

Hypertrophic cardiomyopathy is the most common familial genetic disease of the heart (1/500 to 1/1000), as well as the most common cause of sudden cardiac death in young people and athletes. Because a timely diagnosis may help to prevent sudden death, it is important for internists and general practitioners to be aware of the clinical features of the disease. The morphological and functional features of hypertrophic cardiomyopathy include marked and asymmetric left ventricular hypertrophy, a non-dilated left ventricular cavity, and preserved systolic function. Left ventricular outflow obstruction at rest is present in about 20% of patients.1-3 The clinical course is heterogeneous. Many patients remain asymptomatic throughout life, others develop severe heart failure or atrial fibrillation, and some die suddenly, often at a young age and in the absence of previous symptoms.1-3 We critically re-examine and place in perspective the most appropriate therapeutic strategies for the management of hypertrophic cardiomyopathy.
Sources and search criteria
This review is based on the main studies on hypertrophic cardiomyopathy published in recent years, as well as on the clinical experience of doctors and investigators with particular knowledge of this disease. The treatment strategies reported in the expert consensus document on hypertrophic cardiomyopathy from the American College of Cardiology and the European Society of Cardiology are also incorporated in this article.
Diagnosis
Hypertrophic cardiomyopathy is usually suspected after marked electrocardiographic abnormalities or a heart murmur have been identified during routine clinical evaluation, or because of recent development of dyspnoea or palpitations.1-3 The diagnosis is established by the echocardiographic identification of a hypertrophied and non-dilated left ventricle in the absence of other cardiovascular diseases capable of producing a similar magnitude of hypertrophy1-4 (fig 1). In a minority of patients with electrocardiographic abnormalities suggestive of hypertrophic cardiomyopathy, the echocardiogram may fail to identify left ventricular hypertrophy because of technically inadequate images or wall thickening confined to segments not clearly visualised by the ultrasounds. In such patients, the high resolution images of the heart obtained with magnetic resonance are particularly useful for establishing the diagnosis5,6 (fig 2).
Fig 1.
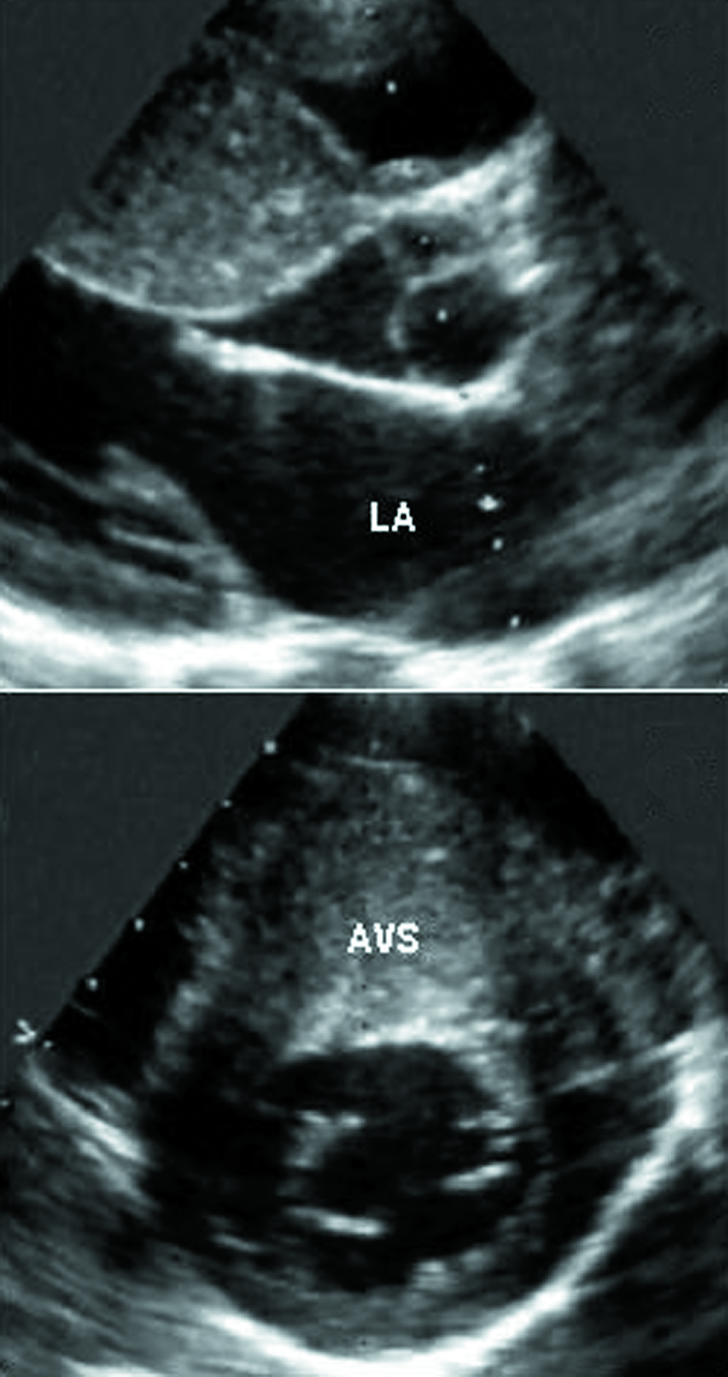
Echocardiographic parasternal long axis (top) and short axis (bottom) views showing marked and asymmetric thickening of the left ventricular wall in a patient with hypertrophic cardiomyopathy. Left ventricular hypertrophy affects principally the anterior ventricular septum (30 mm). AVS=anterior ventricular septum; LA=left atrium
Fig 2.
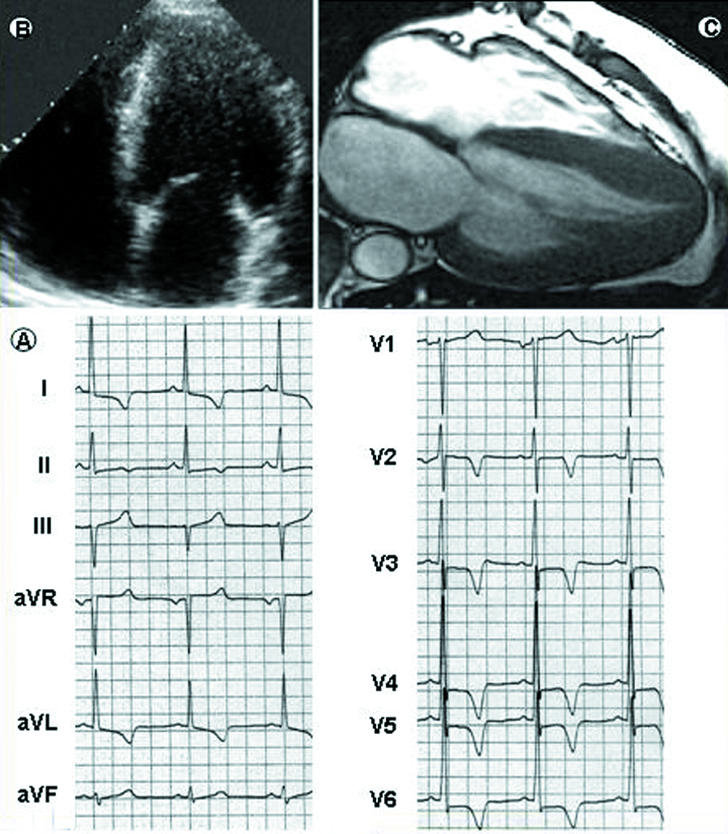
Diagnostic role of cardiac magnetic resonance in hypertrophic cardiomyopathy. In a 33 year old asymptomatic patient, the 12 lead electrocardiogram (bottom left, A) is grossly abnormal, with increased R wave voltages and marked S-T segment alterations in the precordial leads. The two dimensional echocardiogram (top left, B), however, cannot visualise morphological abnormalities and, in particular, does not provide clear images of the apical portion of the left ventricle. Cardiac magnetic resonance (top right, C) shows high resolution images of the heart and marked thickening of the left ventricular wall, which is principally confined to the apical portion of the ventricle. Left ventricular mass is 156 g/m (normal values 83 g/m). The magnetic resonance image is shown courtesy of Massimo Lombardi, MRI Laboratory, Istituto di Fisiologia Clinica, CNR, Pisa, Italy
Summary points
Hypertrophic cardiomyopathy is the most common genetic cardiac disease
It is characterised by marked and asymmetric left ventricular hypertrophy, a non-dilated left ventricular cavity, diastolic impairment, usually preserved systolic function and, in about 20% of patients, left ventricular outflow obstruction at rest caused by mitral-septal contact during systole
Many patients remain asymptomatic throughout life, others develop heart failure or atrial fibrillation, and some die suddenly, often at a young age without previous symptoms
It is the commonest cause of sudden cardiac death in young people and athletes
Stratification of the risk for sudden death is a major management challenge
The cardioverter defibrillator is the only effective treatment for the prevention of sudden death
Medical treatment with β blockers or verapamil improves symptoms of heart failure but has not been shown to modify the clinical course
Patients with outflow obstruction and severe symptoms unresponsive to medical therapy represent about 5% of the patients with hypertrophic cardiomyopathy and are candidates for surgical myectomy or alcohol septal ablation to relieve the outflow obstruction
Paroxysmal or chronic atrial fibrillation develops in an important minority of adult patients
In recent years, DNA analysis has become the definitive method for diagnosis in many genetic diseases. However, hypertrophic cardiomyopathy is caused by mutations in any one of 12 genes encoding proteins of the cardiac sarcomere (http://genetics.med.harvard.edu/~seidman/cg3/).7 Therefore, DNA screening in this disease remains complex, time consuming, mainly confined to a few research laboratories, and is not part of routine clinical evaluation.4 At present, genetic screening identifies the mutation in 50-60% of patients.3,4
Treatment of symptoms
Because hypertrophic cardiomyopathy is a relatively rare condition, and events are uncommon; none of the pharmacological or non-pharmacological treatment strategies is based (and probably will never be based) on randomised controlled trials. Selection of treatment relies principally on retrospective studies and clinical experience.
Pharmacological treatment of heart failure
A large proportion of patients are asymptomatic and may remain free of symptoms throughout life.1-4,8 Since no data indicate that pharmacological therapy may change the course of the disease, treatment is generally not required in low risk asymptomatic patients.1-4 Dyspnoea on exertion represents the most common symptom. Heart failure is caused in large measure by diastolic dysfunction with impaired left ventricular filling, in the presence of preserved systolic function.1-4,9 A dynamic left ventricular outflow obstruction caused by mitral-septal contact during systole, and usually associated with mild or moderate mitral valve regurgitation, is present in about 20% of the patients at rest and develops in a large proportion of patients during effort. Outflow obstruction is associated with an increased risk of heart failure and death.10,11 Chest pain in the absence of coronary atherosclerosis, possibly due to microvascular dysfunction and ischaemia, is also present in an important minority of patients.1-4,12
Pharmacological treatment of heart failure has traditionally been based on the administration of β blockers1-4 (fig 3). By reducing the heart rate, these drugs prolong diastole and improve ventricular filling. β blockers may also decrease the outflow gradient during effort. Verapamil may be used in patients without severe outflow obstruction and is the drug of first choice in patients whose main symptom is chest pain.1-4 In patients with heart failure despite treatment with β blockers or verapamil, the addition of diuretics in low doses is usually effective in alleviating symptoms.1-4 Angiotensin converting enzyme (ACE) inhibitors must be used with caution, because by reducing the afterload may either favour the development or increase outflow obstruction.1-4
Fig 3.

Management of heart failure in hypertrophic cardiomyopathy. (LV=left ventricular; ACE=angiotensin converting enzyme)
About 5% of patients evolve towards the end stage phase of hypertrophic cardiomyopathy, characterised by systolic dysfunction and usually associated with thinning of the left ventricular wall (due to extensive fibrosis) and cavity dilatation.13-15 Such patients are candidates to standard treatment of heart failure secondary to systolic dysfunction, including ACE inhibitors, diuretics, β blockers, and digitalis.1-4,15 Ultimately, many of these patients become candidates for heart transplantation.1-4,15 Because of their high risk for sudden death, a cardioverter defibrillator should be considered as a bridge to transplantation.15
Non-pharmacological treatment of heart failure due to outflow obstruction
Patients with a marked outflow obstruction (outflow gradient 3 50 mm Hg under basal conditions) and severe symptoms (New York Heart Association functional class III or IV) unresponsive to medical therapy represent about 5% of patients with hypertrophic cardiomyopathy and are candidates for either ventricular septal surgical myectomy or percutaneous alcohol septal ablation.1-4,9 In the early 1990s, dual chamber pacing was suggested as a treatment for outflow obstruction. Subsequent randomised studies showed that symptomatic improvement resulted mainly from a placebo effect.2-4
Surgical myectomy has been the gold standard for relieving the outflow gradient over the past 40 years.1,4,9 The operation enlarges the outflow tract through a resection of a small amount of muscle (5-8 g) from the hypertrophied septum. In centres with extensive experience with the procedure, operative mortality is 1-3%.4,9 The operation abolishes the outflow pressure gradient in about 90% of patients and improves symptoms (to functional class I or II) in about 70%.4,9 Two recent and large retrospective studies show that long term mortality in patients who have had the operation is substantially lower than that of patients with outflow obstruction who have not, and similar to that of patients with the non-obstructive form of hypertrophic cardiomyopathy.16,17
Alcohol septal ablation was first developed in the mid 1990s and entails the delivery of alcohol (< 1.5 ml) to the ventricular septum through an angioplasty catheter introduced in a septal branch of the anterior descending artery.4 By causing a myocardial scar in the proximal portion of the septum, the procedure decreases septal thickness and excursion, widening the outflow tract.4,9 Alcohol septal ablation reduces the outflow gradient and improves symptoms in most patients.4,9 Complications include complete atrioventricular block requiring permanent pacemaker implantation (5-20% of patients), large myocardial infarction in areas other than the basal septum, acute mitral valve regurgitation requiring surgery, ventricular fibrillation, and death (2-4% at experienced centres).3,4,9 Therefore, the mortality associated with this technique is not lower than that of surgery. Septal ablation should be performed only at centres with extensive experience with the procedure and hypertrophic cardiomyopathy, to insure a lower rate of complications and proper selection of patients.4 The available follow-up data do not make it possible to rule out that the myocardial scar caused by the procedure may increase the long term risk for sudden death in a myocardium already prone to life threatening tachyarrhythmias. Therefore, it seems prudent to confine the procedure to older patients, or to patients with comorbidities that may substantially increase surgical risk.4
Atrial fibrillation
Paroxysmal or chronic atrial fibrillation develops in about 20% of adult patients with hypertrophic cardiomyopathy and is associated with an increased risk of death due to heart failure.18 From experience in diseases other than hypertrophic cardiomyopathy, amiodarone is considered the most effective drug for prevention of paroxysmal atrial fibrillation.14 β blockers or verapamil, or the two drugs combined, are usually sufficient to control heart rate in patients with chronic atrial fibrillation, but ablation of the atrioventricular node and pacemaker implantation may be necessary in selected patients.14 Experience with catheter ablation of atrial fibrillation is still limited in hypertrophic cardiomyopathy. Oral anticoagulant therapy is indicated in patients with either paroxysmal or chronic atrial fibrillation, because even a single episode of atrial fibrillation is associated with a substantial increase in the risk of systemic embolisation.1-4,18
Risk stratification and prevention of sudden death
Sudden death is the most common modality of death in hypertrophic cardiomyopathy and occurs more commonly in young asymptomatic or only mildly symptomatic patients.1-4 Indeed, hypertrophic cardiomyopathy is the most common cause of sudden cardiac death in athletes.1-4 Therefore, risk stratification has a major role in patient management and has acquired an even greater relevance after the demonstration of the high efficacy of the implantable cardioverter defibrillator in preventing sudden death in this disease.19 The great challenge has thus become to identify the candidates for the device (fig 4). Patients who have survived a cardiac arrest, or experienced one or more episodes of sustained ventricular tachycardia, are considered at high risk and definite candidates to a cardioverter defibrillator for secondary prevention of sudden death.1-4,19-21 These patients, however, represent only a small percentage of the patients with hypertrophic cardiomyopathy.
Fig 4.

Stratification of risk and prevention of sudden death in hypertrophic cardiomyopathy. (LVH=left ventricular hypertrophy; NSVT=non-sustained ventricular tachycardia)
Greater uncertainty persists with regard to the selection of patients for primary prophylactic implantation of a cardioverter defibrillator. Electrophysiological testing has a low prognostic accuracy in hypertrophic cardiomyopathy and does not seem to have a role in risk stratification.4 A large measure of agreement has instead been achieved with regard to several clinical indicators of increased risk. As a general strategy, multiple risk factors are considered to convey a greater likelihood of sudden death.1-4,20 However, a single and strong risk factor may also be an indicator of high risk in selected patients.4
Risk factors
Family history of sudden death
In the United States and most European countries, a family history with two or more premature sudden deaths is considered a justification for implantation of a cardioverter defibrillator.4 Such a family history, however, is uncommon. Much more common is a history of a single premature sudden death. In these patients, the indication to a device is more uncertain, and such a decision is usually based on the identification of additional risk factors. It seems appropriate, however, to inform all the patients with a family history of sudden death regarding the option of the cardioverter defibrillator, as well as the current limitations of risk stratification in hypertrophic cardiomyopathy.
Extreme hypertrophy
Extreme thickness of the left ventricular wall (3 30 mm) is a strong predictor of sudden death in young patients with hypertrophic cardiomyopathy and is associated with an estimated long term risk of sudden death of about 40% at 20 years4,2,2 (fig 1). Because many patients with extreme hypertrophy are young, have no symptoms, and have preserved systolic function, their risk may extend over many decades and prevention of sudden death could allow a near normal duration of life. Therefore, serious consideration should be given to implantation of a cardioverter defibrillator in young patients with extreme hypertrophy, independently of the presence of other risk factors.4,2,2
Unexplained syncope
Syncope represents one of the most challenging clinical presentations, because the mechanisms potentially responsible for syncopal episodes in hypertrophic cardiomyopathy are many, and systematic data regarding the prognostic importance of syncope in this disease are not available. Therefore, management is based on clinical perceptions and experience. In young patients, unexplained (not neurally mediated) syncope at rest or during effort is generally considered a marker of increased risk and a possible indication to a cardioverter defibrillator.1,2,4
Non-sustained ventricular tachycardia
In young patients (≤30 years), brief runs of non-sustained ventricular tachycardia (three or more beats) on Holter monitoring are associated with a significant increase in the risk for sudden death.4,2,3 In such patients, multiple or prolonged runs of non-sustained ventricular tachycardia may be of particular concern and raise the issue of implantation of a cardioverter defibrillator, even in the absence of other risk factors.4
Abnormal blood pressure response to exercise
Hypotensive blood pressure response during upright exercise seems to convey an increased risk for sudden death and may be included in the overall risk profile, particularly in patients younger than 50.4,20
Low clinical risk profile
Patients with mild left ventricular hypertrophy (wall thickness < 20 mm) and without any risk factor can be considered at low risk and have a mean life expectancy similar to that of the general population.4,22
Prophylaxis of infective endocarditis
Infective endocarditis is very uncommon and occurs almost exclusively in patients with outflow obstruction at rest. The vegetations are usually located on the thickened anterior mitral leaflet, less commonly on the aortic valve. The American Heart Association's recommendations for prevention of infective endocarditis should be applied only to patients with obstruction at rest.4,24
Pregnancy
Maternal mortality is very low in hypertrophic cardiomyopathy and seems confined to women with severe heart failure or a high risk for sudden death.4,25 However, in pregnant patients who develop dyspnoea and functional limitation clearly related to hypertrophic cardiomyopathy, treatment with β blockers is advisable, and diuretics may become necessary to limit fluid retention. Most patients with uncomplicated pregnancy may have a normal vaginal delivery.
Lifestyle
Patients with hypertrophic cardiomyopathy should not participate in competitive sports associated with intense exertion or in other strenuous physical activities.26 Patients with a favourable clinical profile—such as no symptoms, a normal or only mildly increased left atrial dimension (< 45 mm), mild left ventricular hypertrophy (< 20 mm), no outflow obstruction under basal conditions, and no risk factors for sudden death—may participate in recreational sports associated with mild to moderate physical activity.27
Additional educational resources for patients and healthcare professionals
http://ww.4hypertrophiccardiomyopathy.org—The website of the Hypertrophic Cardiomyopathy Association (USA), a non-profit organizsation that provides information to patients, their families, and medical providers. No registration is needed
www.mplsheart.com/pages/NewsArticle.asp?ID=3—The website of the Hypertrophic Cardiomyopathy Centre of the Minneapolis Heart Institute Foundation has educational purposes only. No registration is needed
www.cardiomyopathy.org/homepage.htm—This website is designed to provide general information on cardiomyopathies, including hypertrophic cardiomyopathy. No registration is needed
www.mayoclinic.org/hypertrophic-cardiomyopathy—The website of the Mayo Clinic provides general information on hypertrophic cardiomyopathy. It is also a guide for doctors and patients, including a section for appointments. No registration is needed
Sears SF, Shea JB, Conti JB. How to respond to an implantable cardioverter-defibrillator shock. Circulation 2005;111: e380-e382. This educational article reviews patient and family preparations and responses to cardioverter-defibrillator shocks for the purpose of reducing the impact of the device interventions on quality of life.
Family screening
In hypertrophic cardiomyopathy, genetic screening is complex, principally confined to research laboratories, and not part of routine evaluation.4 Clinical screening of first degree relatives is based on electrocardiographic and echocardiographic evaluations and should be encouraged in consideration of the autosomal dominant pattern of disease inheritance. Hypertrophic cardiomyopathy may develop rapidly during adolescence in association with body growth but may first occur also later in life. Therefore, clinical screening should be advised every two years in young family members and about every five years in adults.24
Supplementary Material
 A box on the differential diagnosis of genetic diseases that cause left ventricular wall thickening resembling hypertrophic cardiomyopathy is on bmj.com
A box on the differential diagnosis of genetic diseases that cause left ventricular wall thickening resembling hypertrophic cardiomyopathy is on bmj.com
Contributors: PS and CO worked together in the construction and writing of this review.
Competing interests: None declared.
References
- 1.Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997;336: 775-85. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ. Hypertrophic cardiomyopathy. A systematic review. JAMA 2002;287: 1308-20. [DOI] [PubMed] [Google Scholar]
- 3.Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet 2004;363: 1881-91. [DOI] [PubMed] [Google Scholar]
- 4.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol 2003;42: 1687-13. [DOI] [PubMed] [Google Scholar]
- 5.Moon JCC, McKenna WJ, McCrohon JA, Elliott PM, Smith GC, Pennell DJ. Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J Am Coll Cardiol 2003;41: 1561-7. [DOI] [PubMed] [Google Scholar]
- 6.Carsten R, Norbert MW, Michael JH, Susan AC, Prasad P, Neeta P, et al. Utility of Cardiac Magnetic Resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005;112: 855-61. [DOI] [PubMed] [Google Scholar]
- 7.Seidman JG, Seidman C. The genetic basis for hypertrophic cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 2001;104: 557-67. [DOI] [PubMed] [Google Scholar]
- 8.Spirito P, Chiarella F, Carratino L, Berisso MZ, Bellotti P, Vecchio C. Clinical course and prognosis of hypertrophic cardiomyopathy in an outpatient population. N Engl J Med 1989;320: 749-55. [DOI] [PubMed] [Google Scholar]
- 9.Nishimura RA, Holmes DR Jr. Hypertrophic obstructive cardiomyopathy. N Engl J Med 2004;350: 1320-7. [DOI] [PubMed] [Google Scholar]
- 10.Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348: 295-303. [DOI] [PubMed] [Google Scholar]
- 11.Autore C, Bernabò P, Barillà CS, Bruzzi P, Spirito P. The prognosis importance of left ventricular outflow obstruction in hypertrophic cardiomyopathy varies in relation to the severity of symptoms. J Am Coll Cardiol 2005;45: 1076-80. [DOI] [PubMed] [Google Scholar]
- 12.Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med 2003;349: 1027-35. [DOI] [PubMed] [Google Scholar]
- 13.Harris KM, Spirito P, Maron MS, Zenovich AD, Formisano F, Lesser JR, et al. Prevalence, clinical profile and significance of the end-stage phase in a large hypertrophic cardiomyopathy cohort. Circulation (in press). [DOI] [PubMed]
- 14.Biagini E, Coccolo F, Ferlito M, Perugini E, Rocchi G, et al. Dilated-hypokinetic evolution of hypertrophic cardiomyopathy: prevalence, incidence, risk factors, and prognostic implications in pediatric and adult patients. J Am Coll Cardiol 2005;46: 1543-50. [DOI] [PubMed] [Google Scholar]
- 15.Spirito P, Maron BJ, Bonow RO, Epstein SE. Occurrence and significance of progressive left ventricular wall thinning and relative cavity dilatation in hypertrophic cardiomyopathy. Am J Cardiol 1987;60: 123-9. [DOI] [PubMed] [Google Scholar]
- 16.Woo A, Williams WG, Choi R, Wigle ED, Rozenblyum E, Fedwick K, et al. Clinical and echocardiographic determinants of long-term survival after surgical myectomy in obstructive hypertrophic cardiomyopathy. Circulation 2005;111: 2031-41. [DOI] [PubMed] [Google Scholar]
- 17.Ommen SR, Maron BJ, Olivotto I, Maron MS, Cecchi F, Betocchi S, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005;46: 470-6. [DOI] [PubMed] [Google Scholar]
- 18.Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001;104: 2517-24. [DOI] [PubMed] [Google Scholar]
- 19.Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, Daubert JP, et al. Efficacy of implantable cardioverter defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med 2000;342: 365-73. [DOI] [PubMed] [Google Scholar]
- 20.Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, et al. Sudden death in hypertrophic cardiomopathy: identification of high risk patients. J Am Coll Cardiol 2000;36: 2212-8. [DOI] [PubMed] [Google Scholar]
- 21.Boriani G, Maron BJ, Shen WK, Spirito P. Prevention of sudden death in hypertrophic cardiomyopathy: but which defibrillator for which patient? Circulation 2004,110: 438-42. [DOI] [PubMed] [Google Scholar]
- 22.Spirito P, Bellone P, Harris KM, Bernabò P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med 2000;342: 1778-85. [DOI] [PubMed] [Google Scholar]
- 23.Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J Am Coll Cardiol 2003;42: 873-9. [DOI] [PubMed] [Google Scholar]
- 24.Spirito P, Rapezzi C, Bellone P, Betocchi S, Autore C, Conte MR, et al. Infective endocarditis in hypertrophic cardiomyopathy. Prevalence, incidence, and indications for antibiotic prophylaxis. Circulation 1999;99: 2132-7. [DOI] [PubMed] [Google Scholar]
- 25.Autore C, Conte MR, Piccininno M, Bernabò P, Bonfiglio G, Bruzzi P, et al. Risk associated with pregnancy in hypertrophic cardiomyopathy. J Am Coll Cardiol 2002;40: 1864-9. [DOI] [PubMed] [Google Scholar]
- 26.Maron BJ, Isner JM, McKenna WJ. Bethesda conference: recommendations for determining eligibility for competition in athletes with cardiovascular abnormalities. Task force 3: hypertrophic cardiomyopathy, myocarditis and other myopericardial diseases and mitral valve prolapse. J Am Coll Cardiol 1994;24: 880-5. [DOI] [PubMed] [Google Scholar]
- 27.Maron BJ, Chaitman BR, Ackerman MJ, Bayés de Luna A, Corrado D, Crosson JE, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004;109: 2807-16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.