

Abstract
Type III interferons (IFNs) (interleukin-28/29 or lambda interferon [IFN-λ]) are cytokines with IFN-like activities. Here we show that several classes of viruses induce expression of IFN-λ1 and -λ2/3 in similar patterns. The IFN-λs were—unlike alpha/beta interferon (IFN-α/β)—induced directly by stimulation with IFN-α or -λ, thus identifying type III IFNs as IFN-stimulated genes. In vitro assays revealed that IFN-λs have appreciable antiviral activity against encephalomyocarditis virus (EMCV) but limited activity against herpes simplex virus type 2 (HSV-2), whereas IFN-α potently restricted both viruses. Using three murine models for generalized virus infections, we found that while recombinant IFN-α reduced the viral load after infection with EMCV, lymphocytic choriomeningitis virus (LCMV), and HSV-2, treatment with recombinant IFN-λ in vivo did not affect viral load after infection with EMCV or LCMV but did reduce the hepatic viral titer of HSV-2. In a model for a localized HSV-2 infection, we further found that IFN-λ completely blocked virus replication in the vaginal mucosa and totally prevented development of disease, in contrast to IFN-α, which had a more modest antiviral activity. Finally, pretreatment with IFN-λ enhanced the levels of IFN-γ in serum after HSV-2 infection. Thus, type III IFNs are expressed in response to most viruses and display potent antiviral activity in vivo against select viruses. The discrepancy between the observed antiviral activity in vitro and in vivo may suggest that IFN-λ exerts a significant portion of its antiviral activity in vivo via stimulation of the immune system rather than through induction of the antiviral state.
Type I interferons (IFNs) (alpha/beta interferon [IFN-α/β]) are expressed as a first line of defense against viruses and are known to play a critical role in the antiviral response (38). Type I IFNs combat viruses both directly by inhibiting virus replication in the cells and indirectly by stimulating the innate and adaptive immune responses (38). The direct antiviral activity of type I IFNs is exerted by a number of different mechanisms, e.g., blockage of viral entry into the cell, control of viral transcription, cleavage of RNA, and preventing translation (16, 31, 37). In addition to the direct effects, type I IFNs play immunoregulatory roles and thereby shape the innate and adaptive immune responses. For instance, IFN-α/β induce natural killer cell cytotoxicity and up-regulate expression of major histocompatibility complex class I on most cells and costimulatory molecules on antigen-presenting cells (9, 37). Furthermore, type I IFNs enhance cross-presentation of exogenous antigen in major histocompatibility complex class I and promote T-cell expansion (14, 19, 36).
Type III IFNs (lambda interferon [IFN-λ] or interleukin-28/29 [IL-28/29]), which were discovered recently (20, 32), display IFN-like activities (11, 15, 20, 23, 32), although they exert their action through a receptor complex distinct from the type I IFNs (20, 32). Most of the reports demonstrating antiviral activity of IFN-λ have addressed the issue in an in vitro experimental setup, but one report has shown that a recombinant IFN-λ-expressing vaccinia virus is attenuated in vivo (4), whereas recombinant IFN-λ had no antiviral effect in vivo in the transgenic hepatitis B virus mouse model (30). Thus, we still do not have a clear picture of the antiviral potential of IFN-λ in vivo or of the mechanisms of action.
The IFN-λs have been demonstrated to be induced after stimulation with several single-stranded RNA (ssRNA) viruses, whereas the information on viruses with other genomes (DNA and double-stranded RNA [dsRNA]) is sparse (11). Virtually all cell types are capable of producing type I IFNs in response to viral infections, with the amount of IFN being virus and cell type dependent (7) and with plasmacytoid dendritic cells (pDCs) being the most potent producers of type I IFNs (3). IFN-λs can be produced by a number of cell types, although the pattern of expression has not been elucidated. One report has demonstrated that IFN-λs are produced by pDCs to a greater extent than by monocyte-derived DCs after influenza A virus (IAV) infection, suggesting that pDCs are the primary IFN-λ-producing cells (13). However, this needs to be confirmed for other virus infections.
Here, we have investigated the expression of type I and III IFNs after infection with DNA and RNA viruses in lymphoid, myeloid, and epithelial cell lines, and we have also examined the ability of type I and III IFNs to cross-induce one another. Subsequently, we investigated the antiviral activity of IFN-λ in vitro. In addition, we have evaluated the antiviral activity of recombinant IFN-λ in vivo using three different mouse models for general infection and one model for local infection. Our data show that IFN-λs are induced to variable extents by viruses of all classes of genomes in the cell types examined. Unlike type I IFNs, which do exhibit some degree of cell type-specific expression, with IFN-β being expressed primarily in nonimmune cells and IFN-α primarily in leukocytes, IFN-λ1 and IFN-λ2/3 are expressed in identical patterns in both immune and nonimmune cells. Interestingly, IFN-λ is induced by both type I and type III IFNs; therefore, this class of cytokines belongs to the group of IFN-stimulated genes (ISGs). Finally, we show that recombinant IFN-λ exhibits antiviral activity against the DNA virus herpes simplex virus type 2 (HSV-2), but not the RNA viruses encephalomyocarditis virus (EMCV) and lymphocytic choriomeningitis virus (LCMV), and that IFN-λ treatment enhances HSV-induced expression of IFN-γ, a cytokine known to participate in the response against this virus.
MATERIALS AND METHODS
Reagents.
The growth media used were minimal essential medium (MEM), Dulbecco modified Eagle medium (DMEM), Iscoves medium, and RPMI 1640 medium (all from BioWhittaker), which were supplemented with antibiotics (penicillin [200 IU/ml] and streptomycin [200 μg/ml]) and lipopolysaccharide-free fetal calf serum (FCS; BioWhittaker) at the indicated concentrations. TRIzol was from Invitrogen. Recombinant human IFN-α and IFN-λs were from PBL Biomedical Laboratories and R&D Systems, respectively. Bovine serum albumin and dimethylthiazolyldiphenyltetrazolium bromide (MTT) were from Sigma-Aldrich, Depo-Provera was from Pfizer, and oligodeoxynucleotide (ODN) 1826 was obtained from InvivoGen.
Viruses and cell lines.
The viruses used were HSV-2 (MS and 333 strains), reovirus type 1 (Reo) (Lang strain), Sendai virus (SeV) (Cantrell strain), EMCV (FA strain), LCMV (Armstrong strain), and IAV (strain A/Beijing/353/89 H3N2). The two HSV-2 strains, EMCV, and Reo were grown on monolayers of Vero cells to complete cytopathic effect (HSV-2 and EMCV) or until all cells were affected by the infection as determined by microscopy (Reo) and prepared by two cycles of freezing and thawing, followed by centrifugation for 30 min at 5,000 × g for removal of cellular debris. LCMV strain Armstrong was produced in BHK cells, and cell culture supernatant was harvested 48 h after infection at a low multiplicity of infection (MOI). SeV and IAV were kind gifts from Sampsa Matikainen and Illka Julkunen (National Public Health Institute, Helsinki, Finland). The virus was produced as described previously (29). Given the methods used for preparation of virus stocks, they presumably contained both infectious and defective virions. However, since UV inactivation of the preparations abolished the abilities to induce IFN expression (data not shown), it seems that the IFN response we observe was triggered by the infectious virus particles. The titers of the different virus stocks were as follows: HSV-2 (MS), 3 × 107 PFU/ml; HSV-2 (333), 1 × 107 PFU/ml; EMCV, 4 × 107 PFU/ml; Reo, 3 × 106 PFU/ml; LCMV, 2 × 107 PFU/ml; IAV, 128; and SeV, 6,000 (the last two were determined by hemagglutination titer).
The human cell lines used were hepatocyte carcinoma HepG2, lung carcinoma A549, lymphoma Raji cells, cervix epitheloid carcinoma HeLa cells, and the histiocytic lymphoma cell line U937. HepG2 and A549 cells were cultured in DMEM supplemented with 10% FCS, and HeLa cells were cultured in MEM supplemented with 10% FCS. The adherent cells were passaged by routine trypsinization procedures. Raji and U937 cells were cultured in RPMI 1640 medium supplemented with 10% FCS. The RPMI 1640 medium used for culturing the U937 cells was furthermore supplemented with 0.0025 g/ml glucose and 0.002 mM/ml sodium pyruvate.
Mice, infection, and in vivo treatment.
Female C57BL/6 and IFNAR−/− mice were purchased from Taconic M&B (Denmark) and B&K Universal Ltd. (United Kingdom), respectively. The mice were infected intraperitoneally (i.p.) with 1 × 106 PFU of HSV-2 (strain MS) and 1 × 103 PFU of EMCV, intravenously (i.v.) with 1 × 103 PFU of LCMV, and intravaginally (i.vag.) with 6.7 × 104 PFU HSV-2 (strain 333). For i.p. infection, 3- to 4-week-old animals were used, whereas for i.v. and i.vag. infections, the animals were 6 to 8 weeks old. Six hours prior to systemic infection, mice were injected i.p. (EMCV or HSV-2 infection) or i.v. (LCMV infection) with 10 μg of recombinant murine IFN-α (HyCult Biotechnology; 10,000 IU) or recombinant murine IFN-λ2 (R&D Systems) or with 100 μl of vehicle control phosphate-buffered saline (PBS). Forty-eight hours after infection, the mice were sacrificed, and the livers (HSV-2), hearts (EMCV), or spleens (LCMV) were harvested and frozen at −70°C for subsequent virus titration.
Intravaginal treatment with IFNs, CpG ODN, and HSV-2.
All mice were pretreated by subcutaneous injection of 2 mg Depo-Provera. Four days later and 24 h prior to vaginal HSV-2 infection, one group of mice was anesthetized with Isofluran (Baxter) and received 25 μg of CpG ODN 1826 i.vag. in 20 μl PBS. Six hours prior to infection, three groups of mice were anesthetized and treated i.vag. with either 20 μl of PBS or with 5 μg of recombinant murine IFN-α or murine IFN-λ2 in 20 μl PBS. For HSV-2 infection, all mice were anesthetized with Isofluran and inoculated i.vag. with a lethal dose (6.7 × 104 PFU) of HSV-2 strain 333 in 20 μl Iscoves modified medium while they were placed on their backs and maintained under anesthesia for at least 10 min. Vaginal fluid samples were collected daily after infection (days 1 to 3) by two washes (in each wash, 40 μl of Iscoves medium was pipetted into and out of the vagina 12 to 15 times). The two washes were pooled, and viral titers were determined by plaque assay on Vero cell monolayers.
Inflammation and disease scoring.
Genitally infected mice were examined daily for vaginal inflammation, neurological illness, and death. The severity of disease was graded using the following scores: 0, healthy; 1, genital erythema; 2, moderate genital inflammation; 3, purulent genital lesion and/or generally bad condition; 4, hind limb paralysis; 5, death or sacrifice due to hind limb paralysis.
Antiviral assay.
Antiviral assays were done on HepG2 cells, which were seeded in DMEM supplemented with 10% FCS at a density of 1.5 × 104 in 96-well plates and left to settle. The cells were incubated with successive 10-fold dilutions of IFNs for 24 h before challenge with 10-fold dilutions of EMCV or HSV-2. The cells were incubated with virus for 48 h (EMCV) or 96 h (HSV-2). The medium was removed between each step. The viability of the cells was analyzed by a bioassay based on the dehydrogenase system; this system in intact cells will convert the substrate, MTT, into formazan (blue), which in turn can be measured spectrophotometrically. Briefly, the cells were given MTT and incubated for 2 h. An extraction buffer (containing 6 to 11% sodium dodecyl sulfate and 45% N,N-dimethylformamide) was added to the cells, and the cells were then incubated overnight at 37°C. Subsequently, the absorbance at 570 nm was determined employing the extraction buffer as the blank probe. A570 was directly proportional to antiviral activity (6).
Quantitative reverse transcription-PCR.
Total RNA was extracted with TRIzol according to the recommendations of the manufacturer. Briefly, cells were lysed in TRIzol, and chloroform was added, followed by phase separation by centrifugation. RNA was precipitated with isopropanol and pelleted by centrifugation. Pellets were washed with 80% ethanol and redissolved in RNase-free water. For cDNA generation, 1 μg or 2 μg of RNA was subjected to reverse transcription with oligo(dT) as a primer and Expand reverse transcriptase (both from Roche). Prior to quantitative reverse transcription-PCR, RNA was treated with DNase I (Ambion) to remove any contaminating DNA, the absence of which was confirmed in control experiments in which the reverse transcriptase enzyme was omitted (data not shown). The cDNA was amplified by PCR using the primers shown in Table 1, and the products were measured by the use of SYBR green I (from QIAGEN). The data were normalized to β-actin and presented as the change in induction relative to that of untreated control cells.
TABLE 1.
Primers used for quantitative RT-PCR
| Target gene | Primera | Nucleotide sequence | GenBank accession no. |
|---|---|---|---|
| OAS1 (human) | F | 5′-GCCCTGGGTCAGTTGACTGG-3′ | NM_016816 |
| R | 5′-TGAAGCAGGTGGAGAACTCGC-3′ | ||
| ISG56 (human) | F | 5′-CAGCAACCATGAGTACAAAT-3′ | NM_001548 |
| R | 5′-AAGTGACATCTCAATTGCTC-3′ | ||
| IFN-α (human) | F | 5′-AAATACAGCCCTTGTGCCTGG-3′ | NM_000605 |
| R | 5′-GGTGAGCTGGCATACGAATCA-3′ | ||
| IFN-β (human) | F | 5′-AAGGCCAAGGAGTACAGTC-3′ | NM_002176 |
| R | 5′-ATCTTCAGTTTCGGAGGTAA-3′ | ||
| IFN-λ1 (human) | F | 5′-CGCCTTGGAAGAGTCACTCA-3′ | NM_172140 |
| R | 5′-GAAGCCTCAGGTCCCAATTC-3′ | ||
| IFN-λ2/3 (human) | F | 5′-AGTTCCGGGCCTGTATCCAG-3′b | NM_172138 |
| 5′-GAGCCGGTACAGCCAATGGT-3′b | |||
| 5′-ACTGCAGCCACTCCC-3′c | |||
| 5′-CTCCAGAACCTTCAGCGTCAG-3′c | |||
| PKR (human) | F | 5′-ACACTCGTCTCTGAATCATC-3′ | NM_002759 |
| R | 5′GAGACCATTCATAAGCAACG-3′ | ||
| β-Actin (human) | F | 5′-TCATCACCATTGGCATGAG-3′ | X00351, J00074, and M10278 |
| R | 5′-AGCACTGTGTTGGCGTACAG-3′ | ||
| IRF-7 (murine) | F | 5′-CCCAGACTGCCTGTGTAGACG-3′ | NM_016850 |
| R | 5′-CCAGTCTCCAAACAGCACTCG-3′ | ||
| ISG15 (murine) | F | 5′-AGCAATGGCCTGGGACCTAAA-3′ | NM_015783 |
| R | 5′-AGCCGGCACACCAATCTT-3′ | ||
| β-Actin (murine) | F | 5′-GCTCCCCGGGCTGTATTCC-3′ | NM_007393 |
| R | 5′-CTCTCTTGCTCTGGGCCTCGT-3′ |
The primers are identified as forward (F) and reverse (R).
Used in experiment with A549 cells.
Used in experiments with HeLa, Raji, U937, and HepG2 cells.
Virus yield assay.
Organ samples were weighed, thawed, and homogenized in MEM supplemented with 2% FCS just before use. The homogenates were pelleted by centrifugation at 1,600 × g for 30 min, and supernatants were used for plaque assay. Vaginal washes were thawed and centrifuged for 10 min at 4,500 × g, and the supernatants were used for plaque titration. HSV-2 and EMCV titers were determined on monolayers of Vero cells seeded in MEM supplemented with 5% FCS; HSV-2 was titrated on 20-cm2 tissue culture plates with the cells seeded at a density of 1.0 × 106 cells, whereas EMCV was titrated in 96-well plates with the cells seeded at a density of 1.0 × 104 to 1.4 × 104 per well. The cells were left overnight to settle and infected by incubation for 1 h at 37°C with serial dilutions of organ suspensions and vaginal washes. For HSV-2 titration, the plates were rocked every 15 min to ensure even distribution of virus. After 1 h, 8 ml of medium containing 2% human immunoglobulin was added, and the plates were incubated for 2 days. For liver titration, the organ suspensions were removed before adding immunoglobulin-containing media. After incubation, the plates were stained with 0.03% methylene blue to allow quantification of plaques. For EMCV titration, the first two rows of the 96-well plates containing the most concentrated heart cell suspensions were removed after 1 h and replaced by 100 μl medium. After 4 days, the medium was removed, the cells were fixed with 25 μl of 10% paraformaldehyde for 30 min, and stained with crystal violet for 15 min. The virus titer was determined using the Reed-Muench method. For determinations of LCMV titers in organs, the organs were first homogenized in PBS to yield 10% (vol/wt) organ cell suspensions, and serial 10-fold dilutions were prepared. Each dilution was then plated in duplicate on MC57G cells. Forty-eight hours after infection, infected-cell clusters were detected using rat anti-LCMV (VL-4) monoclonal antibody, peroxidase-labeled goat anti-rat antibody, and o-phenylendiamine (substrate) (5). The numbers of PFU were counted, and results were expressed as PFU/gram of tissue.
Luminex.
Expression of IL-6 and IFN-γ was measured by Luminex Technology, using a 10-plex murine cytokine kit obtained from Biosource. Briefly, the filter plate was washed with assay buffer, and 50 μl of freshly vortexed antibody-conjugated beads was added to each well. The plate was washed with assay buffer, and samples and standards were added. After a brief shake (30 seconds at 1,100 rpm), the plate was incubated at room temperature in the dark for 2 h with light shaking (300 rpm). After one wash step, 25 μl of the detection antibody was added to each well, and the plate was shaken and incubated as described above. Subsequently, the plate was washed and incubated for 30 min with 50 μl of a streptavidin-phycoerythrin solution with shaking (30 s at 1,100 rpm and then 10 min at 300 rpm). Finally, the plate was washed, 125 μl of assay buffer was added to each well, and the plate was shaken for 10 s at 1,100 rpm and read immediately on the Bio-Plex machine.
Statistical analysis.
The data are presented as means ± standard deviations (SD). The statistical significance was estimated with the Wilcoxon rank sum test. P values of less than 0.05 were considered statistically significant.
RESULTS
Characterization of IFN-λ expression during viral infections.
In order to examine the production of IFN-λ in different cell lines in response to viral challenge, we infected the following human cell lines: lung carcinoma A549, cervix epitheloid carcinoma HeLa, lymphoma Raji, and histiocytic lymphoma U937, representing lymphoid cells, myeloid cells, and nonimmune cells. The cells were infected with different viruses representing different types of viruses: HSV-2 (dsDNA), EMCV (plus-strand ssRNA [+ssRNA]), SeV (minus-strand ssRNA [÷ssRNA]), Reo (dsRNA), and IAV (segmented ÷ssRNA). The cells were challenged for 6 or 24 h, and expression of IFN-α, IFN-β, IFN-λ2/3, or IFN-λ1 genes was determined by quantitative PCR. As seen in Fig. 1, the RNA viruses induced a greater expression of IFNs in all the cell lines examined, with SeV being the most potent inducing up to 106-fold induction. This underscores the potent IFN-inducing potential of most RNA viruses, although it ought to be mentioned that such strong induction has been found in some (13), but not all, studies with RNA viruses. The DNA virus HSV-2 did elicit expression of all the IFNs examined but generally not to the same degree as the RNA viruses did. As expected, IFN-β was highly up-regulated in the epithelium-like cell lines at both examination time points, whereas IFN-α was induced to a lower extent in these cells (Fig. 1A and B). We found up-regulation of IFN-α in Raji cells especially at the late time point without observing any significant expression of IFN-β (Fig. 1C), whereas IFN-α and IFN-β were induced to comparable extents by a given virus in the monocyte-like cell line U937 (Fig. 1D). Interestingly, expression of both IFN-λ1 and IFN-λ2/3 was induced in cells primarily producing either IFN-α or IFN-β, and this up-regulation was seen at both time points. These data suggest that unlike type I IFNs, which display some degree of cell type specificity with respect to the pattern of expression, the IFN-λs are more uniformly expressed by all cells in response to virus infection. Moreover, not only were IFN-λs up-regulated in all the cell lines examined, but we also found that IFN-λs were induced to the same degree as IFN-α and IFN-β were.
FIG. 1.

Expression of IFN-β, IFN-α, IFN-λ2/3, and IFN-λ1 in A549 cells (A), HeLa cells (B), Raji cells (C), and U937 cells (D) after stimulation with HSV-2, EMCV, SeV, Reo, and IAV. The cells were seeded and infected with virus at a MOI of 1. Total cellular RNA was extracted 6 or 24 h after viral challenge, and gene expression was analyzed by real-time PCR. For A549 and HeLa cells, which are permissive for HSV-2 and EMCV, results could not be obtained at the late time points due to cytopathic effects. Results are shown as means ± SD (error bars). ND, not done.
Expression of ISGs and IFN-α, IFN-β, or IFN-λs after stimulation with IFN.
IFNs can induce expression of a large number of genes termed ISGs, leading to induction of the antiviral state and other IFN functions (8, 28). We wanted to examine the kinetics of some of the ISGs and at the same time see whether we could detect any cross-induction of the IFNs. To do this, we stimulated HepG2 cells with IFN-α, IFN-λ1, and IFN-λ2/3, and after 2, 4, 6, 8, 12 and 24 h, cells were lysed for RNA purification and quantitative PCR. As seen in Fig. 2A to C, the ISGs, ISG56, serine/threonine protein kinase (PKR), and 2′-5′oligoadenylate synthetase (OAS) were all induced after 2 hours of stimulation, and the levels remained high throughout all the time points examined. We did not detect any significant differences in the levels of induction or in the pattern of induction after stimulation with IFN-α, IFN-λ1, or IFN-λ2.
FIG. 2.

Expression of ISGs and IFNs in HepG2 cells after stimulation with IFN-λ1, IFN-λ2, or IFN-α. The cells were seeded and treated with 125 ng/ml of IFN-λ1, -λ2, or -α (i.e., ∼125 IU/ml IFN-α). Total cellular RNA was extracted 2, 4, 6, 8, 12, or 24 h after stimulation, and expression of (A) ISG56, (B) OAS, (C) PKR, (D) IFN-β, (E) IFN-α, (F) IFN-λ2/3, and (G) IFN-λ1 was analyzed by real-time PCR. For a positive control, RNA was harvested from A549 cells infected for 6 h with SeV. The results are shown as means ± SD (error bars).
When we examined the induction of IFN-α or IFN-β after stimulation with IFN-α, IFN-λ1, or IFN-λ2, no up-regulation at all could be detected (Fig. 2D and E). However, when examining the induction of IFN-λ2/3, we found a 10- to 100-fold induction at the late time points (Fig. 2F). This was observed after stimulation with both IFN-α and IFN-λs. We observed the same phenomenon for IFN-λ1 and with an even stronger induction—up to 1,000-fold (Fig. 2G).
Thus, IFN-λ and IFN-α induce PKR, ISG56, and OAS to the same extent in the cell line examined. However, in contrast to type I IFNs, which are not themselves ISGs, IFN-λ1 and IFN-λ2/3 are cross-induced by both types of IFNs.
Antiviral activity during EMCV and HSV-2 infection in vitro.
As already reported by others, IFN-λ exerts antiviral activity against the +ssRNA virus EMCV as well as other RNA viruses (20, 23, 26, 30, 32); however, there are limited reports concerning the antiviral activity against DNA viruses. Therefore, we stimulated HepG2 cells with different concentrations of IFN-λ1, IFN-λ2/3, or IFN-α, ranging from 0 to 1,000 ng/ml of the cytokine. Subsequently, the cells were challenged with either HSV-2 or EMCV in a dose-dependent manner ranging from 102 to 105 PFU, and prevention of virus-mediated cytotoxicity was evaluated as a measure of IFN activity. As seen in Fig. 3A and confirming previous reports (32), IFN-α, IFN-λ1, and IFN-λ2/3 exert antiviral activity against EMCV challenge in a dose-dependent manner, with IFN-α being more potent than IFN-λ1, which again is more potent than IFN-λ2. When we challenged the cells with HSV-2, we found that IFNs were generally less effective in preventing virus-induced cytotoxicity compared to challenge with EMCV (Fig. 3B). High doses of IFN-α protected the cells after infection with low doses of virus, but this effect was titrated out with increasing doses of virus. IFN-λ1 displayed modest antiviral activity against low doses of HSV-2, and the activity of IFN-λ2 was even lower. Thus, IFN-λ has antiviral activity against EMCV in vitro but appears to be not as efficient as IFN-α, and it is not able to protect cells from virus-induced cytotoxicity after HSV-2 infection. When comparing type I and III IFNs, it should be mentioned that since no internationally agreed scale for specific activity is available for IFN-λ, one should be very cautious not to draw too bold conclusions, since observed differences may be due to differences in specific activities of the preparations.
FIG. 3.

Antiviral activity of type I and III IFNs against EMCV and HSV-2. (A-B) IFN-λ2, IFN-λ1, and IFN-α were added at the indicated doses (for IFN-α, 1 ng is ∼1 IU) to HepG2 cells 24 h prior to challenge with EMCV (A) or HSV-2 (B). Forty-eight and 96 h after infection with EMCV and HSV-2, respectively, cells were assayed for viability with a bioassay. The results are shown as means ± SD (error bars). A570 values were directly proportional to cell viability and therefore antiviral activity of the respective IFNs. IFN-α treatment without viral challenge was used as a baseline of the viability of the cells (n = 4 to 12). (C-D) Virus yield assay. The cells were treated with 103 ng/ml of IFN-λ2, IFN-λ1, and IFN-α and infected 24 h later at a MOI of 0.015 (103 PFU/ml) or a MOI of 0.15 (104 PFU/ml) with EMCV (C) or HSV-2 (D). Cell culture supernatants were harvested 12 h (C) and 24 h (D) later and assayed for infectious virus by titration on Vero cells. The results are shown as mean viral titers ± SD (error bars). (E-F) Antiviral effects of cotreatment with type I and III IFNs. HepG2 cells were seeded and treated with the indicated doses of IFNs 24 h prior to infection with 105 PFU/ml of EMCV (E) and 104 PFU/ml of HSV-2 (F). Forty-eight and 96 h after infection with EMCV and HSV-2, respectively, cells were assayed for viability with a bioassay. The results are shown as means ± SD (error bars).
The MTT assay shown in Fig. 3A and B detects virus-mediated cell death, but we also wanted a direct measure of how the IFNs affected production of infectious progeny virus. Therefore, HepG2 cells were infected with two doses of EMCV or HSV-2, and supernatants were harvested 24 h later for measurement of virus yield. Replication of EMCV was reduced to a level below the limit of detection by IFN-α, whereas the IFN-λs exerted a significant but less pronounced antiviral effect towards EMCV (Fig. 3C). For HSV, we observed that IFN-α totally prevented replication of the virus when infected with a MOI of 0.015 and that it reduced the recovery of virus from about 1 × 105 PFU to about 1 × 103 PFU when infected with a MOI of 0.15 (Fig. 3D). The type III IFNs also reduced virus replication but not to the same extent as type I IFN. At both viral doses, the IFN-λs reduced virus yield about 1 log unit from about 5 × 103 PFU to 5 × 102 PFU (MOI, 0.015) and from about 1 × 105 PFU to 1 × 104 PFU (MOI, 0.15), respectively.
We were also interested in knowing whether type I and III IFNs acted in a redundant, cooperative, or synergistic manner to induce antiviral defense in vitro. Therefore, HepG2 cells were treated with 1 or 10 ng/ml of IFN-α together with increasing doses of IFN-λ1 or -λ2 prior to infection with EMCV or HSV-2. Antiviral activity was evaluated using the MTT assay. Treatment with 10 ng/ml of IFN-α alone gave nearly total protection from EMCV-induced cytotoxicity, whereas 1 ng/ml of IFN-α had only a marginal antiviral effect (Fig. 3E). Cotreatment of the cells receiving 1 ng/ml of IFN-α with increasing doses of IFN-λ resulted in a dose-dependent protection against cell death but with no synergistic or additive antiviral effect compared to cells receiving IFN-λ1 or -λ2 only. For HSV-2, 1 ng/ml and 10 ng/ml of IFN-α exhibited marginal and partial protection against cytotoxicity, respectively (Fig. 3E). At the low dose of IFN-α, we observed a cooperative antiviral effect of cotreatment with IFN-λ1 or -λ2, which was additive rather than synergistic. At the high dose of IFN-α, this phenomenon was observed only for cells receiving 100 ng/ml of IFN-λ1.
IFN-λ protects mice from select systemic viral infections.
In order to evaluate the antiviral activity of IFN-λ in vivo, we turned to the murine system. We used three different mouse models of systemic viral infection to test whether IFN-λ or IFN-α treatment could protect the mice from viral infection: C57BL/6 mice were infected i.p. with EMCV or HSV-2 and i.v. with LCMV. Six hours prior to infection, the mice had been treated with either PBS, IFN-α, or IFN-λ by the same route as used for infection. This treatment led to comparable induction of ISGs by IFN-α and IFN-λ in the target organs (data not shown). IFN-α treatment resulted in a statistically significant reduction of the viral load in organs from animals infected with either EMCV, LCMV, or HSV-2 compared to PBS-treated controls (Fig. 4A and B). In examining the antiviral activity of IFN-λ, we found no significant reduction in viral load in organs from EMCV- or LCMV-infected mice treated with this cytokine. By contrast, in HSV-2-infected mice, we saw a significant, approximately 10-fold, reduction in the hepatic virus titer after IFN-λ treatment compared with PBS controls 48 h postinfection (Fig. 4C). The anti-HSV activity of IFN-λ was not dependent on a functional type I IFN receptor system, since it was also observed in IFNAR−/− mice (Fig. 4D).
FIG. 4.
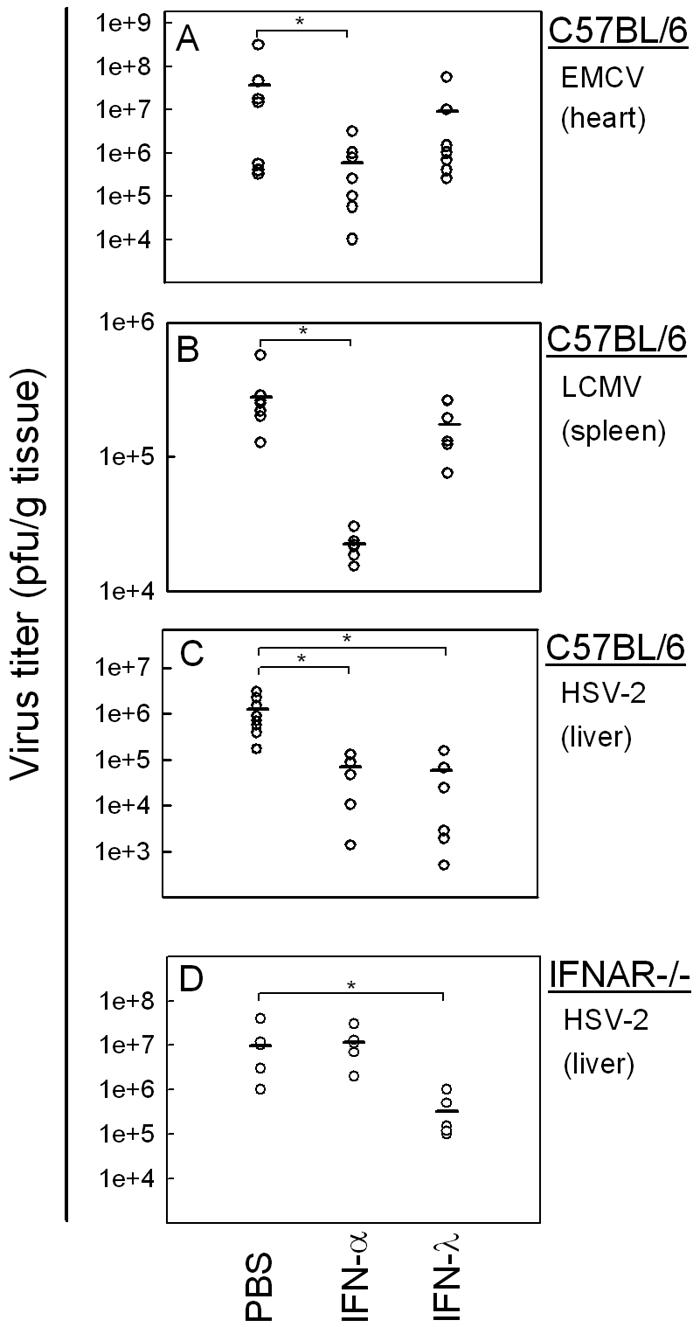
Viral loads in organs from C57BL/6 (A-C) or IFNAR−/− mice (D) after infection with (A) EMCV infection (hearts), (B) LCMV (spleens), and (C-D) HSV-2 (livers). Six hours prior to infection, the animals were treated with either PBS or with 10 μg of IFN-α (10,000 IU) or IFN-λ. The animals were infected i.p with EMCV (1 × 103 PFU) or HSV-2 (1 × 106 PFU) or i.v. with LCMV (1 × 103 PFU). After 48 h of infection, the mice were sacrificed and organs were harvested for determination of viral load. The results are shown as virus titer for individual animals. Statistically significant differences in viral load evoked by IFN treatment (P < 0.05) are marked by asterisks (n = 5 to 8). Mean values are shown as horizontal lines.
We wanted to make sure that a possible antiviral effect of IFN-λ against EMCV in vivo was not overshadowed by endogenous IFN-α/β production. Therefore, IFNAR−/− mice were treated with IFN-α or IFN-λ and infected as described above. However, even in the absence of the type I IFN receptor system, IFN-λ was not able to protect the EMCV-infected animals (data not shown). Taken together, our results show that IFN-λ treatment of mice has antiviral effect against systemic HSV-2, but not EMCV or LCMV, infection.
IFN-λ protects mice from genital herpes infection and disease progression.
To further investigate the anti-HSV-2 activity of IFN-λ in vivo, we used a mouse model of genital herpes infection. CpG DNA has previously been shown to protect mice from genital herpes infection (1, 17) and was used as a positive control. Female C57BL/6 mice were treated i.vag. with CpG DNA 24 h prior to infection or with IFN-α, IFN-λ, or PBS 6 h prior to infection. In accordance with previous work by others (1, 17), CpG DNA totally prevented viral replication (Fig. 5A). IFN-α also displayed antiviral activity but not to the same extent as CpG DNA did. IFN-λ treatment, on the other hand, was as potent as CpG DNA in preventing vaginal virus replication and led to the complete absence of detectable virus in the vaginal mucosa throughout the 3 days of the experiment. Furthermore, IFN-λ-treated mice were entirely protected from disease development up to 20 days after infection (Fig. 5B), whereas mice receiving IFN-α did exhibit some degree of disease development. These results show a highly protective effect of IFN-λ against vaginal herpes, an effect which was comparable to treatment with CpG DNA and superior to IFN-α treatment.
FIG. 5.

Viral load and disease progression after vaginal HSV-2 infection in C57BL/6 mice. Five days prior to infection, the mice were treated with Depo-Provera. Six or 24 h before infection, the mice were treated with 5 μg of IFN-α (5,000 IU), IFN-λ, PBS, or CpG DNA. The animals were infected i.vag. with 6.7 × 104 PFU of HSV-2, and vaginal washes were performed at the indicated time points postinfection. (A) Viral load in vaginal washes 24, 48, and 72 h after infection. The results are shown as mean values ± SD (error bars). (B) Development of inflammation and disease up to 20 days after infection. Disease scores are explained in detail in Materials and Methods, but a disease score of 0 was given to a healthy mouse and a score of 5 was given for death or sacrifice of a mouse due to hind limb paralysis. The results are shown as mean values (n = 5).
IFN-λ treatment elevates the serum levels of IFN-γ during HSV infection.
Experiments in vitro have shown that pretreatment of human macrophages with IFN-α augments the inflammatory response induced by viruses and Toll-like receptor ligands (34), and we wanted to investigate whether IFN-λ may also affect this response. Therefore, we treated mice with PBS, IFN-α, or IFN-λ for 12 h and subsequently challenged the mice i.p. with HSV-2 for another 6 h before harvest of serum. Cytokine levels were measured by Luminex. As seen in Fig. 6A, the IFNs only marginally affected the levels of IL-6, whereas HSV-2 infection led to a significant elevation of IL-6 levels. Pretreatment with IFN-α further elevated production of IL-6, whereas IFN-λ had no effect. By contrast, when we looked for IFN-γ, a cytokine known to play an important role in defense against HSV (18, 22), we observed that IFN-λ, but not IFN-α, augmented the virus-induced levels of this cytokine in serum (Fig. 6B). Therefore, IFN-λ stimulates production of the immunomodulatory cytokine IFN-γ during virus infections in vivo.
FIG. 6.
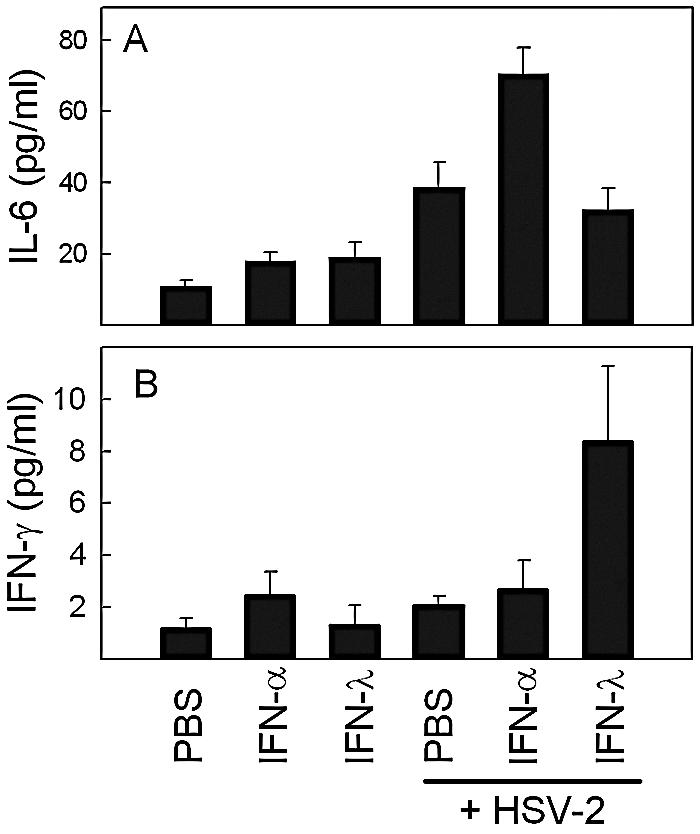
Effect of IFN-α or -λ treatment on HSV-2-induced serum cytokine levels. Mice were treated with PBS or 10 μg of the IFNs (i.e., 10,000 IU in the case of IFN-α) for 12 h before receiving i.p. infection with HSV-2 (1 × 106 PFU). Six hours postinfection, the mice were sacrificed, serum was harvested, and cytokine levels were measured by Luminex. The results are shown as mean values ± SD (error bars) (n = 4).
DISCUSSION
Type III IFNs (also called IL-28/29 or IFN-λ) are a recently identified class of cytokines with IFN-like activities, which have been demonstrated to be expressed in response to certain virus infections and to mediate antiviral activity in vitro (20, 30, 32). However, induction of IFN-λ in response to many other viruses as well as nonviral stimuli remains unresolved, as does the important question of the antiviral activity of type III IFNs in vivo. In this study we show that several classes of viruses representing both DNA as well as different forms of RNA genomes are able to induce IFN-λ expression in vitro in a range of cell lines. IFN-λ1 and IFN-λ2/3 were induced in highly similar patterns after virus infection, and were also—unlike IFN-α and IFN-β—induced directly by stimulation with IFN-α or IFN-λ, thus identifying type III IFNs as ISGs. Moreover, recombinant IFN-λ exhibited potent antiviral activity against HSV-2 in vivo in two different murine models but had no antiviral activity in vivo against EMCV or LCMV. Finally, IFN-λ potentiated expression of the immunomodulatory cytokine IFN-γ during HSV-2 infection in vivo.
Most, if not all, viruses are able to induce production of type I IFNs (7), whereas for type III IFNs, the information is sparse. It has been reported that a number of RNA viruses induce expression of IFN-λ (13, 20, 25, 32, 35), and one report has shown that the DNA virus cytomegalovirus is also able to induce IFN-λ production (10). In this study we examined and compared the ability of viruses with dsDNA, dsRNA, +ssRNA, and ÷ssRNA genomes to induce expression of IFN-λ and found that infection with all classes of viruses triggered expression of type III IFN to various degrees in the cell lines tested and also that IFN-λ was induced to at least the same extent as IFN-α/β was. This is in agreement with a recent report, which has shown that IAV induces comparable levels of type I and type III IFNs in different primary DC subsets in response to IAV infection (13), and we have obtained similar results with HSV in human monocyte-derived macrophages and DCs (24).
When we examined the ability of IFNs to cross-induce one another, we observed that the IFN-λs were in fact induced by both type I and type III IFNs. This finding extends a report showing that IFN-α enhances expression of IFN-λ1 and -λ2/3 in response to Toll-like receptor ligands in human monocyte-derived macrophages (34). Our finding has potential implications for our understanding of IFN-λ biology. First, it shows that the two classes of IFNs, although expressed in very similar patterns, are not regulated through identical mechanisms. IFN-α/β expression is dependent on activation of the transcription factors IFN regulatory factor 3 (IRF-3) and IRF-7, which are activated during viral infections but not by IFNs directly (38). The IFN-inducible nature of IFN-λs, on the other hand, indicates that these genes can be induced directly by IFN-stimulated gene factor 3 or by other IFN-induced transcription factors, such as IRF-1 for example. However, our finding that virus-induced IFN-λ expression occurs faster than IFN-induced IFN-λ expression strongly suggests that viruses can induce expression of the cytokines directly and therefore, that the IFN-λ gene promoters can be activated by a broader set of transcription factors than the type I IFN gene promoters.
Second, the IFN-inducible nature of IFN-λ suggests these cytokines may have the capacity to amplify the IFN response not only in time and magnitude but also in space. Thus, paracrine action of type I and type III IFNs produced by virus-infected cells would stimulate uninfected cells to produce IFN-λ, which could further spread the IFN activity through the uninfected tissue. Testing of these ideas awaits investigation and could be addressed, for instance, by examining whether the antiviral activity of type I IFN is affected in mice unresponsive to IFN-λ.
IFN-α/β can be produced by any nucleated cell with pDCs being the major IFN-producing cell type (2, 7, 12, 33). We showed production of IFN-λ1 and -λ2/3 after viral infection in cell lines of epithelial, lymphoid, and myeloid origin, thus demonstrating that these cytokines can be induced in a range of cell types. However, the question of which cell type is the major IFN-λ-producing cell and whether the cytokine is in fact produced in vivo during different virus infections remain open. Coccia et al. have reported that expression of IFN-α and IFN-λ in pDCs after infection with IAV in vitro followed highly similar kinetics (13) and further found that the cytokines were expressed at similar magnitudes. This could indicate that pDCs are also the primary IFN-λ-producing cells, but this still remains to be conclusively established. Regarding the production of IFN-λ during virus infections in vivo, some data are available. Brand et al. showed up-regulation of IFN-λ2 mRNA in colonic tissue from mice infected with murine cytomegalovirus (10), and Mihm and colleagues (25) found higher levels of IFN-λ1 to -λ3 mRNA expression in chronically hepatitis C virus-infected humans than in healthy controls, even when IFN-α/β levels were not significantly increased. This indicates that IFN-λ is actually produced during viral infection in vivo and also suggests that type I and type III IFNs do not always follow identical patterns of expression.
When we examined the activity of type III IFN in vivo by evaluating the antiviral activity of recombinant IFN-λ2 against three different systemic viral infections, we observed that the cytokine reduced the viral titer of HSV-2, but not EMCV and LCMV, both of which are highly sensitive to type I IFNs (27, 39). Furthermore, when we turned to a model for localized HSV-2 infection, we found that IFN-λ totally prevented virus replication in the vagina and even exceeded the antiviral activity of IFN-α. This was associated with complete protection from virus-induced disease symptoms. HSV is not highly susceptible to type I IFNs, and IFNAR−/− mice display only moderately enhanced viral load after HSV infection compared to wild-type mice (21). Moreover, the observed antiviral activity of IFN-λ against HSV-2, but not EMCV, in vivo contrasts with our findings in vitro, where IFN-λ did exhibit some degree of protection against EMCV, although not to the same extent as IFN-α, but hardly any protection against HSV-2. This could suggest that the high concentrations of IFN-λ required to evoke direct antiviral activity observed in vitro are not established in vivo and that the cytokine is also in possession of other mechanisms to restrict viral replication. We found that IFN-λ augmented virus-induced expression of IFN-γ, a cytokine known to modulate the immune response and to stimulate the antiviral response (18, 22), and our data thus suggest that type III IFNs do have immunomodulatory properties. This idea is supported by one report showing that an IFN-λ2/3-expressing recombinant vaccinia virus was not restricted in vitro in a cell line that expressed the IFN-λ receptor complex but was highly attenuated in vivo (4). Moreover, the recombinant virus enhanced the influx of T lymphocytes into the lung after intranasal infection. Altogether, this suggests that IFN-λ is able to stimulate the antiviral immune response and thus contribute to host defense via mechanisms other than induction of the classical antiviral state. In order to obtain more information on the potential immunomodulatory activity of the IFN-λs and their roles in disease and defense, it will be important to gather knowledge on the distribution of the IFN-λ receptor complex and hence which cell types respond to IFN-λ.
Collectively, in this work we show that type III IFNs are expressed by many cell types in response to widely different classes of viruses as well as to type I and type III IFNs. In addition, the IFN-λs exhibit antiviral activity in vivo to only a subset of viruses susceptible to IFN-α treatment, whereas this class of IFNs equals and even exceeds the antiviral activity of IFN-α in other virus infection models. Therefore, although sharing many features, type I and type III IFNs do show differences both in terms of regulation and biological activity. Future work will reveal the role and mechanism of action of this novel class of cytokines.
Acknowledgments
This work was supported by a donation from Novo Nordisk A/S and by research grants from The Danish Health Science Research Council (grants 22-02-0144 and 22-03-0193). N.A. and H.W. were supported by a fellowship from the Faculty of Health Science, AU, and a scholarship from The Danish Health Science Research Council (grant 22-04-0077). C.B. was the recipient of a postdoctoral fellowship from the Danish Health Science Research Council.
The technical assistance of Kirsten Stadel Petersen and Birthe Søby is greatly appreciated.
REFERENCES
- 1.Ashkar, A. A., S. Bauer, W. J. Mitchell, J. Vieira, and K. L. Rosenthal. 2003. Local delivery of CpG oligodeoxynucleotides induces rapid changes in the genital mucosa and inhibits replication, but not entry, of herpes simplex virus type 2. J. Virol. 77:8948-8956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144-1150. [DOI] [PubMed] [Google Scholar]
- 3.Asselin-Paturel, C., and G. Trinchieri. 2005. Production of type I interferons: plasmacytoid dendritic cells and beyond. J. Exp. Med. 202:461-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartlett, N. W., K. Buttigieg, S. V. Kotenko, and G. L. Smith. 2005. Murine interferon lambdas (type III interferons) exhibit potent antiviral activity in vivo in a poxvirus infection model. J. Gen. Virol. 86:1589-1596. [DOI] [PubMed] [Google Scholar]
- 5.Battegay, M., S. Cooper, A. Althage, J. Banziger, H. Hengartner, and R. M. Zinkernagel. 1991. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J. Virol. Methods 33:191-198. [DOI] [PubMed] [Google Scholar]
- 6.Berg, K., M. B. Hansen, and S. E. Nielsen. 1990. A new sensitive bioassay for precise quantification of interferon activity as measured via the mitochondrial dehydrogenase function in cells (MTT-method). APMIS 98:156-162. [DOI] [PubMed] [Google Scholar]
- 7.Biron, C. A. 1999. Initial and innate responses to viral infections—pattern setting in immunity or disease. Curr. Opin. Microbiol. 2:374-381. [DOI] [PubMed] [Google Scholar]
- 8.Biron, C. A. 2001. Interferons α and β as immune regulators—a new look. Immunity 14:661-664. [DOI] [PubMed] [Google Scholar]
- 9.Biron, C. A., K. B. Nguyen, G. C. Pien, L. P. Cousens, and T. P. Salazar-Mather. 1999. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17:189-220. [DOI] [PubMed] [Google Scholar]
- 10.Brand, S., F. Beigel, T. Olszak, K. Zitzmann, S. T. Eichhorst, J. M. Otte, J. Diebold, H. Diepolder, B. Adler, C. J. Auernhammer, B. Goke, and J. Dambacher. 2005. IL-28A and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am. J. Physiol. Gastrointest. Liver Physiol. 289:G960-G968. [DOI] [PubMed] [Google Scholar]
- 11.Brand, S., F. Beigel, T. Olszak, K. Zitzmann, S. T. Eichhorst, J. M. Otte, B. Goeke, H. Diepolder, B. Adler, C. Auernhammer, and J. Dambacher. 2005. The novel lambda-interferons IL-28A and IL-29 mediate proinflammatory, antiproliferative, and antiviral signals in intestinal epithelial cells. Gastroenterology 129:371. [DOI] [PubMed] [Google Scholar]
- 12.Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5:919-923. [DOI] [PubMed] [Google Scholar]
- 13.Coccia, E. M., M. Severa, E. Giacomini, D. Monneron, M. E. Remoli, I. Julkunen, M. Cella, R. Lande, and G. Uze. 2004. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 34:796-805. [DOI] [PubMed] [Google Scholar]
- 14.Curtsinger, J. M., J. O. Valenzuela, P. Agarwal, D. Lins, and M. F. Mescher. 2005. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174:4465-4469. [DOI] [PubMed] [Google Scholar]
- 15.Dumoutier, L., A. Tounsi, T. Michiels, C. Sommereyns, S. V. Kotenko, and J. C. Renauld. 2004. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-λ1: similarities with type I interferon signaling. J. Biol. Chem. 279:32269-32274. [DOI] [PubMed] [Google Scholar]
- 16.Haller, O., and G. Kochs. 2002. Interferon-induced mx proteins: dynamin-like GTPases with antiviral activity. Traffic 3:710-717. [DOI] [PubMed] [Google Scholar]
- 17.Harandi, A. M., K. Eriksson, and J. Holmgren. 2003. A protective role of locally administered immunostimulatory CpG oligodeoxynucleotide in a mouse model of genital herpes infection. J. Virol. 77:953-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harandi, A. M., B. Svennerholm, J. Holmgren, and K. Eriksson. 2001. Interleukin-12 (IL-12) and IL-18 are important in innate defense against genital herpes simplex virus type 2 infection in mice but are not required for the development of acquired gamma interferon-mediated protective immunity. J. Virol. 75:6705-6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolumam, G. A., S. Thomas, L. J. Thompson, J. Sprent, and K. Murali-Krishna. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202:637-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kotenko, S. V., G. Gallagher, V. V. Baurin, A. Lewis-Antes, M. Shen, N. K. Shah, J. A. Langer, F. Sheikh, H. Dickensheets, and R. P. Donnelly. 2003. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4:69-77. [DOI] [PubMed] [Google Scholar]
- 21.Leib, D. A., T. E. Harrison, K. M. Laslo, M. A. Machalek, N. J. Moorman, and H. W. Virgin. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 189:663-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu, T., K. M. Khanna, B. N. Carriere, and R. L. Hendricks. 2001. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J. Virol. 75:11178-11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meager, A., K. Visvalingam, P. Dilger, D. Bryan, and M. Wadhwa. 2005. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine 31:109-118. [DOI] [PubMed] [Google Scholar]
- 24.Melchjorsen, J., J. Siren, I. Julkunen, S. R. Paludan, and S. Matikainen. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-κB and IRF-3. J. Gen. Virol., in press. [DOI] [PubMed]
- 25.Mihm, S., M. Frese, V. Meier, P. Wietzke-Braun, J. G. Scharf, R. Bartenschlager, and G. Ramadori. 2004. Interferon type I gene expression in chronic hepatitis C. Lab. Investig. 84:1148-1159. [DOI] [PubMed] [Google Scholar]
- 26.Osterlund, P., V. Veckman, J. Siren, K. M. Klucher, J. Hiscott, S. Matikainen, and I. Julkunen. 2005. Gene expression and antiviral activity of alpha/beta interferons and interleukin-29 in virus-infected human myeloid dendritic cells. J. Virol. 79:9608-9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ou, R., S. Zhou, L. Huang, and D. Moskophidis. 2001. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J. Virol. 75:8407-8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pestka, S., C. D. Krause, and M. R. Walter. 2004. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202:8-32. [DOI] [PubMed] [Google Scholar]
- 29.Pirhonen, J., T. Sareneva, M. Kurimoto, I. Julkunen, and S. Matikainen. 1999. Virus infection activates IL-1β and IL-18 production in human macrophages by a caspase-1-dependent pathway. J. Immunol. 162:7322-7329. [PubMed] [Google Scholar]
- 30.Robek, M. D., B. S. Boyd, and F. V. Chisari. 2005. Lambda interferon inhibits hepatitis B and C virus replication. J. Virol. 79:3851-3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sen, G. C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55:255-281. [DOI] [PubMed] [Google Scholar]
- 32.Sheppard, P., W. Kindsvogel, W. Xu, K. Henderson, S. Schlutsmeyer, T. E. Whitmore, R. Kuestner, U. Garrigues, C. Birks, J. Roraback, C. Ostrander, D. Dong, J. Shin, S. Presnell, B. Fox, B. Haldeman, E. Cooper, D. Taft, T. Gilbert, F. J. Grant, M. Tackett, W. Krivan, G. McKnight, C. Clegg, D. Foster, and K. M. Klucher. 2003. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4:63-68. [DOI] [PubMed] [Google Scholar]
- 33.Siegal, F. P., N. Kadowaki, M. Shodell, P. A. Fitzgerald-Bocarsly, K. Shah, S. Ho, S. Antonenko, and Y. J. Liu. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science 284:1835-1837. [DOI] [PubMed] [Google Scholar]
- 34.Siren, J., J. Pirhonen, I. Julkunen, and S. Matikainen. 2005. IFN-α regulates TLR-dependent gene expression of IFN-α, IFN-β, IL-28, and IL-29. J. Immunol. 174:1932-1937. [DOI] [PubMed] [Google Scholar]
- 35.Spann, K. M., K. C. Tran, B. Chi, R. L. Rabin, and P. L. Collins. 2004. Suppression of the induction of alpha, beta, and gamma interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J. Virol. 78:4363-4369. (Erratum, 78:6705.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stager, S., and P. M. Kaye. 2004. CD8+ T-cell priming regulated by cytokines of the innate immune system. Trends Mol. Med. 10:366-371. [DOI] [PubMed] [Google Scholar]
- 37.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227-264. [DOI] [PubMed] [Google Scholar]
- 38.Taniguchi, T., and A. Takaoka. 2002. The interferon-α/β system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr. Opin. Immunol. 14:111-116. [DOI] [PubMed] [Google Scholar]
- 39.Zhou, A., J. M. Paranjape, S. D. Der, B. R. Williams, and R. H. Silverman. 1999. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology 258:435-440. [DOI] [PubMed] [Google Scholar]