

Abstract
Recently we have developed a normal-modes-based algorithm that predicts the direction of protein conformational changes given the initial state crystal structure together with a small number of pairwise distance constraints for the end state. Here we significantly extend this method to accurately model both the direction and amplitude of protein conformational changes. The new protocol implements a multisteps search in the conformational space that is driven by iteratively minimizing the error of fitting the given distance constraints and simultaneously enforcing the restraint of low elastic energy. At each step, an incremental structural displacement is computed as a linear combination of the lowest 10 normal modes derived from an elastic network model, whose eigenvectors are reorientated to correct for the distortions caused by the structural displacements in the previous steps. We test this method on a list of 16 pairs of protein structures for which relatively large conformational changes are observed (root mean square deviation >3 Å), using up to 10 pairwise distance constraints selected by a fluctuation analysis of the initial state structures. This method has achieved a near-optimal performance in almost all cases, and in many cases the final structural models lie within root mean square deviation of 1 ∼ 2 Å from the native end state structures.
INTRODUCTION
To perform biological functions, many proteins undergo complex conformational changes from one functional state (initial state) to another (end state). In many cases, only the initial state is structurally solved whereas the end state structure is not available due to the difficulty of stabilizing it for x-ray crystallography or nuclear magnetic resonance measurements. Therefore it is of great interest to develop computational methods to predict the conformational changes and generate a structural model for the unknown end state.
Direct simulation of protein conformational changes with atomic details poses a formidable challenge to the molecular dynamics that is severely limited in both simulation time and system size. Recent work by a number of researchers has suggested an alternative method to efficiently probe the slow motions of protein complexes: the lowest-frequency normal modes that are computed from a highly simplified elastic network model (ENM) have been shown to give surprisingly good descriptions of the functional dynamics of protein complexes. Many biologically interesting protein conformational changes were found to be dominated by just a handful of lowest-frequency normal modes (1–5). However, finding the relevant modes that contribute to the observed conformational changes with certainty is generally not feasible without any input of structural information for the end state.
Experimentally, pairwise distances between specified atoms of a protein in its native state (in solution) can be measured by NMR. Other techniques are available that utilize fast spectroscopy (for example, site-direct spin labeling combined with electron paramagnetic resonance spectroscopy (6)) to probe atomic pairwise distances of a protein in a transient state. Computationally, it has been found that a small number of pairwise distance constraints (DC) can improve the protein structure modeling significantly (7,8). In the framework of ENM, because functionally interesting conformational changes generally involve only a small number of low-frequency normal modes, it is natural to expect that a small number of pairwise DCs, if chosen properly, would be sufficient for obtaining a good approximation to the conformational changes.
The above idea was first explored in a recent work by us (9), where we proposed a new method based on ENM that predicts the direction of protein conformational changes given the crystal structure for the initial state together with a small number of pairwise DCs for the end state. The predicted conformational change, which is a linear combination of multiple low-frequency normal modes solved from the ENM, is computed as a response displacement induced by a perturbation to the system energy that incorporates the given DCs as restraints. For a list of test cases, we found that the computed response displacement overlaps significantly with the observed conformational changes, when only up to 10 pairwise DCs are used.
Similar studies were conducted by other groups. In one study (10), Tama and co-workers used a linear combination of low-frequency normal modes solved from ENM for flexible fitting of high-resolution structures into low-resolution maps of macromolecular complexes from electron microscopy. Another work by Delarue and co-workers applied this method to the refinement of x-ray crystallography (11). Both studies have demonstrated the efficiency and accuracy of the normal-modes-based method in the modeling of macromolecular structures. There have also been attempts to use ENM-derived normal modes for enhancing the efficiency of MD simulations (12), or to sample the transition pathways between structural states (13,14).
As promising as it has been shown, the method proposed in Zheng and Brooks (9) needs further improvement and extension before it can be used to model the end state structures accurately. First, the amplitude of the conformational change is not predicted by this perturbational method: the amplitude can only be determined with accuracy by a nonperturbational method, such as a search in the conformational space that starts from the initial state conformation (see Methods). Second, as a perturbational method, its accuracy degrades as the conformation deviates further away from the initial state structure, which results in local distortions (for example, incorrect Cα-Cα pseudobond length), therefore a low-energy restraint needs to be enforced to avoid such problems (see Methods). Third, the eigenvectors computed from the initial state structure become less and less accurate as the conformation deviates further away from the initial state, one possible solution is to repeat the NMA periodically (10), which may slow down the computation significantly; to avoid such expensive computing overhead we will develop a much more efficient though approximate technique to correct the inaccurate eigenvectors. The idea is to reorientate the three-dimensional (3D) component of the eigenvectors at each Cα position to correct for the rigid-body movements by the previous moves (see Methods). Last but not least, a well-designed scheme remains to be established to select potentially useful residue pairs whose pairwise distances serve as input of DCs. This task is addressed here using an ENM-based fluctuation analysis of the initial state structures (see Methods).
To summarize, we will go beyond the perturbational method developed in Zheng and Brooks (9) and perform a rapid search in the conformational space that is driven by iteratively minimizing the error of fitting the given DCs while enforcing the restraint of low elastic energy. At each step, the fitting error plus the energy cost term is minimized by linear regression that optimizes the linear combination of the lowest 10 normal modes (after reorientation) as an incremental structural displacement (see Methods). The search starts from the initial state conformation. The goal is to generate a structural model that satisfies the given DCs with low energy cost, which is expected to be a good approximate model for the end state structure.
We will test the above method on a list of 16 test cases of protein structure pairs with relatively large conformational changes (root mean square deviation (RMSD) > 3 Å) between the initial and the end state structures, using up to 10 selected pairwise DCs.
METHODS
Elastic network model
Given the Cα atomic coordinates for a protein's crystal structure (each residue is represented by its Cα atom), we build an elastic network model by using a harmonic potential with a single force constant to account for pairwise interactions between all Cα atoms that are within a cutoff distance (RC = 10 Å). The energy in the elastic network representation of a protein is (1–3):
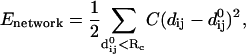 |
(1) |
where dij is the distance between the dynamical coordinates of the Cα atoms i and j, and 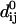 is the distance between Cα atoms i and j, as given in the crystal structure.
is the distance between Cα atoms i and j, as given in the crystal structure.
For the above harmonic Hamiltonian we can perform the standard normal modes analysis (NMA), and using the eigenvectors of the lowest frequency normal modes (starting from mode No. 1 after excluding the six zero-modes for translations and rotations) we can compute the overlaps with the conformational changes between two states with known structures (4). The drastic simplification of representing the complex protein structure by an effective harmonic potential is justified by a study (15) that showed that a single spring constant potential reproduces the slow dynamics that is computed from the normal modes analysis of a complex all-atom potential.
Selection of residue pairs for pairwise DCs
Here we propose a new scheme for predicting and selecting potentially useful residue pairs whose pairwise distances serve as input of the DCs.
For any residue pair (i,j) (excluding the first two residues at the N-terminal and C-terminal ends of the protein, which are usually too flexible), we compute the low-frequency mean square fluctuation for the pairwise distance rij:
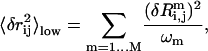 |
(2) |
where 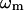 is the eigenvalue of mode m,
is the eigenvalue of mode m, 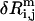 is the perturbational change to rij caused by the structural displacement as described by the eigenvector of mode m. Only the contributions from the lowest M nonzero modes are considered in Eq. 2 (M = 10 as default).
is the perturbational change to rij caused by the structural displacement as described by the eigenvector of mode m. Only the contributions from the lowest M nonzero modes are considered in Eq. 2 (M = 10 as default).
Then we sort all residue pairs (i,j) by 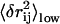 from high to low and select the top N residue pairs (N = 1,…10). We perform the following redundancy check and removing to avoid selecting redundant pairs (residue pairs whose pairwise distance fluctuations are significantly correlated; see below). This ensures that the given DCs are independent of each other and thus provide nonredundant information to maximally constraint the conformational search.
from high to low and select the top N residue pairs (N = 1,…10). We perform the following redundancy check and removing to avoid selecting redundant pairs (residue pairs whose pairwise distance fluctuations are significantly correlated; see below). This ensures that the given DCs are independent of each other and thus provide nonredundant information to maximally constraint the conformational search.
For two residue pairs 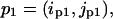 and
and 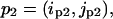 we compute the following pairwise correlation function:
we compute the following pairwise correlation function:
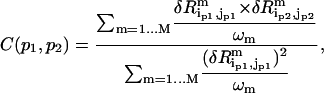 |
(3) |
where 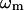 is the eigenvalue of mode m,
is the eigenvalue of mode m, 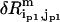 (
(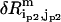 ) is the perturbational change to
) is the perturbational change to 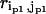 (
(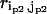 ) caused by the structural displacement as described by the eigenvector of mode m.
) caused by the structural displacement as described by the eigenvector of mode m.
The redundancy removing goes as follows: we start from the residue pair with the highest 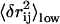 and check each residue pair in order of descending
and check each residue pair in order of descending 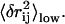 For a residue pair p2, if there exists pair p1 that has been previously selected such that
For a residue pair p2, if there exists pair p1 that has been previously selected such that 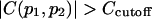 (
(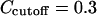 ), then p2 is rejected as redundant; otherwise p2 is selected. We stop when N residue pairs have been selected (N = 1,…10).
), then p2 is rejected as redundant; otherwise p2 is selected. We stop when N residue pairs have been selected (N = 1,…10).
Fitting given DCs with a linear combination of normal modes
To attain the final goal of multisteps iterative fitting to the given DCs, we first study how the fitting is done at a single step.
Suppose we are given N pairwise DCs 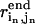 for the end state structure (
for the end state structure (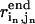 is the pairwise distance between residue in and jn in the end state structure, n = 1, 2, …N). These constraints are incorporated as soft restraints into an error function defined as follows:
is the pairwise distance between residue in and jn in the end state structure, n = 1, 2, …N). These constraints are incorporated as soft restraints into an error function defined as follows:
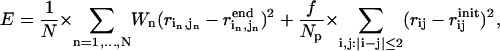 |
(4) |
where the first term enforces the given N DCs, and the second term (elastic energy cost) restraints the pairwise distances between sequentially neighboring residue pairs (i,i + 1) and (i,i + 2), to preserve the local geometry (Cα-Cα pseudobond length and secondary structures) of the initial state structure during the conformational search. By excluding those nonsequential contacts from the energy cost term, we allow interdomain motions and reorganizations of suprasecondary structural elements during the conformational search. For notations, Np is the number of sequentially neighboring residue pairs, f is an overall weight factor for the energy cost term with the default value 100 (f = 100 is empirically found to give generally good results), 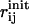 is the distance between residue i and j in the initial state structure,
is the distance between residue i and j in the initial state structure, 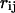 (
(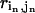 ) is the distance between residue i (in) and j (jn).
) is the distance between residue i (in) and j (jn).
The weight factor for the n'th DC Wn is defined as 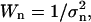 where
where 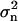 is the mean square error for the n'th DC. When the DCs are derived experimentally,
is the mean square error for the n'th DC. When the DCs are derived experimentally, 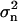 is the experimental error of measuring
is the experimental error of measuring 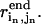 Because we use “simulated” DCs directly obtained from the end-state structures, we simply assume all DCs are equally accurate and set
Because we use “simulated” DCs directly obtained from the end-state structures, we simply assume all DCs are equally accurate and set 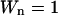 for all of them.
for all of them.
Then we represent the incremental structural displacement 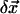 of the ENM as a weighted linear combination of the eigenvectors of the lowest M modes (M = 10 as default):
of the ENM as a weighted linear combination of the eigenvectors of the lowest M modes (M = 10 as default):
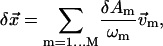 |
(5) |
where 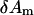 is the coefficient for mode m,
is the coefficient for mode m, 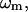 and
and 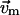 are the eigenvalue and eigenvector for mode m, respectively (after reorientation; see below). Next we want to expand the error function in Eq. 4 around the present conformation in terms of
are the eigenvalue and eigenvector for mode m, respectively (after reorientation; see below). Next we want to expand the error function in Eq. 4 around the present conformation in terms of 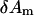 (m = 1,…, M).
(m = 1,…, M).
The perturbational change to the pairwise distance rij is
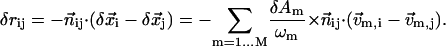 |
(6) |
Here, 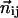 is the unit vector pointing from residue i to j in the present conformation,
is the unit vector pointing from residue i to j in the present conformation, 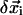 (
(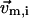 ) is the 3D component of
) is the 3D component of 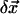 (
(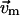 ) at residue i, and
) at residue i, and 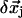 (
(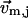 ) is the 3D component of
) is the 3D component of 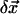 (
(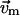 ) at residue j.
) at residue j.
So
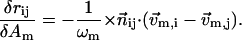 |
(7) |
Therefore the error function in Eq. 4 can be expanded in terms of δAm (m = 1,…, M) as follows,
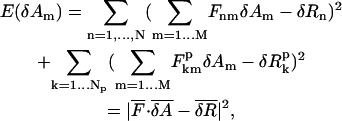 |
(8) |
where
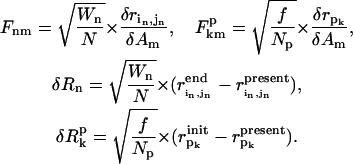 |
(9) |
The line two of Eq. 8 is the compact form of line one, where 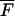 is a (N + Np) × M matrix whose matrix elements are given in Eq. 9,
is a (N + Np) × M matrix whose matrix elements are given in Eq. 9, 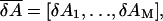
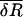 is a (N + Np) dimensional vector whose elements are given in Eq. 9. pk (k = 1,…Np) is an index for each of the Np sequentially neighboring residue pairs included in the energy cost terms (see Eq. 4).
is a (N + Np) dimensional vector whose elements are given in Eq. 9. pk (k = 1,…Np) is an index for each of the Np sequentially neighboring residue pairs included in the energy cost terms (see Eq. 4). 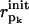 (
(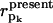 ) is the pairwise distance for pair pk in the initial state structure (present conformation).
) is the pairwise distance for pair pk in the initial state structure (present conformation).
Equation 8 defines a linear-regression (LR) model for minimization and the M variables 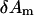 can be solved from the following linear equation:
can be solved from the following linear equation:
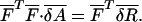 |
(10) |
Finally we plug 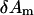 (m = 1,…, M) back into Eq. 5 to solve the optimal incremental structural displacement
(m = 1,…, M) back into Eq. 5 to solve the optimal incremental structural displacement 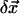 that minimizes the error function in Eq. 4.
that minimizes the error function in Eq. 4.
The above single-step procedure is the key step of our algorithm, which will be iteratively executed in the fitting protocol.
Protocol of iterative fitting to given DCs
The iterative fitting protocol repeats the single-step fitting procedure as detailed in the previous subsection for 100 steps (see Fig. 1 for the flowchart of the entire protocol). Additional explanations are as follows.
FIGURE 1.

Schematic flowchart of the iterative fitting algorithm; see Methods for details.
Amplitude adjustment
At every step, the amplitude of 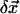 computed from Eq. 5 is adjusted to avoid unwanted distortions. The amplitude is determined such that the fitting error (Eq. 4) is reduced by no more than 10% at first order approximation:
computed from Eq. 5 is adjusted to avoid unwanted distortions. The amplitude is determined such that the fitting error (Eq. 4) is reduced by no more than 10% at first order approximation:
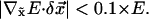 |
(11) |
With the implementation of this amplitude adjustment, the fitting error decays fast in early steps with relatively large amplitude of displacement per step; in later steps when the fitting error becomes small, the amplitude of displacement per step is also reduced to facilitate a finer search. Therefore the amplitude adjustment adaptively allows both a fast convergence at the early stage and a fine search at the later stage of the fitting protocol.
Saturation detection
We stop the iteration and output the conformation 10 steps after detecting a saturation in the logarithm of the fitting error (excluding the energy cost terms); the saturation is found when the rate of decrease in log (fitting error) is less than 1% of the maximal rate recorded in previous steps. The rational is that prolonged fitting after saturation would not be productive because the search has reached a fixed point.
Energy minimization by recruiting higher modes
We recruit all higher modes (excluding the lowest M modes) to further minimize the energy cost (the second term in Eq. 4). At the end of each step, after the incremental displacement is taken, we allow the conformation to relax in the subspace spanned by all modes except the lowest M to further lower its energy. This is implemented by a conjugate gradient minimization procedure (20 steps). This procedure reduces local distortions caused by the use of only the lowest M modes.
Reorientation of eigenvectors after a large displacement
The low-frequency eigenvectors computed for the ENM only give good descriptions of its conformational changes with small amplitude. For the structural displacements with large amplitude, using these eigenvectors to describe them would result in unwanted distortions (for example, distortion of local geometry such as incorrect Cα-Cα pseudobond lengths; see Fig. S1 in Supplementary Material). Here we propose a simple technique that efficiently corrects these distortions by reorientating the old eigenvectors after a relatively large structural displacement without recomputing them.
The basic assumption is that those low-frequency modes describe rigid body collective motions between domains. Therefore we assume that the 3D component of their eigenvectors at each residue within a rigid domain is “attached” to that residue as the ENM undergoes rigid-body movements (see Fig. S1, Supplementary Material), and its orientation relative to its “rigid” neighboring residues is preserved during the movements. For those residues sitting in hinge regions between rigid domains the above assumption may break down, however, those “hinge” residues generally undergo minimal movements so the errors in their moving directions do not result in large distortions.
To make corrections for mode m's eigenvector's 3D component at a given residue n (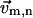 ) after a structural displacement, we do the following:
) after a structural displacement, we do the following:
Form a set of rigid neighbors (SRN). For residue n, examine its neighboring residue i (within 10 Å), then sort them by |δrni |, which is the change of the distance rni after the displacement; finally keep four neighbors with the smallest |δrni | to form the SRN (including residue n itself).
Translate and rotate the old coordinates of the SRN (before the displacement) to minimize the RMSD with its new coordinates (after the displacement) (
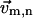 is attached to residue n and undergoes the same translation and rotation as the SRN).
is attached to residue n and undergoes the same translation and rotation as the SRN).Finally translate
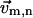 to the new coordinate of residue n.
to the new coordinate of residue n.
To demonstrate its effectiveness in reducing local structural distortions, we plot the distortional elastic energy as a function of the amplitude of the displacement along the direction given by the dominant mode for the observed conformational changes in two cases (see Fig. S2 in Supplementary Material). In both cases, the elastic energy is significantly reduced by this procedure, which implies reduced level of distortions. (The reduction is only slightly better if we exactly recompute the eigenvector as the structure is displaced; see Fig. S2 in Supplementary Material). Therefore, with the same level of tolerance for distortional energy, this procedure allows the conformational search to explore a wider range. This is particularly advantageous for predicting large conformational changes.
Performance evaluation: RMSDeval, RMSDmin, and RMSDlimit
To evaluate the quality of the generated structural models, we compute the RMSD for Cα atoms between the final structural model and the native end state structure, which is denoted as RMSDeval. Unlike the previous study (9), here we do not use the overlap (generalized cosine) to assess the quality of the computed conformational changes as compared with the observed conformational changes, because it only depends on the direction but not the amplitude of conformational changes. We also define RMSDmin to be the minimal RMSD among all models generated during the search. The closeness between RMSDeval and RMSDmin indicates a successfully finished search without serious overfitting (or deviating far away from the end state) that may otherwise result in RMSDeval being significantly larger than RMSDmin.
Next we define a golden standard to evaluate RMSDeval against. Suppose we are given the observed conformational change 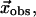 which is obtained by a structural alignment between the initial and end state structures. Then we compute an optimal linear combination of the lowest M modes (denoted as
which is obtained by a structural alignment between the initial and end state structures. Then we compute an optimal linear combination of the lowest M modes (denoted as 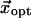 ) to best approximate
) to best approximate 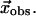 Finally we compute the optimal limit RMSD (denoted as RMSDlimit), which is the RMSD between the native end state structure and the structure obtained by deforming the initial state structure by
Finally we compute the optimal limit RMSD (denoted as RMSDlimit), which is the RMSD between the native end state structure and the structure obtained by deforming the initial state structure by 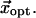 This is essentially the minimal RMSDeval we can ever achieve for a structural model obtained by only using the M lowest modes (assuming no reorientation of eigenvectors). It is given by the following equation:
This is essentially the minimal RMSDeval we can ever achieve for a structural model obtained by only using the M lowest modes (assuming no reorientation of eigenvectors). It is given by the following equation:
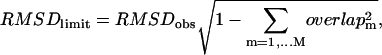 |
(12) |
where overlapm is the overlap between mode m and the observed conformational changes. RMSDobs is the RMSD of the observed conformational changes.
Test of tolerance to errors in given DCs
We introduce randomly generated additive errors up to the maximum of ±2 Å for the value of every DC. For a given set of DCs, we repeat the iterative fitting protocol 10 times for different initial DC errors, then we compute the average and standard deviation of the final model's RMSDeval; the average shows the average performance in the presence of errors, and the standard deviation indicates the level of tolerance to the DC errors. This program will be available to public users upon request.
RESULTS
Test cases
Because the list of test cases used in our previous work is dominated by small conformational changes, we will start by collecting a new list of test cases.
We go through the Molecular Movements Database (http://www.molmovdb.org/) to select suitable pairs of protein movements that satisfy the following three conditions:
The protein has at least 100 residues.
The amplitude of movements falls in the range 3 Å < RMSD < 10 Å.
RMSDlimit (see Methods for definition) for the 10 lowest modes is <3 Å, which guarantees that a good structural model exists.
Finally we collect 16 protein structure pairs with their amplitude of conformational changes ranging from 3.1 to 7.6 Å (Table 1). We will test the fitting protocol on all these cases. For illustrating certain aspects of the protocol, we will use two of them (2LAO→1LST and 1OMP→1ANF) as examples. The results for the remaining 14 cases are given in the Supplementary Material.
TABLE 1.
Test cases information
| PDB1 | PDB2 | Size | RMSDobs (Å) | Overlap | Mode No. | CC |
|---|---|---|---|---|---|---|
| 1bncA | 1dv2A | 433 | 3.86 | 0.906 | 1 | 0.952 |
| 1bp5A | 1a8e | 328 | 6.70 | 0.856 | 2 | 0.829 |
| 1ckmA | 1ckm | 317 | 3.49 | 0.921 | 1 | 0.889 |
| 1e8bA | 1e88A | 160 | 3.46 | 0.826 | 2 | 0.628 |
| 1eps | 1g6sA | 427 | 7.59 | 0.938 | 2 | 0.803 |
| 1ex7A | lex6A | 186 | 3.64 | 0.836 | 1 | 0.787 |
| 1gggA | 1wdnA | 220 | 5.34 | 0.866 | 1 | 0.645 |
| 1omp | 1anf | 370 | 3.77 | 0.676 | 2 | 0.795 |
| 1rkm | 2rkmA | 517 | 3.08 | 0.955 | 1 | 0.808 |
| 1urpA | 2dri | 271 | 4.06 | 0.930 | 2 | 0.696 |
| 2lao | 11st | 238 | 4.70 | 0.886 | 1 | 0.833 |
| 3dapA | 1dapB | 320 | 4.18 | 0.829 | 1 | 0.741 |
| 8ohm | 1cu1A | 435 | 4.39 | 0.955 | 1 | 0.913 |
| 1f3yA | 1jknA | 165 | 3.58 | 0.530 | 1 | 0.645 |
| 1l5bA | 115eA | 101 | 6.51 | 0.544 | 2 | 0.418 |
| 1lfh | 1lfg | 691 | 6.43 | 0.613 | 1 | 0.505 |
The first and second columns are Protein Data Bank (PDB) codes for the protein pairs; the third column is the sequence length of protein PDB1;
the fourth column is the RMSD of the observed conformational changes from PDB1 to PDB2; the fifth column is the overlap and mode number of the mode that dominates the observed conformational changes; and the sixth column is the cross-correlation coefficient (CC) between the observed change in rij and 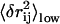 (see Methods).
(see Methods).
We first solve the normal modes from the ENM built from the initial state structures (see Methods). We find that all observed conformational changes are dominated by a single normal mode (the first or second lowest mode), and the overlap between the dominant mode and the observed conformational changes is fairly significant, which ranges between 0.53 and 0.96. This supports the capacity of low-frequency normal modes to capture collective conformational changes widely observed for protein complexes.
Selection of residue pairs for DCs
In our previous study (9), the residue pairs used for the DCs are selected based on the ranking of the observed change in their pairwise distances. The implementation of this selection scheme requires knowing the Cα coordinates of the end state structures, so it is only good for algorithm testing but not for applications where the coordinates of the end state structures are unknown.
Here we propose a new scheme for predicting and selecting those potentially useful residue pairs as the DCs. This new scheme is based solely on a fluctuation analysis of the initial state structures using the low-frequency normal modes (see Methods), which helps experimentalists to decide which pairwise distances they should measure to generate useful DCs for modeling the unknown end state structures.
Following the procedure detailed in Methods, we select 10 residue pairs with the top 10 highest value of 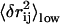 (after redundancy removing; see Methods). Physically, high
(after redundancy removing; see Methods). Physically, high 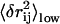 implies large low-frequency fluctuation in rij so it has high probability to undergo large changes during protein conformational changes. In Fig. S3 (see Supplementary Material), we plot the observed change in rij vs.
implies large low-frequency fluctuation in rij so it has high probability to undergo large changes during protein conformational changes. In Fig. S3 (see Supplementary Material), we plot the observed change in rij vs. 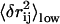 (both are normalized using Z-score; it is obtained by subtracting the average from the raw score then dividing it by the standard deviation). Indeed, we find a positive correlation between them (the correlation coefficient is 0.6 ∼ 0.9 for most cases; see Table 1), which justifies the validity of this selection scheme.
(both are normalized using Z-score; it is obtained by subtracting the average from the raw score then dividing it by the standard deviation). Indeed, we find a positive correlation between them (the correlation coefficient is 0.6 ∼ 0.9 for most cases; see Table 1), which justifies the validity of this selection scheme.
Iterative fitting
The 100-steps iterative fitting is performed for N = 1 ∼ 10 selected DCs. In Fig. 2, we plot the logarithm of the fitting error (excluding the energy cost term), the energy cost term, and RMSDeval (see Methods for definition) as a function of iteration step for two cases. In both cases, the fitting procedure converges fairly fast and smoothly (the fitting error decays roughly exponentially until it saturates well before reaching step 100), and the fitting error is eventually reduced by a factor of 102 ∼ 103. In the meantime, RMSDeval also decreases fast and smoothly, while the energy cost smoothly increases, both of which saturate before the saturation of the fitting error.
FIGURE 2.

The results of our iterative fitting protocol using 10 DCs for: (a) 2LAO → 1LST; (b) 1OMP → 1ANF. In the left panel, we plot the logarithm of the fitting error (+) (excluding the energy cost term), the energy cost term (×), and the RMSDeval (*) as a function of step. In the right panel we plot the fractional contribution from mode No. 1, No. 2, and the remaining eight modes all together to the incremental displacement at every step; mode No. 1 (+), mode No. 2 (×), and the cumulative contribution from mode No. 3–10 (*). Similar results for the remaining 14 cases are shown in Figs. S4–S17.
To reveal the contributions from each of the 10 lowest modes during the fitting procedure, we show in Fig. 2 the fractional contribution from each mode to the incremental structural displacement at every step:
In (2LAO→1LST), mode No. 1 dominates from step 1 to 12 and is thus responsible for the large reduction in RMSDeval from 4.7 to 1.5 Å, which is also consistent with the observed conformational changes being dominated by mode No. 1; after step 12, mode No. 2 and others are also recruited with significant weight by the fitting, when RMSDeval is reduced further down to the minimal value 1.0 Å near step 30.
In (1OMP→1ANF), first mode No. 1 and then mode No. 2 dominates from step 1 to 16 and thus accounts for almost all the reduction in RMSDeval from 3.8 Å to its minimum 1.1 Å near step 20; then, other modes begin to contribute significantly while RMSDeval stays flat.
Therefore, the dominant mode plays a major role in the early stage of fitting process, while other modes are also recruited for refined fitting, particularly in the late stage.
Final structural models
The RMSDeval of the final structural modes for N = 1 ∼ 10 selected DCs are shown in Table S1 (see Supplementary Material) for all test cases. We note that in almost all cases the minimal RMSDeval attained by the final structural models is close to or even lower than the RMSDlimit (see Methods for definition) for the use of 10 lowest modes. Therefore our fitting protocol is near optimal in performance. The results of attaining RMSDeval lower than RMSDlimit are attributed to the use of reoriented eigenvectors.
To assess the quality of the generated structural models, we show the structural alignment between the structural models and the native end state structures (together with the initial state structures aligned with the latter for comparison; see Fig. 3, left panels). The resemblance of the models to the native end state structures as compared with the initial state structures is remarkable. As shown in Fig. 3, right panels, the amplitude of the structural displacement (or difference) between the models and the native end state structures is significantly reduced as compared with the difference between the initial state and the end state structures, except for a few local structural distortions, for example, in the C-terminal of 2LAO, at the tip of a β-hairpin near 173 in 1OMP. In the amplitude plot of the structural differences, those main peaks that indicate large collective movements characteristic of the observed conformational changes are sharply reduced after the fitting. This is because the lowest modes have captured such collective motions.
FIGURE 3.

Quality assessment of the final structural models: (a) 2LAO → 1LST (10 DCs, RMSDeval = 1.0 Å); (b) 1OMP → 1ANF (10 DCs, RMSDeval = 1.1 Å). The left panel shows the backbone conformation of the structural model (denoted as M, colored red), and the initial state structure (denoted as I, colored blue), which are aligned with the native end state structure (denoted as E, colored green). The right panel shows the amplitude of structural displacement/difference between M and E (red curve) versus that between I and E (green curve). Similar results for the remaining 14 cases are shown in Figs. S4–S17.
Dependence on number of DCs
In Fig. 4, we show for each case the dependence of RMSDeval on the number of DCs; in some cases, RMSDeval is reduced to near RMSDlimit for N as small as 1 ∼ 3, while for other cases up to 10 DCs are needed to achieve near-optimal result. In almost all cases, 10 or fewer well-chosen DCs are sufficient to achieve near-optimal performance. This supports the usefulness of this method in applications with experimentally derived DCs that are hard to collect in large numbers.
FIGURE 4.

The dependence of RMSDeval on the number of DCs for all test cases using the lowest M modes for fitting (diamonds, M = 2; circles, M = 10; squares, M = 20).
The minimal number of DCs needed for near-optimal result (RMSDeval ∼ RMSDlimit) is determined by the “effective” number of degrees of freedom for the observed protein conformational changes. For example, for a simple hinge motion between two rigid domains, the “effective” number of degrees of freedom is three (assuming one domain is fixed, the rotation of the second domain is described by three angles). To determine these three variables, up to three DCs are needed (for example: 1ckm, 1eps, 1omp, 1rkm, 1urp, 2lao, 3dap). For more complex conformational changes with more “effective” number of degrees of freedom, more DCs would be needed.
Choice of number of modes used for fitting
The choice of number of modes for fitting is a trade-off between two issues: first, smaller number of modes means smaller search space and thus less computational cost and better chance of finding the global minimum; second, larger number of modes means lower RMSDlimit and therefore potentially better structural models.
We show the results for M = 2, 10, 20 modes in Fig. 4 and Table S1 (see Supplementary Material). A comparison between M = 2 and 10 modes shows that in 10 out of 16 cases a significant reduction in RMSDeval (>0.3 Å) is seen, which is accompanied by a similarly large reduction in RMSDlimit. Therefore, the use of additional modes for fitting besides the dominant mode generally leads to significantly better performance.
To explore the possibility of further improving the performance we test the method for M = 20 modes. We find essentially no significant reduction in RMSDeval from M = 10–20 for almost all cases. This is not surprising because: first, the dependence of RMSDlimit on the number of modes generally reaches a plateau around 10–20 modes, therefore using more modes cannot significantly lower RMSDlimit (see Table S1 of Supplementary Material); and second, more modes means more fitting variables and more difficulty of finding the global minimum, which explains worse results for M = 20 than M = 10 in some cases (see Fig. 4).
Although M = 10 appears to be the best overall among the 3 M values tested here, we still wish to advise future users of this protocol to find the proper number of modes on a case-by-case basis. One useful strategy is to gradually increase the number of modes and see how the resulting structural models change and stop when a “fixed point” is reached.
Tolerance to errors in DCs
We introduce random additive error to the DCs to study its effect on the performance. The results are shown in Table S2 (see Supplementary Material). The variation in RMSDeval is very small for most cases (0.1 ∼ 0.2 Å) despite the errors. This robustness is important when experimentally derived DCs with inherent inaccuracy are used as inputs to this protocol.
Comparison with the old method
For the same list of test cases, we also test the old method that is based on perturbational force pulling (9) (see Table S3 of Supplementary Material). To quantify the improvement, we compute RMSDmin when testing the old method. RMSDmin is the minimal RMSD between the native end state structure and the set of models obtained by displacing the initial state structure along the direction predicted by the old method, which is then compared with RMSDeval obtained by the new method. In almost all cases, the new protocol generates a better model than all possible models produced by the old method (RMSDeval lower than RMSDmin by 0.1 ∼ 1.4 Å). We emphasize that this improvement is underestimated because the old method can only produce a set of models for it cannot predict accurately the amplitude of displacement; the model that achieves RMSDmin cannot be singled out by the old method. It is, however, noted that the old method gives fairly good prediction for the direction of conformational changes (see the high overlap values in Table S3 of Supplementary Material), so it is still useful when only the directional information of protein conformational changes is desired.
DISCUSSIONS
We now further discuss the following issues that are important to the success of our fitting protocol.
Selection of residue pairs for DCs
In this work, we have not only developed a new algorithm that iteratively fits given pairwise DCs, but also designed a new scheme that predicts a small set of potentially useful residue pairs for measuring distance changes by experimentalists. Although our previous work has shown that the results of predicting the directions of protein conformational changes is rather robust to the various selections of residue pairs for DCs (9), the accurate prediction of conformational changes (both their direction and amplitude) does demand a good selection of a set of most informative and nonredundant pairwise DCs. The new scheme proposed here is indeed critical to enable our fitting protocol to achieve near-optimal performance.
Overfitting problem
Overfitting becomes a serious problem in situations where multiple solutions may exist because the “effective” number of constraints (which is smaller than the “nominal” number of constraints because of their redundancies) is not adequate as compared with the number of fitting variables. This problem is particularly relevant here because our fitting protocol tries to fit a small number of DCs (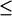 10) with 10 normal modes (as fitting variables). To avoid overfitting, our strategy is to significantly increase the effective number of constraints, which is achieved in the following ways:
10) with 10 normal modes (as fitting variables). To avoid overfitting, our strategy is to significantly increase the effective number of constraints, which is achieved in the following ways:
First, the pairwise DCs selection scheme combined with redundancy removing ensures that the given DCs are indeed independent of each other (or redundancy-free).
Second, the inclusion of the energy cost term in the minimization procedure not only facilitates good local geometry, but also introduces additional distance constraints for sequentially neighboring residue pairs to further increase the effective number of constraints. Indeed, when the number of DCs is smaller than the number of modes, there may exist a large number of structurally diverse conformations that all satisfy those given DCs, so the additional restraint of low energy provides further discriminating power by eliminating many energetically unfavorable solutions.
Computational efficiency
Based on our study of the 16 test cases, we find that the speedup from recomputing normal modes to the eigenvectors reorientation at each step is very significant (10 ∼ 20 times faster). Besides the one-time computing of the eigenvectors of the ENM normal modes, there is virtually no computationally obstacle that limits the potential application of our method to larger proteins.
The fitting protocol runs very fast thanks to the following critical improvements.
The use of only 10 lowest normal modes dramatically reduces the conformational space to search, while maintaining a high accuracy of describing the observed protein conformational changes.
The use of reoriented eigenvectors avoids repeated NMA, so this protocol remains fast even for large proteins and long fitting trajectories.
The linear regression based optimization converges very fast so a relatively small number of steps (say 100) is usually sufficient.
Condition for applications
This method is applicable to the modeling of protein conformational changes that consist of mainly collective motions between rigid-body-like subdomains and are thus well described by a small number of low-frequency normal modes. There are many biologically relevant conformational changes that meet the above criteria, although there also exist protein conformational changes that do not behave like rigid-body motions. Different methods are needed for the analysis of the latter ones.
For future work, we will combine this method with the experimentally derived DCs (for example, from NMR or other fast spectroscopic measurements) to predict unknown protein conformational changes toward transient states.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (National Heart, Lung, and Blood Institute).
References
- 1.Atilgan, A. R., S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, and I. Bahar. 2001. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doruker, P., A. R. Atilgan, and I. Bahar. 2000. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to alpha-amylase inhibitor. Proteins. 40:512–524. [PubMed] [Google Scholar]
- 3.Tama, F., and Y. H. Sanejouand. 2001. Conformational change of proteins arising from normal mode calculations. Protein Eng. 14:1–6. [DOI] [PubMed] [Google Scholar]
- 4.Zheng, W., and S. Doniach. 2003. A comparative study of motor-protein motions by using a simple elastic-network model. Proc. Natl. Acad. Sci. USA. 100:13253–13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng, W., and B. R. Brooks. 2005. Identification of dynamical correlations within the myosin motor domain by the normal mode analysis of an elastic network model. J. Mol. Biol. 346:745–759. [DOI] [PubMed] [Google Scholar]
- 6.Hubbell, W. L., D. S. Cafiso, and C. Altenbach. 2000. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7:735–739. [DOI] [PubMed] [Google Scholar]
- 7.Skolnick, J., A. Kolinski, and A. R. Ortiz. 1997. MONSSTER: a method for folding globular proteins with a small number of distance restraints. J. Mol. Biol. 265:217–241. [DOI] [PubMed] [Google Scholar]
- 8.Debe, D., M. Carlson, J. Sadanobu, S. Chan, and W. Goddard. 1999. Protein fold determination from sparse distance restraints. J. Phys. Chem. B. 103:3001–3008. [Google Scholar]
- 9.Zheng, W., and B. R. Brooks. 2005. Normal-modes-based prediction of protein conformational changes guided by distance constraints. Biophys. J. 88:3109–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tama, F., O. Miyashita, and C. L. Brooks. 2004. Normal mode based flexible fitting of high-resolution structure into low-resolution experimental data from cryo-EM. J. Struct. Biol. 147:315–326. [DOI] [PubMed] [Google Scholar]
- 11.Delarue, M., and P. Dumas. 2004. On the use of low-frequency normal modes to enforce collective movements in refining macromolecular structural models. Proc. Natl. Acad. Sci. USA. 101:6957–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang, Z., Y. Shi, and H. Liu. 2003. Molecular dynamics simulations of peptides and proteins with amplified collective motions. Biophys. J. 84:3583–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maragakis, P., and M. Karplus. 2005. Large amplitude conformational change in proteins explored with a plastic network model: adenylate kinase. J. Mol. Biol. 352:807–822. [DOI] [PubMed] [Google Scholar]
- 14.Kim, M. K., R. L. Jernigan, and G. S. Chirikjian. 2002. Efficient generation of feasible pathways for protein conformational transitions. Biophys. J. 83:1620–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tirion, M. M. 1996. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 77:1905–1908. [DOI] [PubMed] [Google Scholar]