

Abstract
Envelopment of Sindbis virus at the plasma membrane is a multistep process in which an initial step is the association of the E2 protein via a cytoplasmic endodomain with the preassembled nucleocapsid. Sindbis virus is vectored in nature by blood-sucking insects and grows efficiently in a number of avian and mammalian vertebrate hosts. The assembly of Sindbis virus, therefore, must occur in two very different host cell environments. Mammalian cells contain cholesterol which insect membranes lack. This difference in membrane composition may be critical in determining what requirements are placed on the E2 tail for virus assembly. To examine the interaction between the E2 tail and the nucleocapsid in Sindbis virus, we have produced substitutions and deletions in a region of the E2 tail (E2 amino acids 408 to 415) that is initially integrated into the endoplasmic reticulum. This sequence was identified as being critical for nucleocapsid binding in an in vitro peptide protection assay. The effects of these mutations on virus assembly and function were determined in both vertebrate and invertebrate cells. Amino acid substitutions (at positions E2: 408, 410, 411, and 413) reduced infectious virus production in a position-dependent fashion but were not efficient in disrupting assembly in mammalian cells. Deletions in the E2 endodomain (Δ406-407, Δ409-411, and Δ414-417) resulted in the failure to assemble virions in mammalian cells. Electron microscopy of BHK cells transfected with these mutants revealed assembly of nucleocapsids that failed to attach to membranes. However, introduction of these deletion mutants into insect cells resulted in the assembly of virus-like particles but no assayable infectivity. These data help define protein interactions critical for virus assembly and suggest a fundamental difference between Sindbis virus assembly in mammalian and insect cells.
Sindbis virus (SV) is the prototype of the alphaviruses, a family of viruses vectored in nature by mosquitoes and transferred via blood meal to humans and other vertebrates including birds and mammals (48). SV is an icosahedral virus with T=4 architecture composed of three structural proteins: capsid (C), envelope glycoprotein 1 (E1), and E2 (37). There are 240 copies of each of the structural proteins in a mature virus particle in a 1:1:1 stoichiometric arrangement. The outer envelope of the virus is composed of E1 and E2, which form 80 heterotrimeric spikes on the surface of the virus. Sandwiched between this outer shell and the inner shell, or nucleocapsid core (an aggregate of capsid protein and the virus RNA), is a host-derived lipid bilayer. The membrane is traversed by both of the outer envelope proteins E1 and E2. Two amino acids exit the membrane and are exposed on the cytoplasmic side of E1, whereas E2 has a cytoplasmic domain that is 33 amino acids (aa) in length. The integrity of an intact virion is maintained by two distinct interactions between the structural proteins. Lateral E1-E1 protein interactions stabilize the outer shell (1, 39), and an association involving the cytoplasmic domain of E2 and capsid connects the inner and outer shells, holding the particle together (6, 17, 19, 24, 35, 51).
Assembly of the virus particle involves multiple, specific protein-protein interactions. The structural proteins are first translated from 26S subgenomic RNA in the sequence NH2-C-PE2-6K-E1-COOH (20). Capsid is released from the polypeptide via an autoproteolytic activity and subsequently associates with the 49S viral RNA to form an assembled nucleocapsid (10). After capsid release from the polypeptide chain the nascent protein is inserted into the endoplasmic reticulum (ER), where cleavage by signal peptidase releases the 6K protein (20). Prior to export to the Golgi apparatus E1 and PE2 form heterodimers, followed by trimerization of these heterodimers (5, 31, 32). E1 and E2 heterotrimers are exported to the Golgi, where PE2 is processed by furin protease to form E2 releasing the E3 protein (34, 48). E3 is not present in a mature virus particle and is released into the surrounding media. The E1-E2 protein complex is transported to the plasma membrane where the process of virus envelopment takes place (4). At an unknown point in the secretory pathway the endodomain of E2 is pulled through the membrane and exposed to the cytoplasm for nucleocapsid binding (23).
The differences in the chemical and physical properties of membranes between insect and mammalian cells suggest that interactions of virus proteins with these membranes may differ. Insect cells (the natural host for alphaviruses) do not contain significant amounts of cholesterol (<1%) in their membranes (7, 8, 42), whereas mammalian cells have cholesterol as a significant contributor to their structure and function (2). We have previously shown that the difference in the composition of mammalian and insect membranes places different requirements on the transmembrane domains of the virus glycoproteins for proper virus assembly (13-15).
The association of the E2 endodomain with the preformed nucleocapsid is one of the critical events in the envelopment process (10, 17-19, 35, 36). This 33-aa endodomain (aa 391 to 423) is a multifunctional domain that plays a critical role in the integration and processing of the structural proteins in the ER; however, its primary function in assembly is binding to the nucleocapsid (Fig. 1). X-ray crystallographic analysis of the capsid protein revealed a hydrophobic cleft in the protein extending from aa 170 to 250 (19, 46). It has been proposed that this is the site of binding for the E2 endodomain during the process of envelopment.
FIG. 1.

Sequence of the SV E2 endodomain and related alphaviruses. Residues in boldface were deleted, and underlined residues were substituted as described in Materials and Methods. Dots indicate conservation of the sequence of the E2 tail between these alphaviruses.
The E2 endodomain has multiple functions. One is to serve as the signal for the integration of the 6K into the ER membrane and, as such, the E2 endodomain is itself a membrane-spanning protein. It is removed from the membrane and is exposed to the cytoplasm sometime after export from the ER but before it arrives at the plasma membrane and becomes available for capsid binding. Many studies have been directed at determining the nature of the E2-capsid interaction, to identify which residues from each protein are involved in and are critical to binding (11, 24, 35, 36, 44, 49, 50) (Table 1). With two exceptions these studies have been investigations of single amino acid substitutions in the E2 tail (14, 21). Although some of these substitutions significantly reduce infectious virus production, none completely blocks virus production. It is clear from these studies that particular residues such as E2 Y400 play critical roles in the maturation of SV. Two studies in which deletions were made in the E2 endodomain suggested that the length of the tail is critical between the membrane cytoplasmic face and Y400 but is not critical from Y400 to the end, A423. To further elucidate the nature of the interaction of the E2 endodomain with capsid, we have developed a strategy to first identify a region of the E2 tail that may be critical for assembly, in order to make multiple mutations within that region and analyze the effect on SV assembly.
TABLE 1.
Mutations produced in the E2 endodomain
| Position | Mutation | Effect | Reversion | Reference |
|---|---|---|---|---|
| ΔΔK391 | Deletion of K391 | No virus produced in BHK; wild-type virus levels produced in insect cells | None reported | 14 |
| Δ402-406 | Deletion of LAPNA | No effect on virus production | None reported | 21 |
| T398/Y400 | T398A/Y400N | Reduced virus production by 3 logs; no defect in virus assembly | Revertants observed at Y400 but not T398 | 50 |
| 21 | ||||
| P399 | P399G | Released lower than normal levels of virus; produced multicored particles; more heat stable than wild type | None reported | 16 |
| A401 | A401I, A401K | Released lower levels of particles; multicored particles; heat stable | None reported | 16 |
| P404 | P404G | Released lower levels of particles; multicored particles; heat stable | S182N in capsid (no real effect alone); T398M in E2 tail (not studied) | 44 |
| Y400 | Y400F | Reduced particle formation by 3 to 4 logs | None reported | 11 |
| L402 | Y, C, T, G, N, D, and R | Y, 2-log decrease; C and T, 4-log decrease; G, N, and D, 5-log decrease; R, dead; no processing, transport, or translocation defects | None reported | 35 |
| C416 | C416A | Produced virus slower than wild type; produced multicored particles | None reported | 16 |
| C417 | C417A | Highly defective in virus release; produced multicored particles; defective in processing P62-6K | T256M in capsid (alone no affect); S411L in E2 tail (not studied) | 16 |
| S420 | S420C | Affected P62-6K cleavage | None reported | 11 |
MATERIALS AND METHODS
Cells, viruses, and media.
Baby hamster kidney (BHK-21) cells were maintained in Eagle minimal essential medium (MEM) supplemented with 10% fetal bovine serum, 5% tryptose phosphate broth, and 2 mM glutamine as described previously (40). The cells were maintained at 37°C under 5% CO2. The U4.4 cells used were cloned from cells provided by Sonya Buckley (Yale Arbovirus Research Unit, New Haven, CT). This cell line is maintained in Mitsuhashi and Maramorosch medium (29). U4.4 cells are grown at 28°C. The wild-type SV cDNA used in these studies was the SV Toto 1101 (41, 47).
Iodination of capsid protein.
Purified capsid protein from aa 106 to 264 expressed in Escherichia coli (a generous gift from Richard Kuhn, Purdue University) was used as the capsid source. The peptides tested included the full-length 33-aa endodomain, 391-KARRECLTPYALAPNAVIPTSLALLCCVRSANA-423, and truncated peptides from this sequence, 408-IPTSALALLCCVRSANA-423, 416-CCVRSANA-423, 400-YAL-402, 399-PYALAP-404, and 391-KARRECLTPYAL-402, and also a control peptide of an unrelated sequence of moderate hydrophobicity, NH2-ILEPVHGV-COOH. These peptides were synthesized and purified at the California Institute of Technology and were provided by James Strauss. All peptides were resuspended in 100% dimethyl sulfoxide (DMSO) at a concentration of 1 mg/ml since this was the condition required to dissolve the full-length 33mer. The 3mer and 6mer were added in a 1,000-fold molar excess to the capsid protein. All other peptides were added at a 10-fold molar excess. Capsid protein 106-264 (10 μg) was used in all binding reactions. Capsid was bound to peptides by incubation in TEN (50 mM Tris [pH 7.6], 50 mM NaCl, and 1 mM EDTA) buffer. All capsid-peptide binding reactions were adjusted to 40% DMSO, which was the final concentration of DMSO present in the binding reaction with the full-length 33mer. The capsid-peptide binding reaction was allowed to proceed slowly at 4°C overnight. Capsid-peptide complexes were removed from unbound peptide in an Amicon 10 Spin Centricon (Millipore, Bellerica, MA) by four washes each with 2 ml of TEN buffer. As a control for the possibility that the denatured capsid protein could expose Y180 to iodination, C was denatured in 8 M urea. In this reaction 10 μg of C in TEN was added to 8 M urea and incubated at 37°C for 5 h. The denatured C was washed three times with 2 ml of TEN in an Amicon 10 Centricon and brought up to 40 μl with TEN prior to iodination. The iodination reaction was essentially as described by Coombs and Brown (9). In brief, to 40 μl of capsid-peptide complex was added 40 mM glucose, 200 μCi of 125I (1,227 Ci/mM; Perkin-Elmer, Irvine), 0.02 U of glucose oxidase (diluted in phosphate-buffered saline [PBS], 0.9 mM CaCl2, 1.4 mM KH2PO4, 0.5 mM MgCl2 · 6H2O, 136 mM NaCl, 7.9 mM Na2HPO4) (Worthington Biochemicals, Lakewood, NJ), and 0.01 U of lactoperoxidase (diluted in PBS) (Worthington Biochemicals) and brought to a total volume of 63 μl in TEN buffer. Iodination proceeded for 30 min at room temperature and was terminated by the addition of 25 μl of stop buffer consisting of 10 mg of tyrosine/ml, 10% glycerol, and 0.1% xylene cyanol incubated at room temperature for 30 min. Unincorporated label and any labeled free peptide was removed from the protein-peptide complexes by passing the protein through a 5-ml Sephadex G25 column (Sigma, St. Louis, MO). Column matrix was swollen in PBS with 0.5% bovine serum albumin and washed with 10 column volumes of PBS. A V-8 (endoproteinase-Glu-C) digest was made of the capsid-peptide complexes by adding V-8 (dissolved in H2O) to a final concentration of 40 μg/ml. The digest was allowed to proceed for 3 h at 35°C. Peptides generated from the V-8 digest were run on 30-cm Tricine slab gels (25) at 30 mA for 19 h.
Site-directed mutagenesis of Toto 1101 and reverse transcription-PCR.
Using standard megaprimer site-directed mutagenesis protocols (45) and Pfu DNA polymerase, substitution and deletion mutants were generated within the E2 endodomain of SV. Substitutions were made at the following positions in the E2 endodomain: 408I, 410T, 411S, and 413A. Positions 408, 410, and 411 were each mutated to alanine (I408A, T410A, and S411A), whereas the alanine at position 413 was mutated to glycine (A413G). Deletions were made at positions E2 406 to 407, 409 to 411, and 414 to 417. The primer pairs used to create the deletions were as follows: 406-407, sense (5′-GCCCTGGCCCCAAACATCCCAACTTCGCTGGC-3′) and antisense (5′-GCCAGCGAAGTTGGGATGTTTGGGGCCAGGGC-3′), 409-411, sense (5′-CCCAAACGCCGTAATCCTGGCACTCTTGTGC-3′) and antisense (5′-GCACAAGAGTGCCAGGATTACGGCGTTTGGG-3′), and 414-417, sense (5′-CCCAACTTCGCTGGCAGTTAGTAGGTCGG-3′) and antisense (5′-CCGACCTAACTGCCAGCGAAGTTGGG-3′). PCR products from the mutagenized plasmid were placed into the wild-type vector by using unique BssHII and BsiWI sites. After confirmation of the correct sequence throughout the insert, infectious RNA was transcribed in vitro by using SP6 polymerase and introduced into cells by electroporation as described below.
To analyze the mutant viruses produced from transfection for the retention of the original mutations and for the presence of pseudorevertants, reverse transcription-PCR was used. Virus from transfections was pelleted at 40,000 rpm for 1 h. The pellet was then incubated in 100 μl of Tris-EDTA (10 mM Tris [pH 8.0], 1 mM EDTA) and 100 μl of 2× lysis buffer (100 mM Tris-Cl [pH 7.0], 20 mM EDTA, 1% sodium dodecyl sulfate) for 20 min at 37°C (vortexing every 5 min). The lysed virus was then extracted sequentially with phenol twice, phenol-chloroform once, and chloroform once. Extracted RNA was precipitated in RNase-free ethanol overnight at −80°C. RNA was pelleted and resuspended in 10 μl of diethyl pyrocarbonate-treated water. RNA was transcribed by using murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA) under the following conditions. The reaction contained 10× PCR buffer (10 mM Tris-Cl [pH 8.3], 50 mM KCl, 5 mM MgCl2), 20 U of SuperRNasin (Ambion, Austin, TX), 200 μM concentrations each of deoxynucleoside triphosphates (dNTPs) (Roche Diagnostics, Indianapolis, IN), 1.0 μM reverse primer, and 50 U of murine leukemia virus reverse transcriptase in a final volume of 20 μl. Reverse transcription was performed at 42°C for 20 min and 99°C for 5 min. After transcription, the volume of the reaction mixture was increased to 100 μl, and the concentrations of the dNTPs and MgCl2 were adjusted for the increased volume. Sense primer was added to a final concentration of 2.0 μM, and an additional 1.0 μM concentration of antisense primer was added. Taq DNA polymerase was added to a final concentration of 2.5 U/100-μl reaction mixture.
In vitro transcription, RNA transfection, and plaque assay.
Full-length mutant and wild-type cDNAs were linearized by using the enzyme Xho1, treated with proteinase K, phenol extracted, and ethanol precipitated. The templates were transcribed as described previously (15, 41). The infectious RNAs were transfected by electroporation essentially as described by Liljestrom and Garoff (20). Briefly, BHK cells were treated with trypsin, pelleted, and washed in RNase-free PBS (pH 7.0). Washed cells were resuspended to a concentration of 1 × 107 cells/ml. RNA transcripts (20 μl) were added to 400 μl of BHK cells. The electroporation conditions were 1.5 kV, 25 μF, and ∞ resistance. Cells were pulsed once, allowed to sit for 10 min, and then transferred into 10 ml of MEM (no gentamicin) in 25-cm2 tissue culture flasks. Virus was harvested at 24 h posttransfection, flash frozen in liquid nitrogen in 1-ml aliquots, and stored at −80°C. To determine the titer of each virus, plaque assays were carried out as described previously (40). All virus titrations were done on BHK-21 cells.
Transfection of U4.4 (Aedes albopictus) cells.
RNA transcripts of each of the mutants were prepared as described above. Cells were pelleted and washed three times in RNase-free HBS buffer (20 mM HEPES [pH 7.2], 137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 6 mM d-glucose). Washed cells were resuspended to a concentration of 5 × 107 cells/ml. RNA transcripts (20 μl) were added to 400 μl of cells. The electroporation conditions were 1.5 kV, 25 μF, and ∞ resistance. The cells were pulsed once and allowed to sit at room temperature for 10 min. After 10 min the mixture was transferred to a 25-cm2 vented flask, and Mitsuhashi and Maramorosch growth media was added in a dropwise fashion. Transfections were incubated at 28°C under 5% CO2. Virus was harvested at approximately 48 h posttransfection.
Immune precipitation of cells transfected with E2 endodomain deletion mutants.
Anti-whole-virus antibody was produced in rabbits, and SV-specific immunoglobulin G was purified by using a Hi-Trap, protein A column (Amersham Pharmacia Biotech, Piscataway, NJ). Metabolically labeled transfections of BHK-21 cells were carried out as described above. At 6.5 h posttransfection, 5 ml of fresh medium containing 4 μg of actinomycin D (Calbiochem, San Diego, CA)/ml was added to 25-cm2 flasks of cells (∼5 × 106 cells), followed by incubation at 37°C for 1 h. The flasks were then washed with 5 ml of room temperature PBS-D (30 mM KCl, 15.2 mM KH2PO4, 79 mM Na2HPO4, 0.14 NaCl [pH 7.4]), placed into 5 ml of starvation medium (MEM deficient in methionine and cysteine, supplemented with 2 mM glutamine and 3% fetal bovine serum), and returned to 37°C for 1 h. The transfected cells were then labeled with 50 μCi of [35S]methionine-cysteine/ml in 5 ml of starvation medium, followed by incubation at 37°C overnight or until an advanced cytopathic effect was visible. The labeled virus (media) was removed and stored 4°C. The cells were washed once with cold 1× PBS-D. Labeled cell-associated proteins were processed for immune precipitation as described previously (34). A total of 2 μl of anti-whole virus antibody was added to the cell supernatants, followed by rocking at 4°C. To the cell-antibody suspension, 200 μl of protein A-beads was added, followed by rocking overnight at 4°C. Polyacrylamide gel electrophoresis (PAGE) analysis of the supernatant from the precipitation and the labeled media was done to confirm the efficiency of the antibody in removing viral proteins.
TEM.
Samples were analyzed by transmission electron microscopy (TEM) as described previously (38). BHK-21 and Aedes albopictus cell monolayers were transfected with RNA transcribed from either wild type or each of the E2 endodomain deletion mutants (Δ406-07, Δ409-411, and Δ414-417) as described above. At approximately 18 to 20 h posttransfection for BHK cells and 48 h posttransfection for mosquito cells, the media were harvested, and the cell monolayers were scraped from the flasks and pelleted by low-speed centrifugation. Cell pellets were washed twice with PBS-D and fixed with 3% glutaraldehyde (Ladd Research Industries, Williston, VT) in 0.1 M cacodylic acid buffer (pH 7.4; Ladd Research Industries). After the cells were washed three times with 0.1 M cacodylic acid, the cells were stained with 2% osmium tetroxide in cacodylic buffer for 1 h. Cells were then washed as described above and embedded in 2% agarose. The agarose containing the cell sample was then prestained with 1% uranyl acetate (Polaron Instruments, Inc., Hatfield, PA) overnight at 4°C. The samples were washed and carried through ethanol dehydration. Infiltration was done using SPURR compound (LADD Research Industries). Blocks were trimmed on an LKB NOVA Ultrotome (Leica Microsystems, Inc., Deerfield, IL). Ultrathin sections were obtained and stained with 5% uranyl acetate in distilled water for 60 min and in Reynolds lead citrate (pH 12; Mallinkrodt Baker, Inc., Paris, KY) for 4 min. The samples were examined at 80 kV in a JEOL JEM 100S transmission electron microscope.
RESULTS
Protection of capsid protein residue Y180 by E2 endodomain peptides.
SV capsid protein contains four tyrosine residues at positions 162, 180, 189, and 198. One property of tyrosine is its ability to be iodinated by the enzyme lactoperoxidase. In 1987, Coombs and Brown determined that only one of these four tyrosine residues (Y180) is accessible to lactoperoxidase iodination in the intact nucleocapsid and is thus exposed on the surface of the protein in the intact core structure (9). The structure of the capsid protein determined by X-ray crystallography suggested that the tyrosine at position 180 could exist in two conformations (19). In the absence of association with the E2 endodomain, Y180 is exposed on the surface of the protein but in association with the E2 endodomain Y180 is buried in the protein complex. These observations allowed us to develop an assay for the interaction of the E2 endodomain with the capsid protein based on the ability of amino acid sequence derived from the endodomain to cause the conformational change that results in the protection of the tyrosine at position 180 from iodination as described in Materials and Methods. Protection studies were performed with peptides of differing lengths, corresponding to selected regions of the E2 endodomain, and purified capsid protein. Limitations were place on the number and sequences of the peptides used because some peptides could not be purified and sequenced after the removal of protecting groups, probably because of their extreme hydrophobic nature. E2 peptide and capsid protein were mixed, and lactoperoxidase and 125I were added as described in Materials and Methods. The reaction was quenched, incubated with V-8 protease, and then analyzed by Tricine gel electrophoresis as described previously (25). We have previously shown that this treatment releases a peptide containing capsid protein Y180, in the sequence 177-AFTYTSE-183 (9) from purified nucleocapsids. The in vitro experiment revealed that in the presence of a peptide corresponding to the full-length E2 endodomain, capsid protein residue Y180 could not be labeled by lactoperoxidase (protected), while in the absence of E2 peptide it was exposed to labeling by 125I (exposed) as shown in Fig. 2. Table 2 shows the results obtained when this experiment was conducted with a number of peptides representing fragments of the E2 endodomain. This experiment revealed that the conformational change in capsid protein resulting in the protection of capsid Y180 could be obtained with the full-length endodomain. Progressive truncations of the endodomain suggested that the minimum domain needed for protection of Y180 was a peptide containing aa 408 to 415 in the carboxyl portion of the E2 tail. Unfortunately, this sequence could not be tested directly since it could not be produced (see above). Surprisingly, peptides containing the conserved TPY sequence (aa 398 to 400), in the absence of the carboxyl portion of the E2 endodomain, did not produce the conformational change, resulting in the protection of Y180. It has been proposed that the Y400 in the TPY sequence interacted with Y180 and W247 in the capsid and that this interaction was critical for virus assembly and virus infectivity (46).
FIG. 2.
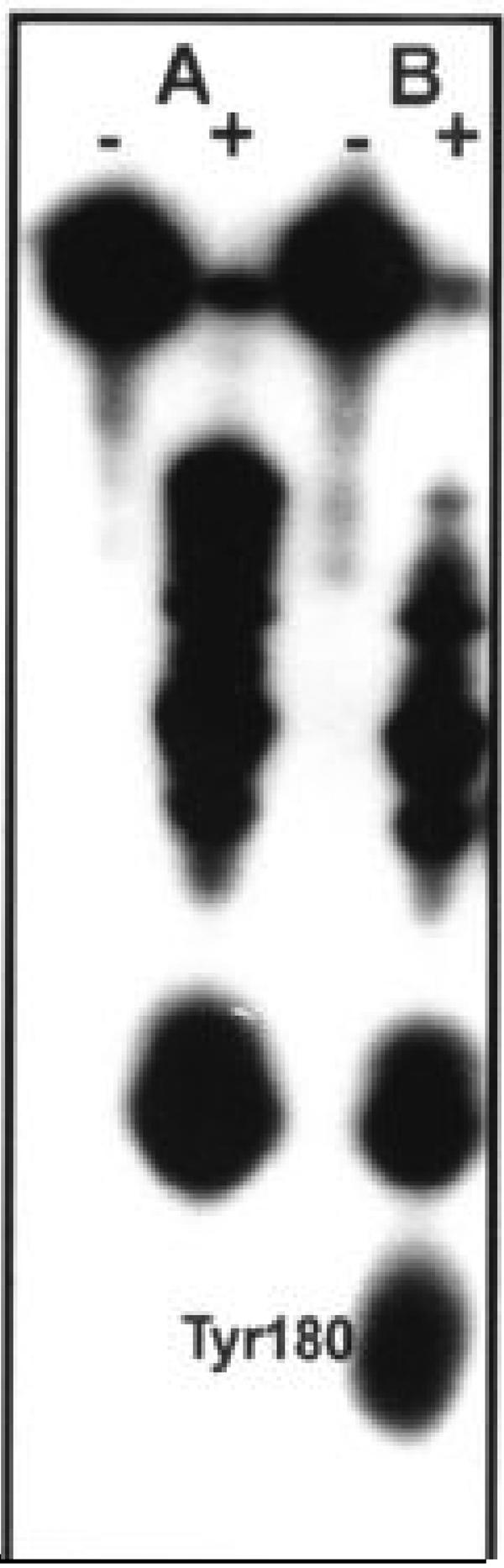
Iodination of capsid residue Y180 as an indicator of E2 protein binding. Capsid protein was iodinated in the presence (A) or absence (B) of a peptide representing the 33-aa E2 endodomain. The reaction was quenched and then treated (+) or not treated (−) with V8 protease to release a peptide containing capsid Y180.
TABLE 2.
Protection of capsid residue Y180 upon association with peptides corresponding to the E2 endodomain
| E2 endodomain peptide sequence | Protection of capsid Y180 |
|---|---|
| 391-KARRECLTPYALAPNAVIPTSLALLCCVRSANA-6K | Yes |
| IPTSLALLCCVRSANA-6K | Yes |
| CCVRSANA-6K | No |
| YAL | No |
| PYALAP | No |
| 391-KARRECLTPYAL | No |
| ILEPVHGV (control) | No |
Construction of E2 endodomain mutants.
The peptide protection experiments, presented above, suggested that the amino acid sequence from aa 408 to 415 in the E2 tail was required for the interaction with capsid protein in the in vitro assay. To further elucidate the role that this sequence plays in virus assembly and function, mutations were created in this region. Both substitution and deletion mutants were produced as described in Materials and Methods. Amino acid substitutions were made at four positions within the targeted domain: I408A, T410A, S411A, and A413G. Deletion mutants were made removing the indicated residues: Δ406-407 (AV), Δ409-411 (PTS), and Δ414-417 (LLCC). The mutants produced cover the entire region of the E2 endodomain identified in the peptide protection experiments (described above). Since the COOH terminus of the E2 endodomain is initially integrated into the ER membrane as the signal sequence for the 6K protein, deletion and substitution mutants in this region were also expected to elucidate the role these amino acids play in this initial event in membrane protein assembly.
Growth of E2 endodomain substitution mutants in BHK cells.
To determine the effect of amino acid substitutions in the E2 endodomain on virus assembly and function, viral RNA was produced from constructs containing the mutations by in vitro transcription and transfected into BHK cells as described in Materials and Methods. Wild-type SV produced an average of 2 × 109 PFU of virus/ml from BHK cells, and the mutants A413G and S411A showed little change in virus production compared to the wild type (Fig. 3A). However, the T410A and I408A mutants showed a reduction in the amount of infectious virus produced. I408A produced 106 PFU/ml, and T410A produced 108 PFU/ml (Fig. 3A). With the exception of the mutant A413G, all of these substitutions introduce changes in the atomic density of the E2 endodomain. The alanine residues that were substituted into the E2 tail are less bulky than the wild-type amino acids that are present: isoleucine (aa 408), threonine (aa 410), and serine (aa 411). The observation that these changes, with the exception of mutant I408A, produce little reduction in infectious virus production and no alteration in protein production (data not shown) suggests that the precise amino acid sequence of this portion of the E2 endodomain is not critical in the assembly of the virion nor does it play a significant role in virus function. It does indicate that as substitutions are made closer to the membrane bilayer, there is a more deleterious effect on the production of infectious virus. In addition, it suggests that position 408 is more critical for infectious virus production than the positions downstream (i.e., 410, 411, and 413) that were also analyzed. Based on these data and findings published by others (11, 16, 35, 43), it seems that many single amino acid substitutions do not modify the endodomain structure to significantly disrupt assembly.
FIG. 3.

Production of infectious virus after transfection of BHK cells with E2 endodomain mutants. (A) Production of infectious virus by E2 endodomain substitution mutants. Virus was harvested 20 to 24 h posttransfection, and titers were determined by plaque assay on BHK cells as described in Materials and Methods. (B) Production of infectious virus by E2 endodomain deletion mutants. Virus was harvested 20 to 24 h posttransfection, and titers were determined by plaque assay on BHK cells as described in Materials and Methods.
Growth of E2 endodomain deletion mutants in mammalian cells.
We have previously demonstrated that the deletion of a single amino acid at the point where the E2 endodomain emerges from the membrane bilayer (E2 K391) blocked virus production from mammalian cells (14). We concluded that this deletion reduced a critical distance between the membrane bilayer and a sequence in the endodomain that interacted with the capsid protein. A second deletion produced in our laboratory that deleted aa 402 to 406 had no effect on virus infection, indicating that a deletion which altered the length of the endodomain after Y400 did not affect virus growth (21). Deletions in other regions of the E2 endodomain have not been examined for their effect on the assembly and growth of SV.
To determine the effect of deletions in the C-terminal portion of the E2 endodomain (as identified by the C-Y180 protection assay) on virus assembly and infectivity, RNA transcripts from the endodomain deletion mutants Δ406-407, Δ409-411, and Δ414-417 were transfected into mammalian cells (Fig. 3B). Virus production from wild-type and mutant constructs was assayed as described above. Mammalian cells transfected with wild-type viral RNA produce a titer of 2 × 109 PFU/ml; however, all three deletion mutants showed a reduction in infectious virus production. The mutant Δ406-407 produced an average of 4 × 107 PFU of virus/ml in BHK cells. This reduction in infectious virus production indicates that positions 406 (alanine) and 407 (valine) could be important for virus assembly and function. It is possible that deletion of these residues changes the position of some downstream amino acids and that this change in position is responsible for the observed phenotypes, The fact that the previously published deletion of aa 402 to 406 has no effect on virus production (21) argues against this possibility. The deletion of aa 406 to 407 is immediately upstream of the I408A substitution, which was also found to be necessary for virus production. The deletion mutations Δ409-411 (deletion of PTS) and Δ414-417 (deletion of LLCC) both completely blocked the production of infectious virus from mammalian cells (Fig. 3B). This result suggests that these motifs are required for assembly and production of infectious SV in BHK cells. It is noteworthy that P409 and aa 415 to 417 (LCC) are conserved among the alphaviruses. Substitutions at P409 and CC 416 and 417 severely limit virus production (11). These data in combination with the observation that virus production was only moderately affected when single amino acids in this region were substituted in the present study indicate that the length of the carboxyl proximal portion of the E2 endodomain and the strictly conserved sequence are critical factors in virus assembly in mammalian cells.
Protein synthesis in BHK cells transfected with E2 endodomain deletion mutants.
We have established that no infectious or noninfectious virus is produced from two of the deletion mutants (Δ409-411 and Δ414-417) and that deletion mutant Δ406-407 produces significantly less infectious virus from vertebrate cells compared to the wild type (Fig. 3B). In order to determine whether defective protein processing is the reason for the complete loss of virus production from BHK cells, cells were transfected with each of the deletion mutants and analyzed for virus protein production, processing, and release.
SV membrane proteins are integrated into the membranes in the ER, where they follow a complex pathway of folding and oligomerization to produce a heterotrimer that is exported from the ER to the plasma membrane (1, 5, 31-33). The ER controls for the production of correctly folded and assembled proteins, and only proteins deemed correctly assembled are shipped to the cell surface. En route to the cell surface the PE2 precursor is proteolytically processed to E2 by furin, a resident protease in the trans-Golgi network. The product of this proteolytic event is the membrane-associated E2 and the 16-kDa E3 protein that is secreted. The production of E3 by a virus or mutant virus indicates that the proteins have passed the quality control of the ER and have been exported to the Golgi apparatus. We examined the proteins secreted from cells transfected with wild-type SV and the deletion mutants to assay the production and export of proteins from the ER.
Virus proteins released into the surrounding medium were analyzed by PAGE with equal volumes of cell supernatant as described in Materials and Methods and as shown in Fig. 4. BHK cells transfected with both wild-type RNA and the E2 deletion mutant Δ406-407 produced a normal complement of virus proteins. Equal volumes of supernatant also revealed equivalent amounts of the E3 glycoprotein produced by the wild type and the double deletion (Fig. 4A). BHK cells transfected with both the Δ409-411 and the Δ414-417 mutants also produced amounts of E3 equal to the wild-type infection; however, neither of these mutants released significant amounts of the virus structural proteins E1 or E2 into the media. The Δ406-407 and Δ409-411 mutants did release capsid protein into the media, but there was little indication of the presence of E1 or E2, whereas mutant Δ414-417 showed little to no release of any structural proteins, including capsid (Fig. 4). The appearance of capsid protein in the absence of significant virus production may be the result of limited cell lysis and the release of some of the large numbers of non-membrane-associated capsids into the media. This conclusion is supported by the electron microscopy analyses described below.
FIG. 4.

Protein synthesis in deletion mutant-transfected cells. (A) Proteins released into the media of BHK cells transfected with E2 endodomain deletion mutants. Equal volumes of media surrounding equal numbers of transfected cells was applied directly to PAGE. Arrows indicate the positions of the virus structural proteins E1, E2, capsid, and E3. (B) Proteins immune precipitated from cells infected with deletion mutant Δ414-417. Lanes: 1, wild-type RNA; 2, mock transfected; 3, transfected with RNA from deletion mutant Δ414-417. PE2, E1, E2, and C are SV structural proteins. Arrows A and B indicate aberrant proteins produced in deletion mutant Δ414-417.
We also examined the cell-associated proteins to determine whether the mutations in the endodomain resulted in aberrant virus structural protein processing. BHK cells were transfected with the three deletion mutants, and proteins were isolated from cells by immune precipitation and analyzed by sodium dodecyl sulfate-PAGE as described in Materials and Methods. The 2- and 3-aa deletions produced protein profiles indistinguishable from that of the wild type (not show). The 4-aa deletion produced two nontypical protein bands that were not recovered in mock-infected or wild-type transfected cells (Fig. 4B). These proteins have estimated molecular masses of 42 and 35 kDa. They are in well-defined compact bands, suggesting that they are not the result of random degradation.
These results suggest that the deletions Δ406-407 and Δ409-411 in the COOH terminus of the E2 endodomain do not affect the insertion of the polyprotein into the ER or processing of the structural proteins (except for Δ414-417). To determine whether mature, functional E1 protein was present at the cell surface, BHK cells transfected with deletion mutants Δ409-411 and Δ414-417 were subjected to analysis by fusion from within. Cell-cell fusion from within (27) was observed for both large deletion mutants, which indicated that functional E1 protein was present at the cell surface (data not shown) in a conformation allowing for low-pH-induced fusion. This further suggests that there are no processing or transport defects in mammalian cells transfected with these deletions (i.e., Δ409-411 and Δ414-417). Collectively, these data suggest that the defect in the E2 deletion mutants, Δ409-411 and Δ414-417, is most likely in the ability of E2 to bind the nucleocapsid.
Electron microscopy of BHK cells transfected with E2 endodomain deletion mutants.
To confirm that the defect in the E2 deletion mutants was in the ability to bind nucleocapsids to modified cell membranes, we analyzed transfected cells by electron microscopy. BHK cells transfected with wild-type SV showed nucleocapsids attached to membranes, nucleocapsids at the plasma membrane, and virus particles budding from the plasma membrane, as is typical in a mammalian cell infection (Fig. 5A). Budding viruses were also observed in cells transfected with the E2 deletion mutant Δ406-407 as expected based on the amount of infectious virus produced. However, a significant number of nucleocapsids were seen free in the cytoplasm with this double deletion mutant (Fig. 5B). This result suggests that the decrease in the amount of infectious virus production from this mutant is due, in part, to a reduced ability of the E2 endodomain to bind nucleocapsids resulting in an accumulation of non-membrane-associated capsids. The electron micrographs of cells transfected with the Δ409-411 (Fig. 5C) and Δ414-417 (Fig. 5D and E) deletion mutants support the conclusions derived from PAGE analysis of proteins secreted from the infected cells. No virus particles were observed budding from the plasma membrane in cells infected with these mutants. The cytoplasm of these cells contained large numbers of nucleocapsids, more than are seen in a wild-type infection. None of the assembled nucleocapsids were attached to internal membranes, and no nucleocapsids were associated with the plasma membrane (electron-dense particles that are membrane associated are smaller than capsids and are most likely ribosomes). These observations support the conclusion presented above that the defects in these deletion mutants are in the ability of the E2 endodomain to bind the preassembled nucleocapsid.
FIG. 5.

Electron micrographs of BHK cells transfected with E2 endodomain deletion mutants. (A) Wild type; (B) Δ406-407 mutant; (C) Δ409-411 mutant; (D) Δ414-417 mutant. (E) Enlargement of an area in panel D. Arrows indicate nucleocapsids. Bars, 200 nm.
Electron microscopy of insect cells transfected with E2 endodomain deletion mutants.
SV is an arbovirus and as such must be capable of assembling virus in cells of both vertebrate and invertebrate origins (3). We have previously shown that deletions in the transmembrane domain of the E2 glycoprotein could restrict the ability of SV to grow in an invertebrate host (14, 15). We suggested that this was a result of the different chemical and physical properties of the cell membranes of the two hosts. These observations suggested that the deletions that prevented the formation of virus particles in mammalian cells may be tolerated to some degree in the invertebrate cells since the targeted domain is also, initially, a transmembrane domain (23). Insect U4.4 cells transfected with wild-type virus produced 5 × 109 PFU/ml. The phenotype of the E2 endodomain deletion mutants in insect cells mimicked the phenotype observed in mammalian cells (Fig. 6). The Δ406-407 mutant produced an average of 108 PFU of virus/ml, and both the Δ409-411 and the Δ414-417 mutants produced no infectious virus from U4.4 (Aedes albopictus) cells. These data are similar to the results obtained in BHK cells (Fig. 3B) and confirm that these sequences are vital for the production of infectious virus in insect cells as well as mammalian cells.
FIG. 6.

Growth of E2 endodomain deletion mutants in insect cells. Aedes albopictus (clone U4.4) cells were transfected with wild type or mutant strains containing virus RNA as described in Materials and Methods.
To determine whether the failure to produce infectious virus in insect cells was also the result of a failure to assemble virus particles as in BHK cells, U4.4 cells transfected with these deletion mutants were analyzed by electron microscopy. We have previously shown that SV maturation in insect cells takes place within internal vesicles rather than at the plasma membrane (3, 12, 28), with some cells displaying large numbers of matured virus particles. The virus contained in the vesicles is released into the medium by a process resembling exocytosis in which the vesicle membrane fuses with the cell membrane, releasing the contents into the surrounding medium (3, 12, 28). In agreement with the data showing the levels of infectious virus production from cells infected with wild-type SV or the double deletion (Δ406-407) mutant, virus particles were seen within vesicles as is typical of infection of insect cells (Fig. 7A, only the wild type is shown).
FIG. 7.

Electron micrographs of Aedes albopictus cells transfected with E2 endodomain deletion mutants. (A) Wild type (arrows indicate virions); (B and C) mutant Δ409-411. Bars, 100 nm.
Transfections of insect cells with the larger deletions (Δ409-411 and Δ414-417) produced a remarkable result. Although no infectivity could be recovered from these transfections, large numbers of virus-like particles were seen within vesicles for both of the deletion mutants (Fig. 7B and C, only the Δ409-411 mutant is shown). Insect cells transfected with wild-type virus or the double mutant contained relatively low amounts of cell-associated virus, whereas cells infected with the triple and quadruple deletions contained very large numbers of cell-associated virus-like particles (Fig. 7B and C). This result may suggest that release of the virus-like particles is impaired in the triple- and quadruple-deletion mutant-infected cells. We have been unable to recover any virus infectivity (as determined by assay on BHK cells), and attempts to release infectious virus from these cells by disrupting the cells also failed. For this reason, we refer to these structures as virus-like particles. These results suggest that the virus particles produced in the triple- and quadruple-mutant-transfected cells is noninfectious for BHK cells. The virus-like particles seen in the insect cells transfected with the triple- and quadruple-deletion mutants was not seen in the wild-type, double-deletion, or mock-transfected cells. Some of these particles are not typical in morphology, although they are consistent in size at 670 to 700 Å in cross sections, as is wild-type SV (Fig. 7B and C). Some of the particles appear to lack the electron-dense centers typical of thin-sectioned SV. More of these virus-like particles were seen in the triple-deletion- than in the quadruple-deletion-transfected cells, suggesting that assembly of the virus-like particles may have been more impaired by the larger deletion. These virus-like particles were found to be very fragile since attempts to purify them from cell homogenates by gradient centrifugation were not successful.
DISCUSSION
Previous studies have indicated that the E2 endodomain specifically interacts with a hydrophobic cleft in the surface of the SV nucleocapsid (6, 17, 19, 24, 35, 51). This interaction is one of the initial steps in the process of virus envelopment at the plasma membrane and is critical in the formation of a mature, infectious virus particle. Attachment of the E2 endodomain to the nucleocapsid is a primary function of this multifunctional domain. Embedded within the E2 endodomain is the signal sequence for insertion of the 6K protein into the ER membrane (20). The C terminus of the E2 endodomain also contains the signalase recognition motif necessary for removal of 6K protein from the E2 protein.
The binding of the E2 endodomain to the preformed nucleocapsid appears to be a two-step event (22). The first step involves binding of the C-terminal portion of the endodomain to the nucleocapsid, followed by a reorientation of the E2 endodomain within the capsid cleft in which E2 Y400 is positioned in close proximity to C Y180 and W247 (19). The mutants analyzed here were targeted to a specific region of the E2 tail identified by in vitro peptide-binding experiments, specifically the domain encompassing residues 408 to 415 within the E2 tail. The protection of the peptide containing capsid Y180 by sequences from the E2 endodomain could result from the conformational change proposed by Lee et al. (19), which causes Y180 to reposition into the hydrophobic cleft in the capsid protein, which is the proposed binding site for the E2 endodomain. Alternatively, the E2 peptides could bind directly to the region containing Y180 and cover it. We favor the first of these explanations since the region of the E2 endodomain around E2 Y400 appears to be the sequence that interacts with capsid Y180 (19) and peptides containing this sequence were not found to produce protection (Table 2). We suggest that the distal region of the E2 endodomain interacts at some site on the capsid protein preparing the hydrophobic cleft in the capsid protein to receive the E2 Y400 region of the endodomain by the conformational change at capsid Y180. This sequence of the E2 endodomain is also the region predicted to be involved in the initial step of capsid binding.
Substitutions of single amino acid residues in the E2 endodomain (I408A, T410A, S411A, and A413G) had little effect on the ability of SV to assemble and produce infectious particles. Two of the substitutions, at positions 408 and 410, had the greatest effect, a reduction of 2 to 3 orders of magnitude and 1 order of magnitude, respectively, in virus production. However, substitutions at positions 411 and 413 had no effect on infectious virus production. In addition, none of these substitution mutants were defective in virus protein production or processing. Despite the fact that three of the four substitutions introduced significant changes in the atomic density of the E2 endodomain, little change in virus production was noted, indicating that the sequence in this region of the E2 endodomain is not a critical factor in the interaction with the nucleocapsid.
If sequence is not a determining factor in the binding of the E2 endodomain, the length of the tail could be a determining factor in binding. The deletion mutants in the present study shorten the length of the E2 tail by 2, 3, and 4 aa (Δ406-407, Δ409-411, and Δ414-417, respectively). It was our hypothesis that these deletions in the E2 endodomain would interfere with the initial step in E2-nucleocapsid binding and that these deletions might also aid in identifying the region of the endodomain which interacted with the ER membrane during the initial stages of polyprotein translocation. The phenotypes of these deletion mutations might also aid in elucidating the role of the composition of the host-derived lipid bilayer in the integration of the polyprotein into the host membrane and how this domain participates in the assembly of infectious virions.
The deletions described here are located in a region of E2 that is predicted to initially be buried in the membrane of the ER when the polyprotein is inserted (23). It is not known at what point in the secretory pathway this domain is extracted from the membrane; however, extraction occurs after export from the ER but prior to arrival at the plasma membrane (23). Extraction of this domain and the proteolytic processing of PE2 produce a mature E2 protein whose primary function is to associate with the preassembled nucleocapsid. We have observed that the larger deletions of 3 and 4 aa expressed in mutants Δ409-411 and Δ414-417, respectively, are lethal mutations. The larger deletion mutants prevent infectious virus production in both mammalian and insect cells, whereas the smaller deletion, Δ406-407, produces infectious virus from both cell types. Electron microscopy has revealed that these mutants do assemble nucleocapsids in mammalian cells; however, they are not capable of binding membranes, and the assembled nucleocapsids are found free in the cytoplasm. It is possible that the defect in capsid binding seen with these deletion mutants is failure to attach nucleocapsids to the truncated endodomain or a failure to extract the E2 tail from the membrane and expose it to the cytoplasm. This hypothesis will be tested by using a clone of SV that allows the extraction of the tail to be monitored (23).
In mammalian cells we observed that nucleocapsids are assembled inside cells and that the E3 protein is released into the media in equivalent amounts for each of the deletion mutants. This indicates that deletions made in the C-terminal portion of the E2 endodomain (specifically between aa 406 and 417) have little effect on the insertion, processing, and transport of the structural proteins of SV in mammalian cells. Collectively, the evidence indicates that aa 406 to 417 in the E2 endodomain are critical specifically for nucleocapsid envelopment in BHK cells.
The deletion mutants described above were found to display a phenotype similar to that seen in mammalian cells for the production of infectious virus when transfected into insect cells. Significant infectious virus production was observed for the double deletion, but in the case of the larger deletions no infectious virus was produced. However, electron microscopy revealed that assembled virus particles or virus-like particles were present within the vesicles of insect cells infected with each of the deletion mutants. This indicates that the larger of these deletion mutants can assemble virus-like particles in the insect cells, a circumstance not seen in mammalian cells. It is possible that the difference in membrane composition between insect and mammalian cells is responsible for this contrastive result. These data suggest that in both cell types, mammalian and insect, the length of the carboxyl-terminal portion of the E2 tail is a critical factor. The sequence within that portion of the tail can be altered with little or no effect on virus assembly and production. However, shortening the tail even by only 2 aa can alter virus production significantly in both mammalian and insect cells.
Mosquito membranes do not contain cholesterol which, among other effects, makes them significantly thinner than cholesterol-rich mammalian cell membranes (2, 8, 26, 30). This feature may allow a shortened membrane-spanning domain to be successfully inserted into the membranes of these cells but not the membranes of mammalian cells. Although virus-like particles can be identified in insect cells infected with the larger deletions, no infectivity can be recovered from these cells, indicating that the assembly process is defective in some important and unknown aspect. Collectively, these observations support the notion that there may be a more fundamental difference in the assembly of SV in insect cells compared to the assembly of SV in mammalian cells than previously thought.
Acknowledgments
This research was supported by a grant from the Foundation for Research, Carson City, Nev., and by the North Carolina Agricultural Research Service.
We thank Richard Kuhn (Purdue University) and James Strauss (California Institute of Technology) for providing the capsid protein and E2 endodomain peptides used in this study.
REFERENCES
- 1.Anthony, R. P., and D. T. Brown. 1991. Protein-protein interactions in an alphavirus membrane. J. Virol. 65:1187-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bretscher, M. S. 1993. Cholesterol and the Golgi apparatus. Science 261:1280-1281. [DOI] [PubMed] [Google Scholar]
- 3.Brown, D. T., and L. D. Condreay. 1986. Replication of alphaviruses in mosquito cells, p. 171-207. In M. J. Schlesinger (ed.), The Togaveriridae and Flaviviridae. Plenum Publishing Corp., New York, N.Y.
- 4.Brown, D. T., M. R. F. Waite, and E. R. Pfefferkorn. 1972. Morphology and morphogenesis of Sindbis virus as seen with freeze-etching techniques. J. Virol. 10:534-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carleton, M., H. Lee, M. Mulvey, and D. T. Brown. 1997. Role of glycoprotein PE2 in the formation and maturation of the Sindbis virus spike. J. Virol. 71:1558-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng, R. H., R. J. Kuhn, N. Olsen, M. G. Rossman, H.-K. Choi, T. J. Smith, and T. S. Baker. 1995. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell 80:621-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clayton, R. B. 1964. The utilization of sterols by insects. J. Lipid Res. 5:3-19. [PubMed] [Google Scholar]
- 8.Cleverley, D., H. Geller, and J. Lenard. 1997. Characterization of cholesterol-free insect cells infectible by baculoviruses: effects of cholesterol on VSV fusion and infectivity and on cytotoxicity induced by influenza M2 protein. Exp. Cell Res. 233:288-296. [DOI] [PubMed] [Google Scholar]
- 9.Coombs, K., and D. T. Brown. 1987. Topological organization of Sindbis virus capsid protein in isolated nucleocapsids. Virus Res. 7:131-149. [DOI] [PubMed] [Google Scholar]
- 10.Ferreira, D. F., R. Hernandez, M. Horton, and D. T. Brown. 2003. Morphological variants of Sindbis virus produced by a mutation in the capsid protein. Virology 307:54-66. [DOI] [PubMed] [Google Scholar]
- 11.Gaedigk-Nitschko, K., and M. J. Schlesinger. 1991. Site-directed mutations in Sindbis virus E2 glycoprotein's cytoplasmic domain and the 6K protein lead to similar defects in virus assembly and budding. Virology 183:206-214. [DOI] [PubMed] [Google Scholar]
- 12.Gliedman, J. B., J. F. Smith, and D. T. Brown. 1975. Morphogenesis of Sindbis virus in cultured Aedes albopictus cells. J. Virol. 16:913-926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernandez, R., D. Ferreira, C. Sinodis, K. Litton, and D. T. Brown. 2005. Single amino acid insertions at the junction of the Sindbis virus E2 transmembrane domain and endodomain disrupt virus envelopment and alter infectivity. J. Virol. 79:7682-7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez, R., H. Lee, C. Nelson, and D. T. Brown. 2000. A single deletion in the membrane-proximal region of the Sindbis virus glycoprotein E2 endodomain blocks virus assembly. J. Virol. 74:4220-4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez, R., C. Sinodis, M. Horton, D. Ferreira, C. Yang, and D. T. Brown. 2003. Deletions in the transmembrane domain of a Sindbis virus glycoprotein alter virus infectivity, stability, and host range. J. Virol. 77:12710-12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivanova, L., and M. J. Schlesinger. 1993. Site-directed mutations in the Sindbis virus E2 glycoprotein identify palmitoylation sites and affect virus budding. J. Virol. 67:2546-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee, H., and D. T. Brown. 1994. Mutations in an exposed domain of Sindbis virus capsid protein result in the production of noninfectious virions and morphological variants. Virology 202:390-400. [DOI] [PubMed] [Google Scholar]
- 18.Lee, H., P. D. Ricker, and D. T. Brown. 1994. The configuration of Sindbis virus envelope proteins is stabilized by the nucleocapsid protein. Virology 204:471-474. [DOI] [PubMed] [Google Scholar]
- 19.Lee, S., K. E. Owen, H. K. Choi, H. Lee, G. Lu, G. Wengler, D. T. Brown, M. G. Rossmann, and R. J. Kuhn. 1996. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure 4:531-541. [DOI] [PubMed] [Google Scholar]
- 20.Liljestrom, P., and H. Garoff. 1991. Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J. Virol. 65:147-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, L. N., H. Lee, R. Hernandez, and D. T. Brown. 1996. Mutations in the endo domain of Sindbis virus glycoprotein E2 block phosphorylation, reorientation of the endo domain, and nucleocapsid binding. Virology 222:236-246. [DOI] [PubMed] [Google Scholar]
- 22.Liu, N., and D. T. Brown. 1993. Phosphorylation dephosphorylation events play critical roles in Sindbis virus maturation. Virology 196:703-711. [DOI] [PubMed] [Google Scholar]
- 23.Liu, N., and D. T. Brown. 1993. Transient translocation of the cytoplasmic (endo) domain of a type I membrane glycoprotein into cellular membranes. J. Cell Biol. 120:877-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez, S., J. S. Yao, R. J. Kuhn, E. G. Strauss, and J. H. Strauss. 1994. Nucleocapsid-glycoprotein interactions required for assembly of alphaviruses. J. Virol. 68:1316-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo, T., and D. T. Brown. 1993. Purification and characterization of a Sindbis virus-induced peptide which stimulates its own production and blocks virus RNA synthesis. Virology 194:44-49. [DOI] [PubMed] [Google Scholar]
- 26.Luukkonen, A., M. Brummer-Korvenkontio, and O. Renkonen. 1973. Lipids of cultured mosquito cells (Aedes albopictus): comparison with cultured mammalian fibroblasts (BHK 21 cells). Biochim. Biophys. Acta 326:256-261. [DOI] [PubMed] [Google Scholar]
- 27.Mann, E., J. Edwards, and D. T. Brown. 1983. Polycaryocyte formation mediated by Sindbis virus glycoproteins. J. Virol. 45:1083-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller, M. L., and D. T. Brown. 1992. Morphogenesis of Sindbis virus in three subclones of Aedes albopictus (mosquito) cells. J. Virol. 66:4180-4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitsuhashi, J., and K. Maramorosch. 1964. Leafhopper tissue culture: embryonic, nymphal, and imaginal tissues from aseptic insects. Contrib. Boyce Thompson Inst. 22:435-460. [Google Scholar]
- 30.Mitsuhashi, J., S. Nakasone, and Y. Horie. 1983. Sterol-free eukaryotic cells from continuous cell lines of insects. Cell Biol. Int. Rep. 7:1057-1062. [DOI] [PubMed] [Google Scholar]
- 31.Mulvey, M., and D. T. Brown. 1996. Assembly of the Sindbis virus spike protein complex. Virology 219:125-132. [DOI] [PubMed] [Google Scholar]
- 32.Mulvey, M., and D. T. Brown. 1994. Formation and rearrangement of disulfide bonds during maturation of the Sindbis virus E1 glycoprotein. J. Virol. 68:805-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mulvey, M., and D. T. Brown. 1995. Involvement of the molecular chaperone BiP in maturation of Sindbis virus envelope glycoproteins. J. Virol. 69:1621-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson, S., R. Hernandez, D. Ferreira, and D. T. Brown. 2005. In vivo processing and isolation of furin protease-sensitive alphavirus glycoproteins: a new technique for producing mutations in virus assembly. Virology 332:629-639. [DOI] [PubMed] [Google Scholar]
- 35.Owen, K. E., and R. J. Kuhn. 1997. Alphavirus budding is dependent on the interaction between the nucleocapsid and hydrophobic amino acids on the cytoplasmic domain of the E2 envelope glycoprotein. Virology 230:187-196. [DOI] [PubMed] [Google Scholar]
- 36.Owen, K. E., and R. J. Kuhn. 1996. Identification of a region in the Sindbis virus nucleocapsid protein that is involved in specificity of RNA encapsidation. J. Virol. 70:2757-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paredes, A. M., D. T. Brown, R. Rothnagel, W. Chiu, R. J. Schoepp, R. E. Johnston, and B. V. Prasad. 1993. Three-dimensional structure of a membrane-containing virus. Proc. Natl. Acad. Sci. USA 90:9095-9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paredes, A. M., D. Ferreira, M. Horton, A. Saad, H. Tsuruta, R. Johnston, W. Klimstra, K. Ryman, R. Hernandez, W. Chiu, and D. T. Brown. 2004. Conformational changes in Sindbis virions resulting from exposure to low pH and interactions with cells suggest that cell penetration may occur at the cell surface in the absence of membrane fusion. Virology 324:373-386. [DOI] [PubMed] [Google Scholar]
- 39.Pletnev, S. V., W. Zhang, S. Mukhopadhyay, B. R. Fisher, R. Hernandez, D. T. Brown, T. S. Baker, M. G. Rossmann, and R. J. Kuhn. 2001. Locations of carbohydrate sites on alphavirus glycoproteins show that E1 forms an icosahedral scaffold. Cell 105:127-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Renz, D., and D. T. Brown. 1976. Characteristics of Sindbis virus temperature-sensitive mutants in cultured BHK-21 and Aedes albopictus (mosquito) cells. J. Virol. 19:775-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rice, C. M., R. Levis, J. H. Strauss, and H. V. Huang. 1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 61:3809-3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rietveld, A., S. Neutz, K. Simons, and S. Eaton. 1999. Association of sterol- and glycosylphosphatidylinositol-linked proteins with Drosophila raft lipid microdomains. J. Biol. Chem. 274:12049-12054. [DOI] [PubMed] [Google Scholar]
- 43.Ryan, C., L. Ivanova, and M. J. Schlesinger. 1998. Effects of site-directed mutations of transmembrane cysteines in Sindbis virus E1 and E2 glycoproteins on palmitylation and virus replication. Virology 249:62-67. [DOI] [PubMed] [Google Scholar]
- 44.Ryan, C., L. Ivanova, and M. J. Schlesinger. 1998. Mutations in the Sindbis virus capsid gene can partially suppress mutations in the cytoplasmic domain of the virus E2 glycoprotein spike. Virology 243:380-387. [DOI] [PubMed] [Google Scholar]
- 45.Sarkar, G., and S. S. Sommer. 1990. The “megaprimer” method of site-directed mutagenesis. BioTechniques 8:404-407. [PubMed] [Google Scholar]
- 46.Skoging, U., M. Vihinen, L. Nilsson, and P. Liljestrom. 1996. Aromatic interactions define the binding of the alphavirus spike to its nucleocapsid. Structure 4:519-529. [DOI] [PubMed] [Google Scholar]
- 47.Strauss, E. G., C. M. Rice, and J. H. Strauss. 1984. Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology 133:92-110. [DOI] [PubMed] [Google Scholar]
- 48.Strauss, J. H., and E. G. Strauss. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weiss, B., U. Geigenmuller-Gnirke, and S. Schlesinger. 1994. Interactions between Sindbis virus RNAs and a 68 amino acid derivative of the viral capsid protein further defines the capsid binding site. Nucleic Acids Res. 22:780-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.West, J., and D. T. Brown. 2006. The role of a conserved tripeptide in the endodomain of Sindbis virus glycoprotein E2 in virus assembly and function. J. Gen. Virol. 87:657-664. [DOI] [PubMed] [Google Scholar]
- 51.Wilkinson, T. A., T. L. Tellinghuisen, R. J. Kuhn, and C. B. Post. 2005. Association of Sindbis virus capsid protein with phospholipid membranes and the E2 glycoprotein: implications for alphavirus assembly. Biochemistry 44:2800-2810. [DOI] [PubMed] [Google Scholar]