosphorylation of Ser-2808 in RyR2 does not result in dissociation of FKBP12.6. FKBP12.6 can bind to both Ser-2808-phosphorylated and non-phosphorylated RyR2 and to the recombinant and native forms of RyR2 (35). The RyR2 mutants at this conserved phosphorylation site (S2808D and S2808A) retain the ability to bind FKBP12.6 (36).
Based upon the above discussion, the disease-causing mutations in RyR2 may alter RyR2 function in ways that do not directly involve FKBP12.6. The three-dimensional location of the central mutation site is in the clamp region, a place where the RyR undergoes significant conformational changes when it switches between the closed state and open states (38, 39): the connected domains 5 and 6 and domains 9 and 10 separate when the RyR transitions from the closed state to the open state, and domain 6 also appears to flip upward (39). The three-dimensional location of a central mutation site suggests that mutations in the central region may alter the conformational changes that normally occur during channel gating (see below).
Mutation-prone N-terminal and Central Regions Interact with One Another
As stated above, disease-linked mutations in RyR1 and RyR2 are concentrated in three regions of the primary sequence. The two mutation hot spots that lie in cytoplasmic regions, viz. the N-terminal and central regions, are well separated in the primary sequence (>1500 amino acids). According to the hypothesis proposed by Ikemoto and Yamamoto (40), these two regions occur in domains that physically interact with each another, and changes in the strength of their interaction affect channel gating. Briefly, the interactions between the N-terminal and central regions (zipping domains) stabilize the RyR in the closed state, and reduction in the affinity of the interaction causes separation of the regions (unzipping) and thereby induces the RyR to open more frequently and to release greater than normal amounts of Ca2+ into the cytoplasm. In healthy muscle, the unzipping action is produced by the activation signals received by the RyR (Ca2+ influx via the cardiac muscle DHPR or conformational changes in the skeletal muscle DHPR upon plasma membrane depolarization). Mutations in the N-terminal or central regions are proposed to weaken the normal interdomain interactions, resulting in unzipping and hyperactive channels.
Here, we have mapped a central mutation site to the clamp region of RyR2. But where is the N-terminal mutation region, and are the N-terminal and central regions in close contact in the three-dimensional structure? Evidence from two studies indicates that the N-terminal mutation region is indeed located in the clamp regions, most likely adjacent to the central mutation region. (i) The N terminus of the brain RyR (known as RyR3) was labeled with GST and was mapped to the clamp region by cryo-EM (23). As shown in Fig. 6b, GST (red) is located in the center of the clamp region, next to the central mutation region (green dots). (ii) Serysheva et al. (41) used homology modeling to predict the three-dimensional structure of RyR1 sequence 216–572, which contains much of the N-terminal mutation region, and they fitted the predicted structure into clamp domain 5 of the three-dimensional architecture of RyR1 (shaded yellow in one clamp structure in Fig. 6b). Domain 5 is adjacent to the central domain GFP insertion that we have mapped. Thus, two independent lines of evidence imply that the N-terminal and central mutation domains are adjacent to one another in the three-dimensional structure of the RyR, a spatial relationship consistent with the domain/domain zipping/unzipping hypothesis of Ikemoto and Yamamoto (40).
The Central Mutation Region in the Skeletal Muscle RyR Is Implicated in Skeletal Muscle-type Excitation-Contraction Coupling
The communication between DHPRs in the plasma membrane and RyRs in the sarcoplasmic reticulum membrane is the key step linking neuron excitation and muscle contraction. In cardiac muscle-type excitation-contraction coupling, a small amount of Ca2+ influx via the DHPR triggers RyR2, a mechanism well known as “calcium-induced calcium release” (12). In skeletal muscle-type excitation-contraction coupling, physical contact between DHPRs and RyRs is essential because conformational changes occurring in DHPRs following plasma membrane depolarization directly induce RyR1 proteins to release Ca2+, a mechanism known as “depolarization-induced calcium release” (42). A region within the cytosolic II–III loop of the DHPR α1-subunit is an important determinant of the interaction between the DHPR and RyR1 (43), but other regions and subunits of the DHPR may also be important (44). However, definitive identification of the regions in the RyR1 sequence that are involved in signaling with the DHPR remains elusive. At least six regions in the sequence of RyR1 have been implicated as contributing to the DHPR interaction or to skeletal muscle-type excitation-contraction coupling; these include residues 1076–1112 (45), residues 1303–1404 (containing divergent region 2) (46), residues 1837–2168 (containing divergent region 3) (47), residues 1924–2446 (containing the central mutation region) (48), residues 2644–3223 (46), and residues 2659–3720 (49).
These data suggest that the DHPR-interacting regions are spread over a wide region of RyR1 primary sequence, encompassing residues 1076–3720. However, it is possible that some or all of these dispersed regions become closely apposed spatially in the three-dimensional structure of RyR1. For example, these diverse regions may all lie in the clamp structures that form the corners of the cytoplasmic assembly, which have been implicated in RyR1/DHPR interactions (42, 50).
We have localized three of six proposed DHPR-interacting regions in the three-dimensional structure of RyR2. Although our mappings were obtained for RyR2, they are applicable to RyR1 because the high degree of sequence identity among the isoforms implies that their tertiary structures should be very similar. Indeed, the three-dimensional reconstructions that have been reported for the three RyR isoforms show strict conservation of the domain architecture of the cytoplasmic region and only a few significant differences at the 2–4-nm resolution level (17, 26, 39). In addition to the central mutation region that we mapped to the bridge between domains 5 and 6 in this study (GFP-modified Ser-2367 in RyR2 corresponds to Arg-2401 in RyR1, which falls within the critical sequence 1924–2446), we have mapped divergent region 2 (Thr-1366 in RyR2 corresponds to Thr-1382 in RyR1 in the critical sequence 1303–1404) to domain 6 (20) and divergent region 3 (Thr-1874 in RyR2 corresponds to Glu-1893 in RyR1 in the critical sequence 1837–2168) to domain 9 (33).
In skeletal muscle, RyR1 proteins self-assemble into two-dimensional ordered arrays at the terminal cisternae membrane of the sarcoplasmic reticulum. The DHPRs on the plasma membrane/transverse tubule system also form ordered arrays such that clusters of four DHPR molecules (“tetrads”) oppose the corners of the RyRs in the adjoining terminal cisternae. Paolini et al. (50) have defined the relative orientation of the two arrays, and we have superimposed onto their structural model our three mappings of the putative regions of RyR1 that are involved in interactions with the DHPR (Fig. 6c). Interestingly, the three regions, the central mutation region (green dots in Fig. 6c) and divergent regions 2 and 3 (red and cyan dots, respectively), all lie in the clamps, near the sites where the DHPR and RyR1 interact in the model of Paolini et al. (50).
Acknowledgments
We thank E. Obeng-Gyimah for help with image processing and Jeff Bolstad for excellent technical assistance. We also thank the Molecular Genetics and Electron Microscopy Core Facilities and the Resource for Visualization of Biological Complexity of the Wadsworth Center.
Footnotes
* This work was supported by American Heart Association Grant 0430076N (to Z. L.), by National Institutes of Health Grant AR40615 (to T. W.), and by research grants from the Canadian Institutes of Health Research and the Heart and Stroke Foundation of Canada, Alberta (to S. R. W. C.). The work performed in the Resource for Visualization of Biological Complexity of the Wadsworth Center was supported by National Institutes of Health Biotechnological Resource Grant RR01219.
The abbreviations used are: RyR, ryanodine receptor; DHPR, dihydropyridine receptor; CPVT, catecholaminergic polymorphic ventricular tachycardia; ARVD2, arrhythmogenic right ventricular dysplasia type 2; GFP, green fluorescent protein; HEK, human embryonic kidney; cryo-EM, cryoelectron microscopy; wt, wild type; PIPES, 1,4-piperazinediethanesulfonic acid; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-pro-panesulfonic acid; GST, glutathione S-transferase.
References
- 1.American Heart Association (2005) Heart Disease and Stroke Statistics–2005 Update, American Heart Association, Dallas, TX
- 2.Chugh SS, Senashova O, Watts A, Tran PT, Zhou Z, Gong Q, Titus JL, Hayflick SJ. J Am Coll Cardiol. 2004;43:1625–1629. doi: 10.1016/j.jacc.2003.11.052. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 4.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K. Circulation. 2001;103:485–490. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 5.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A. Hum Mol Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 6.Bauce B, Rampazzo A, Basso C, Bagattin A, Daliento L, Tiso N, Turrini P, Thiene G, Danieli GA, Nava A. J Am Coll Cardiol. 2002;40:341–349. doi: 10.1016/s0735-1097(02)01946-0. [DOI] [PubMed] [Google Scholar]
- 7.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 8.Laitinen PJ, Swan H, Kontula K. Eur J Hum Genet. 2003;11:888–891. doi: 10.1038/sj.ejhg.5201061. [DOI] [PubMed] [Google Scholar]
- 9.Bagattin A, Veronese C, Bauce B, Wuyts W, Settimo L, Nava A, Rampazzo A, Danieli GA. Clin Chem. 2004;50:1148–1155. doi: 10.1373/clinchem.2003.030734. [DOI] [PubMed] [Google Scholar]
- 10.Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Circulation. 2004;110:2119–2124. doi: 10.1161/01.CIR.0000144471.98080.CA. [DOI] [PubMed] [Google Scholar]
- 11.Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Mayo Clin Proc. 2004;79:1380–1384. doi: 10.4065/79.11.1380. [DOI] [PubMed] [Google Scholar]
- 12.Bers, D. M. (2001) Excitation-Contraction Coupling and Cardiac Contractile Force, Kluwer Academic Publishers Group, Dordrecht, The Netherlands
- 13.McCarty TV, Quane KA, Lynch PJ. Hum Mutat. 2000;15:410–417. doi: 10.1002/(SICI)1098-1004(200005)15:5<410::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 14.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 15.Tiso N, Salamon M, Bagattin A, Danieli GA, Argenton F, Bortolussi M. Biochem Biophys Res Commun. 2002;299:594–598. doi: 10.1016/s0006-291x(02)02689-x. [DOI] [PubMed] [Google Scholar]
- 16.Zhao M, Li P, Li X, Zhang L, Winkfein RJ, Chen SRW. J Biol Chem. 1999;274:25971–25974. doi: 10.1074/jbc.274.37.25971. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Zhang J, Li P, Chen SRW, Wagenknecht T. J Biol Chem. 2002;277:46712–46719. doi: 10.1074/jbc.M208124200. [DOI] [PubMed] [Google Scholar]
- 18.Chen SRW, Li X, Ebisawa K, Zhang L. J Biol Chem. 1997;272:24234–24246. doi: 10.1074/jbc.272.39.24234. [DOI] [PubMed] [Google Scholar]
- 19.Du GG, Imredy JP, MacLennan DH. J Biol Chem. 1998;273:33259–33266. doi: 10.1074/jbc.273.50.33259. [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Zhang J, Wang R, Chen SRW, Wagenknecht T. J Mol Biol. 2004;338:533–545. doi: 10.1016/j.jmb.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 22.Frank, J. (1996) Three-dimensional Electron Microscopy of Macromolecular Assemblies, pp. 54–125, Academic Press, Inc., San Diego, CA
- 23.Liu Z, Zhang J, Sharma M, Li P, Chen SRW, Wagenknecht T. Proc Natl Acad Sci U S A. 2001;98:6104–6109. doi: 10.1073/pnas.111382798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G. Nature. 1988;331:315–319. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- 25.Anderson K, Lai FA, Liu QY, Rousseau E, Erickson HP, Meissner G. J Biol Chem. 1989;264:1329–1335. [PubMed] [Google Scholar]
- 26.Radermacher M, Rao V, Grassucci R, Frank J, Timerman AP, Fleischer S, Wagenknecht T. J Cell Biol. 1994;127:411–423. doi: 10.1083/jcb.127.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagenknecht T, Frank J, Boublik M, Nurse K, Ofengand J. J Mol Biol. 1988;203:753–760. doi: 10.1016/0022-2836(88)90207-0. [DOI] [PubMed] [Google Scholar]
- 28.Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument-Bromage H, Tempst P, Marks AR. J Biol Chem. 1992;267:9474–9477. [PubMed] [Google Scholar]
- 29.Jekakumar LH, Ballester L, Cheng DS, McIntyre JO, Chang P, Olivey HE, Rollins-Smith L, Barnett JV, Murray K, Xin HB, Fleischer S. Biochem Biophys Res Commun. 2001;281:979–986. doi: 10.1006/bbrc.2001.4444. [DOI] [PubMed] [Google Scholar]
- 30.Wehrens XHT, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song L-S, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. X. H. T. [DOI] [PubMed] [Google Scholar]
- 31.Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. J Biol Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- 32.Wagenknecht T, Radermacher M, Grassucci R, Berkowitz J, Xin HB, Fleischer S. J Biol Chem. 1997;272:32463–32471. doi: 10.1074/jbc.272.51.32463. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Liu Z, Masumiya H, Wang R, Jiang D, Li F, Wagenknecht T, Chen SRW. J Biol Chem. 2003;278:14211–14218. doi: 10.1074/jbc.M213164200. [DOI] [PubMed] [Google Scholar]
- 34.George CH, Higgs GV, Lai FA. Circ Res. 2003;93:531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- 35.Xiao B, Sutherland C, Walsh MP, Chen SRW. Circ Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- 36.Stange M, Xu L, Balshaw D, Yamaguchi N, Meissner G. J Biol Chem. 2003;278:51693–51702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
- 37.Zissimopoulos S, Lai FA. J Biol Chem. 2005;280:5475–5485. doi: 10.1074/jbc.M412954200. [DOI] [PubMed] [Google Scholar]
- 38.Orlova EV, Serysheva II, van Heel M, Hamilton SL, Chiu W. Nat Struct Biol. 1996;3:547–552. doi: 10.1038/nsb0696-547. [DOI] [PubMed] [Google Scholar]
- 39.Sharma MR, Jeyakumar LH, Fleischer S, Wagenknecht T. J Biol Chem. 2000;275:9485–9491. doi: 10.1074/jbc.275.13.9485. [DOI] [PubMed] [Google Scholar]
- 40.Ikemoto N, Yamamoto T. Front Biosci. 2002;7:d671–d683. doi: 10.2741/A803. [DOI] [PubMed] [Google Scholar]
- 41.Serysheva II, Hamilton SL, Chiu W, Ludtke SJ. J Mol Biol. 2005;345:427–431. doi: 10.1016/j.jmb.2004.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Protasi F. Front Biosci. 2002;7:d650–d658. doi: 10.2741/A801. [DOI] [PubMed] [Google Scholar]
- 43.Grabner M, Dirksen RT, Suda N, Beam KG. J Biol Chem. 1999;274:21913–21919. doi: 10.1074/jbc.274.31.21913. [DOI] [PubMed] [Google Scholar]
- 44.Ahern CA, Sheridan DC, Cheng W, Mortenson L, Nataraj P, Allen PD, De Waard M, Coronado R. Biophys J. 2003;84:942–959. doi: 10.1016/s0006-3495(03)74911-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leong P, MacLennan DH. J Biol Chem. 1998;273:7791–7794. doi: 10.1074/jbc.273.14.7791. [DOI] [PubMed] [Google Scholar]
- 46.Perez CF, Mukherjee S, Allen PD. J Biol Chem. 2003;278:39644–39652. doi: 10.1074/jbc.M305160200. [DOI] [PubMed] [Google Scholar]
- 47.Proenza C, O’Brien J, Nakai J, Mukherjee S, Allen PD, Beam KG. J Biol Chem. 2002;277:6530–6535. doi: 10.1074/jbc.M106471200. [DOI] [PubMed] [Google Scholar]
- 48.Perez CF, Voss A, Pessah IN, Allen PD. Biophys J. 2003;84:2655–2663. doi: 10.1016/S0006-3495(03)75071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakai J, Sekiguchi N, Rando TA, Allen PD, Beam KG. J Biol Chem. 1998;273:13403–13406. doi: 10.1074/jbc.273.22.13403. [DOI] [PubMed] [Google Scholar]
- 50.Paolini C, Protasi F, Franzini-Armstrong C. J Mol Biol. 2004;342:145–153. doi: 10.1016/j.jmb.2004.07.035. [DOI] [PubMed] [Google Scholar]