

Abstract
We conducted molecular dynamics computer simulations of charged histone tail-DNA interactions in systems mimicking nucleosome core particles (NCP) . In a coarse-grained model, the NCP is modeled as a negatively charged spherical particle with flexible polycationic histone tails attached to it in a dielectric continuum with explicit mobile counterions and added salt. The size, charge, and distribution of the tails relative to the core were built to mimick real NCP. In this way, we incorporate attractive ion-ion correlation effects due to fluctuations in the ion cloud and the attractive entropic and energetic tail-bridging effects. In agreement with experimental data, increase of monovalent salt content from salt-free to physiological concentration leads to the formation of NCP aggregates; likewise, in the presence of MgCl2, the NCPs form condensed systems via histone-tail bridging and accumulation of counterions. More detailed mechanisms of the histone tail-DNA interactions and dynamics have been obtained from all-atom molecular dynamics simulations (including water), comprising three DNA 22-mers and 14 short fragments of the H4 histone tail (amino acids 5–12) carrying three positive charges on lysine+ interacting with DNA. We found correlation of the DNA-DNA distance with the presence and association of the histone tail between the DNA molecules.
INTRODUCTION
In the nucleus of eukaryotic cells, DNA exists mainly as linear domains of uniform DNA-protein complexes, the nucleosomes. The most regular central part of the nucleosome is the nucleosome core particle (NCP), which consists of ∼146–147 bp of DNA wrapped as a 1.75-turn superhelix around a wedge-like octamer of core histones formed by the core-histone protein octamer (1,2). Variable-length double-stranded linker DNA connects the NCPs with each other to form nucleosomal arrays that condense into 30-nm chromatin fibers (3–5). Each of the core histones has unstructured, highly basic N-terminal domains called “histone tails” protruding through the DNA superhelix. The structure of the histone tails largely escapes detection by x-ray crystallography and other experimental methods, implying that they are highly flexible and dynamic (1,6). The histone tails are essential for maintenance of the higher-order compact folded structures of chromatin and for regulation of transcription and replication (7,8). These functions of the histone tails are regulated by covalent modifications of the amino acids, which may change the net charge and distribution of the charged groups in the tails (6,9).
In vitro evidence suggests that folding of the array of nucleosomes into chromatin fibers to attain a compacted secondary 30-nm structure, as well as further interarray oligomerization into tertiary chromatin structure, occurs through two separate salt-dependent condensations (3–5). The fact that the condensed structure of chromatin is induced by high (monovalent) salt and by the presence of divalent ions like Mg2+ and Ca2+ is indicative of an electrostatic mechanism (10–12). Since the basic core histone tails are necessary for both secondary and tertiary condensation (3,4), it is generally presumed that they participate in both intra- and interarray nucleosome-nucleosome interactions. A recent study demonstrated a relocation of the H3 tail domain from mainly intranucleosomal interaction within the fully extended nucleosomal array at low salt to primarily internucleosomal interactions upon the formation of compacted secondary and tertiary structures (13). This suggests that the importance of the tails in chromatin condensation is related to their participation in internucleosome interactions. Furthermore, a recent study demonstrated that all of the different histone tails H2A, H2B, H3, and H4 are involved in the salt-dependent oligomerization of array fibers and that they function independently and additively (14).
Experiments using mainly small-angle x-ray scattering (as well as osmometry and electrophoretic measurements) have shown that isolated (linker-free) NCPs display aggregation as a result of increased concentration of mono- and divalent salt present in solution (15–18), and this compaction seems to be induced by the presence of the charged histone tails, suggested to be due to tail bridging between different particles (15–18). In the NCP, ∼50% of the (−294) negative charge of the DNA polyion is neutralized by positive charges from the histones. The NCP thus has a net negative charge of about −148, and it can be viewed as a highly negatively charged central particle (−236) having eight flexible positively charged chains with a net charge of +88 protruding from it. The details of the mechanism by which these like-charged particles are able to attract each other and facilitate compaction, possibly mediated by bridging through the basic histone tails, is not fully established. Since isolated NCPs display properties qualitatively similar to those of chromatin fibers in terms of the experimentally observed salt-induced compaction, and are more susceptible to biophysical aggregation studies (15–18), this system is a good starting point for theoretical modeling investigations.
Recently, much attention has been focused on understanding the experimentally observed electrostatic association between negative like-charged biopolyelectrolytes such as DNA, F-actin, and filamentous virus particles, induced by the presence of multivalent ions (e.g., Mg2+, Ca2+, Co(NH3)63+, and polyamines) (19–24), and there are several theoretical approaches to explain this behavior (20–22,25,26). Like-charged polyelectrolytes would be expected to repel each other on the basis of a simple mean-field Poisson-Boltzmann treatment of the electrostatics. However, it is now well established that fluctuation-induced dynamic correlation of cations shared by different polyanions gives rise to attractive electrostatic force contributions that explain experimental aggregation (25–28).
Different colloidal systems of negatively charged macroion surfaces with attached (or free) positively charged polyelectrolyte chains also show experimental attraction behavior (29). Such systems have also been studied with computer simulations and theoretical models (30–32). From these studies it is clear that attraction can occur not only due to mobile counterion fluctuation but also because of chain bridging of both electrostatic and entropic origin. The entropic bridging is caused by the gain in entropy that the system incurs by the flexible chains neutralizing not only its own oppositely charged surface, but extending to the charged surfaces of other particles as well. The aggregation behavior of highly negatively charged polyelectrolyte systems bearing grafted positively charged polyion chains thus exhibits considerable principal similarity with the problem of the condensation of NCPs.
Various coarse-grained models have been used in modeling nucleosomal arrays containing linker DNA (33–39). A common feature of these approaches is that the charged flexible histone tails are incorporated either not at all or to a limited extent (in studies by Beard and co-workers (38,39), only one static inflexible H3 tail is present) and that salt dependence is treated highly approximately with effective potentials or within a mean-field Poisson-Boltzmann Debye-Hückel approximation that does not take ion correlation effects into account. Recently, Sun et al. (40) described an improvement of previous work (36,38,39) in their study of the electrostatic mechanism of nucleosomal array folding by computer simulations using an irregular discrete surface-charge optimization (DiSCO) model of the nucleosome core particle including histone tails. This approximation contains the linker DNA and can thus model chromatin-fiber folding, the salt dependence of which was well borne out by these simulations. However, because the histone-tail charges were fixed and the effects of added salt are described by a Debye-Hückel mean-field Poisson-Boltzmann treatment, entropically induced tail bridging and ion-ion correlation attraction mechanisms are not included. In the work described here, we take an alternative approach in modeling the attraction between isolated (linker-free) NCPs mediated by charged histone tails.
Due to the complexity and enormous scale of a full atomic model of several nucleosome core particles with charged histone tails including solvent water, a simplified model to treat NCP aggregation must be invoked. We use a coarse-grained model in which the NCP is described as a negatively charged sphere with attached positively-charged flexible chains whose length and number of charges mimic the real system (Fig. 1). Molecular dynamics (MD) simulations in a dielectric continuum model containing explicit mobile counterions and various amounts of added salt describe the effect of changing experimental conditions. Our simulation results are in agreement with experimentally induced salt dependence of NCP aggregation. They also give insight into the tail-mediated bridging between core particles and are of relevance for the mechanism of secondary and tertiary condensation of nucleosomal arrays. To our knowledge, this is the first theoretical demonstration of nucleosome-nucleosome tail-bridging attraction within a statistical mechanical treatment, which (within the model) gives the equilibrium description (NVT ensemble simulations) for a system whose parameters model the NCP, and which explicitly includes all charged mobile particles, taking into account salt-dependent ion-ion correlation and flexible charged tails.
FIGURE 1.

Initial configuration of the NCP particles of the coarse-grained MD simulations.
To obtain information on the capacity and relevance of histone-tail-mediated DNA-DNA bridging at a detailed molecular level, we complemented the coarse-grained model with full atomic MD simulations with explicit water solvent molecules to study a system of three ordered DNA oligonucleotides in the presence and absence of the fragment containing amino acids 5–12 of the histone tail H4, which contains three charged Lys+ amino acids (Fig. 2 a). We carried out two 30-ns MD simulations for two identical systems with three DNA oligomers and containing 14 tail fragments, but starting from two different initial configurations (Fig. 2 b), as well as one 20-ns simulation for a similar system but with only K+ counterions, without tail fragments. The presence of three DNA 22-mers in the simulation cell allowed direct observation of tail-mediated DNA-DNA contacts. The objective was to add information to the coarse-grained simulations and investigate whether charged lysine residues of such a histone tail fragment could mediate association between DNA molecules, as well as to gain molecular-level insight into this mechanism. The results of the MD simulations do indeed demonstrate histone tail bridging interactions and aggregation of DNA molecules at the molecular level.
FIGURE 2.

(a) Amino acid sequence of the N-terminal fragment of histone H4 studied in this work, consisting of amino acids 5–12. NZ+ atoms of lysine are highlighted by blue balls. (b) Initial configurations for all-atom MD simulations: left, Tail-1; middle, No-Tail (both initial DNA-DNA distance 34.6 Å); right, Tail-2 (initial DNA-DNA distance 22.5 Å).
COMPUTATIONAL METHODS
Coarse-grained dielectric-continuum-model MD simulations of NCP condensation
The NCP was modeled as a spherical particle of effective radius 51 Å with net charge −236 combined with eight strings of linearly connected +1 charges of effective radius 2.5 Å and bond length 7 Å, which represent the histone tails. The charge of the central particle reflects the electrostatic balance between 147 base pairs of the double-stranded DNA (charge −294) and the net positive charge +58 of the amino acids comprising the globular domains of the histones. The central part of the real NCP (the globular part of the histone octamer with 1.75 superhelical turns of 147-bp DNA wrapped around them) is often seen in the textbooks as a short (50–60 Å) cylinder of 100–110 Å diameter. The volume and averaged charge density of the central sphere in our NCP model are roughly equal to these dimensions of the cylinder approximation of the real NCP.
The distance between charged groups of the histone tails (7 Å) was evaluated assuming extended conformation of the polypeptide chain and even distribution of Lys+/Arg+ amino acids. The number of charged groups in each tail was set according to the charges of the real histone tails: H2A, +9; H2B, +12; H3, +13; H4, +10. We used amino acid sequences from Hansen et al. (41) and considered the amino group of the N-terminal protonated (+1). The total charge of histone tails was +88, which, with the charge of the central unit, yielded a total charge of NCP −148.
The interaction potential (force field) of the coarse-grained model consisted of three parts: electrostatic, short-range, and bond potentials. The electrostatic interaction was defined in a standard manner, as a sum of Coulombic potentials from all the charges in the system, in dielectric media with dielectric constant ɛ = 78. In our models, we consider the solvent as a dielectric medium. The interaction potentials thus represent an approximation to the solvent-mediated effective potential between the charged particles in the solution. This means that ɛ is a parameter of the effective ion-ion potential (which sometimes is chosen to be distance-dependent). Existing studies of molecular-ion systems at a molecular level have shown that the effective (mean force) potentials of ions in water usually have one or two oscillations around the Coulomb potential with ɛ = 78 at small (within 8 Å) distances between the ions (42). These oscillations reflect the molecular nature of the solvent. Still, for many applications, including strong polyelectrolytes and high salt concentrations, continuum dielectric models with constant dielectric permittivity have been successful (see, e.g., the review by Pratt et al. (43)). The Ewald summation method (44) was used to compute the long-range part of the electrostatic forces and energies. The short-range potential, acting between any pair of elements of the coarse-grained model (that is, NCP center particles, histone-tail monomers, and ions) has the following form:
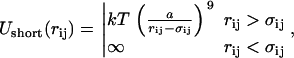 |
(1) |
where rij is the distance between elements i and j, σij = σi + σj is the sum of their hard core radii, and a = 3 Å, a parameter having the sense of the effective thickness of the soft repulsion potential. For hard core radii, we used σ = 47 Å for the NCP central unit and σ = 1 Å for histone tail monomers and ions. The potential in Eq. 1 has effective interaction radius σi + σj + a, which gives an effective size of NCP core of 51 Å and an effective ion (or tail-monomer) diameter of 5 Å. This value fits the position of the first maximum in the −NH3+···Cl− distribution in all-atom simulations of spermine4+ salt solution in water, and reflects the effective size of the charged amino group within the coarse-grained model. Generally the calibration of the force field parameters for the coarse-grained simulations was done on the basis of comparison with all-atom simulations, though no detailed tests have been made. Previously, interaction potential of the ninth inverse power of r has been used in a Monte Carlo simulation of ions around DNA by Montoro and Abascal (45).
The bond potential acting between neighboring monomers of a histone tail has the form
 |
(2) |
with equilibrium distance req =7 Å and characteristic fluctuation Δ = 1 Å. The same kind of potential acts between the NCP core and the first histone monomers of each tail placed outside (req = 55 Å) the central particle of size 47 Å. Some of the first histone monomers of different tails belonging to the same NCP are also bound by the harmonic bond potential to fix positions of their attachment to the central core. Fig. 3 shows the geometry of their connections, with the corresponding equilibrium distances.
FIGURE 3.

Model of the NCP. The three schemes on the righthand side show the connection of the histone tails to the negatively charged core and to each other (two groups of each tail closest to the core are shown as blue spheres). The center of the core particle is drawn as a red ball. The coordinate system has its origin in the center of the core particle with exit points of the H2B and H3 tails forming the x,y plane (z = 0). The middle scheme shows the view from the top along the z axis; top and bottom schemes display projections resulting from respective counterclockwise and clockwise 90° rotations of the scheme in the middle. The lines between particles indicate bonds described by harmonic potential with equilibrium distances in Angstroms indicated in the figure. The three pictures on the lefthand side show views of NCP with all charges in the histone tails and the central core particle in the same projections as the corresponding schemes shown on the righthand side.
The introduced coarse-grained model represents an “idealized symmetrical” approximation of the real structure of the NCP determined by x-ray crystallography (1,6,46) (see Fig. 2 of Wolfe and Hayes (46)). The distance from the first charged group of the tails to the center of the core was set to 55 Å for all tails. Points of exits for the four histone tails (Fig. 3) which protrude through the gears of the DNA superhelix in the real NCP, were set in the x,y plane (z = 0) with 90° between them. The exit points of the H4/1 and H4/2 histone tails were placed 25 Å below and above the x,y plane, opposite to each other and perpendicular to the dyad axis (Fig. 3). Finally, the H2A/1 and H2A/2 tails exit the core particle 25 Å below and above the x,y-plane and are positioned between the H2B/1 and H2B/2 tails.
The constant-temperature molecular dynamics algorithm implementing the Nóse-Hoover thermostat (47), with temperature 300 K and relaxation time 2 ps, was used in the simulations. Since the solvent is not included explicitly, the molecular dynamics should be considered as a tool to generate the canonical (NVT) ensemble, producing the same kind of results as the Monte Carlo simulations. For strongly charged polyelectrolyte systems such as those considered here, the molecular dynamics is a more efficient way to sample the configuration space compared to the Monte Carlo algorithm. In the last decades, constant-temperature molecular dynamics with thermostats of different kinds was often used in polyelectrolyte and other coarse-grained simulations (48–51).
The dynamics in such simulations is artificially accelerated and may be qualitatively reconstructed by scaling the time with some factor. In this work, we do not evaluate this factor and refer to time as it appears in the molecular dynamics equations. For equilibrium canonical properties, the Nóse-Hoover molecular dynamics provides correct results.
Four systems were simulated for 2 × 107 MD steps (200 ns) in a cubic simulation box of side length 400 Å containing 10 NCPs, in the starting configuration shown in Fig.1. The NCP concentration in the box roughly corresponds to the high limit of nucleosome concentration inside the eukaryotic cell nucleus in the interphase, estimated by combined fluorescent correlation spectroscopy and confocal imaging (52). Each system contained a different combination of salt: 1), Salt Free: 1480 K+ ions to neutralize the NCP charge; 2), Low Salt: 320 KCl (8.3 mM) + 1480 K+; 3), Normal Salt: 2520 KCl (65.4 mM) + 1480 K+; 4) Magnesium Salt: 160 MgCl2 (4.15 mM) + 740 Mg2+. The concentration of NCPs in the simulation box was 260 μM and corresponds to 76 mM in DNA phosphate groups for similar systems of real NCPs.
Trajectories for analysis were collected after each 1000 time steps (10 ps). Averages were calculated for the last 25% of the trajectories after achieving convergence in the NCP-NCP radial distribution functions (RDF).
All-atom explicit solvent MD simulation of DNA and histone H4 tail fragments
Three all-atom molecular dynamics simulations were performed in a hexagonal cell containing 9430 water molecules and three 22-base pair B-DNA oligomers with the sequence 5′-d(GATGCAGTCACCGCGAATTGGC)·5′-d(GCCAATTCGCGGTGACTGCATC). Two systems, in addition to DNA and water, contained 14 fragments of the histone H4 (amino acids 5–12, charge +3, Fig. 2 a) and 84 K+ cations. They were simulated for 30 ns. The three simulated systems are abbreviated below as Tail-1, No-Tail, and Tail-2. The “No-Tail” system contained 126 K+ ions to neutralize the DNA charge. It was simulated for 20 ns. The difference between the Tail-1 and Tail-2 systems was in the initial positions of the DNA 22-mers and the histone fragments. In the Tail-1 system, the initial distance between axes of DNA was 34.6 Å, with histone tail fragments distributed between the DNA molecules (Fig. 2 b, left). In the No-Tail simulation, the oligomers were put in the same initial positions as in the Tail-1 system, but no tail fragments were present (Fig. 2 b, middle). In the Tail-2 system, the initial DNA-DNA distance was 22.5 Å, with the tail fragments placed around the “bunch” of the three DNA oligomers (Fig. 2 b, right). The initial appearance of all simulated cells was a hexagon with side dimensions 40 Å, height 86 Å, and volume 357.5 nm3.
We used the CHARMM27 force field (53,54) with the flexible simple-point-charge (SPC) model of water (55) and K+ model from Heinzinger (56). The simulation software was the MDynaMix package (57). Unlike our earlier MD simulations, where we applied “boundary” conditions along the z axis (58,59), in this work DNA oligomers were free to move. The Nóse-Hoover thermostat and barostat (47) were used in all the simulations to keep the temperature at 300 K and the pressure at 1 atm. The pressure was controlled separately in all directions. We implemented a double-time-step algorithm (60), with a short time step of 0.2 fs for fast intramolecular vibrations and short-range (within 5 Å) intermolecular interactions, and a long time step of 2 fs for longer-range interactions (up to 13 Å cutoff). The Ewald method (44) was used to treat the electrostatic interactions. All systems were equilibrated for 360 ps under constant volume/temperature conditions (T = 300 K; 200 ps with DNA and tails fixed + 160 ps with only DNA fixed) followed by release of the DNA constraints and stepwise heating of them (50 K, 40 ps) from 50 to 298 K under constant pressure (1 atm). The switch from constant-volume (NVT) to constant-pressure (NPT) conditions was accompanied by a modest contraction of the cell volume to 332–333 nm3 in the Tail-1 and Tail-2 systems, and 319–320 nm3 in the No-Tail system (density 1.118 g/cm3 in all systems). During the course of the simulations, the concentrations of the components were: DNA, ∼0.015–0.016 M (0.66–0.69 M in nucleotides/L); histone tail fragments, ∼0.07 M; K+, 0.42–0.44 M for Tail-1 and Tail2, and 0.65–0.66 M for No-Tail.
Trajectories were collected with a 0.1-ps time step. Analysis of the MD trajectories was carried out using procedures of the MDynaMix package (57) with a number of additional scripts. To describe DNA-DNA correlations in the simulation cell, we define the distance between the DNA oligomers as the calculated shortest distance for a given point of the DNA axis to the axis of the other DNA The program Curve5 (61) was used to calculate the coordinates of the DNA axes at any moment of the simulation. For each DNA, an array of 65 axis points was calculated with the central 50 points used to determine the distance to the axes of the other two DNAs. All possible pairs of the DNA were scanned. Note that for the hexagonal cell, the distance for perfectly aligned rods is equal to the length of the hexagon side, which fluctuated in the range 37–42 Å during the course of simulations (see Fig. 6). More information about other aspects of the MD simulation setup can be found in previous works (58,59,62–64).
FIGURE 6.

Distribution of the average DNA-DNA distance taken at different time intervals of the MD simulation runs. Shaded boxes display the range of oscillation of the simulation box side length observed during the MD simulations; this value gives the separation between ideal rods evenly placed in the hexagonal cell. Vertical bars indicate the initial DNA-DNA distance in each of the systems. (Averaging in the interval from 5 to 10 ns (4–8 ns for the No-Tail system) is not shown.)
RESULTS
MD simulations of a coarse-grained model show NCP condensation mediated by salt-dependent tail-bridging interactions
Fig. 4 displays snapshots at the end of the simulations in the four systems (Salt Free, Low Salt, Normal Salt, and Mg Salt). Inspection of the trajectories shows that in the beginning of the simulation, the histone tails rapidly collapsed onto their own cores. The mobile cations quickly diffused and associated to the cores. Exchange of mobile cations between the close vicinity of the NCPs and bulk solution, as well as motion of the tails on the negative surface, is rapid compared to the mobility of the core particles. Movement of the NCP particles is the slowest process in the system. However, in all the systems, after ∼5 × 106 steps (50 ns), the NCP-NCP distribution was stabilized. This observation is supported by analysis of the NCP-NCP RDFs taken at different stages of the simulations (data not shown), and by a change in magnitude of the maximum in the RDFs of the histone tails relative to the external cores (see Fig. 5 b, inset).
FIGURE 4.

Snapshots showing distribution of the NCP particles at the end of the four coarse-grained MD simulations with different concentration of K+ (three pictures to the left) and in the presence of Mg2+ (right).
FIGURE 5.

RDFs calculated from the last 25% of the four coarse-grained MD simulations. (a) NCP-NCP correlation function. (b) Intermolecular tail-NCP RDF. Appearance of maximum below 60 Å indicates NCP-NCP aggregation. (Inset) Dependence of the maximum of the RDF on simulation time. (c) Intramolecular tail-NCP RDF. An arrow shows detachment of the histone tails from the negative core caused by interaction with other NCP particles observed in the Normal Salt and Mg Salt systems. (d). RDFs of the counterions (K+ or Mg2+) relative to the center of the NCPs. Arrow in the inset graph indicates a drop in cation concentration on the border between condensed NCP particles and bulk solution observed in the Normal Salt and Mg Salt systems.
In the Salt Free and Low Salt systems, the NCPs repel each other and only rarely form associates consisting of two to three particles. In the Normal Salt system, groups of two to five particles are more stable and usually form “lines.” In the Normal Salt system, we observed slow exchange of the NCPs between aggregates is observed, often seen as a short-distance shift of an NCP from one aggregate to another. Movement of the NCPs in the Mg Salt system is seen as a continuous formation of a single big aggregate that includes all the NCPs. It is formed after ∼100 ns of simulation and remains stable for the rest of the simulation time (with slow movements of the NCPs relative to each other and occasional dissociation of one of the particles).
In Fig. 5, RDFs calculated from the final 25 ns are presented. Pronounced maxima are observed in the NCP-NCP RDF (Fig. 5 a) for the Normal Salt and Mg Salt systems at a distance that only slightly exceeds direct contact between NCPs, whereas no NCP-NCP contacts are seen in the Salt Free and Low Salt systems. Furthermore, aggregation of the NCPs proceeds to a different extent in the Normal Salt and Mg Salt systems. The intensity of the first maximum in the RDF of the Mg Salt system exceeds by almost twice the same peak of the Normal Salt system. Fig. 5 b shows intermolecular, and Fig. 5 c intramolecular, RDFs of the tails relative to the center of the NCPs. The histone tails play a crucial role in formation of the NCP-NCP contacts. The snapshots (Fig. 4) and the intermolecular RDFs (Fig. 5 b) show that the NCP aggregates are maintained by cross-linking. On the other hand, no cross-linking occurs in the Salt Free and Low Salt systems.
Observation of the trajectories and comparison of intramolecular RDFs (Fig. 5 c) reveal that the tails relocate from association with the “host” particle and become associated to the neighboring nucleosome only after NCPs approach each other; i.e., the tails remain close to the host core in isolated nonaggregated NCPs at all salt conditions. In the initial stages of the simulations, intramolecular RDFs of the tails in the Mg Salt and Normal Salt systems are similar to those of the Salt Free and Low Salt systems (not shown). A noticeable presence of the tail outside its own particle at a distance >57 Å in the intramolecular RDF (Fig. 5 c, arrow) appears only after NCPs come in contact and form aggregates. The peak at 55 Å corresponds to the particles closest to the negative sphere, whose movement is restricted by bonds.
The mobile cations (K+ and Mg2+) accumulate near the negative surface of the NCPs (Fig. 5 d). When stable contacts between the NCPs are formed, most of the cations are gathered inside the condensed phase and there is a drop in concentration of the cations between bulk and NCP aggregates (Fig. 5 d, inset, arrow). Interaction of oligocationic tails with chlorine anions is reduced due to the confinement of the tails in the area of negative electrostatic field from the central particle. Although some accumulation of Cl− ions is seen in the vicinity of the tails, the intensities of the maxima in the corresponding RDFs are lower than those of the K+-Cl−- and Mg2+-Cl− RDFs (data not shown).
The results are in excellent agreement with experimental data. Bertin et al. (17) and Mangenot et al. (18) reported a sharp change of the second virial coefficient in solutions of NCPs at salt concentration >50 mM NaCl, which they suggested was due to NCP-NCP interactions via histone-tail cross-linking. Bearing in mind the contribution of the NCPs' own counterions (Donnan salt exclusion effect), the salt concentration used in the Normal Salt system (65.4 mM KCl) corresponds to ionic condition in the real NCP solution of ∼80–150 mM monovalent salt where Mangenot et al. (18) observed a maximal decrease in the second virial coefficient. Similarly, in the presence of a millimolar concentration of Mg2+ and low concentration of monovalent salt, de Frutos and co-workers observed (15) aggregation of the NCPs. Tailless nucleosomes do not show similar properties.
The authors of the cited works argue that increase of the salt concentration above 50 mM leads to the dissociation of the tails from the NCPs, making them capable of bridging with other NCP particles. The concentration of the NCPs in solutions studied by Livolant and co-workers was substantial (∼2 mM or higher in DNA phosphates) (18) and it is reasonable to assume that most of the NCPs were present in the form of “loose” dynamic aggregates with the “maximal tail extension” measured by small-angle x-ray scattering corresponding to the length of the tail domains acting as bridges in the NCP aggregates. The steep increase from 0 to 16–24 Å of the tail extension with increase of NaCl from 0 to 50 mM reported in Bertin et al. (17) and Mangenot et al. (18) would correspond to dissociation from the core of relatively short tail fragments (carrying two to four Lys+/Arg+ charges) and is in agreement with our simulation results (tail RDFs shown in Fig. 5, b and c). We conclude that formation of the NCP aggregates becomes possible when salt conditions allow the existence of a sizeable population of partially dissociated histone tails (i.e., short fragments of tails carrying charge +2–+4). These fragments are sufficient to span the distance between DNA of the neighboring NCPs, as demonstrated by the all-atom MD simulations reported below.
All-atom MD simulations demonstrate histone tail bridging between DNA molecules
In all three simulated systems, NoTail, Tail-1, and Tail-2, DNA preserves the B-form in all features of its structure (base geometry, sugar puckering, torsions, and groove width and depth), with sequence-specific variation and dynamics consistent with the results of most MD studies on B-DNA oligomers reported in the literature (data not shown). A wealth of information is obtained from analysis of the trajectories collected in the all-atom MD simulations. In forthcoming work, more detailed presentation and analysis of the data on DNA hydration and interaction with ions and tails, as well as dynamics of the DNA structure, will be given. The present work concentrates on the description of DNA-DNA interaction and the participation of the histone tail fragments in bridging the neighboring DNA oligomers.
Diffusion parameters (diffusion coefficient, D, and center-of-mass displacement, 〈R〉) of the DNA 22-mer, H4 tail fragments, and K+ were determined for different periods of the simulations, using 5-ns (Tail-1 and Tail-2) and 4-ns (NoTail) windows. Data on diffusion are presented in Table 1. Although the simulation time is too short to obtain quantitatively reliable diffusion coefficients for DNA, the monitoring of this dynamic parameter gives an indication of how the presence of tails affects DNA. The presence of the histone tail fragments leads to a substantial reduction in mobility of the DNA oligomers. The diffusion coefficient of DNA is about two times lower in the Tail-1 and Tail-2 systems compared to the value in the No-Tail system: DDNA ≈ 0.007–0.012 × 10−9 m2/s (〈RDNA〉 ≈ 2.0–2.7 Å/ns) in the Tail-1/Tail-2 system (from 5 to 30 ns of simulation time); compared to DDNA ≈ 0.017–0.027 × 10−9 m2/s (〈RDNA〉 ≈ 3.2–4.1 Å/ns) in the No-Tail system (from 4 to 20 ns). In all systems, K+ ions and water are mobile, with no significant differences in diffusion parameters in the beginning or during the course of simulation; DK ≈ 0.68–1.08 × 10−9 m2/s (〈RK〉 ≈ 20.3 ÷ 25.5 Å/ns); Dwat ≈ 1.6–1.8 × 10−9 m2/s (〈Rwat〉 ≈ 31.0–32.8 Å/ns). The mobility of water is slightly higher in the No-Tail system compared to the Tail-1/Tail-2 systems where additional solute (14 histone tail fragments) is present. Diffusion parameters of the tails vary in the range Dtail ≈ 0.18–0.68 × 10−9 m2/s (〈Rtail〉 ≈ 3.4–6.4 Å/ns) during the simulation, showing faster mobility at the initial stage of the MD run (from 0 to 5 ns).
TABLE 1.
Diffusion parameters of the DNA oligomers, fragments of the H3 histone tails, and K+ calculated for different intervals of the MD simulation runs
| Time interval (ns) | No-Tail | Tail-1 | Tail-2 |
|---|---|---|---|
| 0–5 ns | 0.0278 (4.10) DNA | 0.0173 (3.24) DNA | 0.0103 (2.52) DNA |
| (0-4 ns for No-Tail) | 1.012 (24.7) K+ | 0.0558 (5.8) H43+ | 0.0680 (6.40) H43+ |
| 0.850 (22.6) K+ | 0.686 (20.3) K+ | ||
| 5–10 ns | 0.0210 (3.56) DNA | 0.0116 (2.66) DNA | 0.0119 (2.69) DNA |
| (4-8 ns for No-Tail) | 1.080 (25.5) K+ | 0.0268 (4.03) H43+ | 0.0405 (4.94) H43+ |
| 0.785 (21.7) K+ | 0.720 (20.8) K+ | ||
| 10–15 ns | 0.0243 (3.85) DNA | 0.0116 (2.67) DNA | 0.0109 (2.58) DNA |
| 0.994 (24.4) K+ | 0.0296 (4.24) H43+ | 0.0316 (4.38) H43+ | |
| (8–12 ns for No-Tail) | 0.855 (22.7) K+ | 0.748 (21.2) K+ | |
| 15–20 ns | 0.0261 (3.97) DNA | 0.0105 (2.55) DNA | 0.0103 (2.52) DNA |
| (12–16 ns for No-Tail) | 0.973 (24.2) K+ | 0.0307 (4.30) H43+ | 0.0270 (4.00) H43+ |
| 0.915 (23.5) K+ | 0.690 (20.4) K+ | ||
| 20–25 ns | 0.0171 (3.23) DNA | 0.0095 (2.40) DNA | 0.0093 (2.36) DNA |
| (16–20 ns for No-Tail) | 0.910 (23.4) K+ | 0.0350 (4.66) H43+ | 0.0330 (4.46) H43+ |
| 0.840 (22.5) K+ | 0.730 (20.9) K+ | ||
| 25–30 ns | – | 0.0108 (2.55) DNA | 0.0071 (2.07) DNA |
| 0.0292 (4.20) H43+ | 0.0187 (3.36) H43+ | ||
| 0.853 (22.7) K+ | 0.797 (21.9) K+ |
The first number is the diffusion coefficient (in m2/s multiplied by 109), the number in parentheses is the mean center-of-mass displacement, 〈R〉, in Å/ns. Due to the expensive memory requirements, trajectories of water molecules were not collected over the entire simulation; instead, for each system, two intervals of 4–5 ns each were recorded. Diffusion of water is fast, D = 1.8 × 10−9 m2/s with 〈R〉 = 32.8 Å/ns in the No-Tail system, and 1.6 × 10−9 m2/s with 〈R〉 = 31.0 Å/ns in the Tail-1 and Tail-2 systems.
The movement of DNA is slow, but for the present purpose of investigating tail-mediated DNA contacts for a compact DNA system, the timescale of the simulation should be sufficient. The mean-square displacement of DNA molecules is ∼15 Å (12–16 Å for No-Tail from 4 to 20 ns, and 10–14 Å for Tail-1/Tail-2 from 5 to 30 ns) during the simulation run which, in combination with the ability of the oligonucleotides to rotate and slide relative to each other, gives a lot of flexibility to adjust to tail interactions. The tails display a mean-square displacement of ∼15–30 Å, and there is a large number of them present (14 molecules) for interaction with three different DNA molecules, which improves the statistics. Thus, the ability of tails to induce various DNA-DNA contacts should be adequate. Furthermore, the simulation of two systems with very different initial positioning of the tails relative to DNA, to a certain extent enables testing of the effectiveness of tail presence for inducing DNA-DNA interactions.
It is important that the ion distribution is converged to have a reliable description of the electrostatic interactions (65,66). On the average, each K+ ion travels ∼100 Å during the simulation time in all the systems. Recently Varnai and Zakrzewska demonstrated that for a single oligonucleotide system of smaller volume than ours (180 nm3 compared to 330 nm3), potassium ions sampled >90% of the MD simulation box volume after 10 ns (65), indicating that the present simulation time of 30 ns should be satisfactory in this respect.
In Fig. 6, we compare the No-Tail system with the Tail-1 and Tail-2 systems with respect to the averaged distance between axes of pairs of DNA molecules in the simulation cell (the calculation of which is defined in Computational Methods) taken with 4-ns (No-Tail) and 5-ns (Tail-1 and Tail-2) windows. In the presence of tails, the most populated DNA-DNA distances are in the range 24–34 Å (Fig. 6 middle, right). On the other hand, without tails the distribution is shifted to larger distances around 33–48 Å (Fig. 6 left). The system without tails thus expands and tends to distribute the DNA as far as possible from each other close to the “ideal” packing of ∼40 Å.
Although the curves show some structure, due to the short time of averaging, the range of DNA-DNA distances is similar for the Tail-1 and Tail-2 systems. This demonstrates that despite the different initial positioning of DNA and tails in Tail-1 and Tail-2, there is a statistical appearance of close association of DNA oligonucleotides when the prehistory of the “abnormal” Tail-2 starting configuration should have relaxed. In the No-Tail system, DNA oligomers repel each other. After the release of DNA molecules in the beginning of the MD run, the oligonucleotides sample distances larger than the initial separation of 34 Å. The prehistory of the Tail-2 system makes it less mobile compared to the Tail-1 system. However, after ∼10 ns, we observe that in both systems, DNA-DNA distances varies in the same region, between 22 and 38 Å.
The difference in DNA-DNA interaction is also very clearly demonstrated in the snapshots of the three DNA molecules in the simulation cell for the cases of the No-Tail and Tail-1 systems in Fig. 7. In Fig. 7 a, a snapshot of close DNA-DNA association from the Tail-1 system taken at t = 21.92 ns is shown. Close association at the upper parts of all three DNA oligomers mediated by histone tails is visible. For comparison a similar snapshot for the No-Tail (t = 16.00 ns) system is shown in Fig. 7 b. For this configuration, one of the three DNA molecules is somewhat tilted compared to the other two. In the Tail-1 system a total of six histone fragments (with four of them inside “the three-DNA bundle”) are located in the region of close DNA-DNA approach, thus reducing the repulsion and forming DNA-DNA bridges.
FIGURE 7.

Snapshots from the all-atom MD simulations (view from top of the simulation cell along the DNA axes; pink spheres are K+). (a) Tail-1 system, t = 21.92 ns; H4 histone tail fragments shown in sticks with NZ atoms of Lys+ as blue spheres. The two top DNA molecules (yellow and beige) lie with tails approximately parallel to each other, with axis-axis distance in the range 27–29 Å; several histone fragments bridge between these molecules. (b) No-Tail system, t = 16.00 ns. DNA 22-mers repel each other and are separated by distances expected for evenly distributed rods (see also Figs. 5 and 7). PDB files with coordinates of all (excluding water) atoms of the snapshots shown in the figure are given in Supplementary Material.
Next we investigate whether correlation exists between DNA-DNA distance and association of cations to areas of close DNA-DNA contacts (Fig. 8). For this purpose, we use a kind of three-dimensional distribution function. The cation density (K+ or NZ+ atom of Lys) is calculated as a function of DNA-DNA separation (ordinate in Fig. 8) and the distance of the charged group to the middle point connecting DNA axes at the location of the closest DNA-DNA distance. This approach is similar to the method used by Hamelberg et al. (67) and in our earlier work (64) to describe correlations between cation binding and the width of the minor groove of DNA. The data comparing the No-Tail and the Tail-1 systems, which had the same starting positions of DNA, clearly shows that in the presence of tails, cations tend to accumulate with a preference for configurations of close contact between DNA molecules. The maxima of the tail NZ+ atom distribution is correlated with positions for which there is high probability of short DNA-DNA axis distance. This demonstrates the statistical effect of how the presence of tails is related to close DNA-DNA contacts. We also see that the potassium counterion distribution is more spread out in the Tail-1 system, indicating the replacement of K+ counterions at electronegative sites close to DNA by the charged tail groups. The average distance distribution data for the Tail-2 system is similar to that of the Tail-1 system and shows that the result is not heavily dependent on initial positions of DNA and tails. Clear correlation between association of Lys+ residues and close DNA-DNA distances also can be seen from the three-dimensional distribution of NZ atoms averaged over shorter (5-ns) time periods at the end of the 30-ns simulation runs (data not shown).
FIGURE 8.

(a) Distribution of DNA-DNA distances: Tail-1 (green, solid line) and Tail-2 (blue, dashed line) systems were averaged between 5 and 30 ns, and the No-Tail (red, dot-dashed line) between 4 and 20 ns. (b–d). Densities of ions as a function of DNA-DNA distance (y axis) and relative to the distance between the ion and the midpoint of the DNA-DNA separation (x axis). Darker color corresponds to higher ion density. (b) NZ atoms of Lys+. (c) K+ in the Tail-1 system. (d) K+ in the No-Tail system.
Close DNA-DNA contacts correlate with positioning of charged tail groups. When there is a space between the DNA double helices (at a distance >30 Å), Lysine+ residues prefer to stay directly between the contacting DNA molecules (see peaks at x = 0 in Fig. 8, b and c). When the DNA-DNA contacts are so close that there is insertion of sugar-phosphate backbone of one DNA into the groove of the other DNA, the tails mediate interaction by binding to the electronegative sites of both DNA molecules, which are in close proximity (see NZ+ peak in Fig. 8 b at DNA-DNA distance ∼27 Å and x = 5–10 Å). This configuration is observed in the Tail-1 system and is also illustrated by the snapshot in Fig. 7 a.
Generally, the MD results show that the histone tail fragments interact with DNA in a dynamic manner. It is hardly possible to identify well-defined structures even for the most frequently observed tail-DNA contacts. Some few events of long-lived (several nanoseconds) contact of the charged amino group of Lys+ with DNA were recorded (the longest contact, 7.5 ns, was with one O4* atom). This dynamic picture is in agreement with NMR studies of NCP solutions showing that the tails are very mobile (68,69).
DISCUSSION
The simulations described here clearly demonstrate electrostatically salt-induced condensation of NCPs mediated by tail bridging for isolated (linker-free) NCP within a computer simulation model based on a coarse-grained electrostatic continuum description containing the NCP modeled as a charged sphere, mobile counterions, and flexible charged tails. The model contains several approximations, necessitated by the complexity of the system. The continuum description of solvent water has the effect of neglecting solvent-mediated effects, but is consistent within the level of a coarse-grained model. Solvent hydration of charged ions is approximately accounted for by an effective ion diameter. The core particle shape is approximated by a sphere, whereas the real NCP is more in the form of a flat cylinder, and thus excluded volume and packing effects are approximate in this respect. Furthermore, the NCP electrostatic potential is described by a charged sphere (In the simulations it is introduced by a fixed point charge at the center of the sphere, but due to Gauss' law, this potential is the same as that generated by a sphere with a surface charge density equivalent to that of the point charge.) This description is based on the rationale of using a model for which the electrostatic problem can be solved without further approximations and with a method (Nóse-Hoover NVT ensemble simulations) that generates the correct (within that model) equilibrium properties. In this way, we incorporate attractive ion-ion correlation effects due to fluctuations in the ion cloud and the attractive entropic and energetic tail bridging. We believe that these two physical mechanisms are absolutely crucial in the description of NCP interactions, because it is well established that for polyelectrolytes in general and for DNA in particular, fluctuation-induced dynamic correlation of cations shared by different polyanions gives rise to attractive electrostatic force contributions that explain experimental aggregation (27,28,70). The effect is more pronounced in the presence of multivalent ions. Thus, any model of the aggregation of NCPs, where the high negative charge originates from the DNA wrapped around the histone core, particularly in the presence of divalent ions, should incorporate this mechanism. Furthermore, experimental data on isolated (linker-free) NCPs show that maximal extension of the tails at high salt coincides with the emergence of attraction between the particles (16,18). In addition, experimental results on nucleosomal arrays indicate that tails relocate from mainly intranucleosome interaction with their own core particle in the extended array at low salt, to primarily bridging-like internucleosomal interactions upon formation of condensed secondary and tertiary chromatin aggregates (13,71). To describe such a physical mechanism, a model with flexible charged tails is necessary. Very recently, a related coarse-grained MD simulation model was published, which described tail-bridging attraction between two charged spherical NCPs with grafted charged flexible tails (but within a Debye-Hückel model for the effects of salt) (72).
The present model is mainly relevant for comparison with and interpretation of experimentally monovalent salt-induced aggregation of isolated (linker-free) NCPs (15) and the agreement of the results with data such as those described above is encouraging, indicating that the main mechanism of this aggregation is captured in the simulations. For the system containing isolated nucleosome core particles and Mg2+ ions, data available in the literature (15) agree with the simulations. The ability of divalent cations at millimolar concentration to promote compaction of nucleosomal arrays is well established (4) and this study gives physical insight relevant to these results. Even though linkers are absent in the model, similar tail bridging may be present in nucleosomal arrays. Furthermore, the interarray oligomerization of different arrays where nucleosomes are already condensed is expected to be dominated by the nucleosome-nucleosome interactions, since linkers have already adjusted in the folding of array monomers. The recent works (13,14) performed on nucleosomal arrays are in accordance with the present theoretical picture of tail-mediated nucleosomal interactions. Gordon et al. (14) demonstrated the independent (with some exceptions for the H4 tail) and additive (related to the charge on the tails) character of the Mg2+-induced chromatin interarray oligomerization. Using radiolabeled H3 tails, Zheng et al. (13) showed by ultraviolet cross-linking in low to moderate monovalent salt, where the nucleosomal array is extended, that the tails are associated with the DNA of their own nucleosome core, whereas upon formation of condensed secondary and tertiary arrays compacted by addition of Mg2+, the tails interact with the DNA of the other nucleosomes.
All-atom MD simulations of three DNA oligonucleotides with and without the presence of a histone H4 tail fragment with charge +3 demonstrated a bridging association of DNA induced by the tail peptide fragments and participation of the positive lysine side chains. The system of close DNA molecules with charged tail-peptide fragments between them should have some bearing on the possible bridging events that can take place in real aggregated NCPs. The separation of ∼33–35 Å between DNA of different nucleosomes in the recently solved x-ray structure of an NCP tetramer (including linker DNA) (73), indicates that the distance range captured in DNA association events of our simulations is realistic. Furthermore, a recent structural investigation, using small-angle x-ray scattering of compacted DNA in the presence of polylysine, showed a lattice spacing of 30.4 Å between DNA molecules (74), which is in good agreement with the distribution of average DNA-DNA distances observed in the MD simulations.
Future comparison of MD simulations for unmodified and modified tails mimicking reduced positive charge corresponding to transcriptionally active states of chromatin, could help in understanding the physical basis and molecular mechanisms behind epigenetic control of transcriptional regulation.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
Note added in proof: We thank the Nanyang Technological University (NTU) overseas attachment program for enabling A.L. to stay at NTU, and gratefully acknowledge the NTU Bioinformatics Research Centre for computer resources. We are thankful to Dr. Helmut Schiessel for sharing his work before publication (72).
This work was supported by a Singapore Ministry of Education University Research Committee grant and a grant from the Singapore Agency for Science Technology and Research through the Biomedical Research Council to L.N., and by the Swedish Research Council to A.L.
References
- 1.Luger, K., A. W. Mader, R. K. Richmond, D. F. Sargent, and T. J. Richmond. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 389:251–260. [DOI] [PubMed] [Google Scholar]
- 2.Davey, C. A., D. F. Sargent, K. Luger, A. W. Maeder, and T. J. Richmond. 2002. Solvent mediated interactions in the structure of nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319:1097–1113. [DOI] [PubMed] [Google Scholar]
- 3.Woodcock, C. L., and S. Dimitrov. 2001. Higher-order structure of chromatin and chromosomes. Curr. Opin. Genet. Dev. 11:130–135. [DOI] [PubMed] [Google Scholar]
- 4.Hansen, J. C. 2002. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 31:361–392. [DOI] [PubMed] [Google Scholar]
- 5.Luger, K., and J. C. Hansen. 2005. Nucleosome and chromatin fiber dynamics. Curr. Opin. Struct. Biol. 15:188–196. [DOI] [PubMed] [Google Scholar]
- 6.Luger, K., and T. J. Richmond. 1998. The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 8:140–146. [DOI] [PubMed] [Google Scholar]
- 7.Horn, P. J., and C. L. Peterson. 2002. Chromatin higher order folding: wrapping up transcription. Science. 297:1824–1827. [DOI] [PubMed] [Google Scholar]
- 8.Wolffe, A. P. 1998. Chromatin: Structure and Function, 3rd ed. Academic Press, San Diego.
- 9.Ren, Q., and M. A. Gorovsky. 2003. The nonessential H2A N-terminal tail can function as an essential charge patch on the H2A.Z variant N-terminal tail. Mol. Cell. Biol. 23:2778–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Holde, K. E. 1989. Chromatin. Springer-Verlag, New York.
- 11.Clark, D. J., and T. Kimura. 1990. Electrostatic mechanism of chromatin folding. J. Mol. Biol. 211:883–896. [DOI] [PubMed] [Google Scholar]
- 12.van Holde, K., and I. Zlatanova. 1996. What determine the folding of the chromatin fiber? Proc. Natl. Acad. Sci. USA. 93:10548–10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng, C., X. Lu, J. C. Hansen, and J. J. Hayes. 2005. Salt-dependent intra- and inter-nucleosomal interactions of the H3 tail domain in a model oligonucleosomal array. J. Biol. Chem. 280:33552–33557. [DOI] [PubMed] [Google Scholar]
- 14.Gordon, F., K. Luger, and J. C. Hansen. 2005. The core histone N-terminal tail domains function independently and additively during salt-dependent oligomerization of nuclesomal arrays. J. Biol. Chem. 280:33701–33706. [DOI] [PubMed] [Google Scholar]
- 15.de Frutos, M., E. Raspaud, A. Leforestier, and F. Livolant. 2001. Aggregation of nucleosomes by divalent cations. Biophys. J. 81:1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mangenot, S., A. Leforestier, P. Vachette, D. Durand, and F. Livolant. 2002. Salt-induced conformation and interaction changes of nucleosome core particles. Biophys. J. 82:345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertin, A., A. Leforestier, D. Durand, and F. Livolant. 2004. Role of histone tails in the conformation and interaction of nucleosome core particles. Biochemistry. 43:4773–4780. [DOI] [PubMed] [Google Scholar]
- 18.Mangenot, S., E. Raspaud, C. Tribet, L. Belloni, and F. Livolant. 2002. Interactions between isolated nucleosome core particles. A tail bridging effect? Eur. Phys. J. E. 7:221–231. [Google Scholar]
- 19.Wilsons, R. W., and V. A. Bloomfield. 1979. Counterion-induced condensation of deoxyribonucleic acid. A light-scattering study. Biochemistry. 18:2192–2196. [DOI] [PubMed] [Google Scholar]
- 20.Bloomfield, V. A. 1996. DNA condensation. Curr. Opin. Struct. Biol. 6:334–341. [DOI] [PubMed] [Google Scholar]
- 21.Lyubartsev, A. P., J. X. Tang, P. A. Janmey, and L. Nordenskiöld. 1998. Electrostatically induced polyelectrolyte association of rod like virus particles. Phys. Rev. Lett. 81:5465–5468. [Google Scholar]
- 22.Tang, J. X., P. A. Janmey, A. P. Lyubartsev, and L. Nordenskiöld. 2002. Metal ion-induced lateral aggregation of filamentous viruses fd and M13. Biophys. J. 83:566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angelini, T. E., H. Liang, W. Wriggers, and G. C. L. Wong. 2003. Like-charge attraction between polyelectrolytes induced by counterion charge density waves. Proc. Natl. Acad. Sci. USA. 100:8634–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai, Y., R. Das, I. S. Millett, D. Herschlag, and S. Doniach. 2005. Probing counterion modulation repulsion and attraction berween nucleic acid duplexes in solution. Proc. Natl. Acad. Sci. USA. 102:1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyubartsev, A. P., and L. Nordenskiöld. 1995. Monte Carlo simulation study of ion distribution and osmotic pressure in hexagonally oriented DNA. J. Phys. Chem. 99:10373–10382. [Google Scholar]
- 26.Gronbech-Jensen, N., R. J. Mashl, R. F. Bruinsma, and W. M. Gelbart. 1997. Counterion-induced attraction between rigid polyelectrolytes. Phys. Rev. Lett. 78:2477–2480. [Google Scholar]
- 27.Ray, J., and G. S. Manning. 1997. Effect of counterion valence and polymer charge density on the pair potential of two polyions. Macromolecules. 30:5739–5744. [Google Scholar]
- 28.Gelbart, W. M., R. F. Bruinsma, P. A. Pincus, and V. A. Parsegian. 2000. DNA-inspired electrostatics. Phys. Today. 53:38–44. [Google Scholar]
- 29.Claesson, P. M., E. Poptoshev, E. Blomberg, and A. Dedinaite. 2005. Polyelectrolyte-mediated surface interactions. Adv. Colloid Interface Sci. 114–115:173–187. [DOI] [PubMed] [Google Scholar]
- 30.Miklavic, S. J., C. E. Woodward, B. Jönsson, and T. Åkesson. 1990. Interaction of charged surfaces with grafted polyelectrolytes: a Poisson-Boltzmann and Monte Carlo study. Macromolecules. 23:4149–4157. [Google Scholar]
- 31.Rescic, J., and P. Linse. 2000. Charged colloidal solutions with short flexible counterions. J. Phys. Chem. B. 104:7852–7857. [Google Scholar]
- 32.Podgornik, R. 2004. Polyelectrolyte-mediated bridging interactions. J. Polym. Sci. [B]. 42:3539–3556. [Google Scholar]
- 33.Katritch, V., C. Bustamante, and W. K. Olson. 2000. Pulling chromatin fibers: computer simulations of direct physical micromanipulations. J. Mol. Biol. 295:29–40. [DOI] [PubMed] [Google Scholar]
- 34.Martino, J. A., and W. K. Olson. 1998. Modeling chain folding in protein-constrained circular DNA. Biophys. J. 74:2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martino, J. A., V. Katritch, and W. K. Olson. 1999. Influence of nucleosome structure on the three-dimensional folding of idealized minichromosomes. Struct. Fold. Des. 7:1009–1022. [DOI] [PubMed] [Google Scholar]
- 36.Beard, D. A., and T. Schlick. 2001. Computational modeling predicts the structure and dynamics of chromatin fiber. Structure. 9:105–114. [DOI] [PubMed] [Google Scholar]
- 37.Wademann, G., and J. Langowski. 2002. Computer simulation of the 30-nanometer chromatin fiber. Biophys. J. 82:2847–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beard, D. A., and T. Schlick. 2001. Modeling salt-mediated electrostatics of macromolecules: the discrete surface charge optimization algorithm and its application to the nucleosome. Biopolymers. 58:106–115. [DOI] [PubMed] [Google Scholar]
- 39.Zhang, Q., D. A. Beard, and T. Schlick. 2003. Constructing irregular surfaces to enclose macromolecular complexes for mesoscale modeling using the discrete surface charge optimization (DiSCO) algorithm. J .Comput .Chem. 24:2063–2074. [DOI] [PubMed] [Google Scholar]
- 40.Sun, J., Q. Zhang, and T. Schlick. 2005. Electrostatic mechanism of nucleosomal array folding revealed by computer simulation. Proc. Natl. Acad. Sci. USA. 102:8180–8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen, J. C., C. Tse, and A. P. Wolffe. 1998. Structure and function of the core histone N-termini: more than meets the eye. Biochemistry. 37:17637–17641. [DOI] [PubMed] [Google Scholar]
- 42.Lyubartsev, A. P., and A. Laaksonen. 1997. Osmotic and activity coefficient from effective potentials for hydrated ions. Phys.Rev.E. 55:5689–5696. [Google Scholar]
- 43.Pratt, L. R., G. Hummer, and A. E. Garcia. 1994. Ion-pair potentials-of-mean-force in water. Biophys. Chem. 51:147–165. [Google Scholar]
- 44.Allen, M. P., and D. J. Tildesley. 1987. Computer Simulations of Liquids. Clarendon, Oxford, UK.
- 45.Montoro, J. C. G., and J. L. F. Abascal. 1995. Ionic distribution around simple DNA models. I. Cylindrically averaged properties. J. Chem. Phys. 103:8273–8284. [Google Scholar]
- 46.Wolffe, A. P., and J. J. Hayes. 1999. Chromatin disruption and modification. Nucleic Acids Res. 27:711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martyna, G. J., D. J. Tobias, and M. L. Klein. 1994. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101:4177–4189. [Google Scholar]
- 48.Stevens, M. J., and K. Kreker. 1995. The nature of flexible linear polyelectrolytes in salt free solution: a molecular dynamics study. J. Chem. Phys. 103:1669–1690. [Google Scholar]
- 49.Limbach, H. J., and C. Holm. 2001. End effects of strongly charged polyelectrolytes: a molecular dynamics study. J. Chem. Phys. 114:9674–9682. [Google Scholar]
- 50.Stevens, M. J. 2004. Coarse grained simulations of lipid bilayers. J. Chem. Phys. 121:11942–11948. [DOI] [PubMed] [Google Scholar]
- 51.Lyubartsev, A. P. 2005. Multiscale modeling of lipids and lipid bilayers. Eur. Biophys. J. 35:53–61. [DOI] [PubMed] [Google Scholar]
- 52.Weidemann, T., M. Wachsmuth, T. Knoch, G. Muller, W. Waldeck, and J. Langowski. 2003. Counting nucleosomes in living cells with a combination of fluorescent correlation spectroscopy and confocal imaging. J. Mol. Biol. 334:229–240. [DOI] [PubMed] [Google Scholar]
- 53.Foloppe, N., and A. D. MacKerell. 2000. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J.Comput. Chem. 21:86–104. [Google Scholar]
- 54.MacKerell, A. D., and N. Banavali. 2000. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J.Comput. Chem. 21:105–120. [Google Scholar]
- 55.Toukan, K., and A. Rahman. 1985. Molecular-dynamics study of atomic motions in water. Phys. Rev. B. 31:2643–2648. [DOI] [PubMed] [Google Scholar]
- 56.Heinzinger, K. 1985. Computer simulations of aqueous electrolyte solutions. Physica B. 131:196–216. [Google Scholar]
- 57.Lyubartsev, A. P., and A. Laaksonen. 2000. M.DynaMix: a scalable portable parallel MD simulation package for arbitrary molecular mixtures. Comput. Phys. Commun. 128:565–589. [Google Scholar]
- 58.Korolev, N., A. P. Lyubartsev, L. Nordenskiöld, and A. Laaksonen. 2001. Spermine: an “invisible” component in the crystals of B-DNA. A grand canonical Monte Carlo and molecular dynamics simulation study. J. Mol. Biol. 308:907–917. [DOI] [PubMed] [Google Scholar]
- 59.Korolev, N., A. P. Lyubartsev, A. Laaksonen, and L. Nordenskiöld. 2003. A molecular dynamics simulation study of oriented DNA with polyamine and sodium counterions: diffusion and averaged binding of water and cations. Nucleic Acids Res. 31:5971–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tuckerman, M., B. Berne, and G. J. Martyna. 1992. Reversible multiple time scale molecular-dynamics. J. Chem. Phys. 97:1990–2001. [Google Scholar]
- 61.Stofer, E., and R. Lavery. 1994. Measuring the geometry of DNA grooves. Biopolymers. 34:337–346. [DOI] [PubMed] [Google Scholar]
- 62.Korolev, N., A. P. Lyubartsev, A. Laaksonen, and L. Nordenskiöld. 2002. On the competition between water, sodium ions, and spermine in binding to DNA. A molecular dynamics computer simulation study. Biophys. J. 82:2860–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korolev, N., A. P. Lyubartsev, A. Laaksonen, and L. Nordenskiöld. 2004. Molecular dynamics simulation study of oriented polyamine- and Na-DNA: Sequence specific interactions and effects on DNA structure. Biopolymers. 73:542–555. [DOI] [PubMed] [Google Scholar]
- 64.Korolev, N., A. P. Lyubartsev, A. Laaksonen, and L. Nordenskiöld. 2004. A molecular dynamics simulation study of polyamine- and sodium-DNA. Interplay between polyamine binding and DNA structure. Eur. Biophys. J. 33:671–682. [DOI] [PubMed] [Google Scholar]
- 65.Varnai, P., and K. Zakrzewska. 2004. DNA and its counterions: a molecular dynamics study. Nucleic Acids Res. 32:4269–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ponomarev, S. Y., K. M. Thayer, and D. L. Beveridge. 2004. Ion motion in molecular dynamics simulations on DNA. Proc. Natl. Acad. Sci. USA. 101:14771–14775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hamelberg, D., L. D. Williams, and W. D. Wilson. 2001. Influence of the dynamic positions of cations on the structure of the DNA minor groove: sequence-dependent effects. J. Am. Chem. Soc. 123:7745–7755. [DOI] [PubMed] [Google Scholar]
- 68.Hilliard, P. R., R. M. Smith, and R. L. Rill. 1986. Natural abundance carbon-13 nuclear magnetic resonance studies of histone and DNA dynamics in nucleosome cores. J. Biol. Chem. 261:5992–5998. [PubMed] [Google Scholar]
- 69.Smith, R. M., and R. L. Rill. 1989. Mobile histone tails in nucleosomes. Assignment of mobile segments and investigations of their role in chromatin folding. J. Biol. Chem. 264:10574–10581. [PubMed] [Google Scholar]
- 70.Oosawa, F. 1968. Interaction between parallel rodlike macroions. Biopolymers. 6:1633–1647. [Google Scholar]
- 71.Dorigo, B., T. Schalch, K. Bystricky, and T. J. Richmond. 2003. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 327:85–96. [DOI] [PubMed] [Google Scholar]
- 72.Muhlbacher, F., C. Holm, and H. Schiessel. 2006. Controlled DNA compaction within chromatin: the tail-bridging effect. Europhys. Lett. 73:135–141. [Google Scholar]
- 73.Schalch, T., S. Duda, D. F. Sargent, and T. J. Richmond. 2005. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 436:138–141. [DOI] [PubMed] [Google Scholar]
- 74.DeRouchey, J., R. R. Netz, and J. Rädler. 2005. Structural investigations of DNA-polycation complexes. Eur. Phys. J. E. 16:17–28. [DOI] [PubMed] [Google Scholar]