

Abstract
We have used the interface between a nanochannel and a microchannel as a tool for applying controlled forces on a DNA molecule. A molecule, with a radius of gyration larger than the nanochannel width, that straddles such an interface is subject to an essentially constant entropic force, which can be balanced against other forces such as the electrophoretic force from an applied electric field. By controlling the applied field we can position the molecule as desired and observe the conformation of the molecule as it stretches, relaxes, and recoils from the nanochannel. We quantify and present models for the molecular motion in response to the entropic, electrophoretic, and frictional forces acting on it. By determining the magnitude of the drag coefficients for DNA molecules in the nanostructure, we are able to estimate the confinement-induced recoil force. Finally, we demonstrate that we can use a controlled applied field and the entropic interfacial forces to unfold molecules, which can then be manipulated and positioned in their simple extended morphology.
INTRODUCTION
The dynamics of DNA in nanochannels is a relevant topic considering the current efforts toward the development of micro- and nanofluidic devices for biomolecular analysis. In previous years, such devices have been studied for their ability to isolate, manipulate, and investigate single biomolecules. Among the technologies tested have been entropic trap arrays (1–4), micro- and nanopillar arrays (5–9), nanopores (10–14), and nanochannels (15–18). Each has shown promise as either a tool for the analysis of statistical mechanical properties of long biopolymers or as a tool for sorting biomolecules according to their length. For example, the contour lengths of molecules that were driven into a 100-nm channel have been measured by Tegenfeldt et al. (16). They were able to determine the length of a single 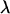 -DNA strand to within 400 bp in 1 min. Using nanochannels it is also possible to observe the action of an enzyme on a single DNA molecule and consequently map its restriction sites (17).
-DNA strand to within 400 bp in 1 min. Using nanochannels it is also possible to observe the action of an enzyme on a single DNA molecule and consequently map its restriction sites (17).
It has been demonstrated that DNA molecules inserted partly into a nanopillar array with an applied electric field experience a confinement-induced entropic force, causing them to recoil from the array, when the field is turned off (8). This effect, termed entropic recoil, was used to sort two DNA populations of different lengths with a resolution of 11% by length (7). The resolution of pillar arrays is blurred by the fact that DNA molecules entering the array at a distinct point may enter the array diagonally, wrap around pillars, or even form sideways loops. All these different conformations probably have different friction and lead to different recoil times. We have therefore investigated entropic recoil from nanochannels, a geometry that allows only one direction of insertion. Nanochannels also simplify the optical analysis (17) and allow a better comparison to well-known theoretical models like de Gennes' blob model for confined polymers (19).
Here, for the first time, we investigate the motion and conformation of single DNA molecules in 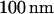 channels while they are manipulated with both electric fields and confinement-induced entropic forces. Fig. 1 A illustrates the recoil experiment. In addition to the basic recoil process we investigate several new types of motion in nanochannels. Sometimes as shown in Fig. 1 B, when a molecule is electrophoretically driven into a nanochannel, its leading end is folded over on itself. We show that in our experiments the looped DNA strands can be unfolded by the recoil process. Note that folding of DNA has already been observed as quantized resistance changes during DNA translocation through artificial nanopores (12,20). The term “folded DNA” just refers to a transient loop formation and should not be confused with protein folding, for example.
channels while they are manipulated with both electric fields and confinement-induced entropic forces. Fig. 1 A illustrates the recoil experiment. In addition to the basic recoil process we investigate several new types of motion in nanochannels. Sometimes as shown in Fig. 1 B, when a molecule is electrophoretically driven into a nanochannel, its leading end is folded over on itself. We show that in our experiments the looped DNA strands can be unfolded by the recoil process. Note that folding of DNA has already been observed as quantized resistance changes during DNA translocation through artificial nanopores (12,20). The term “folded DNA” just refers to a transient loop formation and should not be confused with protein folding, for example.
FIGURE 1.

Types of motion and conformations studied. Relaxed DNA molecules in nanochannels have an equilibrium extension length. (A) Extraction of a relaxed strand from a nanochannel. The entropic force driving the extraction is due to the difference in configuration space on either side of the nanochannel/microchannel interface. This process of self-extraction, which only occurs for molecules straddling the interface, is called a recoil process. (B) In some cases molecules are inserted in the nanochannels with their leading end folded over on itself, creating a loop. During the recoil process these molecules unfold, which means the looped section of DNA straightens. (C) A molecule electrophoretically driven into the nanochannel stretches as the entropic and electric forces pull on the molecule in opposite directions. After the molecule has entirely entered the nanochannel, the electric field is switched off and it is allowed to relax to its equilibrium extension length 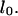 (D) If a molecule is partially driven into a nanochannel and immediately allowed to recoil, it will show a combination of unstretching and recoiling.
(D) If a molecule is partially driven into a nanochannel and immediately allowed to recoil, it will show a combination of unstretching and recoiling.
The entropic recoil force was shown to be mathematically distinct from an elastic restoring force (8). A molecule undergoing entropic recoil speeds up over time. In contrast, one expects that the contraction due to entropic elasticity would slow down over time. Here we cause molecules to undergo either elastic contraction or entropic recoil. Fig. 1 C illustrates the contraction of a stretched molecule inside a nanochannel. As shown in Fig. 1 D, we also demonstrate that under certain initial conditions it is possible for a molecule to undergo both the contraction and entropic recoil processes simultaneously.
A basic theory (8) can be derived by neglecting the self-avoidance effects of the persistent DNA. A molecule at the interface has the entropy per unit length  inside the nanochannel and entropy per unit length
inside the nanochannel and entropy per unit length 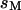 in the microchannel. Thus the entropy is
in the microchannel. Thus the entropy is 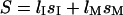 where
where  and
and  are the lengths in the two regions. The length of the molecule that resides in the microchannel can be rewritten as
are the lengths in the two regions. The length of the molecule that resides in the microchannel can be rewritten as 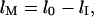 where
where 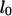 is the full extended length of the DNA strand in the nanochannel. The entropic force
is the full extended length of the DNA strand in the nanochannel. The entropic force 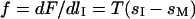 results from the free energy
results from the free energy  and is found to be independent of the inserted length
and is found to be independent of the inserted length 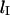 inside the nanochannel. The molecule retraction at velocity
inside the nanochannel. The molecule retraction at velocity 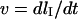 is only hindered by the hydrodynamic drag force
is only hindered by the hydrodynamic drag force 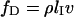 inside a confined environment (21). Here
inside a confined environment (21). Here 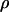 is the hydrodynamic drag per unit extended length in the nanochannel. Solving the resulting differential equation
is the hydrodynamic drag per unit extended length in the nanochannel. Solving the resulting differential equation  under the condition
under the condition 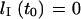 leads to:
leads to:
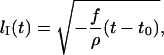 |
(1) |
where 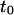 is the time of complete extraction.
is the time of complete extraction.
MATERIALS AND METHODS
Device fabrication
Devices were patterned on a mirror-polished fused silica wafer with a thickness of 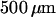 (MarkOptics, Santa Ana, CA) using a combination of electron beam and optical lithography. Initially, a layer of gold,
(MarkOptics, Santa Ana, CA) using a combination of electron beam and optical lithography. Initially, a layer of gold, 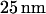 thick, was evaporated onto poly(methyl-methacrylate) (PMMA) electron-beam resist to help draw current during the electron-beam process. Negative patterns of the nanochannel regions were exposed using a JBX-9300FS electron beam lithography system (JEOL, Peabody, MA). After removal of gold and PMMA development, the patterns were transferred to a chrome mask by evaporation and lift off. Microchannel patterns were then added to the mask using optical lithography and the same lift-off process. Both micro- and nanochannels were etched simultaneously using a Plasmalab 80Plus REI (Oxford Instruments, Eynsham, UK) with a CHF3/O2 mixture at 50 Watts for 20 min. Access holes were created by alumina powder blasting from the backside of the wafer. Finally a
thick, was evaporated onto poly(methyl-methacrylate) (PMMA) electron-beam resist to help draw current during the electron-beam process. Negative patterns of the nanochannel regions were exposed using a JBX-9300FS electron beam lithography system (JEOL, Peabody, MA). After removal of gold and PMMA development, the patterns were transferred to a chrome mask by evaporation and lift off. Microchannel patterns were then added to the mask using optical lithography and the same lift-off process. Both micro- and nanochannels were etched simultaneously using a Plasmalab 80Plus REI (Oxford Instruments, Eynsham, UK) with a CHF3/O2 mixture at 50 Watts for 20 min. Access holes were created by alumina powder blasting from the backside of the wafer. Finally a 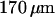 fused silica cover wafer (MarkOptics) was touch bonded and annealed at 1050°C to the device wafer, enclosing the channels. Nanoports (Upchurch Scientific, Oak Habor, WA) were sealed to the access holes forming buffer reservoirs. A schematic of the completed device can be seen in Fig. 2. Several electron micrographs of the nanostructures before bonding are shown in Fig. 3.
fused silica cover wafer (MarkOptics) was touch bonded and annealed at 1050°C to the device wafer, enclosing the channels. Nanoports (Upchurch Scientific, Oak Habor, WA) were sealed to the access holes forming buffer reservoirs. A schematic of the completed device can be seen in Fig. 2. Several electron micrographs of the nanostructures before bonding are shown in Fig. 3.
FIGURE 2.

Schematic of the nanochannel array device. (A) Cross section of device consisting of two bonded fused silica wafers (a, b) with the upper one containing the structure. The microchannel was contacted from the top of the device and fluid reservoirs (c) were attached. Electrical connections to the channel were made by platinum electrodes (d). (B) Close-up of the nanochannel array in the upper wafer. DNA molecules have been drawn in the loading zone (a), as they enter a nanochannel (b), and in an elongated equilibrium conformation in a nanochannel (c).
FIGURE 3.

Electron micrograph of nanochannel array etched into 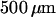 fused silica wafer. (A) Top-down view showing the interface between the microchannel and the array of nanochannels. Both were etched
fused silica wafer. (A) Top-down view showing the interface between the microchannel and the array of nanochannels. Both were etched 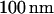 deep. (B) Entrance to two nanochannels. The channels are
deep. (B) Entrance to two nanochannels. The channels are  wide. The wall separating the two channels is
wide. The wall separating the two channels is  wide. (C) Closer view of a nanochannel entrance. Floor roughness is
wide. (C) Closer view of a nanochannel entrance. Floor roughness is 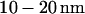 and is attributed to the etching process.
and is attributed to the etching process.
DNA and buffer preparation
T4-bacteriophage DNA molecules (Wako, Richmond, VA) were stained with the bis-intercalating dye YOYO-1 (Molecular Probes, Eugene, OR) and used at a concentration of 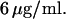 The contour length of single T4-DNA (169 kbp) can be calculated from the basepair spacing of
The contour length of single T4-DNA (169 kbp) can be calculated from the basepair spacing of  to
to 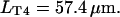 But recent studies have shown that the dye TOTO-1, which is similar to YOYO-1, increases the contour length
But recent studies have shown that the dye TOTO-1, which is similar to YOYO-1, increases the contour length 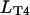 by 30–35% at a dye to basepair ratio of 1:4 (21,22). Thus at our dye ratio of 1:5
by 30–35% at a dye to basepair ratio of 1:4 (21,22). Thus at our dye ratio of 1:5 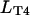 is expected to rise by 23% to
is expected to rise by 23% to 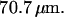 Although in the past, the persistence length of the DNA was assumed to increased from
Although in the past, the persistence length of the DNA was assumed to increased from  when stained (21,22), recent investigations indicate that staining reduces the persistence length to
when stained (21,22), recent investigations indicate that staining reduces the persistence length to  (23). The two protonated amino and two protonated imino groups of YOYO-1 also decrease the average charge of the negative DNA backbone. Given our buffer concentrations, we assume a counterion shielding of 60% (24,25) and calculate a charge per unit length of
(23). The two protonated amino and two protonated imino groups of YOYO-1 also decrease the average charge of the negative DNA backbone. Given our buffer concentrations, we assume a counterion shielding of 60% (24,25) and calculate a charge per unit length of 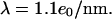 As changes in the properties are all less than an order of magnitude, we expect that the behavior of unstained DNA would be similar to the observed dynamics of the DNA/dye complex. The buffer consisted of 445 mM Tris-borate and 10 mM EDTA (5×TBE, pH 8.3, Sigma, St. Louis, MO) with 5% (v/v) β-mercaptoethanol (Sigma) as an antiphotobleaching agent and 2.5% (w/w) poly(n-vinylpyrrolidone) (PVP; molecular weight = 10,000; Sigma) to reduce both electroosmotic flow and unspecific binding of DNA to channel walls (18,26,27). Electrical contact was made with platinum electrodes inserted into the reservoirs.
As changes in the properties are all less than an order of magnitude, we expect that the behavior of unstained DNA would be similar to the observed dynamics of the DNA/dye complex. The buffer consisted of 445 mM Tris-borate and 10 mM EDTA (5×TBE, pH 8.3, Sigma, St. Louis, MO) with 5% (v/v) β-mercaptoethanol (Sigma) as an antiphotobleaching agent and 2.5% (w/w) poly(n-vinylpyrrolidone) (PVP; molecular weight = 10,000; Sigma) to reduce both electroosmotic flow and unspecific binding of DNA to channel walls (18,26,27). Electrical contact was made with platinum electrodes inserted into the reservoirs.
Microscopy
The individual molecules were observed with an IX70 inverted microscope (Olympus, Melville, NY), which was equipped with a 100×/0.93 NA oil immersion objective (Olympus) and illuminated by a 100-W mercury arc lamp. An XF100 filter set (Omega Optical, Brattleboro, VT) was used for fluorescence imaging. Images were acquired using an ICCD-350F camera (Videoscope, Dulles, VA) connected to a DVD recorder at a rate of 29.97 frames per second. DNA molecule movement was extracted from the videos by using a combination of thresholding and morphological algorithms programmed in MATLAB (The Mathworks, Natick, MA).
Experimental protocol
DNA molecules were driven from the reservoirs into the microchannel with a bias of 30 V. To drive a molecule from the microchannel into a nanochannel, a 3-V bias was always applied. This bias resulted in  in nanochannels and
in nanochannels and  in the loading channel. Any further manipulations within the nanochannel were conducted under electric fields of either
in the loading channel. Any further manipulations within the nanochannel were conducted under electric fields of either 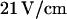 or
or  Three types of experiments were conducted for each molecule that we observed. First, the molecules were driven entirely into the nanochannel, and the field was then switched off. Their relaxation to equilibrium extension length in the nanochannel was studied. Once they had contracted, they were slowly driven back down the nanochannel until a small portion of the molecule had reached the microchannel. At this point the field was again switched off and the molecules were observed to undergo a pure recoil process. Finally, after exiting the nanochannel, molecules were driven back in electrophoretically. This time, however, the field was switched off before the DNA strands had completely entered the nanochannel. As a result, they were observed to both recoil and unstretch simultaneously.
Three types of experiments were conducted for each molecule that we observed. First, the molecules were driven entirely into the nanochannel, and the field was then switched off. Their relaxation to equilibrium extension length in the nanochannel was studied. Once they had contracted, they were slowly driven back down the nanochannel until a small portion of the molecule had reached the microchannel. At this point the field was again switched off and the molecules were observed to undergo a pure recoil process. Finally, after exiting the nanochannel, molecules were driven back in electrophoretically. This time, however, the field was switched off before the DNA strands had completely entered the nanochannel. As a result, they were observed to both recoil and unstretch simultaneously.
RESULTS AND DISCUSSION
For each molecule, three basic investigations were performed: stretching with ensuing relaxation, relaxed recoil, and stretched recoil. An example of a complete experimental time course can be seen in Fig. 4 as a color-coded intensity graph. The plot shows the normalized intensity along the channel axis plotted against time.
FIGURE 4.

Intensity time trace of the three basic manipulations performed with a single T4 DNA molecule. The plot shows the normalized intensity along the channel (x axis) versus the time 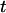 (dotted line = channel entrance). At
(dotted line = channel entrance). At 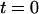 the molecule is stretched while being driven with 21 V/cm from the microchannel (x < 0) into the nanochannel (x > 0). It is then allowed to sit in the nanochannel and contract until it reaches its equilibrium length. At
the molecule is stretched while being driven with 21 V/cm from the microchannel (x < 0) into the nanochannel (x > 0). It is then allowed to sit in the nanochannel and contract until it reaches its equilibrium length. At 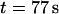 a 0.5 V/cm pulse moves the molecule to the channel entrance (a). A small part straddles the interface and the molecule begins to recoil from the nanochannel into the microchannel. At
a 0.5 V/cm pulse moves the molecule to the channel entrance (a). A small part straddles the interface and the molecule begins to recoil from the nanochannel into the microchannel. At 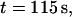 DNA is driven partially into channel with a
DNA is driven partially into channel with a 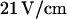 pulse (b). When the field is turned off, it begins to both contract and recoil simultaneously. The difference between the profiles of the different recoil processes can clearly be seen.
pulse (b). When the field is turned off, it begins to both contract and recoil simultaneously. The difference between the profiles of the different recoil processes can clearly be seen.
Molecule relaxation
We investigated the relaxation of stretched molecules in a nanochannel. The DNA strands were electrophoretically driven from the microchannel into a nanochannel. Stretching was due to the electric force pulling the molecules into the nanochannel against a resistance at the entrance. The resistance at the entrance is probably due to the entropic interface force and friction for molecules encountering the entrance edges. Note that this method of stretching is completely different than those previously used such as hooking around pillars (21) or stretching by microbead techniques (28). Upon fully entering a nanochannel, a molecule begins to relax and finally reaches its equilibrium extension length inside the channel. Though the dynamics of this contraction in nanochannels has not yet been modeled, as far as the authors are aware, DNA contraction in nanoslits was tested by Bakajin et al. (21) and treated numerically by Stigter (24). We assume that the end-to-end length  of a contracting molecule in a nanochannel can be roughly characterized with an exponential fitting function:
of a contracting molecule in a nanochannel can be roughly characterized with an exponential fitting function:
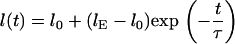 |
(2) |
Here 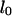 is the equilibrium length in the channel,
is the equilibrium length in the channel, 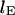 the measured initial extended length, and
the measured initial extended length, and 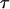 is the time constant of contraction. The contraction of several T4 DNA strands was fit with this function fixing
is the time constant of contraction. The contraction of several T4 DNA strands was fit with this function fixing 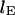 to the measured length at
to the measured length at 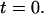 The results were
The results were  and
and 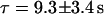 with n = 8, where n is the number of measurements. The ratio of extension was
with n = 8, where n is the number of measurements. The ratio of extension was  An example of one of these fits is plotted in Fig. 5. The equilibrium extended length of
An example of one of these fits is plotted in Fig. 5. The equilibrium extended length of 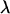 -DNA molecules in
-DNA molecules in 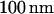 channels was measured to be
channels was measured to be 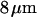 by Tegenfeldt et al. (16). They also found that the extended length
by Tegenfeldt et al. (16). They also found that the extended length 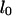 scales linearly with the contour length
scales linearly with the contour length  which leads to an expected length of
which leads to an expected length of 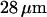 for a T4 molecule. This is in close agreement with our measured value. In all experiments molecules drifted at
for a T4 molecule. This is in close agreement with our measured value. In all experiments molecules drifted at 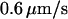 inside the nanochannels. This was compensated for either mathematically or with an electric field offset of
inside the nanochannels. This was compensated for either mathematically or with an electric field offset of 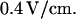 The origin of the drift might be an electrochemical or osmotic gradient due to different DNA or dye concentrations in the fluid reservoirs.
The origin of the drift might be an electrochemical or osmotic gradient due to different DNA or dye concentrations in the fluid reservoirs.
FIGURE 5.

Plot of end-to-end length  versus time
versus time 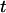 for a contracting T4 molecule in a nanochannel. The molecule was stretched out by driving it electrophoretically into the channel. At
for a contracting T4 molecule in a nanochannel. The molecule was stretched out by driving it electrophoretically into the channel. At 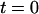 the field was turned off and the molecule began to contract. To quantify this, the function in Eq. 2 was fit to the data. Setting
the field was turned off and the molecule began to contract. To quantify this, the function in Eq. 2 was fit to the data. Setting  to
to  which is the measurement value at
which is the measurement value at 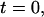 results in
results in 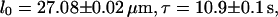 and
and 
Relaxed recoil
After the molecules had completely relaxed to their equilibrium length inside the nanochannels, they were driven electrophoretically to the entrance of the channels. Once the tip of the molecule was straddling the interface, the voltage was turned off and the molecules were observed to completely recoil from the nanochannel. Because molecules were allowed to reach equilibrium before beginning to recoil, this process was driven purely by the entropic recoil force and unaffected by elastic restoration. The insertion length of the molecules in the nanochannel plotted versus time shows a square root behavior that was fitted by Eq. 1. The results for the ratio of the entropic force to the drag coefficient 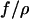 were
were 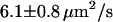 (n = 23) and
(n = 23) and 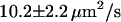 (n = 18) for molecules recoiling from the two sides of the nanochannel array. The ambient drift, responsible for the difference between the two values, can be compensated for by averaging the two, resulting in a corrected value of
(n = 18) for molecules recoiling from the two sides of the nanochannel array. The ambient drift, responsible for the difference between the two values, can be compensated for by averaging the two, resulting in a corrected value of 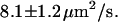 In addition, a set of experiments with an electrical offset correction resulted in
In addition, a set of experiments with an electrical offset correction resulted in  Fig. 6 shows this set of experiments as well as a fit to all of the curves. The theory accurately describes the data. Our values for
Fig. 6 shows this set of experiments as well as a fit to all of the curves. The theory accurately describes the data. Our values for  are three times higher than the value of
are three times higher than the value of 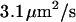 determined by Turner et al. (8) for DNA recoil from nanopillar arrays. This is probably due to an increased entropic force
determined by Turner et al. (8) for DNA recoil from nanopillar arrays. This is probably due to an increased entropic force 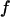 due the higher confinement in 100-nm nanochannels compared to nanopillar arrays with 125-nm spacing.
due the higher confinement in 100-nm nanochannels compared to nanopillar arrays with 125-nm spacing.
FIGURE 6.

Plot of insertion length  over time
over time 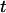 for 14 T4 DNA molecules recoiling from the nanochannel. All curves are fit together with Eq. 1. From this fit, the ratio of the entropic force to the drag coefficient was determined to be
for 14 T4 DNA molecules recoiling from the nanochannel. All curves are fit together with Eq. 1. From this fit, the ratio of the entropic force to the drag coefficient was determined to be 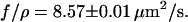
Stretched recoil
As in the stretching experiments, molecules were electrophoretically driven from the microchannel into the nanochannels. But this time the strands were only inserted partly before the field was switched off. Under these circumstances the molecules were stretched during their entrance but were not given the opportunity to contract before the recoil process had begun. A combined contraction and recoil was observed. A comparison between one of these combined processes and a pure recoil process can be seen in Fig. 4. The difference between the two can clearly be distinguished.
To formulate a recoil theory that accommodates contracting, we introduce a stretching factor 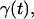 which describes the degree to which the inserted part of the molecule is stretched compared to the equilibrium extension inside our nanochannels. As in Eq. 2, we assume an exponential dependence on the time
which describes the degree to which the inserted part of the molecule is stretched compared to the equilibrium extension inside our nanochannels. As in Eq. 2, we assume an exponential dependence on the time 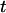 :
:
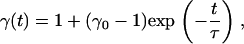 |
(3) |
with the contraction time constant  and the initial stretching factor
and the initial stretching factor  at the beginning of the recoil. Therefore we may write, for the insertion length
at the beginning of the recoil. Therefore we may write, for the insertion length 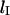 of a molecule that begins to both recoil and contract at the instant the electric field is turned off,
of a molecule that begins to both recoil and contract at the instant the electric field is turned off,
 |
(4) |
which is the model that we have used to fit the data. The model treats contraction and recoiling as decoupled processes, which is a valid assumption for recoil events that last much longer than the contraction time constant  Fig. 7 compares a stretched recoil process with a relaxed recoil process for the same molecule in the same channel. Each has been fit using the appropriate model. As a result we obtain
Fig. 7 compares a stretched recoil process with a relaxed recoil process for the same molecule in the same channel. Each has been fit using the appropriate model. As a result we obtain 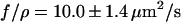 for an offset corrected set of experiments (n = 13), which is close to the value we obtained for relaxed recoil experiments. This indicates that the modified recoil model, which accounts for the entropic force as well as the stretching, is a good first approximation.
for an offset corrected set of experiments (n = 13), which is close to the value we obtained for relaxed recoil experiments. This indicates that the modified recoil model, which accounts for the entropic force as well as the stretching, is a good first approximation.
FIGURE 7.

Demonstration of the difference between a process driven purely by the entropic recoil force and a process driven by both entropic recoil and entropic contraction. The insertion length  is plotted against time
is plotted against time 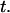 Both experiments are done with the same molecule. The curves were fit with the appropriate model (Eq. 1 or Eq. 3). As a result we obtain
Both experiments are done with the same molecule. The curves were fit with the appropriate model (Eq. 1 or Eq. 3). As a result we obtain 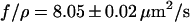 for the pure recoil event and
for the pure recoil event and 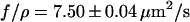 for the combined process. The strand was stretched by 18% in the combined process and relaxed with a time constant of
for the combined process. The strand was stretched by 18% in the combined process and relaxed with a time constant of 
DNA unfolding by recoil
The technique of plotting normalized fluorescence intensity along the channel axis over time (17) is a powerful tool for investigating the unfolding behavior of DNA molecules. This is important for us as many of the recoil experiments showed a folded conformation upon entry, two examples of which are shown in Fig. 8. Folded portions of DNA molecules are brighter than unfolded portions in the time trace plots. The possibility that the brighter portion of the molecule has simply been compressed can be ruled out by observing its evolution through time. We would expect compressed regions to decompress and reach equilibrium with the rest of the molecule over a period of time on par with the timescale of the observed contraction processes. Instead, the border between the bright and the dim sections does not become less defined through time. Only the observed lengths of the bright and dim portions change during the recoil process. Finally, when a certain threshold is reached the molecule suddenly becomes uniform in intensity and slightly longer. This event may correspond to the final act in an unfolding process where the energy required to bend the end of the DNA molecule is released. Note that molecules that appeared to be folded were discarded from all pure recoil and stretch-recoil measurements.
FIGURE 8.

Intensity time traces of different types of molecule folding during recoil processes. (dotted lines = channel entrance). (A) A T4 DNA molecule is driven into nanochannel with a 21 V/cm pulse. It immediately begins to contract and recoil. In this case, however, the front end is looped (a) and unfolds over time. The molecule straightens when the folded end finally flops open (b). (B) Similar to the molecule in panel A, this molecule contracts, recoils, and unfolds (c) simultaneously. It also shows the rare case of a knot (d) in the middle of the strand that does not unfold. Because any loose folds would straighten out during the recoil process, this part of the molecule must be entangled.
From a theoretical point of view, folded DNA molecules should be more stretched than unfolded ones due to volume exclusion effects. This implies that, for a molecule with an unfolded equilibrium extension length of  the observed length will be larger than
the observed length will be larger than 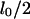 when it is folded in half. Therefore the folded configuration is entropically unfavorable in comparison with the straight configuration, so that given enough time any folded molecules should spontaneously unfold. For molecules with a significant degree of folding (more than several microns), there were no observable decreases in length of the folded segment of the molecule during our experiments. We therefore assume that spontaneous unfolding must occur over a much larger timescale than that of the unfolding process that we induce using the recoil force.
when it is folded in half. Therefore the folded configuration is entropically unfavorable in comparison with the straight configuration, so that given enough time any folded molecules should spontaneously unfold. For molecules with a significant degree of folding (more than several microns), there were no observable decreases in length of the folded segment of the molecule during our experiments. We therefore assume that spontaneous unfolding must occur over a much larger timescale than that of the unfolding process that we induce using the recoil force.
DNA friction determination
To determine hydrodynamic drag of DNA in the nanochannels, molecules were repeatedly driven by an electric field of 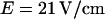 in both directions. Fig. 9 shows a portion of two time traces demonstrating molecules driven in both directions. Folded molecules could easily be distinguished, and only unfolded molecules were used for velocity measurements. Velocities were measured by a linear fit of the molecule position versus time. Fig. 10 displays the distribution of the measurement values as well as two Gaussian fits, one for each direction. The average velocity of all measurements was
in both directions. Fig. 9 shows a portion of two time traces demonstrating molecules driven in both directions. Folded molecules could easily be distinguished, and only unfolded molecules were used for velocity measurements. Velocities were measured by a linear fit of the molecule position versus time. Fig. 10 displays the distribution of the measurement values as well as two Gaussian fits, one for each direction. The average velocity of all measurements was  which corresponds to a mobility of
which corresponds to a mobility of 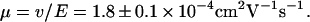 This value is slightly higher than
This value is slightly higher than 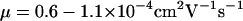 measured for
measured for 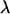 -DNA in silicon/polydimethylsiloxane nanochannels (29). In these measurements an overall linear relation between velocity and electric field was measured. This linearity was also confirmed for DNA translocation through nanopores at high electric fields of the order of
-DNA in silicon/polydimethylsiloxane nanochannels (29). In these measurements an overall linear relation between velocity and electric field was measured. This linearity was also confirmed for DNA translocation through nanopores at high electric fields of the order of 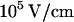 (20). This is in agreement with theoretical models of confined polymers (30) and confined DNA (21) that predict hydrodynamic friction coefficients that are dependent on channel diameter and viscosity but not on the electric field.
(20). This is in agreement with theoretical models of confined polymers (30) and confined DNA (21) that predict hydrodynamic friction coefficients that are dependent on channel diameter and viscosity but not on the electric field.
FIGURE 9.

Intensity time traces for two T4 DNA molecules driven electrophoretically within a nanochannel. The molecules have already completely relaxed and do not change their extended length. (A) Straightened molecule. First the molecule is driven downward (a) and then upward (b) by 21 V/cm pulses. (B) Molecule with the upper end folded. This molecule is also driven downwards (c) and then upwards (d). The response was the same as in panel A. No unfolding was observed.
FIGURE 10.

Histogram showing the distribution of velocities that were obtained for molecules driven through nanochannels under an electric field of 21 V/cm in both directions. The distributions for the two directions are fit with Gaussians. The results are 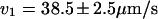 and
and 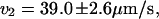 which are equal to within the measurement error.
which are equal to within the measurement error.
The hydrodynamic friction force on a molecule is 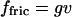 with the viscous drag coefficient
with the viscous drag coefficient  As the channel diameter is on the order of the persistence length
As the channel diameter is on the order of the persistence length  most of the hydrodynamic interactions between molecule segments are screened (21). Thus, the drag coefficient is assumed to be
most of the hydrodynamic interactions between molecule segments are screened (21). Thus, the drag coefficient is assumed to be 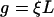 with the friction coefficient per unit length
with the friction coefficient per unit length 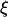 and the contour length
and the contour length 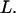 For a nonaccelerating DNA strand of charge
For a nonaccelerating DNA strand of charge 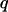 and charge per unit contour length
and charge per unit contour length 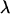 the friction force and the electrical force
the friction force and the electrical force 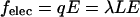 are in equilibrium. Therefore, the friction coefficient per unit contour length is
are in equilibrium. Therefore, the friction coefficient per unit contour length is  Assuming a charge per unit length of
Assuming a charge per unit length of 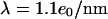 we calculate a friction coefficient of
we calculate a friction coefficient of 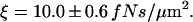 This is five times higher than the pure hydrodynamic friction coefficient
This is five times higher than the pure hydrodynamic friction coefficient 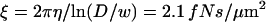 of a solid tube with the DNA diameter
of a solid tube with the DNA diameter  inside another PVP solution-filled tube with diameter
inside another PVP solution-filled tube with diameter 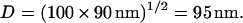 The calculation is a lower bound as it neglects the average DNA segment tilt in the channel as well as frictional interactions with the channel walls. Additionally, although electroosmotic backflow was minimized by the dynamic PVP coating, it may not have been completely eliminated. Future studies with different surface coatings may isolate the relative contributions of hydrodynamic friction, DNA-wall interactions, and electroosmotic backflow to the overall friction. The friction coefficient can be converted to drag per unit extended length
The calculation is a lower bound as it neglects the average DNA segment tilt in the channel as well as frictional interactions with the channel walls. Additionally, although electroosmotic backflow was minimized by the dynamic PVP coating, it may not have been completely eliminated. Future studies with different surface coatings may isolate the relative contributions of hydrodynamic friction, DNA-wall interactions, and electroosmotic backflow to the overall friction. The friction coefficient can be converted to drag per unit extended length 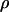 by scaling
by scaling 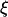 with the previously determined extended length
with the previously determined extended length 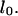 The result is
The result is 
Calculation of entropic recoil force
From the determined drag coefficient 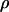 and the fitted ratio of the entropic force to the drag coefficient
and the fitted ratio of the entropic force to the drag coefficient 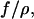 we calculate an entropic force of
we calculate an entropic force of 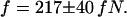 Note that this force is specific for DNA molecules at the interface between a nanochannel and the microchannel in our device. To obtain a theoretical estimate for comparison with the measured entropic recoil force, we calculated the free energy difference between a DNA strand in a 100-nm channel and in free solution. This was done using Schaefer's polymer model for persistent polymers (31). Inside the nanochannel, this model was combined with de Gennes' “blob” model (19) as has been described elsewhere in detail (15,16,32). Dividing the free energy change by half the extended length, which is approximately the distance the center of mass moves during recoil, leads to an estimate for the entropic recoil force of
Note that this force is specific for DNA molecules at the interface between a nanochannel and the microchannel in our device. To obtain a theoretical estimate for comparison with the measured entropic recoil force, we calculated the free energy difference between a DNA strand in a 100-nm channel and in free solution. This was done using Schaefer's polymer model for persistent polymers (31). Inside the nanochannel, this model was combined with de Gennes' “blob” model (19) as has been described elsewhere in detail (15,16,32). Dividing the free energy change by half the extended length, which is approximately the distance the center of mass moves during recoil, leads to an estimate for the entropic recoil force of 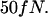 The difference between the measured and estimated values for force might be due to the uncertainty of the shielding of the DNA backbone charge which influences the calculation of the drag. A better theoretical estimate might also be achieved by using a more sophisticated numerical polymer model such as the worm-like chain model, though such a calculation would require numerical simulations. Note also that our approach attributes the free energy change to the difference in configuration space accessible to a molecule, and assumes that electrical interaction between DNA and the walls is negligible. This is a valid assumption considering that the Debye screening length at the buffer concentration used is less than
The difference between the measured and estimated values for force might be due to the uncertainty of the shielding of the DNA backbone charge which influences the calculation of the drag. A better theoretical estimate might also be achieved by using a more sophisticated numerical polymer model such as the worm-like chain model, though such a calculation would require numerical simulations. Note also that our approach attributes the free energy change to the difference in configuration space accessible to a molecule, and assumes that electrical interaction between DNA and the walls is negligible. This is a valid assumption considering that the Debye screening length at the buffer concentration used is less than 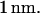
CONCLUSION
We have demonstrated several ways of manipulating single DNA molecules in nanochannels. The entropic force was used to stretch molecules, to retract molecules from the nanochannels, and to straighten folded strands. In the case of molecule retraction, we were able to demonstrate the difference between those processes driven purely by the entropic recoil force and those processes for which elastic restoration also plays a role. Both kinds of molecule retraction could be interpreted quantitatively. By combining the results with friction measurements we were able to estimate the entropic force acting on DNA molecules at the entrance to our nanochannels.
This work is relevant for both the statistical physics and the applied biophysics fields. From a statistical mechanics perspective, further insight into the nature of the entropic force might be gained by investigating single molecule retraction from nanochannels of different widths. Stretching and relaxation experiments using molecules of different sizes could lead to a better understanding of the elastic forces for DNA strands, as theory suggests that longer polymer molecules have smaller spring constants (33). As we have shown, elongation of molecules in nanochannels enables researchers to optically visualize molecule folding. Further studies in this area may lead to a better understanding of DNA packaging into bacteriophage heads or chromatin (34), which is an important topic in biology. Devices combining nanochannels with nanopore-like constrictions will give us the opportunity to combine optical and electrical DNA investigations. The entropically driven process of DNA recoil from nanopatterned geometries has already proven its potential to separate DNA by length (7). Our work will lead to an improved device geometry to achieve higher separation resolution for DNA molecules in the kilobase to megabase pair range.
Acknowledgments
This work was supported by the National Human Genome Research Institute, as well as the Nanobiotechnology Center (NBTC), which is funded by the STC Program of the National Science Foundation under agreement No. ECS-9876771 and the New York State Office of Science, Technology and Academic Research (NYSTAR). Device fabrication was performed in part at the Cornell NanoScale Science and Technology Facility (CNF) with the assistance of Rob Ilic. CNF is a member of the National Nanotechnology Infrastructure Network, which is supported by the National Science Foundation, as well as in the Cornell Center for Materials Research (CCMR).
J. T. Mannion and C. H. Reccius contributed equally to this publication.
References
- 1.Han, J., and H. G. Craighead. 2000. Separation of long DNA molecules in a microfabricated entropic trap array. Science. 288:1026–1029. [DOI] [PubMed] [Google Scholar]
- 2.Han, J. Y., and H. G. Craighead. 2002. Characterization and optimization of an entropic trap for DNA separation. Anal. Chem. 74:394–401. [DOI] [PubMed] [Google Scholar]
- 3.Slater, G. W., Y. Gratton, M. Kenward, L. McCormick, and F. Tessier. 2003. Deformation, stretching, and relaxation of single-polymer chains: fundamentals and examples. Soft Matter. 1:365–391. [Google Scholar]
- 4.Tessier, F., J. Labrie, and G. W. Slater. 2002. Electrophoretic separation of long polyelectrolytes in submolecular-size constrictions: a Monte Carlo study. Macromolecules. 35:4791–4800. [Google Scholar]
- 5.Huang, L. R., J. O. Tegenfeldt, J. J. Kraeft, J. C. Sturm, R. H. Austin, and E. C. Cox. 2002. A DNA prism for high-speed continuous fractionation of large DNA molecules. Nat. Biotechnol. 20:1048–1051. [DOI] [PubMed] [Google Scholar]
- 6.Kaji, N., Y. Tezuka, Y. Takamura, M. Ueda, T. Nishimoto, H. Nakanishi, Y. Horiike, and Y. Baba. 2004. Separation of long DNA molecules by quartz nanopillar chips under a direct current electric field. Anal. Chem. 76:15–22. [DOI] [PubMed] [Google Scholar]
- 7.Cabodi, M., S. W. P. Turner, and H. G. Craighead. 2002. Entropic recoil separation of long DNA molecules. Anal. Chem. 74:5169–5174. [DOI] [PubMed] [Google Scholar]
- 8.Turner, S. W. P., M. Cabodi, and H. G. Craighead. 2002. Confinement-induced entropic recoil of single DNA molecules in a nanofluidic structure. Phys. Rev. Lett. 88:128103. [DOI] [PubMed] [Google Scholar]
- 9.Bakajin, O., T. A. J. Duke, J. Tegenfeldt, C. F. Chou, S. S. Chan, R. H. Austin, and E. C. Cox. 2001. Separation of 100-kilobase DNA molecules in 10 seconds. Anal. Chem. 73:6053–6056. [DOI] [PubMed] [Google Scholar]
- 10.Chang, H., F. Kosari, G. Andreadakis, M. A. Alam, G. Vasmatzis, and R. Bashir. 2004. DNA-mediated fluctuations in ionic current through silicon oxide nanopore channels. Nano Lett. 4:1551–1556. [Google Scholar]
- 11.Heng, J. B., C. Ho, T. Kim, R. Timp, A. Aksimentiev, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp. 2004. Sizing DNA using a nanometer-diameter pore. Biophys. J. 87:2905–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li, J. L., M. Gershow, D. Stein, E. Brandin, and J. A. Golovchenko. 2003. DNA molecules and configurations in a solid-state nanopore microscope. Nat. Mater. 2:611–615. [DOI] [PubMed] [Google Scholar]
- 13.Storm, A. J., C. Storm, J. H. Chen, H. Zandbergen, J. F. Joanny, and C. Dekker. 2005. Fast DNA translocation through a solid-state nanopore. Nano Lett. 5:1193–1197. [DOI] [PubMed] [Google Scholar]
- 14.Li, J., D. Stein, C. McMullan, D. Branton, M. J. Aziz, and J. A. Golovchenko. 2001. Ion-beam sculpting at nanometre length scales. Nature. 412:166–169. [DOI] [PubMed] [Google Scholar]
- 15.Reisner, W., K. J. Morton, R. Riehn, Y. M. Wang, Z. N. Yu, M. Rosen, J. C. Sturm, S. Y. Chou, E. Frey, and R. H. Austin. 2005. Statics and dynamics of single DNA molecules confined in nanochannels. Phys. Rev. Lett. 94:196101. [DOI] [PubMed] [Google Scholar]
- 16.Tegenfeldt, J. O., C. Prinz, H. Cao, S. Chou, W. W. Reisner, R. Riehn, Y. M. Wang, E. C. Cox, J. C. Sturm, P. Silberzan, and R. H. Austin. 2004. The dynamics of genomic-length DNA molecules in 100-nm channels. Proc. Natl. Acad. Sci. USA. 101:10979–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riehn, R., M. C. Lu, Y. M. Wang, S. F. Lim, E. C. Cox, and R. H. Austin. 2005. Restriction mapping in nanofluidic devices. Proc. Natl. Acad. Sci. USA. 102:10012–10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueda, M., T. Hayama, Y. Takamura, Y. Horiike, T. Dotera, and Y. Baba. 2004. Electrophoresis of long deoxyribonucleic acid in curved channels: the effect of channel width on migration dynamics. J. Appl. Phys. 96:2937–2944. [Google Scholar]
- 19.de Gennes, P. G. 1979. Scaling Concepts in Polymer Physics. Cornell University Press, Ithaca, NY.
- 20.Chen, P., J. J. Gu, E. Brandin, Y. R. Kim, Q. Wang, and D. Branton. 2004. Probing single DNA molecule transport using fabricated nanopores. Nano Lett. 4:2293–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bakajin, O. B., T. A. J. Duke, C. F. Chou, S. S. Chan, R. H. Austin, and E. C. Cox. 1998. Electrohydrodynamic stretching of DNA in confined environments. Phys. Rev. Lett. 80:2737–2740. [Google Scholar]
- 22.Perkins, T. T., D. E. Smith, R. G. Larson, and S. Chu. 1995. Stretching of a single tethered polymer in a uniform-flow. Science. 268:83–87. [DOI] [PubMed] [Google Scholar]
- 23.Sischka, A., K. Toensing, R. Eckel, S. D. Wilking, N. Sewald, R. Ros, and D. Anselmetti. 2005. Molecular mechanisms and kinetics between DNA and DNA binding ligands. Biophys. J. 88:404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stigter, D. 2002. Wall effects on DNA stretch and relaxation. Biophys. Chem. 101:447–459. [DOI] [PubMed] [Google Scholar]
- 25.Schellman, J. A., and D. Stigter. 1977. Electrical double layer, zeta potential, and electrophoretic charge of double-stranded DNA. Biopolymers. 16:1415–1434. [DOI] [PubMed] [Google Scholar]
- 26.Gao, Q. F., and E. S. Yeung. 1998. A matrix for DNA separation: genotyping and sequencing using poly(vinylpyrrolidone) solution in uncoated capillaries. Anal. Chem. 70:1382–1388. [DOI] [PubMed] [Google Scholar]
- 27.Kang, S. H., M. R. Shortreed, and E. S. Yeung. 2001. Real-time dynamics of single-DNA molecules undergoing adsorption and desorption at liquid-solid interfaces. Anal. Chem. 73:1091–1099. [DOI] [PubMed] [Google Scholar]
- 28.Smith, S. B., L. Finzi, and C. Bustamante. 1992. Direct mechanical measurements of the elasticity of single DNA-molecules by using magnetic beads. Science. 258:1122–1126. [DOI] [PubMed] [Google Scholar]
- 29.Campbell, L. C., M. J. Wilkinson, A. Manz, P. Camilleri, and C. J. Humphreys. 2004. Electrophoretic manipulation of single DNA molecules in nanofabricated capillaries. Lab Chip. 4:225–229. [DOI] [PubMed] [Google Scholar]
- 30.Brochard, F., and P. G. de Gennes. 1977. Dynamics of confined polymer-chains. J. Chem. Phys. 67:52–56. [Google Scholar]
- 31.Schaefer, D. W., J. F. Joanny, and P. Pincus. 1980. Dynamics of semiflexible polymers in solution. Macromolecules. 13:1280–1289.
- 32.Reccius, C. H., J. T. Mannion, J. D. Cross, and H. G. Craighead. 2005. Compression and free expansion of single DNA molecules in nanochannels. Phys. Rev. Lett. 95:268101. [DOI] [PubMed]
- 33.Bueche, F. 1962. Physical Properties of Polymers. Interscience, New York, NY.
- 34.Smith, D. E., S. J. Tans, S. B. Smith, S. Grimes, D. L. Anderson, and C. Bustamante. 2001. The bacteriophage phi 29 portal motor can package DNA against a large internal force. Nature. 413:748–752. [DOI] [PubMed] [Google Scholar]