

Abstract
All 13C NMR studies of brain glycogen to date relied on observing the incorporation of 13C label into glycogen, and thus interpretation was potentially affected by changes in 13C label turnover rates. The goal of this study was to quantify total brain glycogen concentration under conditions of hypoglycemia or normoglycemia using biochemical methods. Rats were sacrificed using a focused microwave fixation device. The results showed that metabolism of brain glycogen was Glc- and insulin-sensitive and that insulin-induced hypoglycemia promoted a gradual glycogenolysis. Moreover, we show that there are very mild effects of isoflurane and α-chloralose anesthesia on brain glycogen concentration. Altogether these result show that total brain glycogen serves as a substantial source of glucosyl units during insulin-induced moderate hypoglycemia and therefore may be neuroprotective. Finally we also conclude that previous interpretation of 13C NMR spectroscopy data accurately reflected the changes in total brain glycogen content.
INTRODUCTION
Glucose (Glc) is the main substrate for brain energy metabolism. Although the brain relies on a continuous supply of Glc for normal function, it has been proposed based on studies using 13C NMR spectroscopy in conjunction with infusion of 13C labeled glucose (Choi et al., 2000; Choi et al., 1999), that brain glycogen can serve as a substantial source of Glc equivalents during acute insulin-induced hypoglycemia (Choi et al., 2003; Gruetter, 2003). Brain glycogen is known to exert a neuroprotective effect (Brown et al., 2003). However, the ability of brain glycogen to provide Glc equivalent to brain energy metabolism has been questioned: Whereas it has been stated that brain cannot store more than a few minutes’ supply as glycogen (reviewed in Cryer et al., 2003), we suggested that glycogen stores are not completely depleted after 2 hours of hypoglycemia (Choi et al., 2003). Recent studies using 13C NMR spectroscopy have shown that brain glycogen metabolism was very slow both in light α-chloralose anesthetized rats (Choi et al., 1999) and in human subjects (Oz et al., 2003), and that the total concentration of brain glycogen in the awake, normoglycemic rat was 3.3 ± 0.8 μmol/g (Choi and Gruetter, 2003). All NMR studies of brain glycogen to date were performed in conjunction with observing the incorporation 13C label into brain glycogen. The interpretation of 13C NMR data was potentially complicated by the fact that only the labeled part of the molecule is measured and therefore potentially suffers from the limitation related to 13C label turnover. On the other hand, using biochemical methods following brain extraction to measure metabolically labile compounds such as glycogen needs to address postmortem artifacts (Swanson and Choi, 1993). It is difficult to quantitatively account for all of the possible metabolic products that might be quickly generated due to rapid post-mortem degradation (Cruz and Dienel, 2002).
Brain glycogen is located almost entirely in astrocytes, the most numerous cells in the brain, which are distributed throughout the brain (Savchenko et al., 2000) (Sloane et al., 2000). In culture, it has been shown that both hyperglycemia (Swanson et al., 1989) and insulin increased glycogen content (Dringen and Hamprecht, 1992). In vivo, the concentration of brain glycogen can be influenced by glucose (Goldberg and O’toole, 1969; Nelson et al., 1968) and insulin seems to promote glycogenesis as well in the brain (Choi et al., 2003; Daniel et al., 1977; Strang and Bachelard, 1971). These early studies were performed in the presence of rather high concentrations of plasma glucose. Therefore, it was not clear whether these effects of insulin on glycogen content were only achieved in supraphysiologic hyperglycemic conditions (Daniel et al., 1977) or if brain glycogen might be regulated by both insulin-dependent and insulin-independent mechanisms. Therefore, determining the relative contribution of each may have great relevance to type-1 diabetes (reviewed in Brown, 2004; Gruetter, 2003). Concentrations of brain glycogen can be further influenced by neurotransmitters (Magistretti et al., 1986; Pellerin and Magistretti, 1994) and anesthetics (Nelson et al., 1968; Nordstrom and Siesjo, 1978). Apparently, the degree of brain glycogen increase seems to be related to the depth, duration, and type of anesthesia used, with pentobarbital producing greater increases in glycogen content than, for example, ether (Nelson et al., 1968).
The goal of this study was to measure total brain glycogen concentration during different glycemic states, including hypoglycemia, using microwave fixation and biochemical methods (Cruz and Dienel, 2002) in rats under light anesthesia.
METHODS
Groups of animals studied:
Table 1 summarizes the 12 different experimental groups. In groups A, B and D, blood was collected by tail bleed. The two groups of rats that underwent hypoglycemia (groups K and L) were treated as previously described (Choi et al., 2001). Hypoglycemia was achieved by infusing 12 insulin Units/hr/kg and adjusting plasma Glc levels between 1 to 2 mmoles/L.
Table 1:
Description of the experimental groups studied.
| Gr. of rats | “Identiflers” | n | Prior Anesthesia | Time under isoflurane anesthesia | Time under α - chloralose anesthesia |
|---|---|---|---|---|---|
| A | Fasted only | 6 | Overnight fasted | ∼ 10 min isoflurane only. No preparation. | — |
| B | 48 hrs 10% Glc | 7 | Overnight fasted & then fed ad lib. for 48 hrs with 10% Glc sol. Only | ∼ 10 min isoflurane only. No preparation. | — |
| C | Preparation only | 4 | Overnight fasted | Rat preparation (1 hr 47 min ± 5 min) | — |
| D | 1h45′ under isoflurane | 4 | Overnight fasted & then fed ad lib. for 48 hrs with 10% Glc sol. Only | 1 hr 45 min ± 13 min under isoflurane. No preparation. | — |
| E | 8 hrs α-chloralose | 5 | Overnight fasted | Rat preparation (1 hr 42 min ± 8 min) | I.V. infusion of α-chloralose ONLY (8 hrs 8 min ± 3 min) |
| F | 5 hrs α-chloralose | 5 | Overnight fasted | Rat preparation (1 hr 36 min ± 4 min) | I.V. infusion of α-chloralose ONLY (5 hrs ± 1 min) |
| G | Insulin (6 I.U./kg/hr) & 5 mM plasma Glc | 5 | Overnight fasted | Rat preparation (1 hr 25 min ± 13 min) | 5 hrs 4 min ± 5 min |
| H | Insulin (6 I.U./kg/hr) & 10 mM plasma Glc | 5 | Overnight fasted | Rat preparation (1 hr 53 min ± 4 min) | 4 hrs 38 min ± 8 min |
| I | Somatostatin (0.75 μg/kg/min) & 10 mM plasma Glc | 5 | Overnight fasted | Rat preparation (1 hr 52 min ± 5 min) | 4 hr 59 min ± 6 min |
| J | Somatostatin (0.75 μg/kg/min) & 20 mM plasma Glc | 5 | Overnight fasted | Rat preparation (1hr 50 min ± 7 min) | 4 hrs 54 min ± 5 min |
| K | Short acute hypoglycemia | 5 | Overnight fasted | Rat preparation (1 hr 47 min ± 4 min) | I.V. inf. of Glc (plasma Glc: 13.9 ± 0.7 mM) during 3 hrs followed by 30 min hypoglycemia. Time under α-chloralose anesthesia: 5 hrs 1 min ± 3 min |
| L | Long acute hypoglycemia | 6 | Overnight fasted | Rat preparation (1 hr 58 min ± 17 min) | I.V. inf. of Glc (plasma Glc: 13.7 ± 0.7 mM during 3 hrs followed by 2 hrs hypoglycemia. Time under α-chloralose anesthesia: 5 hrs 54 min ± 28 min |
Data are shown as average ± SEM. Abbreviations: ad lib., ad libitum; Glc, glucose; n, number of rats; inf., infusion; I.U., insulin Unit; I.V., intravenous; sol., solution.
Animal preparation
The study was performed according to the guidelines for the care and use of laboratory animals at the University of Minnesota and was approved by the Institutional Animal Care and Use Committee (IACUC).
Male Sprague-Dawley rats (271 ± 4 g, Harlan, Madison, WI, USA) were fasted overnight with free access to water before studies. They were anesthetized using isoflurane (isoflurane, Halocarbon laboratories, 5% for induction and 1.8-2.0% for maintenance) in a 70:35 mixture of nitrous oxide (N2O) and oxygen (O2) gases for preparation. Then, rats were intubated and ventilated with a pressure-driven ventilator (Kent Scientific, Litchfield, CT, U.S.A.) during the rest of the experiment (Table 1: groups C and E-L). End tidal CO2, respiratory rate and pattern were monitored during rat preparation and experiments with a capnometer (SurgiVet, Waukesha, WI, USA). Preparation consisted of inserting catheters into both femoral arteries for blood gases and Glc analysis, and into both femoral veins for intravenous (I.V.) infusion of α-chloralose (Sigma), Glc (Mallinckrodt), insulin (Humulin R, Eli Lilly and Company, Indianapolis, IN, USA) and somatostatin (octreotide acetate [Sandostatin], Novartis).
Animal anesthesia
Immediately after rat preparation, anesthesia was switched to α-chloralose, which was administered I.V. as follows: 30 minutes after a 40 mg/kg bolus injection, a continuous I.V. infusion was started (26.7 mg/kg/hr) to maintain a light anesthesia state according to previous report (Ueki et al., 1992). All solutions used for I.V. infusion were diluted in Dulbecco’s Phosphate-Buffered Saline (dPBS without calcium chloride and without magnesium chloride, BioWhittaker, Maryland, USA).
Rat body temperature was maintained at 37.0 ± 0.5°C with a heating plate system (LAB-line slide warmer) based on a feedback obtained from a rectal temperature probe (Cole Palmer, Vernon Hills, IL, U.S.A.).
Arterial blood was withdrawn from the femoral artery through a polyethylene tubing line (PE50, Becton Dickinson) at least every 30 min. About 0.05 ml of blood was kept to measure physiological parameters with a pH/blood gas analyzer (model 248, RapidLab, BAYER) and the rest of the blood (∼0.15 ml) was immediately centrifuged after sampling to obtain plasma. Plasma Glc concentration was measured using the glucose oxidase method in a Glc analyzer (GM7 Micro-stat, Analox instruments, London, UK). Physiologic parameters (respiration rate, breathing volume and cutoff) were adjusted throughout the experiments to maintain pCO2 and pH within normal physiological ranges (Table 2).
Table 2:
Physiologic parameters.
| “Identifiers” | n | Plasma Glc | pH | PCO2 | PO2 | Body temp. |
|---|---|---|---|---|---|---|
| All groups during rat preparation (i.e. C, E, F, G, H, I, J, K, and L) | 36 | 10.6 ± 0.3 | 7.49 ± 0.01 | 32.1 ± 0.8 | 149.0 ± 6.3 | 37.0 ± 0.1 |
| E | 5 | 7.1 ± 0.1 | 7.41 ± 0.01 | 37.1 ± 0.1 | 146.2 ± 6.8 | 37.1 ± 0.1 |
| F | 5 | 7.5 ± 0.4 | 7.41 ± 0.01 | 35.1 ± 1.8 | 152.1 ± 12.4 | 37.1 ± 0.0 |
| G | 5 | 4.9 ± 0.1 | 7.41 ± 0.01 | 38.0 ± 0.7 | 145.1 ± 5.8 | 37.1 ± 0.0 |
| H | 5 | 10.2 ± 0.5 | 7.35 ± 0.01 | 42.4 ± 0.9 | 135.6 ± 5.2 | 37.2 ± 0.0 |
| I | 5 | 10.4 ± 0.1 | 7.40 ± 0.01 | 39.9 ± 1.0 | 130.0 ± 5.9 | 37.1 ± 0.0 |
| J | 5 | 20.8 ± 0.2 | 7.38 ± 0.00 | 41.7 ± 0.9 | 146.0 ± 8.2 | 37.2 ± 0.1 |
| K | 4 | 1.7 ± 0.1 | 7.31 ± 0.01 | 41.5 ± 1.3 | 159.7 ± 11.5 | 37.0 ± 0.2 |
| L | 6 | 1.7 ± 0.0 | 7.34 ± 0.02 | 37.9 ± 1.6 | 191.0 ± 32.0 | 37.0 ± 0.2 |
Data are shown as mean ± SEM. Values are averaged during the last hours of experiments (ex.: during insulin or somatostatin infusion etc...). Therefore, these values do not include the values obtained during rat preparation (except for the first line). Abbreviations: Glc, glucose; temp., temperature.
The infusion rate of Glc (20% weight/volume in dPBS) was continuously adjusted based on the measured plasma Glc concentrations to maintain target plasma glycemic levels.
Animal sacrifice and brain extraction
At the end of each experiment, rats were sacrificed using a focused microwave fixation device (by applying 4kW onto a 2 ml volume for 1.4 sec) (Gerling Applied Engineering, Inc.) which inactivates most brain enzymes before extraction or digestion, thereby minimizing possible in vitro glycogen loss (Kong et al., 2002). The anterior parts of the brain (i.e. excluding cerebellum) were dissected, and immediately placed into liquid nitrogen and manually reduced to powder with a mortar and a pestle. Brain powder was stored for less than 6 weeks at -80°C.
Glycogen Assay
Biochemical measurement of brain glycogen was performed on the brain extracts as previously described (Cruz and Dienel, 2002). Briefly, 0.03N HCl (500 μl) was carefully added to frozen brain powders (∼200 mg) in tubes on dry ice. Brain powders were homogenized (ultrasonic processor, Cole-Parmer Instruments, Vernon Hills, Illinois, USA), heated to 90°C for 45 minutes and homogenized again.
For the glycogen assay, sodium acetate (Sigma) was added to 200 μl of ice-cold samples in 0.03N HCl to attain a final concentration of 0.1 mM sodium acetate. Well-mixed portions of each sample were incubated at 37°C for at least 2 hours with gentle mixing (Reciprocal water bath shaker, model R76, New Brunswick Scientific, Edison, NJ, USA) in two parallel aliquots, one containing amylo-α-1,4-α-1,6-glucosidase (24 μg/mL, Roche) to convert glycogen to Glc and the other without amylo-α-1,4-α-1,6-glucosidase. The samples were centrifuged subsequently to remove particulate matter, and Glc levels were determined in measured aliquots of the supernatant fraction using the same Analox instrument as above. Glycogen levels per gram wet weight were calculated as Glc released by amylo-α-1,4-α-1,6-glucosidase, that is, Glc level after incubation with amyloglucosidase minus that without the enzyme (Ghajar et al., 1982).
Lactate assay
To evaluate potential post-mortem glycogenolysis and glucose breakdown, tissue lactate levels were assessed in parallel in the supernatant fraction. These measurements were performed using a highly specific method measuring oxygen consumption necessary for the oxidation of L-lactate into pyruvate by an oxygen reductase (GMRD-090 lactate reagent kit, Analox Instruments, USA) on the Analox reader (same as above). Measurements were excluded when brain lactate concentration exceeded 3.0 μmol/g.
Statistical analysis
The statistical analysis was performed using the SAS package (SAS Institute Inc., Cary, NC). Brain glycogen concentrations of different groups of rats were compared using the analysis of variance, followed by the Tukey test for multiple comparisons between means (GLM procedure). The linear regression model was used to check the correlation between brain glycogen and brain glucose measurements (considered as covariable) and to compare the slope and intercepts of the regression lines in different groups.
RESULTS
First, we compared brain glycogen concentrations obtained under different levels and types of anesthesia. Comparing brain glycogen concentrations between the groups of rats that were shortly induced with isoflurane (Table 1 and fig. 1: groups A & B) with the rats that underwent ∼1 hour 45 min anesthesia under isoflurane (Table 1 and fig. 1: group C) before being sacrificed indicated that ∼1 hour 45 min of isoflurane anesthesia caused a small increase in brain glycogen concentration of ∼0.6 μmol/g/hr (Fig. 1: compare group A or B with C). 8 additional hours of α-chloralose anesthesia produced no discernable change in brain glycogen concentration (Fig. 1: compare group C with E), although after 5 hours of α-chloralose anesthesia (Table 1: group F, average brain glycogen ± SEM: 5.8 ± 0.2 μmol/g) brain glycogen was slightly higher than the concentration observed in rats that only underwent a preparation under isoflurane anesthesia (Table 1: group C, average brain glycogen ± SEM: 4.6 ± 0.2 μmol/g) (two tailed t-test assuming equal variance, P<0.01, not shown). Taken together these results show that there were very mild effects of isoflurane and light α-chloralose anesthesia (26.7 mg/kg/hr I.V.) on brain glycogen concentration.
Figure 1:
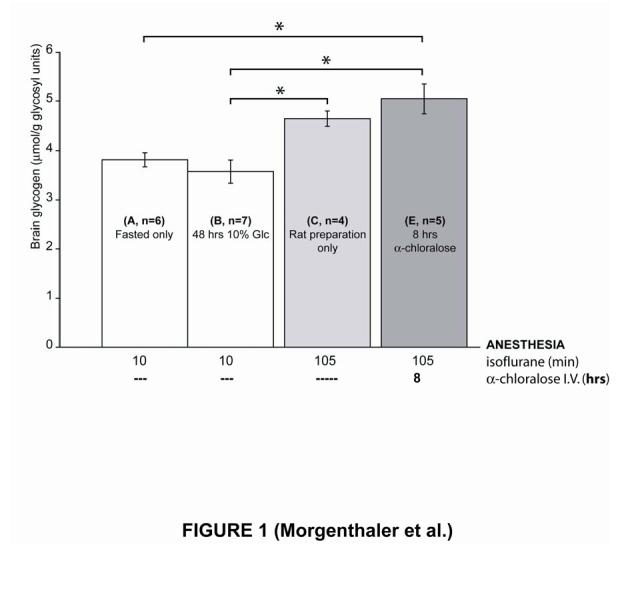
Effects of anesthetics on brain glycogen concentration. ∼1 hour 45 min under isoflurane anesthesia result in an increase in brain glycogen (compare group C with groups A and B). However, 8 additional hours under α-chloralose anesthesia do not significantly increase brain glycogen concentration further (compare E with C). Overnight fasting (compare group A with B) did not substantially affect brain glycogen. Error bars indicate SEM. Significant differences between groups (with Tukey test, P < 0.05) are indicated by *. Abbreviation: Glc, glucose.
Second, we studied whether overnight fasting affected brain glycogen. Comparing the rats which were fasted the night prior sacrifice (Table 1 and fig. 1: group A) with the rats which were fasted two nights before and then fed for 48 hrs with a 10% Glc solution (Table 1 and fig. 1: group B) showed that neither overnight fasting nor the 48 hours Glc feeding regime substantially affected brain glycogen content. Moreover, the preparation itself neither affected brain glycogen concentration as judged from comparison of groups C with D (average brain glycogen ± SEM: group C: 4.6 ± 0.2 μmol/g and group D: 4.8 ± 0.1μmol/g). Finally, the biochemical measurements of brain glycogen in the two groups of rats that were fed with Glc 10% for 48 hours (averaged brain glycogen concentration ± SEM: 3.6 ± 0.2 μmol/g in group B with short isoflurane anesthesia and 4.8 ± 0.1 μmol/g in group D after ∼1 hour 45 min under isoflurane anesthesia) were in excellent agreement with previous 13C NMR measurements of total brain glycogen which reported a brain glycogen concentration ± SEM of 3.3 ± 0.3 μmol/g (Choi and Gruetter, 2003).
Then, we assessed the transport kinetics of cerebral glucose by plotting biochemical postmortem measurements of brain glucose concentration as a function of plasma glucose concentrations measured just prior to sacrifice (Fig. 2). As previously reported in α-chloralose lightly anesthetized rats, the brain Glc concentration was a linear function of the plasma Glc concentration (Choi et al., 2001; Choi et al., 2002). There was no significant difference in the distribution of brain Glc concentration in the isoflurane anesthetized rats (Fig. 2: open squares) compared to α-chloralose anesthetized rats (Fig. 2: solid triangles). Moreover, the biochemical measurements presented here were in agreement with the linear relation proposed before when we measured the concentration of brain Glc by 13C NMR methods in living rats (Choi et al., 2001) (Fig. 2). No significant difference between the slopes of the previous and present regression line was found. In both cases, at plasma Glc levels lower than 2 mmoles/L, the brain Glc concentration approached zero (Fig. 2, see also Fig. 4). Taken together, these results show that the distribution of brain Glc presented no substantial differences between isoflurane and α-chloralose anesthetized rats.
Figure 2:

Comparison of brain Glc, as a function of plasma Glc, under isoflurane (2%) and α-chloralose anesthesia (26.7 mg/kg/hr). The concentration of brain glucose measured by biochemical assay in postmortem rat brain samples is plotted as a function of plasma glucose concentration. The solid line, which represents the best fit of the reversible Michaelis-Menten model of glucose transport described previously (Choi et al., 2001; Gruetter et al., 1998), is in agreement with the biochemical measurements presented here. Abbreviation: Glc, glucose.
Figure 4:

insulin-induced hypoglycemia (< 2 mmoles/L plasma Glc) promoted gradual glycogenolysis. First group (K) was measured after 30 min. of hypoglycemia and second group (L) after 120 min. Control group (F) is shown as mean ± 2 SD (95% confidence interval) in the shaded top band. Whereas, plasma Glc represents the averaged values measured during 30 min (K) and 120 min (L) of hypoglycemia, brain Glc and glycogen are postmortem measurements. Errors bars indicate SEM. Abbreviation: Glc, glucose. Table 1: Description of the experimental groups studied. Table 2: Physiologic parameters.
We also studied whether total brain glycogen content can be influenced by Glc and insulin. To assess the effects of Glc and insulin separately, as well as administered together, rats from group F (maintained for 5 hours under α-chloralose anesthesia after preparation) were used as control (see table 1). As previously observed (Choi et al., 2003), endogenous insulin secretion was suppressed by somatostatin infusion as suggested from the greatly reduced glucose infusion rate required to maintain constant plasma glucose concentration (groups I and J). The infusion of Glc, either alone (i.e. infused with somatostatin, Fig. 3: compare group I and J) or administered together with insulin (Fig. 3: compare group G and H), had a significant effect on brain glycogen concentration (P=0.02). This analysis was achieved by including the correlation between brain glycogen and brain glucose (Pearson R=0.42, P=0.03) in a general linear model with brain Glc as covariable. The results show a significant effect of brain Glc on brain glycogen and no significant difference was found between the slopes of the regression lines in the two groups. Moreover, by eliminating the influence of brain Glc concentration, our results also show a significant effect (P=0.03) of insulin on brain glycogen concentration. This difference between the two experimental groups (with or without somatostatin administration) with respect to the insulin concentration is explained by the significant (P=0.03) difference of the intercepts of the corresponding regression lines. Taken together, these results show that there was a significant synergistic effect of Glc and insulin in increasing brain glycogen concentrations when administered together for 4 hours.
Figure 3:

Significant synergistic effect of brain glucose and insulin on brain glycogen content. The groups of rats presented here were infused for 4 hrs either in the presence of insulin (6U/kg/hr) or somatostatin (0.75 mg/kg/min) and their glycemia was kept close to 5 mM (group G), 10 mM (groups H and I) and 20 mM (group J). In the control group (F), there was no infusion of Glc (average plasma Glc ± SEM: 7.3 ± 0.3 mM), and no exogenous administration of insulin or somatostatin. Error bars indicate SEM. The slopes of regression lines are similar and the intercepts are significantly different. Abbreviation: Glc, glucose.
The comparison between the measurements obtained here using biochemical methods with previous 13C NMR measurements of total brain glycogen was assessed. Considering the rats that were infused for 4 hours with 6 I.U./kg/hr together with Glc to maintain plasma Glc level close to 10 mmoles/L (Table 1 and fig. 3: group H), there was an excellent agreement between biochemical and previous 13C NMR measurements (average brain glycogen concentration ± SEM: biochemical meas.: 6.2 ± 0.3 μmol/g and 13C NMR meas.: 5.8 ± 0.2 μmol/g) (Choi et al., 2003). However, for the group of rats that were infused with somatostatin together with Glc to maintain plasma Glc level close to 10 mmoles/L (Table 1 and fig. 3: group I), brain glycogen concentration obtained previously by 13C NMR was significantly lower than the value obtained here using biochemical methods (average brain glycogen concentration ± SEM: biochemical meas.: 5.3 ± 0.3 μmol/g and 13C NMR meas.: 2.1 ± 0.2 μmol/g) (Choi et al., 2003).
Finally, the effect of hypoglycemia on brain glycogen metabolism was assessed in the last groups of rats (Table 1: groups K and L). At plasma Glc levels lower than 2 mmoles/L (mean ± SEM: 1.7 ± 0.0 mmoles/L), the brain Glc concentration was 0.1 ± 0.1 μmol/g (Fig. 4, see also fig. 2). Although the decrease of brain glycogen after only 30 min of hypoglycemia was already significant (P<0.05, fig. 4: compare group K with control group F), after 1.5 more hour of hypoglycemia, brain glycogen concentration was significantly lower (P< 0.01, fig. 4: compare group L with K), but not yet completely depleted. Brain glycogen concentration decreased from 3.1 ± 0.4 μmol/g (group K) to 0.9 ± 0.6 μmol/g (group L) during 1.5 hour, thus allowing to evaluate a glycogenolysis rate of ∼1.5 μmol/g/hr. These results show that insulin-induced moderate hypoglycemia promoted gradual glycogenolysis even though brain Glc was similar in both hypoglycemic groups.
DISCUSSION
Although 13C NMR is the only method to measure brain glycogen non invasively, measurements of brain glycogen by 13C NMR are affected by in vivo turnover rates (Choi and Gruetter, 2003; Choi et al., 2003; Choi et al., 1999; Oz et al., 2003). Quantitative determination of glycogen content using biochemical methods is challenging due to rapid postmortem degradation of brain glycogen and involves the study of a higher number of animals (Cruz and Dienel, 2002; Kong et al., 2002). Here, to minimize postmortem degradation of this labile compound, rats were sacrificed using a focused microwave fixation device. In addition, tissue lactate concentration was measured to ensure that postmortem metabolism had a negligible impact on the interpretation of the data. Overall total brain glycogen concentrations obtained in the present study were in agreement with previous in vivo studies using 13C NMR (Choi and Gruetter, 2003; Choi et al., 2001; Choi et al., 2003) suggesting that 13C NMR allows for a correct interpretation of glycogen concentration changes even in the presence of label turnover.
When investigating the effects of anesthesia on brain glycogen we noted a small increase in brain glycogen concentration after ∼1 hour 45 min under isoflurane anesthesia compared to the rats that were only induced with isoflurane. However, continuing under α-chloralose anesthesia for 5-8 hours resulted in minimal change in brain glycogen concentration (fig. 1). Clearly, a 2% isoflurane or light α-chloralose anesthesia at 26.7 mg/kg/hr I.V. does not result in the same kinds of massive glycogen synthesis reported for pentobarbital anesthesia (Choi et al., 2002; Nelson et al., 1968). During deep pentobarbital anesthesia, it has been shown that the increase in brain glycogen appeared to be related to the great diminution of neuronal activity (Phelps, 1972). Likewise, anesthetics only increase glycogen when applied in vivo, and not in astrocyte cultures (Swanson et al., 1989). In the present study, the anesthetics used allowed to maintain a light anesthesia state (Ueki et al., 1992) and therefore produced a very small increase in glycogen content.
The similarity of brain glycogen concentrations under isoflurane and α-chloralose suggested a similar reduction in brain energy metabolism for these 2 anesthetics. To confirm this, we measured the effects of anesthesia on brain Glc which has been shown to be sensitive to the levels of anesthesia (Choi et al., 2002). The absence of a significant difference in the distribution of brain Glc concentration as a function of plasma Glc in the isoflurane compared to α-chloralose anesthetized rats (fig. 2) is additional evidence suggesting similar rates of brain energy metabolism. This is unlike previously reported in deep pentobarbital anesthesia where much higher brain Glc and glycogen levels were observed (Choi et al., 2002). It further suggests the notion of important control of glycogen content by astrocytic glucose content. The observed linear relationship between plasma and brain glucose is a consequence of using the reversible Michaelis-Menten model of glucose transport to describe glucose content (Choi et al., 2001; Choi et al., 2002; Gruetter et al., 1998) and it is in excellent agreement with previous 13C NMR methods in rats under α-chloralose anesthesia (Choi et al., 2001).
The hypothesis that brain Glc might exert a control over glycogen levels was further supported by increased brain Glc resulting in increased brain glycogen concentration (fig. 3). Reciprocally, fasting did not affect Glc levels and therefore did not substantially affect brain glycogen (fig. 1), consistent with brain Glc not being rate-limiting for metabolism in fasting. As increased levels of Glc would raise insulin secretion, we also tested the effects of insulin on brain glycogen. We showed that insulin had an impact similar to Glc on increasing brain glycogen concentration (fig. 3). Moreover, there was a significant synergistic effect of Glc and insulin in increasing brain glycogen concentrations when administered together for 4 hrs. It has already been suggested that the concentration of glycogen may reflect the combined effect of elevated plasma glucose and insulin to promote net glycogen synthesis in the brain (Choi and Gruetter, 2003; Daniel et al., 1977; Nelson et al., 1968; Strang and Bachelard, 1971). Here, we showed in rats under physiologic plasma Glc concentrations that brain glycogen might be regulated by Glc-dependant (Fig. 3, compare groups F, G and H, or compare groups I and J) and Glc-independent (Fig. 3, compare groups H and I), as well as insulin-dependent (Fig. 3, compare groups H and I) and insulin-independent mechanisms (Fig. 3, compare groups I and J). These findings are in excellent agreement with previous 13C NMR measurements (Choi et al., 2003), although the experiments with somatostatin resulted in lower 13C incorporation and the estimation of lower glycogen concentration (Choi et al., 2003) suggesting an effect of low concentration of insulin mainly on basal glycogen turnover rates. The effect of Glc on brain glycogen was further studied in acute moderate hypoglycemia (fig. 4). When plasma Glc levels were below 2 mM, the brain Glc concentration was close to zero, consistent with Glc transport becoming rate limiting for whole brain metabolism during hypoglycemia. This moderate hypoglycemia promoted a gradual glycogenolysis at a rate of ∼1.5 μmol/g/hr. This value is in agreement with the glycogenolytic flux of 0.038 ± 0.005 μmol/g/min previously estimated by 13C NMR spectroscopy (Choi et al., 2003). The persistence of glycogen during hypoglycemia can be explained by a partial Glc supply deficit (Gruetter, 2003). Thus, the brain can store more than a few minutes’ supply of Glc equivalent as glycogen, and brain glycogen is likely to play a significant role during hypoglycemia.
Altogether, these results suggest that total brain glycogen serves as a viable glucose reservoir during extended periods of moderate hypoglycemia. Glycogen may be neuroprotective due to increased glycogen breakdown to Glc-6-phospate during hypoglycemia (Swanson and Choi, 1993). One possible neuroprotective mechanism of glycogen is to provide ATP to allow continuous uptake of excitotoxic glutamate from the synaptic cleft (Gruetter, 2003) and to provide lactate as fuel for neurons (Magistretti and Pellerin, 1996). Such neuroprotection might interfere with proper recognition of hypoglycemia by interfering with glucose-sensing neurons. It has been proposed that brain glucose is the signal that triggers defense mechanisms (for example, cerebral blood flow increase and glycogen utilization) aimed at improving glucose equivalent delivery to the brain during hypoglycemia (Choi et al., 2001). It is also well known that type-1 diabetics who experience a single hypoglycemic episode are more susceptible to succeeding periods of hypoglycemia. The mechanism is unknown but in addition to the sympathetic nervous system, Glc (Criego et al., 2005) and glycogen (Choi et al., 2003) may also be involved. Therefore, glycogen might be implicated in the mechanism of hypoglycemia unawareness syndrome observed clinically in patients with type I diabetes, and may play a much more important neuroprotective role in many disorders, such as sleep deprivation, than previously assumed.
ACKNOWLEDGEMENTS
We wish to thank Tianwen K. Yue for his help with the animal preparation. This work was supported by NIH R01NS42005 and JDRF 1-2002-722.
Footnotes
- ATP
- Adenosine Tri-Phosphate
- Glc
- glucose
- I.U.
- insulin unit
- I.V.
- intravenous
- NMR
- nuclear magnetic resonance
REFERENCES
- Brown AM. Brain glycogen re-awakened. Journal of Neurochemistry. 2004;89:537–552. doi: 10.1111/j.1471-4159.2004.02421.x. [DOI] [PubMed] [Google Scholar]
- Brown AM, Tekkok SB, Ransom BR. Glycogen regulation and functional role in mouse white matter. J Physiol. 2003;549:501–512. doi: 10.1113/jphysiol.2003.042416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IY, Gruetter R. In vivo 13C NMR assessment of brain glycogen concentration and turnover in the awake rat. Neurochem Int. 2003;43:317–322. doi: 10.1016/s0197-0186(03)00018-4. [DOI] [PubMed] [Google Scholar]
- Choi IY, Lee SP, Kim SG, Gruetter R. In vivo measurements of brain glucose transport using the reversible Michaelis-Menten model and simultaneous measurements of cerebral blood flow changes during hypoglycemia. J Cereb Blood Flow Metab. 2001;21:653–663. doi: 10.1097/00004647-200106000-00003. [DOI] [PubMed] [Google Scholar]
- Choi IY, Lei H, Gruetter R. Effect of deep pentobarbital anesthesia on neurotransmitter metabolism in vivo: on the correlation of total glucose consumption with glutamatergic action. J Cereb Blood Flow Metab. 2002;22:1343–1351. doi: 10.1097/01.WCB.0000040945.89393.46. [DOI] [PubMed] [Google Scholar]
- Choi IY, Seaquist ER, Gruetter R. Effect of hypoglycemia on brain glycogen metabolism in vivo. J Neurosci Res. 2003;72:25–32. doi: 10.1002/jnr.10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IY, Tkac I, Gruetter R. Single-shot, three-dimensional “non-echo” localization method for in vivo NMR spectroscopy. Magn Reson Med. 2000;44:387–394. doi: 10.1002/1522-2594(200009)44:3<387::aid-mrm8>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Choi IY, Tkac I, Ugurbil K, Gruetter R. Noninvasive measurements of [1-(13)C]glycogen concentrations and metabolism in rat brain in vivo. J Neurochem. 1999;73:1300–1308. doi: 10.1046/j.1471-4159.1999.0731300.x. [DOI] [PubMed] [Google Scholar]
- Criego AB, Tkac I, Kumar A, Thomas W, Gruetter R, Seaquist ER. Brain glucose concentrations in patients with type 1 diabetes and hypoglycemia unawareness. J Neurosci Res. 2005;79:42–47. doi: 10.1002/jnr.20296. [DOI] [PubMed] [Google Scholar]
- Cruz NF, Dienel GA. High glycogen levels in brains of rats with minimal environmental stimuli: implications for metabolic contributions of working astrocytes. J Cereb Blood Flow Metab. 2002;22:1476–1489. doi: 10.1097/01.WCB.0000034362.37277.C0. [DOI] [PubMed] [Google Scholar]
- Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care. 2003;26:1902–1912. doi: 10.2337/diacare.26.6.1902. [DOI] [PubMed] [Google Scholar]
- Daniel PM, Love ER, Pratt OE. The influence of insulin upon the metabolism of glucose by the brain. Proc R Soc Lond B Biol Sci. 1977;196:85–104. doi: 10.1098/rspb.1977.0031. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hamprecht B. Glucose, insulin, and insulin-like growth factor I regulate the glycogen content of astroglia-rich primary cultures. J Neurochem. 1992;58:511–517. doi: 10.1111/j.1471-4159.1992.tb09750.x. [DOI] [PubMed] [Google Scholar]
- Ghajar JB, Plum F, Duffy TE. Cerebral oxidative metabolism and blood flow during acute hypoglycemia and recovery in unanesthetized rats. J Neurochem. 1982;38:397–409. doi: 10.1111/j.1471-4159.1982.tb08643.x. [DOI] [PubMed] [Google Scholar]
- Goldberg ND, O’Toole AG. The properties of glycogen synthetase and regulation of glycogen biosynthesis in rat brain. J Biol Chem. 1969;244:3053–3061. [PubMed] [Google Scholar]
- Gruetter R. Glycogen: the forgotten cerebral energy store. J Neurosci Res. 2003;74:179–183. doi: 10.1002/jnr.10785. [DOI] [PubMed] [Google Scholar]
- Gruetter R, Ugurbil K, Seaquist ER. Steady-state cerebral glucose concentrations and transport in the human brain. J Neurochem. 1998;70:397–408. doi: 10.1046/j.1471-4159.1998.70010397.x. [DOI] [PubMed] [Google Scholar]
- Kong J, Shepel PN, Holden CP, Mackiewicz M, Pack AI, Geiger JD. Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J Neurosci. 2002;22:5581–5587. doi: 10.1523/JNEUROSCI.22-13-05581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Hof PR, Martin JL. Adenosine stimulates glycogenolysis in mouse cerebral cortex: a possible coupling mechanism between neuronal activity and energy metabolism. J Neurosci. 1986;6:2558–2562. doi: 10.1523/JNEUROSCI.06-09-02558.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L. Cellular bases of brain energy metabolism and their relevance to functional brain imaging: evidence for a prominent role of astrocytes. Cereb Cortex. 1996;6:50–61. doi: 10.1093/cercor/6.1.50. [DOI] [PubMed] [Google Scholar]
- Nelson SR, W. SD, V. PJ, H. LO. Control of glycogen levels in brain. Journal of Neurochemistry. 1968;15:1271–1279. doi: 10.1111/j.1471-4159.1968.tb05904.x. [DOI] [PubMed] [Google Scholar]
- Nordstrom CH, Siesjo BK. Effects of phenobarbital in cerebral ischemia. Part I: cerebral energy metabolism during pronounced incomplete ischemia. Stroke. 1978;9:327–335. doi: 10.1161/01.str.9.4.327. [DOI] [PubMed] [Google Scholar]
- Oz G, Henry PG, Seaquist ER, Gruetter R. Direct, noninvasive measurement of brain glycogen metabolism in humans. Neurochem Int. 2003;43:323–329. doi: 10.1016/s0197-0186(03)00019-6. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps CH. Barbiturate-induced glycogen accumulation in brain. An electron microscopic study. Brain Res. 1972;39:225–234. doi: 10.1016/0006-8993(72)90797-4. [DOI] [PubMed] [Google Scholar]
- Savchenko VL, McKanna JA, Nikonenko IR, Skibo GG. Microglia and astrocytes in the adult rat brain: comparative immunocytochemical analysis demonstrates the efficacy of lipocortin 1 immunoreactivity. Neuroscience. 2000;96:195–203. doi: 10.1016/s0306-4522(99)00538-2. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Hollander W, Rosene DL, Moss MB, Kemper T, Abraham CR. Astrocytic hypertrophy and altered GFAP degradation with age in subcortical white matter of the rhesus monkey. Brain Res. 2000;862:1–10. doi: 10.1016/s0006-8993(00)02059-x. [DOI] [PubMed] [Google Scholar]
- Strang RH, Bachelard HS. Effect of insulin on levels and turnover of intermediates of brain carbohydrate metabolism in vivo. J Neurochem. 1971;18:1799–1807. doi: 10.1111/j.1471-4159.1971.tb09585.x. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Choi DW. Glial glycogen stores affect neuronal survival during glucose deprivation in vitro. J Cereb Blood Flow Metab. 1993;13:162–169. doi: 10.1038/jcbfm.1993.19. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Yu AC, Sharp FR, Chan PH. Regulation of glycogen content in primary astrocyte culture: effects of glucose analogues, phenobarbital, and methionine sulfoximine. J Neurochem. 1989;52:1359–1365. doi: 10.1111/j.1471-4159.1989.tb09180.x. [DOI] [PubMed] [Google Scholar]
- Ueki M, Mies G, Hossmann KA. Effect of alpha-chloralose, halothane, pentobarbital and nitrous oxide anesthesia on metabolic coupling in somatosensory cortex of rat. Acta Anaesthesiol Scand. 1992;36:318–322. doi: 10.1111/j.1399-6576.1992.tb03474.x. [DOI] [PubMed] [Google Scholar]