

Abstract
Tumor cyclooxygenase-2 (COX-2) expression is known to be associated with enhanced tumor invasiveness. In the present study, we evaluated the importance of the COX-2 product prostaglandin E2 (PGE2) and its signaling through the EP4 receptor in mediating non-small cell lung cancer (NSCLC) invasiveness. Genetic inhibition of tumor COX-2 led to diminished matrix metalloproteinase (MMP)-2, CD44, and EP4 receptor expression and invasion. Treatment of NSCLC cells with exogenous 16,16-dimethylprostaglandin E2 significantly increased EP4 receptor, CD44, and MMP-2 expression and matrigel invasion. In contrast, anti-PGE2 decreased EP4 receptor, CD44, and MMP-2 expression in NSCLC cells. EP4 receptor signaling was found to be central to this process, because antisense oligonucleotide-mediated inhibition of tumor cell EP4 receptors significantly decreased CD44 expression. In addition, agents that increased intracellular cAMP, as is typical of EP4 receptor signaling, markedly increased CD44 expression. Moreover, MMP-2-AS treatment decreased PGE2-mediated CD44 expression, and CD44-AS treatment decreased MMP-2 expression. Thus, PGE2-mediated effects through EP4 required the parallel induction of both CD44 and MMP-2 expression because genetic inhibition of either MMP-2 or CD44 expression effectively blocked PGE2-mediated invasion in NSCLC. These findings indicate that PGE2 regulates COX-2-dependent, CD44- and MMP-2-mediated invasion in NSCLC in an autocrine/paracrine manner via EP receptor signaling. Thus, blocking PGE2 production or activity by genetic or pharmacological interventions may prove to be beneficial in chemoprevention or treatment of NSCLC.
Cyclooxygenase is the rate-limiting enzyme for the production of prostaglandins and thromboxanes from free arachidonic acid (1). Two isoenzymes of cyclooxygenase (COX)1 have now been described: a constitutive enzyme COX-1 present in most cells and tissues and an inducible enzyme COX-2 (also referred to as PGHS-2). COX-2 is known to be up-regulated in response to cytokines, growth factors, and other stimuli (1, 2). Mounting evidence documents elevated expression of COX-2 in a variety of malignancies including colon, gastric, esophageal, prostate, pancreatic, breast, and lung carcinomas (3–12). Overexpression of tumor COX-2 may be important in tumor invasion (13, 14), angiogenesis (15–18), resistance to apoptosis (15, 19–21), and suppression of host immunity (12, 22). We reported previously that COX-2 is overexpressed in human NSCLC, and the resultant high level PGE2 production mediates dysregulation of host immunity by altering the balance of interleukins 10 and 12 (12). Accordingly, specific inhibition of COX-2 or PGE2 led to significant in vivo tumor reduction in murine lung cancer models (22). Recently, other investigators have expanded on and corroborated these observations, indicating the importance of COX-2 expression in lung cancer (23–28).
Elevated expression of COX-2 has been shown to increase tumor invasiveness and enhance metastatic potential (3, 29). The complex events associated with tumor cell invasion include the active movement of cells across the extracellular matrix and spread to distant organ sites (30). CD44 is a cell surface receptor for hyaluronate, a major glycosaminoglycan component of extracellular matrix. Adhesion to extracellular matrix, a critical initial step in the metastatic process, has been found to be CD44-dependent in several malignancies (31–34). Our previous findings indicate that tumor COX-2 expression is a critical determinant of CD44 expression and invasion in NSCLC (35). In concert with CD44, matrix metalloproteinase (MMP) expression is important for tumor invasion. Cell surface CD44 induces co-clustering of MMPs, thereby promoting MMP activity, tumor invasion, and angiogenesis (31, 36, 37). Our current studies focus on the role of tumor-derived PGE2 in modulating COX-2-dependent NSCLC invasion.
PGE2, produced at heightened levels in COX-2-overexpressing tumor cells, affects target cells through interaction with G-protein-coupled EP receptors of at least four distinct subtypes. In this study, we addressed the pathways whereby autocrine/paracrine production of PGE2 could impact the invasive phenotype via EP receptor signaling in NSCLC.
Here, we report that antibody-mediated blockade of tumor-derived PGE2 decreased both CD44 and MMP-2 expression as well as invasion. In addition, exposure of NSCLC cells to 16,16-dimethyl-prostaglandin E2 (dmPGE2) up-regulated CD44, EP4 receptor, and MMP-2 expression and potently enhanced invasion. The parallel increase in both CD44 and MMP-2 expression appears to be required to mediate invasiveness; genetic inhibition of either CD44 or MMP-2 expression abrogated PGE2-mediated invasion. This is the first report indicating an important autocrine/paracrine role of PGE2 in the regulation of CD44 and MMP-2-dependent invasion in human NSCLC.
EXPERIMENTAL PROCEDURES
Reagents
Forskolin, 3-isobutyl-1-methylxanthine (IBMX), dibutyryl cAMP, dibutyryl cGMP, and cholera toxin were obtained from Sigma. NS-398 (N-(2-cyclohexyloxy-4 nitrophenyl) methasulfonamide) and dmPGE2 were purchased from Cayman Chemicals (Ann Arbor, MI). Anti-PGE2 (monoclonal antibody 2B5) and isotype-matched control mouse IgG1 (MOPC21) were generously provided by Pharmacia, Peapack, NJ. Pertussis toxin was obtained from Calbiochem.
Stably Transduced COX-2 Cell Lines
NSCLC cells, A549 (human lung adenocarcinoma), and H157 (squamous cell carcinoma) were obtained from American Type Culture Collection (Manassas, VA) and NCI, National Institutes of Health, respectively. A 2.0-kb cDNA fragment of human COX-2 (generously provided by Dr. Harvey Herschman, UCLA) was cloned into the PmeI site of the retroviral vector pLNCX (Clontech, Palo Alto, CA), which contains the cytomegalovirus promoter for controlling transcription of the cDNA insert and the neomycin-resistant gene for G418 (Invitrogen) selection. Sense-oriented (COX-2-S) and antisense-oriented (COX-2-AS) expression vectors were prepared as previously described (35). For virus production, 70% confluent 293T cells were transfected with COX-2-S, COX-2-AS, and pLNCX (vector alone). Tumor cells were then transduced with high titer-producing COX-2-S, COX-2-AS, or pLNCX virus. Following transduction, the tumor cells were characterized as previously described (35). Briefly, COX-2-S and COX-2-AS NSCLC cells were confirmed by PCR, and PCR-positive clones were then screened by Western blot and EIA analysis for COX-2 expression and PGE2 production, respectively. For each cell line, an approximate 10-fold higher level of COX-2 expression and PGE2 production was noted in COX-2-S compared with parental or vector controls (35). In contrast, COX-2-AS produced 4-fold less COX-2 and PGE2. These cells were then expanded for further studies.
Flow Cytometry Analysis
For the analysis of CD44 and EP4 receptor expression, the cells from each treatment group were harvested and labeled with either fluorescein isothiocyanate-conjugated anti-CD44 (Dako Corp., Carpinteria, CA) for 1 h or anti-EP4 receptor antibody (Cayman Chemical) for 90 min. For evaluation of EP4 receptor expression, cells were washed with phosphate-buffered saline plus 2% fetal bovine serum and then incubated for 1 h with secondary fluorescein isothiocyanate-conjugated goat anti-rabbit serum (Jackson ImmunoResearch Laboratory, West Grove, PA). The fluorescein isothiocyanate-labeled cells were then analyzed by flow cytometry using a Becton Dickinson FACScan. Controls included cells stained with fluorescein isothiocyanate-conjugated mouse IgG1 (Dako Corp.) or with secondary antibody alone.
Gelatin Zymography
Serum-free tumor supernatants were analyzed for the level of MMP-2 using SDS-polyacrylamide gel zymography. Briefly, 5 × 105 cells from each group were plated in six-well plates. Complete medium was replaced with serum-free medium after 24 h. Following incubation at 37 °C for 24 h, the serum-free medium from each well was collected and analyzed by 10% zymogram-ready gels (Bio-Rad) according to the manufacturer’s protocol. Following electrophoresis, the gels were soaked in 2.5% Triton X-100 on a shaker for 1 h, changing the solution after 30 min to eliminate SDS. After overnight incubation in zymogen activation buffer (50 mm Tris-HCl (pH 7.5), containing 5 mm CaCl2, 200 mm NaCl and 0.02% Brij-35) at 37 °C, the gels were rinsed in distilled water and stained for 2–3 h with Phastgel Blue R stain as previously described (38).
Invasion Assay
The membrane invasion assay was performed in Matrigel-coated invasion chambers (Becton Dickinson Labware, Franklin Lakes, NJ) as previously described (35). Control and COX-2 transfectants were cultured in RPMI 1640 supplemented with 10% fetal bovine serum. Tumor cell-conditioned medium was generated as previously described (35) and was added in the lower chamber as a chemoattractant. 5 × 104 cells were plated in the upper chamber in both control and test conditions. Following an 18-h incubation at 37 °C in a humidified 5% CO2 atmosphere, the cells in the upper chamber and on the Matrigel were mechanically removed with a cotton swab. The cells adherent to the outer surface of the membrane were stained with Diff-Quick stain (Dade Behring Inc., Newark, DE) according to the manufacturer’s instructions. The invading cells were examined, counted, and photographed by microscopy (Nikon Labphot-2 Microscope with an attached Spot Digital Camera, A. G. Heinz, Lake Forest, CA) at μ50 magnification. Six fields were counted per filter in each group, and the experiment was repeated five separate times.
CD44, MMP-2, and EP4 Receptor Phosphorothioate Antisense Oligonucleotides
Human CD44, MMP-2, and EP4 receptor antisense and control phosphorothioate-modified DNA oligonucleotides were synthesized by the Molecular Biology Institute and UCLA Jonsson Comprehensive Cancer Center. The sequence of the CD44-specific antisense oligonucleotides was 5′-TATTTGAAGACGTAC, and that of control oligonucleotides was 5′-TATGTTAAGGACATC. The sequence of the MMP-2-specific antisense oligonucleotides was 5′-CACATCTTTCCGTCA, and that of control oligonucleotides was 5′-CGTCCCTATACGACC. The sequence of the EP4 receptor-specific antisense oligonucleotides was 5′-AGGTGTGAGGCTGTG, and that of control oligonucleotides was 5′-AGGGTTGAGCGTGGT. Briefly, in all studies, NSCLC cells were cultured to 50% confluence. The regular tissue culture medium was replaced with serum-free medium. For MMP-2 blocking studies, control and MMP-2 phosphorothioate antisense oligonucleotides (5 μm) were added to the wells. Following a 30-h incubation, serum-free medium (SFM) was collected and analyzed for MMP-2 by gelatin zymograph. In order to evaluate the role of MMP-2 in PGE2-induced CD44 expression, tumor cells were treated with control and MMP-2 phosphorothioate antisense oligonucleotides (5 μm) for 30 h and were further incubated with dmPGE2 for 3 h. The treated cells were then analyzed for CD44 expression by flow cytometry. For EP4 receptor blocking and CD44 expression studies, control and EP4 phosphorothioate antisense oligonucleotides (2 μm) were added to tumor cells. Following a 48-h incubation, cells were treated with dmPGE2 for 3 h and then analyzed for EP4 receptor and CD44 expression by flow cytometry. For CD44 blocking and MMP-2 expression studies, control and CD44 phosphorothioate antisense oligonucleotides (4 μm) were added to tumor cells. Following a 72-h incubation, cells were incubated with dmPGE2 for 3 h, and SFM was collected and analyzed for MMP-2 by gelatin zymography.
RESULTS AND DISCUSSION
Experimental data support the relationship between COX-2, its synthesized product PGE2, and neoplastic transformation of epithelial cells (39, 40). Transcription of COX-2 is constitutively up-regulated in several different malignancies including lung cancer (4–9, 11, 12). Our previous studies documented a tumor COX-2-dependent regulation of CD44 and invasion in NSCLC (35). Here, we investigated the pathways by which COX-2 promotes tumor cell invasion. We tested whether the COX-2 enzyme product PGE2 was responsible, in an autocrine/paracrine manner, for mediating the increase in CD44 and MMP-2. In order to determine the effect of PGE2 on CD44 expression, NSCLC cells were treated with dmPGE2. A dose-dependent increase in NSCLC cell CD44 expression was detected by flow cytometry analysis (Fig. 1A). IL-1β, a potent inducer of COX-2, was found to increase CD44 expression in a PGE2-dependent manner (Fig. 1B). These findings suggest that the induction of CD44 expression in NSCLC is dependent, at least in part, on tumor-derived PGE2.
Fig. 1.

A, PGE2 augments CD44 expression in NSCLC. A549 cells (106 cells/ml) were cultured in complete medium. Following overnight incubation, the cells were treated with dmPGE2 (2.5, 5.0, and 10.0 μg/ml) or vehicle (control) for 4 h. Compared with control, a 3-fold increase in CD44 expression is seen at 5.0 (*, p < 0.01) and 10.0 μg/ml dmPGE2 (*, p < 0.01). MFI, mean fluorescence intensity. The data represent the mean ± S.D. of triplicate determinations in one representative experiment of three separate experiments. B, anti-PGE2 inhibits IL-1β induced CD44 expression. A549 cells (106 cells/ml) were cultured in the presence or absence of IL-1β (280 units/ml) for 24 h. For antibody treatment, the cells were incubated with control or anti-PGE2 antibody prior to the addition of IL-1β. Compared with the control, a 2.5-fold increase in CD44 expression is seen in IL-1β treated cells (p < 0.05). Anti-PGE2 significantly reduced the capacity of IL-1β to induce CD44 (*, p < 0.05). The data represent the mean ± S.D. of triplicate determinations in one representative experiment of three separate experiments. C, agents that impact G-coupled protein receptor signaling and cAMP level modulate CD44 expression. A549 cells (106 cells/ml) were cultured in complete medium with various agents for 24 h. CD44 expression was analyzed by flow cytometry. Anti-PGE2 significantly decreased constitutive CD44 expression. Agents that increase intracellular cAMP levels, including dmPGE2 (10 μg/ml), forskolin (100 μm), IBMX (100 μm), dibutyryl cAMP (100 μm), and cholera toxin (CTX; 1 μg/ml) were potent inducers of CD44 expression. Dibutyryl cGMP (200 μm) served as a negative control and did not induce CD44 expression. Pertussis toxin (PTX; 200 ng/ml) treatment significantly decreased constitutive as well as PGE2-induced CD44 expression. Asterisks denote a statistically significant increase or decrease compared with control values (*, p < 0.01; **, p < 0.05). The data represent the mean ± S.D. of triplicate determinations in one representative experiment of three separate experiments.
PGE2 elicits its autocrine/paracrine effects on target cells through interaction with prostaglandin E series receptors of four distinct subtypes, designated EP1, EP2, EP3, and EP4. EP1 causes influx of Ca2+ and activation of protein kinase C; receptors EP2 and EP4 activate adenylate cyclase, which increases cellular cyclic AMP levels and activates protein kinase A; and EP3 signals primarily through an inhibitory G protein to decrease intracellular cyclic AMP levels (41).
PGE2 has the capacity to bind EP2 and/or EP4 receptors, thereby increasing intracellular cAMP, which in turn activates the protein kinase A pathway (42–44). To determine the role of cAMP in the PGE2-mediated induction of CD44 expression, we tested the capacity of forskolin, cholera toxin, dibutyryl cAMP, IBMX, and dibutyryl cGMP to induce CD44 expression. Forskolin and cholera toxin were chosen because they activate adenylate cyclase activity, thereby increasing cAMP in a manner similar to PGE2 (45, 46). Dibutyryl cAMP was used as an exogenous source of cAMP capable of attaining cellular entry. IBMX is a phosphodiesterase inhibitor and thus causes an increase in intracellular cAMP levels (45). Dibutyryl cAMP (but not dibutyryl cGMP), forskolin, cholera toxin, and IBMX significantly induced CD44 expression (Fig. 1C). In contrast to the action of cholera toxin, pertussis toxin treatment of NSCLC cells significantly suppressed CD44 expression (Fig. 1C). Anti-PGE2 and increased intracellular cGMP resulted in decreased CD44 expression (Fig. 1C). Similar results were seen with H157 NSCLC cells (data not shown). These findings provide evidence that G-protein-coupled receptors regulate the eicosanoid-cAMP signal transduction pathway in NSCLC and thus up-regulate cell surface CD44 expression and tumor cell invasion.
The roles of the different prostaglandin receptors, their divergent intracellular signaling pathways, and their target genes involved in mediating the effects of PGE2 on normal or neoplastic cells remain to be fully elucidated. Based on previous studies implicating EP4 receptor signaling in PGE2-mediated, G-coupled protein-dependent events in human cancer cells (47, 48), we evaluated the regulation of EP4 in response to increased COX-2 and PGE2. Overexpression of COX-2 increased tumor cell EP4 receptor expression and thus may increase sensitivity to the autocrine and paracrine effects of PGE2 itself. The results presented here indicate an increased expression of EP4 receptor in COX-2-overexpressing (COX-2-S) cells and a relative decreased expression in COX-2 antisense (COX-2-AS) cells compared with vector-transduced controls (Fig. 2A). We next sought to determine whether PGE2 itself played a role in the COX-2-mediated regulation of EP4 receptor expression. Whereas PGE2 induced EP4 receptor expression, anti-PGE2 antibody significantly down-regulated constitutive A549 EP4 receptor expression (Fig. 2B). This suggests that the COX-2-mediated increase in EP4 receptor expression involves autocrine signaling by tumor-derived PGE2 (Fig. 2B). In addition, these findings suggest that COX-2-overexpressing lung cancer cells not only produce increased PGE2 but also have heightened sensitivity to PGE2-mediated effects due to overexpression of the EP4 receptor (Fig. 2B). These data are consistent with the previous reports indicating that PGE2 may regulate EP receptor expression in certain cell types (49). The fact that anti-PGE2 monoclonal antibody did not significantly decrease constitutive EP4 receptor expression in H157 cells suggests that other COX-2-dependent mediators may be operative in maintaining EP4 receptor expression in these cells (Fig. 2B). Further studies will be required to define these additional products.
Fig. 2. EP4 receptor expression in NSCLC is COX-2-dependent.
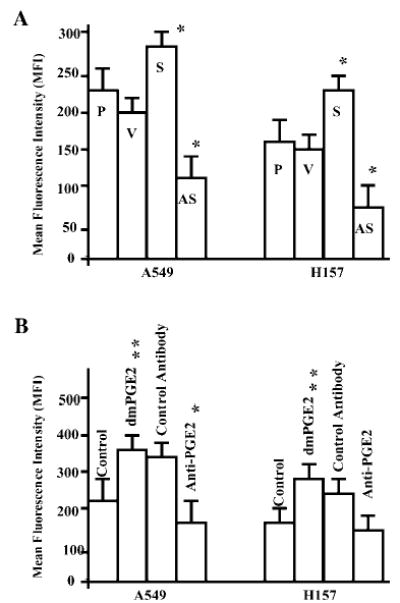
A, A549 cells (106 cells/ml) including parental (P), empty vector (V), COX-2-S (S), and COX-2-AS (AS) transfectants were evaluated for EP4 receptor expression by flow cytometry. Compared with parental and vector controls, MFI was augmented in COX-2-S cells (*, p < 0.05) but diminished in COX-2-AS cells (*, p < 0.05). B, treatment of A549 and H157 parental cells with 10 μg/ml dmPGE2 increased EP4 receptor fluorescence intensity. Anti-PGE2 significantly blocked EP4 expression in A549 cells. Asterisks denote a statistically significant increase or decrease compared with control values (**, p < 0.001; *, p < 0.05). The data represent the mean ± S.D. of triplicate determinations in one representative experiment out of three separate experiments.
In order to determine the role of EP4 receptor-dependent signaling in regulating CD44 expression in COX-2-S cells, specific inhibition of EP4 receptor expression was accomplished with antisense oligonucleotides. As anticipated, EP4 receptor-specific antisense oligonucleotides used in these studies significantly blocked PGE2-mediated elevated expression of the EP4 receptor in NSCLC cells (data not shown). The PGE2-mediated up-regulation of CD44 expression in NSCLC cells and the constitutive high level expression of CD44 in COX-2-S cells were significantly decreased by specific genetic inhibition of the EP4 receptor (Table I). Thus, EP4 receptor-dependent signaling plays a direct role in PGE2-mediated CD44 expression in NSCLC cells as well as the constitutively elevated levels of CD44 seen in COX-2-S cells.
Table I. EP4 receptor-specific antisense oligonucleotides inhibit CD44 expression.
Cells from each group were incubated with EP4-AS or control 15-mer oligonucleotides (2 μm) for 48 h. CD44 expression was determined by flow cytometry analysis. A significant decrease in CD44 expression was noted following EP4-antisense treatment. The data represent the mean ± S.D. of triplicate determinations in one representative experiment of three separate experiments. Medium alone controls showed CD44 expression that was not significantly different from oligonucleotide control values (data not shown).
| Mean fluorescence intensity
|
||
|---|---|---|
| Experimental conditions | Control oligonucleotides | EP4-antisense oligonucleotides |
| A549-vector | 87.5 ± 0.7 | 50 ± 1.4a |
| A549-vector + dmPGE2 | 181.5 ± 4.9 | 65.5 ± 7.7a |
| A549-sense | 138 ± 4.9 | 49 ± 4.2a |
| H157-vector | 57.5 ± 11 | 45 ± 0.0b |
| H157-vector + dmPGE2 | 156 ± 1.4 | 58.5 ± 0.7a |
| H157-sense | 488 ± 4.9 | 255 ± 5.6a |
p < 0.01 compared with control oligonucleotide.
p < 0.05 compared with control oligonucleotide.
Degradation of extracellular matrix and penetration of the basement membrane are required for tumor invasion and metastasis (50). The MMP family is a group of proteolytic enzymes that have been associated with invasion and metastasis (50, 51). The major component of the basement membrane, type IV collagen, serves as a substrate for MMP-2 and MMP-9 (72- and 92-kDa type IV collagenases, respectively). Several reports have correlated neoplastic epithelial invasion and metastasis with overexpression of these MMPs (52). We focused on MMP expression because of reports indicating an increase in MMP-2 expression and metastatic potential in COX-2-overexpressing colon cancer cells (29) and because of the involvement of MMPs in metastasis of NSCLC (53, 54). Furthermore, CD44 serves to induce co-clustering with MMPs and can therefore promote MMP activity, tumor invasion, and angiogenesis (31, 36). Here we find that the level of MMP-2 is increased in COX-2-overexpressing NSCLC cells but decreased in COX-2 antisense cells (Fig. 3A). Thus, overexpression of tumor COX-2 elevates MMP-2 and facilitates invasion in NSCLC. The addition of anti-PGE2 antibody to COX-2-S cells significantly blocked MMP-2 production in NSCLC cells (Fig. 3B), indicating that the autocrine/paracrine influence of PGE2 regulates MMP-2 expression in these cells. Attiga et al. (55) have reported the inhibition of MMP-2 and MMP-9 by COX-2 inhibitors in prostate cancer. Consistent with these previous findings, the treatment of COX-2-overexpressing cells (A549-S) with a COX-2-specific inhibitor showed decreased levels of MMP-2 (Fig. 3C). However, MMP-9 was not regulated in A549 or H157 cell lines (data not shown).
Fig. 3.
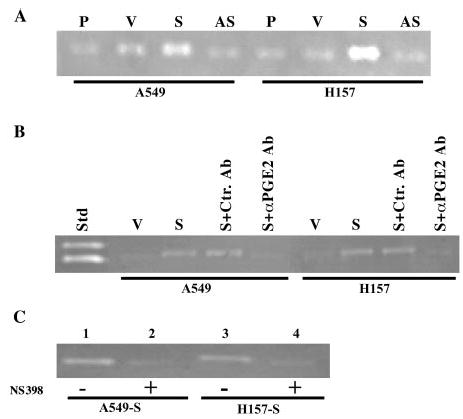
A, overexpression of COX-2 increases MMP-2 in NSCLC. A549 and H157 cells including parental (P), empty vector (V), COX-2-S (S), and COX-2-AS (AS) transfectants were cultured in SFM. After 24 h, supernatants were collected and run on gelatin zymogram gels. Compared with parental, vector, and COX-2-AS cells, COX-2-S cells showed a higher level of MMP-2 activity. B, anti-PGE2 inhibits MMP-2 activity in COX-2-overexpressing NSCLC cells. COX-2-S cells were incubated with anti-PGE2 for 24 h, and SFM was collected and analyzed by zymography. Std, standard; V, vector control (A549 or H157); S, A549 or H157 sense cells; S + Ctrl. Ab, A549 or H157 sense cells plus control antibody; S + α PGE2, A549 or H157 cells plus anti-PGE2. Both A549 and H-157 sense (S) show enhanced level of MMP-2 activity. An inhibition of MMP-2 level was seen following anti-PGE2 treatment (S + α PGE2). C, COX-2-specific inhibitors decrease MMP-2 activity. SFM was collected from A549 and H157 sense cells treated with COX-2 inhibitor, and MMP-2 activity was assessed by zymography. Lanes 1 and 3, no inhibitors; lanes 2 and 4, following NS398 (1 μm) exposure. MMP-2 levels were decreased in A549-S (lane 2) and H157-S (lane 4) cells in the presence of the COX-2 inhibitor.
In order to define the pathways responsible for COX-2-dependent induction of MMP-2, we determined the level of tumor MMP-2 in the presence of exogenous PGE2. The addition of PGE2 increased the level of MMP-2 and invasion in NSCLC (Fig. 4, A and B, lower left panel). Decreases in MMP-2 level and invasion were observed when MMP-2-AS oligonucleotides were utilized in PGE2-treated cells (Fig. 4, A and B, lower right panel). Similar results were also obtained in H157-S cells (data not shown). MMP-2-AS oligonucleotides were also shown to block PGE2-mediated CD44 expression in NSCLC cell (Table II). In activated human monocytes, MMPs are known to be regulated through a PGE2-cAMP-dependent pathway (56). More recently, CD44 has been implicated in the regulation of MMPs in human cancer cells (57, 58), which suggests a fundamental role for CD44 in regulating MMP-2 expression and the invasive phenotype. Previously, we have reported the inhibition of invasion in COX-2-S cells using anti-CD44 antibody. Here we demonstrate the inhibition of PGE2-mediated expression of MMP-2 and NSCLC cell invasion with CD44-specific antisense oligonucleotides (Fig. 5, A and B, lower right panel). Thus, blocking CD44 expression inhibits the expression of MMP-2 and invasion in NSCLC cells.
Fig. 4.
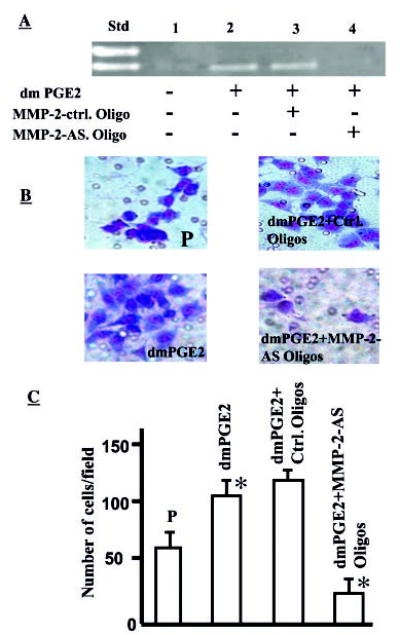
A, PGE2 augments MMP-2 activity in NSCLC cells. SFM was collected from A549-P (P; 106 cells/ml) in the absence or presence of dmPGE2 (10 μg/ml) and run on gelatin zymogram gels. Std, standard; lane 1, A549-P cells in medium alone without PGE2; lane 2, A549-P cells with dmPGE2 (10 μg/ml); lane 3, A549-P cells with dmPGE2 (10 μg/ml) plus MMP-2 control oligonucleotides; lane 4, A549-P cells with dmPGE2 (10 μg/ml) plus MMP-2 antisense oligonucleotides. An increase in MMP-2 level was observed in PGE2-treated cells (lane 2). This PGE2-mediated increase in MMP-2 was effectively blocked by MMP-2-AS oligonucleotides (lane 4). B, PGE2-induced NSCLC invasion is MMP-2-dependent. A549-P cells cultured in medium alone (upper left panel), in the presence of 10 μg/ml dmPGE2 (lower left panel), dmPGE2 plus MMP-2 control oligonucleotide (upper right panel), and dmPGE2 plus MMP-2 antisense oligonucleotide (lower right panel) were used for Matrigel matrix assays. Compared with control (upper left panel), significantly more cells invaded in the presence of dmPGE2 (lower left panel) and fewer cells invaded in the presence of MMP-2 antisense oligonucleotide (lower right panel). C, bar graph representing the number of cells invaded in each group. A 2.5-fold increase in the number of cells invading the matrix is seen in dmPGE2-treated cells (p > 0.05), whereas significantly fewer cells invaded in MMP-2 antisense oligonucleotide-treated cells (p < 0.05).
Table II. MMP-2-specific antisense oligonucleotides (oligos) inhibit PGE2-mediated CD44 expression.
dmPGE2-treated cells were incubated with MMP-2-AS (AS) or control (Ctrl.) 15-mer oligonucleotides (5 μm) for 30 h. CD44 expression was determined by flow cytometry analysis. PGE2 significantly increased CD44 expression. A significant decrease in CD44 expression was noted following MMP-2-antisense treatment. The data represent the mean ± S.D. of triplicate determinations in one representative experiment of three separate experiments.
| Experimental conditions | Mean fluorescence intensity |
|---|---|
| A549-vector | 166 ± 6.3 |
| A549-vector + dmPGE2 | 235 ± 1.4a |
| A549-vector + dmPGE2 + Ctrl. oligos | 225 ± 22.6 |
| A549-vector + dmPGE2 + AS-oligos | 168 ± 2.1b |
p < 0.001 compared with A549-vector control.
p < 0.001 compared with control oligonucleotide.
Fig. 5.
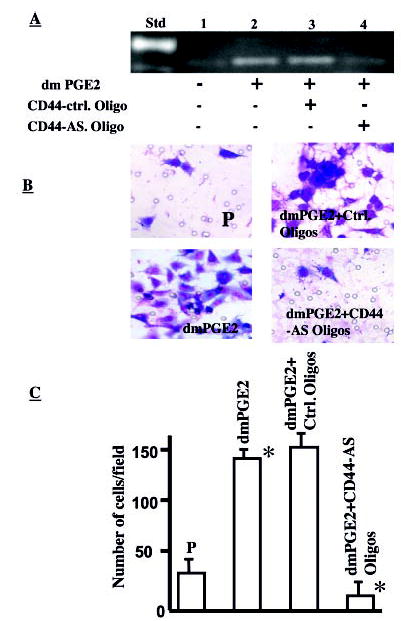
A, PGE2-induced MMP-2 activity is CD44-dependent in NSCLC cells. SFM was collected from A549 (P, 106 cells/ml) in the absence or presence of dmPGE2 (10 μg/ml) and run on gelatin zymogram gels. Std, standard; lane 1, A549-P cells in medium alone without PGE2; lane 2, A549-P cells with dmPGE2 (10 μg/ml); lane 3, A549-P cells with dmPGE2 (10 μg/ml) plus CD44 control oligonucleotides; lane 4, A549-P cells with dmPGE2 (10 μg/ml) plus CD44 antisense oligonucleotide. An increase in MMP-2 level was observed in PGE2-treated cells (lanes 2 and 3). PGE2-mediated increase in MMP-2 was effectively blocked by CD44 antisense oligonucleotide (lane 4). B, PGE2-induced NSCLC invasion is CD44-dependent. A549 cells cultured in medium alone (upper left panel) in the presence of 10 μg/ml dmPGE2 (lower left panel), dmPGE2 plus control oligonucleotide (upper right panel), and dmPGE2 plus CD44 antisense oligonucleotide (lower right panel) were used for Matrigel matrix assays. Compared with control (upper left panel), significantly more cells invaded in the presence of dmPGE2 (lower left panel), and fewer cells invaded in the presence of CD44 antisense oligonucleotide (lower right panel). C, bar graph representing the number of cells invaded in each group. A 6-fold increase in the number of cells invading the matrix is seen in dmPGE2-treated cells (p < 0.05), whereas significantly fewer cells invaded in CD44 antisense oligonucleotide-treated cells (p < 0.05).
Here, for the first, time we document the autocrine/paracrine role of PGE2 in up-regulating the expression of CD44 and MMP-2 in human lung cancer cells. In collaboration with CD44, MMP-2 expression is also responsible for NSCLC cell invasion. Thus, our studies and reports in the literature support the hypothesis that CD44 and MMP-2 act in concert to promote tumor invasion (59, 60). Clear understanding of the regulatory pathways whereby COX-2 overexpression leads to CD44- and MMP-2-dependent invasion in NSCLC may promote the development of specific targeted therapies that are based on the molecular pathogenesis of the disease.
Footnotes
This work was supported by National Institutes of Health Grants P50 CA90388 and RO1 CA71818, the American Lung Association, Merit Review Research Funds from the Department of Veterans Affairs, the Tobacco-related Disease Research Program of the University of California, and the Research Enhancement Award Program in Cancer Gene Medicine.
The abbreviations used are: COX, cyclooxygenase; MMP, matrix metalloproteinase; NSCLC, nonsmall cell lung cancer; PGE2, prostaglandin E2; IBMX, 3-isobutyl-1-methylxanthine; dmPGE2, 16,16-dimethyl-prostaglandin E2; IL, interleukin; SFM, serum-free medium.
References
- 1.Herschman H. Biochim Biophys Acta. 1996;1299:125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 2.Herschman HR. Cancer Metastasis Rev. 1994;13:241–256. doi: 10.1007/BF00666095. [DOI] [PubMed] [Google Scholar]
- 3.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. J Clin Invest. 2000;105:1589–1594. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, Koki AT. Cancer. 2000;89:2637–2645. doi: 10.1002/1097-0142(20001215)89:12<2637::aid-cncr17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 5.Yip-Schneider MT, Barnard DS, Billings SD, Cheng L, Heilman DK, Lin A, Marshall SJ, Crowell PL, Marshall MS, Sweeney CJ. Carcinogenesis. 2000;21:139–146. doi: 10.1093/carcin/21.2.139. [DOI] [PubMed] [Google Scholar]
- 6.Shao J, Sheng H, Inoue H, Morrow JD, DuBois RN. J Biol Chem. 2000;275:33951–33956. doi: 10.1074/jbc.M002324200. [DOI] [PubMed] [Google Scholar]
- 7.Kirschenbaum A, Klausner AP, Lee R, Unger P, Yao S, Liu X, Levine AC. Urology. 2000;56:671–676. doi: 10.1016/s0090-4295(00)00674-9. [DOI] [PubMed] [Google Scholar]
- 8.Shamma A, Yamamoto H, Doki Y, Okami J, Kondo M, Fujiwara Y, Yano M, Inoue M, Matsuura N, Shiozaki H, Monden M. Clin Cancer Res. 2000;6:1229–1238. [PubMed] [Google Scholar]
- 9.Molina MA, Sitja-Arnau M, Lemoine MG, Frazier ML, Sinicrope FA. Cancer Res. 1999;59:4356–4362. [PubMed] [Google Scholar]
- 10.Chan G, Boyle JO, Yang EK, Zhang F, Sacks PG, Shah JP, Edelstein D, Soslow RA, Koki AT, Woerner BM, Masferrer JL, Dannenberg AJ. Cancer Res. 1999;59:991–994. [PubMed] [Google Scholar]
- 11.Dannenberg AJ, Zakim D. Semin Oncol. 1999;26:499–504. [PubMed] [Google Scholar]
- 12.Huang M, Stolina M, Sharma S, Mao J, Zhu L, Miller P, Wollman J, Herschman H, Dubinett S. Cancer Res. 1998;58:1208–1216. [PubMed] [Google Scholar]
- 13.Niki T, Kohno T, Iba S, Moriya Y, Takahashi Y, Saito M, Maeshima A, Yamada T, Matsuno Y, Fukayama M, Yokota J, Hirohashi S. Am J Pathol. 2002;160:1129–1141. doi: 10.1016/s0002-9440(10)64933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kakiuchi Y, Tsuji S, Tsujii M, Murata H, Kawai N, Yasumaru M, Kimura A, Komori M, Irie T, Miyoshi E, Sasaki Y, Hayashi N, Kawano S, Hori M. Cancer Res. 2002;62:1567–1572. [PubMed] [Google Scholar]
- 15.Liu XH, Kirschenbaum A, Yao S, Lee R, Holland JF, Levine AC. J Urol. 2000;164:820–825. doi: 10.1097/00005392-200009010-00056. [DOI] [PubMed] [Google Scholar]
- 16.Masferrer JL, Leahy KM, Koki AT. Curr Med Chem. 2000;7:1163–1170. doi: 10.2174/0929867003374336. [DOI] [PubMed] [Google Scholar]
- 17.Masferrer JL, Leahy KM, Koki AT, Zweifel BS, Settle SL, Woerner BM, Edwards DA, Flickinger AG, Moore RJ, Seibert K. Cancer Res. 2000;60:1306–1311. [PubMed] [Google Scholar]
- 18.Uefuji K, Ichikura T, Mochizuki H. Clin Cancer Res. 2000;6:135–138. [PubMed] [Google Scholar]
- 19.Tsujii M, Dubois R. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 20.Ding XZ, Tong WG, Adrian TE. Anticancer Res. 2000;20:2625–2631. [PubMed] [Google Scholar]
- 21.Tanji N, Kikugawa T, Yokoyama M. Anticancer Res. 2000;20:2313–2319. [PubMed] [Google Scholar]
- 22.Stolina M, Sharma S, Lin Y, Dohadwala M, Gardner B, Luo J, Zhu L, Kronenberg M, Miller PW, Portanova J, Lee JC, Dubinett SM. J Immunol. 2000;164:361–370. doi: 10.4049/jimmunol.164.1.361. [DOI] [PubMed] [Google Scholar]
- 23.Hida T, Leyton J, Makheja A, Ben-Av P, Hla T, Martinez A, Mulshine J, Malkani S, Chung P, Moody T. Anticancer Res. 1998;18:775–782. [PubMed] [Google Scholar]
- 24.Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A. Cancer Res. 1998;58:4997–5001. [PubMed] [Google Scholar]
- 25.Hida T, Yatabe Y, Achiwa H, Muramatsu H, Kozaki K-i, Nakamura S, Ogawa M, Mitsudomi T, Sugiura T, Takahashi T. Cancer Res. 1998;58:3761–3764. [PubMed] [Google Scholar]
- 26.Watkins DN, Lenzo JC, Segal A, Garlepp MJ, Thompson PJ. Eur Respir J. 1999;14:412–418. doi: 10.1034/j.1399-3003.1999.14b28.x. [DOI] [PubMed] [Google Scholar]
- 27.Ochiai M, Oguri T, Isobe T, Ishioka S, Yamakido M. Jpn J Cancer Res. 1999;90:1338–1343. doi: 10.1111/j.1349-7006.1999.tb00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hida T, Kozaki K, Muramatsu H, Masuda A, Shimizu S, Mitsudomi T, Sugiura T, Ogawa M, Takahashi T. Clin Cancer Res. 2000;6:2006 –2011. [PubMed] [Google Scholar]
- 29.Tsujii M, Kawano S, DuBois R. Proc Natl Acad Sci U S A. 1997;94:3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohn EC, Liotta LA. Cancer Res. 1995;55:1856–1862. [PubMed] [Google Scholar]
- 31.Yu Q, Stamenkovic I. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamb RF, Hennigan RF, Turnbull K, Katsanakis KD, MacKenzie ED, Birnie GD, Ozanne BW. Mol Cell Biol. 1997;17:963–976. doi: 10.1128/mcb.17.2.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartolazzi A, Peach R, Aruffo A, Stamenkovic I. J Exp Med. 1994;180:53–66. doi: 10.1084/jem.180.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seiter S, Arch R, Reber S, Komitowski D, Hofmann M, Ponta H, Herrlich P, Matzku S, Zoller M. J Exp Med. 1993;177:443–455. doi: 10.1084/jem.177.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dohadwala M, Luo J, Zhu L, Lin Y, Dougherty G, Sharma S, Huang M, Pold M, Batra R, Dubinett S. J Biol Chem. 2001;276:20809–20812. doi: 10.1074/jbc.C100140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Q, Stamenkovic I. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 37.Isacke CM, Yarwood H. Int J Biochem Cell Biol. 2002;34:718–721. doi: 10.1016/s1357-2725(01)00166-2. [DOI] [PubMed] [Google Scholar]
- 38.Leber TM, Balkwill FR. Anal Biochem. 1997;249:24–28. doi: 10.1006/abio.1997.2170. [DOI] [PubMed] [Google Scholar]
- 39.Sheng H, Shao J, Morrow J, Beauchamp R, DuBois R. Cancer Res. 1998;58:362–366. [PubMed] [Google Scholar]
- 40.Subbaramaiah K, Altorki N, Chung WJ, Mestre JR, Sampat A, Dannenberg AJ. J Biol Chem. 1999;274:10911–10915. doi: 10.1074/jbc.274.16.10911. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Hughes-Fulford M. Br J Cancer. 2000;82:2000–2006. doi: 10.1054/bjoc.2000.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang M, Sharma S, Mao JT, Dubinett SM. J Immunol. 1996;157:5512–5520. [PubMed] [Google Scholar]
- 43.Jabbour HN, Milne SA, Williams AR, Anderson RA, Boddy SC. Br J Cancer. 2001;85:1023–1031. doi: 10.1054/bjoc.2001.2033. [DOI] [PubMed] [Google Scholar]
- 44.Coleman RA, Smith WL, Narumiya S. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- 45.Panettieri R, Lazaar A, Pure E, Albelda S. J Immunol. 1995;154:2358–2365. [PubMed] [Google Scholar]
- 46.Dhanasekaran N, Tsim ST, Dermott JM, Onesime D. Oncogene. 1998;17:1383–1394. doi: 10.1038/sj.onc.1202242. [DOI] [PubMed] [Google Scholar]
- 47.Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi K, Ohuchida S, Sugimoto Y, Narumiya S, Sugimura T, Wakabayashi K. Cancer Res. 2002;62:28–32. [PubMed] [Google Scholar]
- 48.Sheng H, Shao J, Washington MK, DuBois RN. J Biol Chem. 2001;276:18075–18081. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 49.Southall MD, Vasko MR. J Biol Chem. 2001;276:16083–16091. doi: 10.1074/jbc.M011408200. [DOI] [PubMed] [Google Scholar]
- 50.Kleiner DE, Stetler-Stevenson WG. Cancer Chemother Pharmacol. 1999;43(suppl):42–51. doi: 10.1007/s002800051097. [DOI] [PubMed] [Google Scholar]
- 51.Stetler-Stevenson WG. Invasion Metastasis. 1994;14:259–268. [PubMed] [Google Scholar]
- 52.Bramhall SR. Int J Pancreatol. 1997;21:1–12. doi: 10.1007/BF02785914. [DOI] [PubMed] [Google Scholar]
- 53.Brown PD, Bloxidge RE, Stuart NS, Gatter KC, Carmichael J. J Natl Cancer Inst. 1993;85:574–578. doi: 10.1093/jnci/85.7.574. [DOI] [PubMed] [Google Scholar]
- 54.Iizasa T, Fujisawa T, Suzuki M, Motohashi S, Yasufuku K, Yasukawa T, Baba M, Shiba M. Clin Cancer Res. 1999;5:149–153. [PubMed] [Google Scholar]
- 55.Attiga FA, Fernandez PM, Weeraratna AT, Manyak MJ, Patierno SR. Cancer Res. 2000;60:4629–4637. [PubMed] [Google Scholar]
- 56.Corcoran ML, Stetler-Stevenson WG, DeWitt DL, Wahl LM. Arch Biochem Biophys. 1994;310:481–488. doi: 10.1006/abbi.1994.1196. [DOI] [PubMed] [Google Scholar]
- 57.Takahashi K, Eto H, Tanabe KK. Int J Cancer. 1999;80:387–395. doi: 10.1002/(sici)1097-0215(19990129)80:3<387::aid-ijc9>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Thant AA, Machida K, Ichigotani Y, Naito Y, Hiraiwa Y, Senga T, Sohara Y, Matsuda S, Hamaguchi M. Cancer Res. 2002;62:3962–3965. [PubMed] [Google Scholar]
- 59.Sun LK, Beck-Schimmer B, Oertli B, Wuthrich RP. Kidney Int. 2001;59:190–196. doi: 10.1046/j.1523-1755.2001.00479.x. [DOI] [PubMed] [Google Scholar]
- 60.Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, Seiki M. J Cell Biol. 2001;153:893–904. doi: 10.1083/jcb.153.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]