

Abstract
Papillomaviruses are a group of small non-enveloped DNA tumor viruses whose infection usually causes benign epithelial lesions (warts). Certain types of HPVs, such as HPV-16, HPV-18, and HPV-31, have been recognized as causative agents of cervical cancer and anal cancer and their infections, which arise via sexual transmission, are associated with more than 95% of cervical cancer. Papillomaviruses infect keratinocytes in the basal layer of stratified squamous epithelia and replicate in the nucleus of infected keratinocytes in a differentiation-dependent manner. Viral gene expression in infected cells depends on cell differentiation and is tightly regulated at the transcriptional and post-transcriptional levels. A noteworthy feature of all papillomavirus transcripts is that they are transcribed as a bicistronic or polycistronic form containing two or more ORFs and are polyadenylated at either an early or late poly(A) site. In the past ten years, remarkable progress has been made in understanding how this complex viral gene expression is regulated at the level of transcription (such as via DNA methylation) and particularly post-transcription (including RNA splicing, polyadenylation, and translation). Current knowledge of papillomavirus mRNA structure and RNA processing has provided some clues on how to control viral oncogene expression. However, we still have little knowledge about which mRNAs are used to translate each viral protein. Continuing research on post-transcriptional regulation of papillomavirus infection will remain as a future focus to provide more insights into papillomavirus-host interactions, the virus life-cycle, and viral oncogenesis.
Keywords: papillomaviruses, gene expression, RNA splicing, RNA polyadenylation, posttranscriptional regulation
2. INTRODUCTION
Papillomaviruses are a group of small non-enveloped DNA tumor viruses with a virion size of ∼55 nm in diameter. This group of viruses infects various animals from birds to mammals, including humans. Papillomaviruses usually cause benign tumor but sometimes also cause malignancies. To date, more than one hundred human and animal papillomavirus genotypes (types) have been completely sequenced. It is likely that additional types will be added as additional papillomavirus genomes are cloned and sequenced. Recently, the International Committee on the Taxonomy of Viruses (ICTV) has accepted that papillomaviruses are a distinct taxonomic family, the Papillomaviridae, and are unrelated to the polyomaviruses and SV40. The taxonomic status of papillomavirus types, subtypes, and variants is based on the sequence of their L1 genes which differ from each other by at least 10%, 2-10%, and maximally 2%, respectively [1]. The bovine papillomavirus type 1 (BPV-1) and human papillomavirus type 1a (HPV-1a) genomes were the first papillomavirus genomes to be completely sequenced [2;3]. For the past two decades, BPV-1 has served as a prototype for studies of the molecular biology of papillomaviruses.
Papillomaviruses replicate and assemble exclusively in the nucleus. Virus infects the keratinocytes in the basal layers of a stratified squamous epithelium. However, viral gene expression and replication proceed in a tightly controlled fashion regulated by keratinocyte differentiation. Although the mechanism on how keratinocyte differentiation regulates HPV gene expression is not fully understood, there is general agreement that viral gene expression leads to the expression of six nonstructural viral regulatory proteins (E1, E2, E4, E5, E6 and E7) from the early region of the viral genome in undifferentiated or intermediately differentiated keratinocytes and two structural viral capsid proteins (L1 and L2) from the late region of the genome in keratinocytes undergoing terminal differentiation (Fig. 1). The E4 protein continues to be expressed in the terminally differentiated keratinocytes. E1 and E2 are involved in viral DNA replication and the regulation of early transcription. E4 expressed in a productive infection associates with cytokeratin filament collapse. E5, E6, and E7 are viral oncogenes and their expression induces cell immortalization and transformation. In particular, E6 and E7 are two viral oncoproteins that inactivate, respectively, p53 and pRb, two cellular tumor suppressor proteins.
Fig. 1.
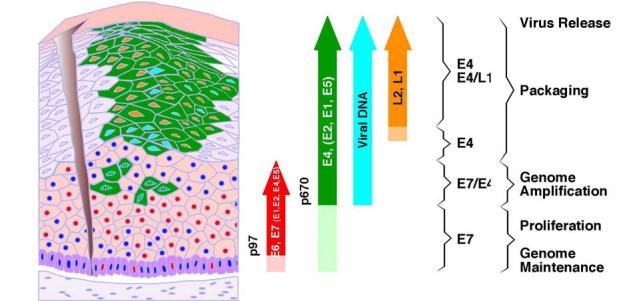
Orchestrated viral gene expression in productive papillomavirus infection. The differentiated epithelium is represented diagrammatically on the left, and the markers expressed are represented as arrows on the right. Following access to the basal layer, the virus is thought to establish itself as a low_copy_number episome. The E6 and E7 proteins (red nuclei) are expressed from the early promoter, P97 in HPV-16, in the lower epithelial layers (red arrow). Dark blue circles represent nuclei of uninfected or nonpermissive cells. The expression of E4, and presumably that of other proteins (green) whose genes lie downstream of the differentiation_dependent late promoter (P670 in HPV-16) (green arrow), is triggered before the expression of E6 and E7 ceases. The E4_expressing cells (green) that also express E7 are predicted to contain all the early transcription products to facilitate genome amplification. Expression of the virus capsid proteins (L1 and L2) (orange arrow) follows the completion of genome amplification and occurs in a subset of cells that express E4. The precise stage in the virus life cycle supported at any particular point is apparent from the combination of markers expressed (modified with permission from John Doobar [151]).
Although papillomavirus infections usually result in benign lesions, human papillomavirus (HPV) infection sometimes progresses to the development of malignant lesions. Certain types of HPVs, such as HPV-16, HPV-18, and HPV-31, which are designated “high-risk” or “oncogenic,” have been recognized as causative agents of cervical and anal cancers. These sexually transmitted viruses are associated with more than 95% of cervical cancer. Among those high-risk HPVs, HPV-16 alone is responsible for development of 58.9% of cervical cancer[4]. This knowledge has led to the development not only of HPV vaccines [5;6], but also of cervical cancer screening strategies which incorporate HPV testing[7;8]. A characteristic consequence of cancer associated with persistent infection by these high-risk HPVs is that viral genomes are commonly found integrated into the cancer cell genome in such a way as to disrupt the viral E2 gene. Other types of HPVs, designated “low-risk” or “non-oncogenic,” such as HPV-6 and HPV-11, induce benign condylomata acuminata and are very rarely found in genital malignancies. E6 and E7 from low-risk HPVs, inactivate cellular p53 and pRb tumor suppressor proteins less efficiently than do E6 and E7 from high-risk HPVs[9].
3. PAPILLOMAVIRUS GENOME STRUCTURE, GENE TRANSCRIPTION AND TRANSCRIPTION MAP
All papillomaviruses contain a double-stranded, circular DNA genome approximately 8 kb in size that can be divided, in general, into three major regions: early, late, and a long control region (LCR or noncoding region [NCR]). The three regions in all papillomaviruses are separated by two polyadenylation (pA) sites: early pA (AE) and late pA (AL) sites (Fig. 2). The early region of papillomavirus genomes occupies over 50% of the virus genome from its 5′ half and encodes six common open reading frames (E1, E2, E4, E5, E6 and E7) [3] that translate individual proteins as briefly described above. Two other ORFs, E3 and E8, were also assigned to this region initially, but only the E8 ORF in BPV-1 [10-12] and HPV-31 [13;14] has been proven to encode a protein, a spliced E8^E2C fusion protein, which functions as a negative regulator of viral transcription and replication. Unlike BPV-1 and HPV-31, E8 in several rabbit papillomaviruses have been characterized as an oncogene, with features similar to those of the E5 of both BPV-1 and several HPVsHPV-31[15;16]. The late region of all papillomavirus genomes, covering almost 40% of the virus genome, lies downstream of the early region and encodes L1 and L2 ORFs for translation of a major (L1) and a minor (L2) capsid protein. The LCR region, a segment of about 850 bp (10% of the HPV genome), has no protein-coding function, but bears the origin of replication as well as multiple transcription factor binding sites that are important in regulation of RNA polymerase II-initiated transcription from viral early as well as late promoters. How these transcription factor binding sites are involved in regulation of papillomavirus gene expression has been summarized in a recent comprehensive review [17]
Fig. 2.

Genome structure and transcription map of HPV-16. The bracket line in the middle of the panel represents a linear form of the virus genome for better presentation of head-to-tail junction, promoters (arrows), and early (AE) and late (AL) polyadenylation sites. The open reading frames (ORFs) (open boxes) are diagramed above the bracket and the numbers above each ORF are the nucleotide positions of the first nucleotide of the start codon and last nucleotide of the stop codons in the HPV-16 genome. The E4 ORF spans two exons and formation of the intact E4 ORF requires RNA splicing (dashed lines). LCR indicates a long control region. Below the bracket line are the reported RNA species derived from alternative promoter usage and alternative splicing. Exons (heavy lines) and introns (thin lines) are illustrated on each species of the RNA, with splice site positions being numbered by nucleotide positions in the virus genome. Although all early transcripts driven by early promoter P97 are illustrated with a 5′-end from nt 97, multiple transcription starting sites in W12 cells have been mapped by 5′ RACE at nt 83 (frequency[F] 3), 91(F2), 93 (F2), 94(F1), 95(F4), 96(F8) (Carl C. Baker, unpublished data). Similarly, all Late transcripts driven by differentiation-inducible promoter P670 are shown for convenience with a 5′-end from nt 670, but the real 5′ ends in W12 cells have been mapped by 5′ RACE to nt 682(F4), 713(F5), and 774(F3) (Carl C. Baker, unpublished data).
In general, each ORF in a papillomavirus genome is often referred to as a gene. However, a gene, in molecular terms, is defined as the entire nucleic acid sequence that is necessary for the synthesis of a functional transcript. According to this definition, a gene is not equivalent to an ORF. In eukaryotes and many viruses, a gene usually contains exons and introns. An ORF encoding a polypeptide is usually spread across multiple exons from various parts of the genome which are combined into a full-length ORF through RNA splicing. This is particularly true in papillomaviruses even though only one ORF (E1^E4) in papillomaviruses spans two separate exons. In fact, extensive mRNA mapping and analysis of BPV-1 transformed C127 cells as well as productively infected bovine fibropapillomas has led to the conclusion that each transcript from the BPV-1 genome could be bicistronic, tricistronic, or even polycistronic with two or more ORFs. On the other hand, a particular ORF could exist in multiple mRNAs transcribed from different promoters. Until today, the transcription map of BPV-1 has been the best known map for our understanding of the complexity of papillomavirus gene expression and has been summarized in the HPV compendium[18]. However, to determine which transcripts encode which viral proteins has been a challenge and we still have only a very limited knowledge of which proteins are translated from each transcript. Recent HPV-16 transcript mapping and gene expression studies have shed some light on this problem.
Although there has been some recent progress on characterization of new promoters and minor transcripts from HPV-11 and HPV-31[19-22], this review will emphasize HPV-16 and update the HPV-16 transcription map. The HPV-16 genome contains two major promoters. The P97 promoter lies upstream of the E6 ORF and is responsible for almost all early gene expression [23]. The P670 promoter lies within the E7 ORF region and is responsible for late gene expression[24]. Although other minor promoters in the early regions of the genome[25] have been described, their activities in the context of an episomal HPV genome remains unknown. The HPV-16 P97 promoter, equivalent to P99 in HPV-31 [26;27] and P105 in HPV-18 [28-30], is very potent and tightly controlled, primarily by upstream cis-elements in the LCR. These cis-elements, including four consensus E2-binding sites (E2-BSs), ACC(N6)GGT [31-33], interact with cellular transcription factors and the viral transactivator/repressor E2 and regulate the transcription of P97 from undifferentiated basal cells to highly differentiated keratinocytes[17]. However, recent studies indicate that E2 functions as a repressor for P97 transcription at steps after TBP or TFIID binding [34] and its transcriptional repression only occurs in cells harboring integrated, but not episomal HPV-16 DNA [35]. The resulting early primary transcripts (pre-mRNAs) all have three exons and two introns, which undergo alternative RNA splicing and are polyadenylated at nt 4215. As shown in the figure 2, each intron of the early transcripts could be spliced out through utilization of three alternative 3′ splice sites at nt 409, 526 or 742 in intron 1 [36-38], and three other alternative 3′ ss at nt 2582, 2709, 3358 in intron 2[39-41], leading to the production of at least 14 species of mRNA transcripts with various coding potential.
The HPV-16 P670 promoter[24] is a late-promoter. Its activity can be induced only in differentiated keratinocytes. The similar activity of late promoter P742 in HPV-31[26;27] could be induced during vegetative replication of HPV-31 in raft cultures. In other DNA viruses, the late promoter activation always requires viral lytic DNA replication [42;43]. Although transcription of late promoter P742 in HPV-31 has been related to the protein kinase C pathway [44;45], it remains puzzling on how the late promoter is activated. A recent study suggests that cell differentiation signals alone are sufficient to activate transcription from the late promoter P742, although DNA amplification is also required for maximal activity of the late promoter [45]. A cis element located in the E6 and E7 coding region appears to regulate the late promoter [45;46]. Nevertheless, both the late P670 promoter in HPV-16 and P742 in HPV-31 are positioned in the E7 coding region and transcription from the late promoter has to bypass the early pA site to allow expression of the late region. As a result, a true late pre-mRNA is a chimeric transcript of early and late regions, with the early region in its 5′ half and the late region in its 3′ half. This late pre-mRNA can be processed into early region transcripts which are cleaved and polyadenylated at the early poly(A) site (transcripts P and Q) and late region transcripts which are cleaved and polyadenylated at the late poly(A) site (transcripts R, S, and T) (Fig. 2). In contrast to early transcripts, the late region transcripts are detectable only in differentiated keratinocytes with vegetative DNA replication[47].
4. DNA METHYLATION IN PAPILLOMAVIRUS GENE EXPRESSION
DNA methylation regulates cellular and viral gene expression since introduction of methyl groups to cytosine residues at CpG dinucleotides leads to transcriptional silencing. This is mainly because DNA methylation in a promoter region prevents certain transcription factors from accessing their binding sites. Alternatively, methylated CpG dinucleotides interact with methyl CpG binding proteins which recruit histone deacetylase in association with chromatin. Consequently, deacetylation of histone leads to changes in the chromatin configuration that are refractory to transcription.
Methylation of papillomavirus DNA was described some 20 years ago [48-50], but significance of the DNA methylation in papillomavirus gene expression and carcinogenesis was not fully understood[51-54]. It appears that CpG methylation in HPV-16 and HPV-18 genomes occurs more often in LCR regions and part of L1 ORF region than in any other parts of the virus genome in cervical cancer cell lines[55;56]. These are the regions that lie upstream of the P97 promoter and are therefore not transcribed when the HPV DNA is integrated within the early region. CpG methylation in E2BS prevents E2 protein binding in vitro[54] and modulates E2 protein function in cells in transcription activation[57]. Interestingly, methylation of integrated HPV DNA in cervical cancer cell lines and HPV-associated primary cancers prevents most viral DNA from transcription activation and leaves only one or at most two transcription center (s) per cell being active in the perinucleolar region[58].
CpG methylation appears in correlation to HPV pathogenesis and an efficient methylation of HPV DNA might associate with progression of cervical cancer development. CpG methylation was found to be more common in carcinomas than in dysplasias [59]. This important observation has been extended to cells of anal epithelia infected with HPV-16. In low-grade anal intraepithelial neoplasia, methylation is high in CpGs overlapping the viral enhancer, but rare in the promoter. In high-grade anal intraepithelial neoplasia, CpG methylation is high in promoter regions [60]. However, DNA methylation in cancers appears not to be specific for HPV DNA, rather occurs as a frequent event throughout the host genome. The frequency of hypermethylation of many cellular genes has been found being increased significantly with increasing severity of neoplasia[61;62].
5. RNA SPLICING IN PAPILLOMAVIRUS GENE EXPRESSION
RNA splicing regulates gene expression at the posttranscriptional level and plays an important role during the papillomavirus life cycle. RNA splicing to remove an intron and to coordinately ligate individual exons involves five small U RNAs (U1, U2, U4, U5, and U6) and many splicing factors. Classical intronic splicing signals consist of three cis-elements: a 5′ splice site (5′ss), a branch site, and a 3′ splice site (3′ss) containing a run of 15 to 40 pyrimidines (usually U′s) called a polypyrimidine tract (PPT). Defining the exon-intron boundary is the first step in the accurate recognition of an intron 5′ ss and an intron 3′ ss. This involves interaction of the 5′ ss with U1 snRNP, of the branch site with U2 snRNP, and of the 3′ ss with U2AF65 and U2AF35 (a heterodimer of U2 auxiliary factors), modulated by many cellular splicing factors including serine/arginine-rich (SR) proteins. SR proteins are a growing family of structurally related and highly conserved cellular splicing factors that are characterized by the presence of an RNA-recognition motif (RRM) and Ser/Arg (SR) dipeptides. Today, more than 11 members have been included in the family (62a). Since the majority of viral and mammalian RNA introns are suboptimal, lacking consensus sequences in their splice sites, how these suboptimal splice sites are selected correctly during RNA splicing has been a major focus of splicing research in the past decade. This effort led to the discovery of exon recognition elements, which were later renamed exonic splicing enhancers (ESEs). Subsequently, exonic splicing suppressors or silencers (ESS) were also identified. Intronic splicing enhancers and silencers, in addition to conventional splicing signals, were also described. These have been summarized extensively in two recent reviews [63;64].
As described above, almost all papillomavirus transcripts are polycistronic, with multiple exons and introns. These polycistronic transcripts feature suboptimal splicing signals that undergo extensive alternative RNA splicing. Papillomaviruses encode multiple proteins in a very compact genome and take advantage of alternative splicing to differentially express these proteins in a cell differentiation-specific and temporal manner. Regulation of alternative 3′ ss selection by ESEs and ESSs has been extensively studied in the BPV-1 late pre-mRNA (Fig. 3). The primary transcripts expressed from the viral late promoter have a common late leader 5′ ss, but use a proximal (at nt 3225) or a distal (at nt 3605) 3′ ss for RNA splicing. Splicing at the proximal 3′ ss leads to the production of E4 mRNAs polyadenylated at the early poly(A) site and L2 mRNAs polyadenylated at the late poly(A) site, whereas splicing at the distal 3′ss triggers splicing of a downstream intron containing the early pA site and results in the production of late L1 mRNA. In situ hybridization studies demonstrated that the distal 3′ ss is utilized only in the granular cell layer of the epidermis, while selection of the proximal 3′ ss occurs in both the granular and spinous layers[65]. This differentiation-specific alternative 3′ ss selection is related to the suboptimal features of both the proximal and distal 3′ss and is regulated by at least five viral cis-elements; three ESEs and two ESSs have been identified to date. Among these elements, three cis-acting elements, SE1, SE2, and ESS1, are positioned between the proximal and the distal 3′ ss, with ESS1 immediately downstream of SE1 and 122 nts upstream of SE2 (Fig. 3). These elements regulate the selection of the proximal 3′ ss for RNA splicing[66-70], whereas two other elements, SE4 and ESS2, are located downstream of the distal 3′ ss and control selection of the distal 3′ ss[71]. Thus, the five cis-elements interact in a coordinate fashion to specify which 3′ ss should be used. Any subtle changes that disrupt this coordinate balance can provoke a switch from one 3′ ss to another. Multiple cellular splicing factors have been found to interact with those ESEs and ESSs as shown in Fig. 3 and to play important roles in governing the selection of alternative 3′ ss. It is our belief that regulation of the usage of two alternative 3′ ss in the splicing of BPV-1 late pre-mRNAs is an intricate network of viral exonic cis-elements which is governed closely by cellular splicing factors in accordance with the stage of keratinocyte differentiation.
Fig. 3.

Positive and negative regulation of alternative splice site selection by ESE and ESS elements. (A and B) Production of BPV-1 and HPV-16 L1 and L2 are regulated by alternative RNA splicing through multiple cis-elements: exonic splicing enhancers SE1, SE2 or SE4 in BPV-1 [66;67;69-71;152] or ESE in HPV-16 [72] and exonic splicing silencers ESS1 or ESS2 in BPV-1 [66;68;70;71] or ESS in HPV-16 [73]. These cis-elements function in regulation of RNA splicing through their interactions with different trans-acting factors. It should be noted that many viral sequences, including the L2 ORF, can serve as both exon and intron (heavy lines). (C) Alternative 3′ splice site selection in expression of HPV-16 early transcripts. Diagram shows each intron has three alternative 3′ splice sites in a HPV-16 early pre-mRNA that contain all early ORFs (see their relative positions in a pre-mRNA in Fig. 2). The cap-dependent splicing of intron 1 has been documented biochemically[36]. Numbers above or below the RNA diagrams A, B, and C are nucleotide positions of either RNA 5′ ends, 5′ splice sites, 3′ splice sites or polyadenylation sites. A slash between two numbers indicates a splice junction.
Regulation of splicing of HPV-16 late pre-mRNAs also appears to utilize splicing enhancers and silencers. HPV-16 late pre-mRNAs also have three exons and two introns as well as two potential poly(A) sites. The second intron is an alternative intron/exon for both BPV-1 and HPV-16. The late leader 5′ ss preferentially splices to the nt 3358 3′ ss to remove intron 1 and this step of splicing appears to be promoted through a 65-nt AC-rich ESE residing approximately 100 nts downstream of the nt 3358 3′ ss in exon 2[72]. Efficient recognition of the nt 3358 3′ ss might also be promoted by exon 2 definition (via binding of the U1 snRNP and other factors to the 5′ splice site at nt 3632). However, the exon definition-mediated splicing enhancement of the nt 3358 3′ ss appears to be neutralized by the presence of a splicing silencer positioned approximately 45-nts upstream of the 3632 5′ ss[72]. The presence of an ESS immediately upstream of the nt 3632 5′ ss may be important to counteract U1 recognition of the nt 3632 5′ss and enhance recognition of the early pA site at nt 4215 by the cellular polyadenylation machinery (see more discussion below). As diagramed, intron 2 retention is essential for L2 production and might be further enhanced by an hnRNP A1-binding ESS in the 3′ terminal exon of the late transcripts[73]. In spite of these observations, many questions remain. How do the ESE and ESS in exon 2 function in selection of the nt 3358 3′ ss for the intron 1 splicing and suppression of the nt 3632 5′ ss utilization for intron 2 retention? What roles do the early pA site and any putative intronic cis-elements play in splicing of the late pre-mRNAs? Does the identified ESS in the 3′-terminal exon of the late transcripts inhibit selection of the nt 5639 3′ ss by neutralizing a putative ESE’s function? And perhaps most critically, how are all these elements regulated during keratinocyte differentiation? Nevertheless, these initial reports have laid the groundwork for more mechanistic studies.
Fewer studies have been done on the regulation of the papillomaviral early gene expression at the level of RNA splicing. Early studies suggested that RNA splicing might be important for E7 expression in HPV-16[74;75] and E1 production in HPV-11[22] and HPV-31 [76]. However, what regulates or controls splicing of the viral early transcripts during papillomavirus infection has not been examined until recently. Recent studies suggest splicing of HPV-16 early transcripts are subject to regulation by capping machinery and ESEs. Unlike BPV-1 early transcripts, each of which might be transcribed from separate promoters[18], high-risk HPV early transcripts are predominantly transcribed from a single early promoter, P97 in HPV-16 and P99 in HPV-31. Each pre-mRNA consists of three exons and two introns, with three alternative 3′ ss in each intron (Fig. 3C). For example, HPV-16 E6 and E7 pre-mRNAs are transcribed from the same P97 promoter as bicistronic E6E7 pre-mRNAs, and have an intron in the E6 coding region with one 5′ splice site and three alternative 3′ splice sites. Splicing of E6E7 pre-mRNAs by alternative utilization of these three 3′ splice sites produce E6*I, E6*II, and E6^E7 mRNAs, respectively (Fig. 2). If this intron remains unspliced, the resulting E6E7 mRNA expresses oncogenic full length E6. It has been shown that the E6E7 pre-mRNA is efficiently spliced in vitro only when capped and that cellular cap-binding factors are involved in the splicing. The cap-dependent splicing of the E6E7 pre-mRNA is extremely efficient in cervical cancer-derived cells, producing mostly E6*I, but inefficient in cells infected with a common retroviral expression vector, pLXSN16E6E7, due to the expanded length of exon 1 in the pre-mRNAs expressed from this vector, suggesting that efficient splicing of the E6E7 pre-mRNA depends on the distance of the cap-proximal intron from the RNA 5′ cap[36]. Interestingly, splicing of the E6E7 RNA was shown to provide more E7 RNA templates and to promote E7 translation, whereas a lack of RNA splicing was found to produce a low level of E7 translation. It appears that HPV-16 E6E7 pre-mRNA takes advantage of its small cap-proximal exon to confer efficient splicing for better E7 expression[36]. However, overly efficient splicing of E6E7 transcripts would prevent expression of functional E6 protein. Obviously, the optimal ratio of E6 and E7 proteins depends on finely balanced E6* splicing. Whether E6* splicing is regulated during the viral life cycle remains to be determined. Preliminary studies looking for viral or cellular factors that might interact with E6E7 pre-mRNAs and prevent E6 splicing has demonstrated that viral early proteins E2 and E6 could interact preferentially with the intron 1 region of the E6E7 pre-mRNAs and prevent E6E7 splicing in vitro (Bodaghi and Zheng, unpublished data). These studies suggest that efficient intron 1 splicing of early transcripts could be regulated through viral early proteins. However, further extensive in vivo studies are needed to verify the observation and to determine whether any viral cis-elements or other cellular or viral trans-acting factors are also involved in selection of three alternative 3′ ss in intron 1. A search for mechanisms that control intron 2 splicing of the early transcripts has led to the discovery of an ESE in the 3′ terminal exon (E4 exon) of early transcripts. The importance of the newly characterized ESE in nt 3385 3′ ss selection has been verified by deletion of the ESE, which prevents usage of the nt 3358 3′ ss and polyadenylation at the early pA site [72]. However, how this ESE inhibits late gene expression remains in question and requires more careful studies.
6. RNA POLYADENYLATION IN PAPILLOMAVIRUS GENE EXPRESSION
RNA polyadenylation plays an important role in control of viral and eukaryotic gene expression. The maturation of the 3′ end of most pre-mRNAs involves cleavage of the nascent transcript and addition of a poly(A) tail (polyadenylation) with 150-200 adenylate residues. Cleavage and polyadenylation are tightly coupled events that are triggered through recognition of three RNA signals by the cellular polyadenylation machinery. These RNA signal sequences include a highly conserved hexanucleotide signal, AAUAAA, a cleavage site generally positioned 11-23 nts downstream of the AAUAAA, and a GU-rich element 10-30 nts further downstream of the cleavage site [77]. Biochemical studies have demonstrated at least five cellular factors involved in RNA polyadenylation: poly(A) polymerase (PAP), cleavage and polyadenylation specificity factor (CPSF), cleavage stimulation factor (CstF), and cleavage factors I and II (CFI and CFII). CPSF interacts with the AAUAAA signal sequence. CstF recognizes the GU-rich downstream elements in the pre-mRNA and stabilizes the binding of CPSF and enhances polyadenylation efficiency. CPSF-73, a subunit of CPSF with a putative Zn2+-dependent endonuclease domain, has recently been shown to contact the cleavage site and is thought to execute 3′ cleavage[78].
Expression of L1 and L2, the virus capsid proteins, is restricted to cells undergoing differentiation in the stratified epithelium. As discussed above, this could be partially because of cell differentiation/viral DNA replication-dependent late promoter activation[24;26;27]. However, L1 and L2 proteins are produced in the most terminally differentiated keratinocytes in the upper layers of the epithelium, while pre-mRNA containing L1 and L2 sequences can be detected in the nuclei of less-differentiated cells, strongly suggesting that late gene expression is restricted largely by post-transcriptional processes[79]. Post-transcriptional regulation of papillomavirus late gene expression is quite complex. The late transcription unit overlaps at least part of the early transcription unit and contains two polyadenylation sites (early and late) that are potentially in competition with each other (see Figure 2). In addition, the early poly(A) site lies within the second intron of the L1 pre-mRNA, so splicing and early polyadenylation compete with each other and are mutually exclusive events. Thus, an early to late switch in papillomavirus gene expression could be effected by changes in either splicing or polyadenylation, or both.
Early studies on the post-transcriptional regulation of papillomavirus late gene expression revealed negative cis-elements in the 3′ untranslated regions (UTR) of BPV-1 and HPV-16 late transcripts[80;81]. Characterization of the 51-nt BPV-1 inhibitory cis-element showed that a consensus cryptic 5′ splice site is responsible for the inhibitory activity and that binding of the U1 snRNP is essential for this function[82]. Further studies demonstrated that this element inhibits polyadenylation at the late poly(A) site through a direct interaction between the U1 70K protein and poly(A) polymerase (Fig. 4)[82;83]. This interaction is mediated by a poly(A) polymerase regulatory domain (PRD) in U170K which is shared with several other RNA processing factors such as U1A, SRp75, and U2AF65 [84]. Surprisingly, the distance from the cryptic 5′ splice site to the polyadenylation signal is not critical for inhibition as long as the 5′ splice site is positioned in the terminal exon of the pre-mRNA[85].
Fig. 4.

Suppression of late poly(A) site selection by a cryptic 5′ splice site in a 3′-terminal exon. A. Diagram shows the mechanism of how a cryptic 5′ splice site in the 3′ UTR of BPV-1 late pre-mRNA interacts with the U1 snRNA 5′ end for base-pairing and how this interaction inhibits late polyadenylation (pA late). Diagram shows that U1 snRNP suppresses RNA polyadenylation through the interaction of its U1 70K subunit with poly(A) polymerase (PAP) [83]. U1A, SRp75, and U2AF65 also contain a PAP regulatory domain and efficiently inhibit PAP in vitro and in vivo through their interactions with PAP [84]. Heavy line in the middle represents papillomavirus late transcripts, with U1 snRNP shown above the line base-pairing with a cryptic 5′ splice site (see the sequence AAG/GUAAGU below the line) in the 3′ UTR region of the late pre-mRNA. Figure is modified with permission from reference [83]. B. Four weak 5′ splice sites (diamonded on the RNA and highlighted sequences by underlines 1 to 4) that are resided in a negative regulatory element (colored) in the 3′-end of HPV-16 late pre-mRNAs are essential for repression of polyadenylation at the late poly(A) site with the 2nd 5′ splice site most suppressive[82;86].
The HPV-16 negative regulatory element (NRE) is more complex than the BPV-1 element and maps to a 79-nt sequence spanning from the 3′ end of the L1 ORF into the late 3′ UTR [81]. This element is composed of 5′ and 3′ sub-elements that can function independently. The 5′ portion is similar to the BPV-1 element and contains four weak consensus 5′ splice sites that also bind a U1 snRNP-like complex that contains the U1A protein but not the U170K protein[82;86]. The 3′ portion is GU-rich and is a less potent inhibitor. The NRE binds multiple RNA processing factors, including U2AF65, CstF-64, HuR, and SF2/ASF[86-89]. The closely related HPV-31 has an even more complex late negative regulatory element composed of a 130-nt major element upstream of the late poly(A) site and a minor 110-nt downstream element[90]. The major upstream element contains a single weak consensus 5′ splice site and also binds U2AF65, CstF-64, and HuR. The exact functions of the HPV-16 and HPV-31 NREs are not fully understood. Two of the NRE-binding proteins (U1A and U2AF65) contain known PRDs that may inhibit polyadenylation[84]. The NRE may also compete with the downstream CstF-binding polyadenylation element for CstF, leading to a less stable polyadenylation complex and reduced polyadenylation efficiency[90]. Fully processed NRE-containing HPV-16 late transcripts have also been shown to be retained in the nucleus of undifferentiated W12 cells[88].
At early stages of the viral life cycle, viral pre-mRNAs are polyadenylated almost exclusively at the early poly(A) site. Therefore the early to late switch in processing of papillomavirus pre-mRNAs requires not only the release of inhibition of the late poly(A) site, but also downregulation of polyadenylation at the early poly(A) site. In BPV-1-transformed C127 cells, about 90% of the transcripts were found to be terminated in the late region, suggesting that transcription termination might prevent late gene expression[91]. However, more recent studies suggest that termination is mechanistically linked to polyadenylation [92]. This would indicate that poly(A) site choice precedes transcription termination within the late region. Surprisingly, mutation of the early AAUAAA polyadenylation signal only minimally affects early mRNA levels, although it can significantly increase read-through into the late region. In BPV-1, essentially all mutant mRNAs are polyadenylated at an alternative site approximately 100 nt upstream of the wild type early poly(A) site which is controlled by the nonconsensus UAUAUA polyadenylation element[93]. Similar observations have been made for HPV-31 and HPV-16 [38;94], suggesting that there are very strong regulatory elements that direct polyadenylation to this region of the viral genome. In HPV-31, the early polyadenylation region contains a single consensus hexanucleotide sequence (AAUAAA) and three weak CstF binding sites, whereas the late poly(A) site contains AAUAAA and a single high affinity CstF binding site [95]. Differentiation of keratinocytes led to an approximately 50% increase in read-through accompanied by a 40% decrease of CstF protein, suggesting that CstF may play a critical role in regulating poly(A) site choice during the viral life cycle [95]. Additional studies identified a major negative regulatory element within the first 800 nts of the L2 ORF which appears to be required for the preferential use of the early poly(A) site in undifferentiated cells [94].
Recent studies demonstrated that early polyadenylation of HPV-16 pre-mRNAs is up-regulated by two distinct cis-elements flanking the early poly(A) site. Deletion of either of these elements increases read-through into the late region [38;96]. The early 3′UTR contains a 57-nt U-rich polyadenylation enhancer which binds multiple factors including hFip1, CstF-64, hnRNP C1/C2, and polypyrimidine tract binding proteins (PTB) [38]. hFip1 is a subunit of CPSF which binds to U-rich sequences and stimulates poly(A) polymerase[97]. PTB is a known regulator of splicing and has recently been shown to regulate the cleavage/polyadenylation of a terminal exon[98]. A second cis-element is located in the L2 ORF 174-nts downstream of the early poly(A) site and features six GGG motifs that bind hnRNP H and enhance use of the early poly(A) site[96;99]. Interestingly, hnRNP H levels are highest in the lower layers of cervical epithelium where use of the early polyadenylation site would predominate.
Polyadenylation and splicing are both cotranscriptional processes and can influence each other. A 65-nt AC-rich splicing enhancer located approximately 100 nts downstream of the HPV-16 nt 3358 3′ splice site both stimulates splicing at the nt 3358 3′ splice site and up-regulates polyadenylation of the early transcripts at the early poly(A) signal [72]. This leads to expression of predominantly E1^E4 early transcripts. However, L1 and L2 expression also requires splicing to the nt 3358 3′ splice site, but without activation of the early poly(A) signal. How activation of the early poly(A) site is avoided during the processing of L2 mRNA is unknown. However, for L1 mRNAs, splicing of the nt 3632 5′ splice site to the nt 5639 3′ splice site would compete with early polyadenylation since these two events are mutually exclusive. A 40-nts exonic splicing silencer has been identified immediately downstream of the 65-nt AC-rich splicing enhancer in the E4 coding region and was shown to inhibit recognition of the nt 3632 5′ splice site[72]. In addition, an hnRNP A1 binding site immediately downstream of the nt 5639 3′ splice site has been shown to negatively regulate selection of this 3′ splice site[73]. Thus, these two elements likely regulate the competition between splicing and polyadenylation.
7. RNA INSTABILITY, CODON USAGE, AND TRANSLATIONAL INITIATION CONTROL IN PAPILLOMAVIRUS GENE EXPRESSION
Many mature cellular mRNAs are translated efficiently and constitutively. However, this is not the case for most papillomavirus mRNAs. Currently, there are at least three hypotheses to explain the limited efficiency of viral protein production in papillomavirus infection: RNA instability, rare codon usage, and translational initiation control.
RNA instability was initially proposed to regulate HPV-16 late gene expression when a negative cis-element flanking the HPV-16 late poly(A) site was identified and shown to destabilize late transcripts in an in vitro mRNA decay assay[100]. However, this mRNA destabilization activity has never been confirmed in vivo and subsequent work has suggested that this element inhibits RNA polyadenylation (see above). This work triggered a series of studies that sought to identify similar RNA instability element in other papillomaviruses, leading to the identification of several additional elements which appeared to negatively regulate late gene expression. However, almost all of these elements have recently been shown to regulate RNA polyadenylation or RNA splicing rather than directly regulating late mRNA stability. For example, a 57-nt, AU-rich element in the 3′ UTR of the HPV-1 late transcripts [101;102] and a cis-element in the first 514-nts of HPV-16 L1 ORF [103] were initially identified as inhibitory elements that appear to target mRNAs for degradation and block HPV-1 and HPV-16 L1 expression. The inhibitory effects of these elements can be overcome by HIV Rev and RRE (Rev-responsive element) or CTE (constitutive transport element) of simian retrovirus type 1 (SRV-1)[101;104], suggesting a blockade of RNA export-related events instead. Subsequently, the HPV-16 negative element has been characterized to be an exonic splicing silencer that inhibits splicing of HPV-16 late pre-mRNAs [38]. Another example is the finding of two distinct negative RNA elements in the HPV-16 L2 transcript, one at the 5′ end and another in the middle region of the L2 ORF, that reduce L2 mRNA levels and inhibit the use of the mRNA for translation in vitro [105-107]. In contrast to the L1 inhibitory element, the inhibitory function of the two negative elements in the L2 transcript can not be overridden by the SRV-1 CTE [107]. Recently, the element at the 5′ end of L2 was found to be a polyadenylation element containing multiple triple G motifs that interacts with hnRNP H and promote early polyadenylation[96].
Inefficient translation of some papillomavirus transcripts in mammalian cells has been partially attributed to the presence of rare codons in those papillomavirus ORFs. Abundance of the corresponding tRNAs in those cells or tissues was thought to be a limiting factor for the translational of these proteins. This hypothesis was initially proposed in modulation of HPV-16 E7 translation in cell-free translation systems [108] and carefully examined in expression of BPV-1 late transcripts[109] by two different approaches and received some experimental support. First, optimization of codon usage in both the L1 and L2 ORFs of BPV-1 significantly improved L1 and L2 protein production in COS-1 cells. Second, addition of exogenous aminoacyl-tRNAs from yeast or bovine liver to rabbit reticulocyte lysates dramatically improved translation of the wt L1 sequence[109]. In particular, co-transfection of the wt L1 gene with a gene expressing tRNA(Ser)(CGA) also significantly enhanced L1 protein expression [110]. Data indicate that matching of tRNA availability to codon usage is one determinant that restricts expression of papillomavirus late genes to differentiated epithelia. A recent report shows that mouse primary keratinocytes substantially change their tRNA profiles upon differentiation. Aminoacyl_tRNAs from differentiated mouse keratinocytes, but not those from undifferentiated mouse keratinocytes, could enhance the translation of authentic wt L1 mRNA, suggesting that differentiation_associated change to tRNA profiles enhances L1 expression in differentiated keratinocytes [111]. Codon usage and tRNA profiles also appear to correlate with translation of keratin 14 and involucrin mRNAs in undifferentiated and differentiated keratinocytes, respectively [111]. Today, the rare codon usage has been found in all eight ORFs from 79 HPV genomes[112]. Codon modification has been applied as a basic tool to improve production of HPV-16 E5 [113], E7[114;115], L1 and L2[107;116;117] as well as HPV-11 L1 [118].
Translation initiation control of papillomavirus gene expression restricts papillomavirus gene expression. As stated above, almost all papillomavirus transcripts are bicistronic or polycistronic. Initiation of protein translation on a downstream ORF requires specialized mechanisms. Considering that ribosomes scan an mRNA in a linear fashion starting from the RNA 5′ end, how ribosomes bypass an upstream ORF for translation of the second ORF remains as a question for most papillomavirus transcripts. Translation re-initiation, leaky scanning, ribosome shunting (jumping), and internal ribosome entry are possible mechanisms for translation of downstream ORFs in a bicistronic or polycistronic mRNA. The first three possibilities have been proposed for the protein translation of papillomavirus transcripts and each has some limited experimental support[75;119;120]. Our recent studies show that both HPV-16 and HPV-18 E7 are translational products of spliced E6*I through translation re-initiation[36](Tang, Tao, McCoy, and Zheng, in submission). The unspliced bicistronic E6E7 mRNAs mainly serve for translation of E6, rather than E7, due to the limited distance (only 2 nts) between the upstream E6 ORF and the downstream E7 ORF. Ribosomes would not be able to efficiently re-initiate E7 translation in an unspliced full-length, bicistronic E6E7 mRNA because translation termination and re-initiation would have to occur in within the two nts that separate the E6 stop codon and the E7 start codon. Splicing of the E6*I mRNAs creates a pre-mature stop codon immediately downstream of the splice junction and increases the distance between the E6*I ORF and the E7 ORF to approximately 130-nts, a better condition for a scanning ribosome to re-initiate E7 transation (Tang, Tao, McCoy, and Zheng, in submission). Other studies also show that RNA splicing plays an important role in E1 production and viral DNA replication of HPV-31 and HPV-11[22;76], but the mechanisms of this enhancement might be different from HPV-16 or 18 E7 production. Human cytokeratin 7 and rabbit α1-globin protein share a sequence motif SEQIKA that interacts with HPV-16 E7 mRNA. This interaction promotes E7 mRNA stability, but results in suppression of E7 translation[121].
8. RNA-BASED CONTROL OF VIRAL GENE EXPRESSION IN PAPILLOMAVIRUS INFECTION
Multiple RNA-based approaches, including antisense, ribozyme, trans-splicing, and recent RNAi technology, have been developed to block or exploit papillomavirus gene expression. Although all of these approaches show some promising results in vitro and in vivo, the RNA-based gene therapy of HPV infections, like any other gene therapy attempts, is facing some obstacles of delivery and stability issues of therapeutic nucleic acids. The first approach of the endeavor was an antisense RNA approach in blocking HPV-18 E6 and E7 expression in C4-1 cells[122]. Subsequently, the HPV E6E7 antisense RNA was found to inhibit tumorigenicity of cervical cancer cells in nude mice[123]. Antisense DNA oligonucleotides followed as an alternative small molecule against HPV-18 and HPV-16 E6 and E7 expression and function by inducing RNase H-mediated digestion of the DNA-RNA hybrids in the targeted region and/or blocking translation of the targeted mRNA[124;125]. Today, antisense technology has been experimentally used to block HPV-6 and -11 E1-mediated viral DNA replication[126], HPV-11 E6 and E7-mediated papilloma progression[127], and inhibit growth of HPV-16+ mouse tumor[128].
In contrast to antisense DNA oligo approach, ribozymes are RNA molecules containing an enzymatic activity that cleaves the targeted RNA, once base-paired with the target through their flanking sequences. Two ribozymes were tested in 1990s to inhibit papillomavirus oncogene expression. Hammerhead ribozymes which target any RNA containing the triplet 5′-NUH-3′ (N is any nt; H is C, U, or A)[129] were initially applied to block CRPV E7[130], HPV-16[131;132] and HPV-18 E6 and E7 expression[133] and showed some promising results. Hairpin ribozymes that cleave an RNA with a sequence motif 5′-C(U)NGU(G/A)C(A)-3′[134;135] were designed later to silence HPV-16 E6 and E7 expression and inhibit the growth of HPV-16 containing cells[135;136]. Compared with hammerhead ribozymes, which are smaller in size, hairpin ribozymes have not been explored widely for their potential use for papillomavirus therapy.
RNA interference (RNAi) mediated by small interfering RNA (siRNA) duplex of about 21 nts can sequence-specifically target and degrade an mRNA. Use of a synthetic, double-stranded, short RNAs as siRNAs has been widely applied to the study of gene function[137]. RNAi mediated by introduced synthetic siRNA duplexes has been used to silence oncogene expression of HPV-16[138-140] and HPV-18[141](Tang, Tao, McCoy, and Zheng, in submission) in cervical cancer cells. Silencing of E6 and E7 expression from HPV-positive cervical cancer cells results in cell cycle arrest at G1, cell growth retardation[140](Tang, Tao, McCoy, and Zheng, in submission) and slow tumor formation of HPV-16+ cervical cancer cells in NOD/SCID mice[142]. However, desensitization by repeat siRNA treatment of HPV-16+ cervical cancer cells or stable expression of E6 or E7 siRNA in HPV-16+ cervical cancer cells could make the cells resistant to siRNA function[140].
Spliceosome-mediated RNA Trans-splicing (SMaRT™) has been exploited as a way to specifically target the expression of a therapeutic molecule to cervical cancers that universally express HPV pre-mRNAs[143;144]. This technology expresses a pre-trans-splicing molecule (PTM) consisting of an antisense binding domain, strong splicing signals, and an exon encoding a therapeutic molecule. Tethering of the PTM to the target HPV pre-mRNA through base-pairing interactions promotes splicing of the pre-mRNA to the therapeutic exon through trans-splicing. Ideally the therapeutic molecule is expressed only in cells expressing the target pre-mRNA which in this case would be the cervical cancer cells. Trans-splicing efficiencies in excess of 50% have been achieved by adenoviral delivery of an optimized PTM to SiHa cervical cancer cells.
9. REMARKS AND PERSPECTIVES
Almost twenty-five years have passed since assignment of individual ORFs on the first two completely sequenced papillomavirus genomes, HPV-1a and BPV-1[2;3]. Ever since, understanding of each ORF′s coding potential and its corresponding protein function from different types of HPVs, especially high-risk HPVs, has greatly revolutionized our knowledge of papillomavirus biology and many important principles of virus-cell interactions and viral oncogenesis. Remarkable progress has been made in achieving our ultimate goal to prevent high-risk papillomavirus-induced cervical cancer by HPV vaccine which may receive licensure from regulatory agencies in the near future[145-147]. However, we know remarkably little about how each of these viral proteins is translated from a native mRNA. Ironically, we don’t even know in most cases which transcripts are true mRNAs used for protein translation during papillomavirus infection. This is mainly because papillomaviruses produce no monocistric mRNA. The principle of one gene encoding one mRNA with one ORF for translation of one protein does not apply to any of the papillomaviruses. Single gene knock-out in the context of the whole virus genome without affecting the expression of other viral genes would not work for papillomavirus study. As we have reviewed, papillomaviruses use a very economic way to express their genes from a very compact genome and the resulting transcripts are either bicistronic or polycistronic, with multiple ORFs in a single message. Alternative RNA splicing plays a critical role in determining which ORFs will be translated from these multicistronic transcripts. As translation of papillomavirus proteins from polycistronic mRNAs can be blocked by the presence of the upstream ORFs, splicing of these pre-mRNAs to disrupt an upstream ORF will facilitate scanning by the ribosome from the RNA 5′ end to more efficiently access the downstream ORF for its translation initiation. To do so, papillomaviruses have evolved complex mechanisms to regulate splicing of individual viral pre-mRNAs in a cell differentiation-specific manner as evidenced by the presence of multiple regulatory cis-elements in each species of viral pre-mRNA (Fig. 3). Although we have learned a great deal in characterization of regulated splicing of BPV-1 late pre-mRNA, finding of other similar cis-elements and elucidation of their function in individual HPV pre-mRNA remains an important research priority.
There are many intriguing questions remaining to be addressed. In high-risk HPVs, several important ORFs, including E6, E1 and L2, bear an intron in their coding regions (Fig. 2); splicing of the individual intron will disrupt the ORF and abolish production of the full-length protein. How do these introns escape recognition while other introns in the same pre-mRNA are removed by the cellular splicing machinery? More strikingly, how are mRNAs with a retained intron efficiently exported from the nucleus to the cytoplasm before their nuclear degradation since an intron-containing RNA is generally not exportable? We are particularly baffled by the mechanisms required for L2 expression from a polycistronic mRNA. The L2 pre-mRNA contains two introns and two poly(A) signals. Retention of intron 2 from the L2 coding region and suppression of early poly(A) site recognition are required to make an mRNA with an intact L2 ORF. However, we know little about what regulates the two events. Together, to explore the mechanism involving a selective intron retention and polyadenylation will be a prospective focus in papillomavirus post-transcriptional research. Although papillomavirus late polyadenylation appears to be suppressed by a cryptic 5′ splice site(s) in the late 3′ terminal exon, early polyadenylation might be controlled by a totally different mechanism.
Post-transcriptional regulation of papillomavirus gene expression is tightly coupled to cell-differentiation. This might be attributable to participation of cell differentiation-associated factors and some viral proteins. HPV E2 appears to be involved in regulation of RNA splicing[148] and enhancement of SF2/ASF expression[89]. Identification of differentially expressed cellular factors and viral proteins that contribute to the post-transcription regulation will definitely shed light on this complex orchestration of papillomavirus gene expression. Codon modification to improve viral protein translation has been very useful, but the exact mechanism(s) underlying this enhancement remain a question. Availability of the tRNA pool might not be the only explanation. Since an mRNA with optimized codons tends to be more abundant than its native counterpart[111], the codon optimization might result in disruption of negative or generation of positive regulatory elements in an RNA. In addition, the codon optimization might disrupt the binding sites of differentiation-specific microRNAs, abundant tiny RNA molecules (∼22-nts in size) recently discovered to be involved in the regulation of a large number of transcripts at the translational level[149;150].
10. ACKNOWLEDGEMENTS
We thank Douglas Lowy for his critical reading of the manuscript, John Doorbar for Fig. 1 for modification, Samuel Gunderson for his permission of Fig. 4A modification, and Hans-Ulrich Bernard for sharing his unpublished review. This research was supported by the intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
11. REFERENCES
- 1.de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur HH. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 2.Chen EY, Howley PM, Levinson AD, Seeburg PH. The primary structure and genetic organization of the bovine papillomavirus type 1 genome. Nature. 1982;299:529–534. doi: 10.1038/299529a0. [DOI] [PubMed] [Google Scholar]
- 3.Danos O, Katinka M, Yaniv M. Human papillomavirus 1a complete DNA sequence: a novel type of genome organization among papovaviridae. EMBO J. 1982;1:231–236. doi: 10.1002/j.1460-2075.1982.tb01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJ, Meijer CJ. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 5.Schiller JT, Davies P. Delivering on the promise: HPV vaccines and cervical cancer. Nat. Rev. Microbiol. 2004;2:343–347. doi: 10.1038/nrmicro867. [DOI] [PubMed] [Google Scholar]
- 6.Lowy DR, Frazer IH. Chapter 16: Prophylactic human papillomavirus vaccines. J. Natl. Cancer Inst. Monogr. 2003:111–116. doi: 10.1093/oxfordjournals.jncimonographs.a003472. [DOI] [PubMed] [Google Scholar]
- 7.Cuschieri KS, Cubie HA. The role of human papillomavirus testing in cervical screening. J.Clin.Virol. 2005;32(Suppl 1):S34–S42. doi: 10.1016/j.jcv.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Molijn A, Kleter B, Quint W, van Doorn LJ. Molecular diagnosis of human papillomavirus (HPV) infections. J. Clin. Virol. 2005;32(Suppl 1):S43–S51. doi: 10.1016/j.jcv.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89:213–228. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 10.Lambert PF, Spalholz BA, Howley PM. A transcriptional repressor encoded by BPV-1 shares a common carboxy-terminal domain with the E2 transactivator. Cell. 1987;50:69–78. doi: 10.1016/0092-8674(87)90663-5. [DOI] [PubMed] [Google Scholar]
- 11.Hubbert NL, Schiller JT, Lowy DR, Androphy EJ. Bovine papilloma virus-transformed cells contain multiple E2 proteins. Proc. Natl. Acad. Sci.; U.S.A. 1988. pp. 5864–5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choe J, Vaillancourt P, Stenlund A, Botchan M. Bovine papillomavirus type 1 encodes two forms of a transcriptional repressor: structural and functional analysis of new viral cDNAs. J.Virol. 1989;63:1743–1755. doi: 10.1128/jvi.63.4.1743-1755.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stubenrauch F, Hummel M, Iftner T, Laimins LA. The E8E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J.Virol. 2000;74:1178–1186. doi: 10.1128/jvi.74.3.1178-1186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stubenrauch F, Zobel T, Iftner T. The E8 domain confers a novel long-distance transcriptional repression activity on the E8E2C protein of high-risk human papillomavirus type 31. J.Virol. 2001;75:4139–4149. doi: 10.1128/JVI.75.9.4139-4149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harry JB, Wettstein FO. Transforming properties of the cottontail rabbit papillomavirus oncoproteins Le6 and SE6 and of the E8 protein. J.Virol. 1996;70:3355–3362. doi: 10.1128/jvi.70.6.3355-3362.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han R, Cladel NM, Reed CA, Christensen ND. Characterization of transformation function of cottontail rabbit papillomavirus E5 and E8 genes. Virology. 1998;251:253–263. doi: 10.1006/viro.1998.9416. [DOI] [PubMed] [Google Scholar]
- 17.Bernard HU. Gene expression of genital human papillomaviruses and considerations on potential antiviral approaches. Antivir.Ther. 2002;7:219–237. [PubMed] [Google Scholar]
- 18.Baker CC, Calef C. Maps of papillomavirus mRNA transcripts. In: Los Alamos National Laboratory, editor. “The human papillomaviruses compendium”. Los Alamos National Laboratory; 1995. [Google Scholar]
- 19.Ozbun MA, Meyers C. Two novel promoters in the upstream regulatory region of human papillomavirus type 31b are negatively regulated by epithelial differentiation. J.Virol. 1999;73:3505–3510. doi: 10.1128/jvi.73.4.3505-3510.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozbun MA. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J.Virol. 2002;76:11291–11300. doi: 10.1128/JVI.76.22.11291-11300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen E, Alam S, Meyers C. Genetic and biochemical analysis of cis regulatory elements within the keratinocyte enhancer region of the human papillomavirus type 31 upstream regulatory region during different stages of the viral life cycle. J.Virol. 2004;78:612–629. doi: 10.1128/JVI.78.2.612-629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng W, Jin G, Lin BY, Van Tine BA, Broker TR, Chow LT. mRNA splicing regulates human papillomavirus type 11 E1 protein production and DNA replication. J.Virol. 2003;77:10213–10226. doi: 10.1128/JVI.77.19.10213-10226.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smotkin D, Wettstein FO. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc. Natl. Acad. Sci.; U.S.A. 1986. pp. 4680–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grassmann K, Rapp B, Maschek H, Petry KU, Iftner T. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J.Virol. 1996;70:2339–2349. doi: 10.1128/jvi.70.4.2339-2349.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenstierne MW, Vinther J, Hansen CN, Prydsoe M, Norrild B. Identification and characterization of a cluster of transcription start sites located in the E6 ORF of human papillomavirus type 16. J.Gen.Virol. 2003;84:2909–2920. doi: 10.1099/vir.0.19332-0. [DOI] [PubMed] [Google Scholar]
- 26.Ozbun MA, Meyers C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J.Virol. 1997;71:5161–5172. doi: 10.1128/jvi.71.7.5161-5172.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hummel M, Hudson JB, Laimins LA. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J.Virol. 1992;66:6070–6080. doi: 10.1128/jvi.66.10.6070-6080.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider-Gadicke A, Schwarz E. Different human cervical carcinoma cell lines show similar transcription patterns of human papillomavirus type 18 early genes. EMBO J. 1986;5:2285–2292. doi: 10.1002/j.1460-2075.1986.tb04496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thierry F, Heard JM, Dartmann K, Yaniv M. Characterization of a transcriptional promoter of human papillomavirus 18 and modulation of its expression by simian virus 40 and adenovirus early antigens. J.Virol. 1987;61:134–142. doi: 10.1128/jvi.61.1.134-142.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romanczuk H, Thierry F, Howley PM. Mutational analysis of cis elements involved in E2 modulation of human papillomavirus type 16 P97 and type 18 P105 promoters. J.Virol. 1990;64:2849–2859. doi: 10.1128/jvi.64.6.2849-2859.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Androphy EJ, Lowy DR, Schiller JT. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature. 1987;325:70–73. doi: 10.1038/325070a0. [DOI] [PubMed] [Google Scholar]
- 32.Hawley-Nelson P, Androphy EJ, Lowy DR, Schiller JT. The specific DNA recognition sequence of the bovine papillomavirus E2 protein is an E2-dependent enhancer. EMBO J. 1988;7:525–531. doi: 10.1002/j.1460-2075.1988.tb02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sousa R, Dostatni N, Yaniv M. Control of papillomavirus gene expression. Biochim. Biophys. Acta. 1990;1032:19–37. doi: 10.1016/0304-419x(90)90010-x. [DOI] [PubMed] [Google Scholar]
- 34.Hou SY, Wu SY, Zhou T, Thomas MC, Chiang CM. Alleviation of human papillomavirus E2-mediated transcriptional repression via formation of a TATA binding protein (or TFIID)-TFIIB-RNA polymerase II-TFIIF preinitiation complex. Mol.Cell Biol. 2000;20:113–125. doi: 10.1128/mcb.20.1.113-125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bechtold V, Beard P, Raj K. Human papillomavirus type 16 E2 protein has no effect on transcription from episomal viral DNA. J.Virol. 2003;77:2021–2028. doi: 10.1128/JVI.77.3.2021-2028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng ZM, Tao M, Yamanegi K, Bodaghi S, Xiao W. Splicing of a Cap-proximal Human Papillomavirus 16 E6E7 Intron Promotes E7 Expression, but can be Restrained by Distance of the Intron from its RNA 5′ Cap. J.Mol.Biol. 2004;337:1091–1108. doi: 10.1016/j.jmb.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 37.Ordonez RM, Espinosa AM, Sanchez-Gonzalez DJ, Armendariz-Borunda J, Berumen J. Enhanced oncogenicity of Asian-American human papillomavirus 16 is associated with impaired E2 repression of E6/E7 oncogene transcription. J.Gen.Virol. 2004;85:1433–1444. doi: 10.1099/vir.0.19317-0. [DOI] [PubMed] [Google Scholar]
- 38.Zhao X, Oberg D, Rush M, Fay J, Lambkin H, Schwartz S. A 57-nucleotide upstream early polyadenylation element in human papillomavirus type 16 interacts with hFip1, CstF-64, hnRNP C1/C2, and polypyrimidine tract binding protein. J.Virol. 2005;79:4270–4288. doi: 10.1128/JVI.79.7.4270-4288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley M, Crawford L. Detection of novel splicing patterns in a HPV-16-containing keratinocyte cell line. Virology. 1990;178:254–262. doi: 10.1016/0042-6822(90)90401-c. [DOI] [PubMed] [Google Scholar]
- 40.Sherman L, Alloul N, Golan I, Durst M, Baram A. Expression and splicing patterns of human papillomavirus type-16 mRNAs in pre-cancerous lesions and carcinomas of the cervix, in human keratinocytes immortalized by HPV 16, and in cell lines established from cervical cancers. Int.J.Cancer. 1992;50:356–364. doi: 10.1002/ijc.2910500305. [DOI] [PubMed] [Google Scholar]
- 41.Sherman L, Alloul N. Human papillomavirus type 16 expresses a variety of alternatively spliced mRNAs putatively encoding the E2 protein. Virology. 1992;191:953–959. doi: 10.1016/0042-6822(92)90271-p. [DOI] [PubMed] [Google Scholar]
- 42.Tang S, Yamanegi K, Zheng ZM. Requirement of a 12-base-pair TATT-containing sequence and viral lytic DNA replication in activation of the Kaposi’s sarcoma-associated herpesvirus K8.1 late promoter. J.Virol. 2004;78:2609–2614. doi: 10.1128/JVI.78.5.2609-2614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wade EJ, Spector DH. The human cytomegalovirus origin of DNA replication (oriLyt) is the critical cis-acting sequence regulating replication-dependent late induction of the viral 1.2-kilobase RNA promoter. J.Virol. 1994;68:6567–6577. doi: 10.1128/jvi.68.10.6567-6577.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hummel M, Lim HB, Laimins LA. Human papillomavirus type 31b late gene expression is regulated through protein kinase C-mediated changes in RNA processing. J.Virol. 1995;69:3381–3388. doi: 10.1128/jvi.69.6.3381-3388.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodily JM, Meyers C. Genetic analysis of the human papillomavirus type 31 differentiation-dependent late promoter. J.Virol. 2005;79:3309–3321. doi: 10.1128/JVI.79.6.3309-3321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spink KM, Laimins LA. Induction of the human papillomavirus type 31 late promoter requires differentiation but not DNA amplification. J.Virol. 2005;79:4918–4926. doi: 10.1128/JVI.79.8.4918-4926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barksdale SK, Baker CC. Differentiation-specific expression from the bovine papillomavirus type 1 P2443 and late promoters. J.Virol. 1993;67:5605–5616. doi: 10.1128/jvi.67.9.5605-5616.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danos O, Katinka M, Yaniv M. Molecular cloning, refined physical map and heterogeneity of methylation sites of papilloma virus type 1a DNA. Eur.J.Biochem. 1980;109:457–461. doi: 10.1111/j.1432-1033.1980.tb04815.x. [DOI] [PubMed] [Google Scholar]
- 49.Wettstein FO, Stevens JG. Shope papilloma virus DNA is extensively methylated in non-virus-producing neoplasms. Virology. 1983;126:493–504. doi: 10.1016/s0042-6822(83)80007-5. [DOI] [PubMed] [Google Scholar]
- 50.Burnett TS, Sleeman JP. Uneven distribution of methylation sites within the human papillomavirus la genome: possible relevance to viral gene expression. Nucleic Acids Res. 1984;12:8847–8860. doi: 10.1093/nar/12.23.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosl F, Arab A, Klevenz B, zur HH. The effect of DNA methylation on gene regulation of human papillomaviruses. J.Gen.Virol. 1993;74(Pt 5):791–801. doi: 10.1099/0022-1317-74-5-791. [DOI] [PubMed] [Google Scholar]
- 52.Choo KB, Liew LN, Liew SJ, Cheng WT. Hypermethylation of the human papillomavirus transgenome in transgenic mice. Proc. Natl. Sci. Counc. Repub.; China. 1993. pp. 159–163. [PubMed] [Google Scholar]
- 53.List HJ, Patzel V, Zeidler U, Schopen A, Ruhl G, Stollwerk J, Klock G. Methylation sensitivity of the enhancer from the human papillomavirus type 16. J.Biol.Chem. 1994;269:11902–11911. [PubMed] [Google Scholar]
- 54.Thain A, Jenkins O, Clarke AR, Gaston K. CpG methylation directly inhibits binding of the human papillomavirus type 16 E2 protein to specific DNA sequences. J.Virol. 1996;70:7233–7235. doi: 10.1128/jvi.70.10.7233-7235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Badal V, Chuang LS, Tan EH, Badal S, Villa LL, Wheeler CM, Li BF, Bernard HU. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J.Virol. 2003;77:6227–6234. doi: 10.1128/JVI.77.11.6227-6234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Badal S, Badal V, Calleja-Macias IE, Kalantari M, Chuang LS, Li BF, Bernard HU. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology. 2004;324:483–492. doi: 10.1016/j.virol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 57.Kim K, Garner-Hamrick PA, Fisher C, Lee D, Lambert PF. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J.Virol. 2003;77:12450–12459. doi: 10.1128/JVI.77.23.12450-12459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Tine BA, Kappes JC, Banerjee NS, Knops J, Lai L, Steenbergen RD, Meijer CL, Snijders PJ, Chatis P, Broker TR, Moen PT, Jr., Chow LT. Clonal selection for transcriptionally active viral oncogenes during progression to cancer. J.Virol. 2004;78:11172–11186. doi: 10.1128/JVI.78.20.11172-11186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Lie K, Barrera-Saldana HA, Wiley DJ, Bernard HU. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J.Virol. 2004;78:12762–12772. doi: 10.1128/JVI.78.23.12762-12772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wiley DJ, Huh J, Rao JY, Chang C, Goetz M, Poulter M, Masongsong E, Chang CI, Bernard HU. Methylation of human papillomavirus genomes in cells of anal epithelia of HIV-infected men. J. Acquir. Immune. Defic. Syndr. 2005;39:143–151. [PubMed] [Google Scholar]
- 61.Feng Q, Balasubramanian A, Hawes SE, Toure P, Sow PS, Dem A, Dembele B, Critchlow CW, Xi L, Lu H, McIntosh MW, Young AM, Kiviat NB. Detection of hypermethylated genes in women with and without cervical neoplasia. J.Natl.Cancer Inst. 2005;97:273–282. doi: 10.1093/jnci/dji041. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Martins CR, Fansler ZB, Roemer KL, Kincaid EA, Gustafson KS, Heitjan DF, Clark DP. DNA methylation in anal intraepithelial lesions and anal squamous cell carcinoma. Clin.Cancer Res. 2005;11:6544–6549. doi: 10.1158/1078-0432.CCR-05-0374. [DOI] [PubMed] [Google Scholar]
- 62a.Sanford JR, Ellis J, Caceres JF. Multiple roles of arginine/serine-rich splicing factors in RNA splicing. Biochem Soc Trans. 2005;33:443–446. doi: 10.1042/BST0330443. [DOI] [PubMed] [Google Scholar]
- 63.Zheng ZM. Regulation of alternative RNA splicing by exon definition and exon sequences in viral and Mammalian gene expression. J.Biomed.Sci. 2004;11:278–294. doi: 10.1159/000077096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ladd AN, Cooper TA. Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol. 2002;3:reviews0008. doi: 10.1186/gb-2002-3-11-reviews0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barksdale S, Baker CC. Differentiation-specific alternative splicing of bovine papillomavirus late mRNAs. J.Virol. 1995;69:6553–6556. doi: 10.1128/jvi.69.10.6553-6556.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng ZM, He P, Baker CC. Selection of the bovine papillomavirus type 1 nucleotide 3225 3′ splice site is regulated through an exonic splicing enhancer and its juxtaposed exonic splicing suppressor. J.Virol. 1996;70:4691–4699. doi: 10.1128/jvi.70.7.4691-4699.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng ZM, He PJ, Baker CC. Structural, functional, and protein binding analyses of bovine papillomavirus type 1 exonic splicing enhancers. J.Virol. 1997;71:9096–9107. doi: 10.1128/jvi.71.12.9096-9107.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng ZM, Huynen M, Baker CC. A pyrimidine-rich exonic splicing suppressor binds multiple RNA splicing factors and inhibits spliceosome assembly. Proc. Natl. Acad. Sci.; U.S.A. 1998. pp. 14088–14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng ZM, He PJ, Baker CC. Function of a bovine papillomavirus type 1 exonic splicing suppressor requires a suboptimal upstream 3′ splice site. J.Virol. 1999;73:29–36. doi: 10.1128/jvi.73.1.29-36.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng ZM, Quintero J, Reid ES, Gocke C, Baker CC. Optimization of a weak 3′ splice site counteracts the function of a bovine papillomavirus type 1 exonic splicing suppressor in vitro and in vivo. J.Virol. 2000;74:5902–5910. doi: 10.1128/jvi.74.13.5902-5910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng ZM, Reid ES, Baker CC. Utilization of the bovine papillomavirus type 1 late-stage-specific nucleotide 3605 3′ splice site is modulated by a novel exonic bipartite regulator but not by an intronic purine-rich element. J.Virol. 2000;74:10612–10622. doi: 10.1128/jvi.74.22.10612-10622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rush M, Zhao X, Schwartz S. A splicing enhancer in the E4 coding region of human papillomavirus type 16 is required for early mRNA splicing and polyadenylation as well as inhibition of premature late gene expression. J.Virol. 2005;79:12002–12015. doi: 10.1128/JVI.79.18.12002-12015.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao X, Rush M, Schwartz S. Identification of an hnRNP A1-dependent splicing silencer in the human papillomavirus type 16 L1 coding region that prevents premature expression of the late L1 gene. J.Virol. 2004;78:10888–10905. doi: 10.1128/JVI.78.20.10888-10905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smotkin D, Prokoph H, Wettstein FO. Oncogenic and nononcogenic human genital papillomaviruses generate the E7 mRNA by different mechanisms. J.Virol. 1989;63:1441–1447. doi: 10.1128/jvi.63.3.1441-1447.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sedman SA, Barbosa MS, Vass WC, Hubbert NL, Haas JA, Lowy DR, Schiller JT. The full-length E6 protein of human papillomavirus type 16 has transforming and trans-activating activities and cooperates with E7 to immortalize keratinocytes in culture. J.Virol. 1991;65:4860–4866. doi: 10.1128/jvi.65.9.4860-4866.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hubert WG, Laimins LA. Human papillomavirus type 31 replication modes during the early phases of the viral life cycle depend on transcriptional and posttranscriptional regulation of E1 and E2 expression. J.Virol. 2002;76:2263–2273. doi: 10.1128/jvi.76.5.2263-2273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen F, MacDonald CC, Wilusz J. Cleavage site determinants in the mammalian polyadenylation signal. Nucleic Acids Res. 1995;23:2614–2620. doi: 10.1093/nar/23.14.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ryan K, Calvo O, Manley JL. Evidence that polyadenylation factor CPSF-73 is the mRNA 3′ processing endonuclease. RNA. 2004;10:565–573. doi: 10.1261/rna.5214404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stoler MH, Wolinsky SM, Whitbeck A, Broker TR, Chow LT. Differentiation-linked human papillomavirus types 6 and 11 transcription in genital condylomata revealed by in situ hybridization with message-specific RNA probes. Virology. 1989;172:331–340. doi: 10.1016/0042-6822(89)90135-9. [DOI] [PubMed] [Google Scholar]
- 80.Furth PA, Baker CC. An element in the bovine papillomavirus late 3′ untranslated region reduces polyadenylated cytoplasmic RNA levels. J.Virol. 1991;65:5806–5812. doi: 10.1128/jvi.65.11.5806-5812.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kennedy IM, Haddow JK, Clements JB. Analysis of human papillomavirus type 16 late mRNA 3′ processing signals in vitro and in vivo. J.Virol. 1990;64:1825–1829. doi: 10.1128/jvi.64.4.1825-1829.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Furth PA, Choe WT, Rex JH, Byrne JC, Baker CC. Sequences homologous to 5′ splice sites are required for the inhibitory activity of papillomavirus late 3′ untranslated regions. Mol.Cell Biol. 1994;14:5278–5289. doi: 10.1128/mcb.14.8.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gunderson SI, Polycarpou-Schwarz M, Mattaj IW. U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol.Cell. 1998;1:255–264. doi: 10.1016/s1097-2765(00)80026-x. [DOI] [PubMed] [Google Scholar]
- 84.Ko B, Gunderson SI. Identification of new poly(A) polymerase-inhibitory proteins capable of regulating pre-mRNA polyadenylation. J.Mol.Biol. 2002;318:1189–1206. doi: 10.1016/s0022-2836(02)00240-1. [DOI] [PubMed] [Google Scholar]
- 85.Fortes P, Cuevas Y, Guan F, Liu P, Pentlicky S, Jung SP, Martinez-Chantar ML, Prieto J, Rowe D, Gunderson SI. Inhibiting expression of specific genes in mammalian cells with 5′ end-mutated U1 small nuclear RNAs targeted to terminal exons of pre-mRNA. Proc. Natl. Acad. Sci.; U.S.A. 2003. pp. 8264–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cumming SA, McPhillips MG, Veerapraditsin T, Milligan SG, Graham SV. Activity of the human papillomavirus type 16 late negative regulatory element is partly due to four weak consensus 5′ splice sites that bind a U1 snRNP-like complex. J.Virol. 2003;77:5167–5177. doi: 10.1128/JVI.77.9.5167-5177.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dietrich-Goetz W, Kennedy IM, Levins B, Stanley MA, Clements JB. A cellular 65-kDa protein recognizes the negative regulatory element of human papillomavirus late mRNA. Proc. Natl. Acad. Sci.; U.S.A. 1997. pp. 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koffa MD, Graham SV, Takagaki Y, Manley JL, Clements JB. The human papillomavirus type 16 negative regulatory RNA element interacts with three proteins that act at different posttranscriptional levels. Proc. Natl. Acad. Sci.; U.S.A. 2000. pp. 4677–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McPhillips MG, Veerapraditsin T, Cumming SA, Karali D, Milligan SG, Boner W, Morgan IM, Graham SV. SF2/ASF binds the human papillomavirus type 16 late RNA control element and is regulated during differentiation of virus-infected epithelial cells. J.Virol. 2004;78:10598–10605. doi: 10.1128/JVI.78.19.10598-10605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cumming SA, Repellin CE, McPhillips M, Radford JC, Clements JB, Graham SV. The human papillomavirus type 31 late 3′ untranslated region contains a complex bipartite negative regulatory element. J.Virol. 2002;76:5993–6003. doi: 10.1128/JVI.76.12.5993-6003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baker CC, Noe JS. Transcriptional termination between bovine papillomavirus type 1 (BPV-1) early and late polyadenylation sites blocks late transcription in BPV-1-transformed cells. J.Virol. 1989;63:3529–3534. doi: 10.1128/jvi.63.8.3529-3534.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Buratowski S. Connections between mRNA 3′ end processing and transcription termination. Curr.Opin.Cell Biol. 2005;17:257–261. doi: 10.1016/j.ceb.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 93.Andrews EM, DiMaio D. Hierarchy of polyadenylation site usage by bovine papillomavirus in transformed mouse cells. J.Virol. 1993;67:7705–7710. doi: 10.1128/jvi.67.12.7705-7710.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Terhune SS, Hubert WG, Thomas JT, Laimins LA. Early polyadenylation signals of human papillomavirus type 31 negatively regulate capsid gene expression. J.Virol. 2001;75:8147–8157. doi: 10.1128/JVI.75.17.8147-8157.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Terhune SS, Milcarek C, Laimins LA. Regulation of human papillomavirus type 31 polyadenylation during the differentiation-dependent life cycle. J.Virol. 1999;73:7185–7192. doi: 10.1128/jvi.73.9.7185-7192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oberg D, Fay J, Lambkin H, Schwartz S. A downstream polyadenylation element in human papillomavirus type 16 L2 encodes multiple GGG motifs and interacts with hnRNP H. J.Virol. 2005;79:9254–9269. doi: 10.1128/JVI.79.14.9254-9269.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaufmann I, Martin G, Friedlein A, Langen H, Keller W. Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly(A) polymerase. EMBO J. 2004;23:616–626. doi: 10.1038/sj.emboj.7600070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Le Sommer C, Lesimple M, Mereau A, Menoret S, Allo MR, Hardy S. PTB regulates the processing of a 3′-terminal exon by repressing both splicing and polyadenylation. Mol.Cell Biol. 2005;25:9595–9607. doi: 10.1128/MCB.25.21.9595-9607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arhin GK, Boots M, Bagga PS, Milcarek C, Wilusz J. Downstream sequence elements with different affinities for the hnRNP H/H’ protein influence the processing efficiency of mammalian polyadenylation signals. Nucleic Acids Res. 2002;30:1842–1850. doi: 10.1093/nar/30.8.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kennedy IM, Haddow JK, Clements JB. A negative regulatory element in the human papillomavirus type 16 genome acts at the level of late mRNA stability. J.Virol. 1991;65:2093–2097. doi: 10.1128/jvi.65.4.2093-2097.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tan W, Schwartz S. The Rev protein of human immunodeficiency virus type 1 counteracts the effect of an AU-rich negative element in the human papillomavirus type 1 late 3′ untranslated region. J.Virol. 1995;69:2932–2945. doi: 10.1128/jvi.69.5.2932-2945.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sokolowski M, Zhao C, Tan W, Schwartz S. AU-rich mRNA instability elements on human papillomavirus type 1 late mRNAs and c-fos mRNAs interact with the same cellular factors. Oncogene. 1997;15:2303–2319. doi: 10.1038/sj.onc.1201415. [DOI] [PubMed] [Google Scholar]
- 103.Collier B, Oberg D, Zhao X, Schwartz S. Specific inactivation of inhibitory sequences in the 5′ end of the human papillomavirus type 16 L1 open reading frame results in production of high levels of L1 protein in human epithelial cells. J.Virol. 2002;76:2739–2752. doi: 10.1128/JVI.76.6.2739-2752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tan W, Felber BK, Zolotukhin AS, Pavlakis GN, Schwartz S. Efficient expression of the human papillomavirus type 16 L1 protein in epithelial cells by using Rev and the Rev-responsive element of human immunodeficiency virus or the cis-acting transactivation element of simian retrovirus type 1. J.Virol. 1995;69:5607–5620. doi: 10.1128/jvi.69.9.5607-5620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sokolowski M, Tan W, Jellne M, Schwartz S. mRNA instability elements in the human papillomavirus type 16 L2 coding region. J.Virol. 1998;72:1504–1515. doi: 10.1128/jvi.72.2.1504-1515.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Collier B, Goobar-Larsson L, Sokolowski M, Schwartz S. Translational inhibition in vitro of human papillomavirus type 16 L2 mRNA mediated through interaction with heterogenous ribonucleoprotein K and poly(rC)-binding proteins 1 and 2. J.Biol.Chem. 1998;273:22648–22656. doi: 10.1074/jbc.273.35.22648. [DOI] [PubMed] [Google Scholar]
- 107.Oberg D, Collier B, Zhao X, Schwartz S. Mutational inactivation of two distinct negative RNA elements in the human papillomavirus type 16 L2 coding region induces production of high levels of L2 in human cells. J.Virol. 2003;77:11674–11684. doi: 10.1128/JVI.77.21.11674-11684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.De Pasquale C, Kanduc D. Modulation of HPV-16 E7 translation by tRNAs in eukaryotic cell-free translation systems. Biochem. Mol. Biol. Int. 1998;45:1005–1009. doi: 10.1002/iub.7510450518. [DOI] [PubMed] [Google Scholar]
- 109.Zhou J, Liu WJ, Peng SW, Sun XY, Frazer I. Papillomavirus capsid protein expression level depends on the match between codon usage and tRNA availability. J.Virol. 1999;73:4972–4982. doi: 10.1128/jvi.73.6.4972-4982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gu W, Li M, Zhao WM, Fang NX, Bu S, Frazer IH, Zhao KN. tRNASer(CGA) differentially regulates expression of wild-type and codon-modified papillomavirus L1 genes. Nucleic Acids Res. 2004;32:4448–4461. doi: 10.1093/nar/gkh748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhao KN, Gu W, Fang NX, Saunders NA, Frazer IH. Gene codon composition determines differentiation-dependent expression of a viral capsid gene in keratinocytes in vitro and in vivo. Mol.Cell Biol. 2005;25:8643–8655. doi: 10.1128/MCB.25.19.8643-8655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao KN, Liu WJ, Frazer IH. Codon usage bias and A+T content variation in human papillomavirus genomes. Virus Res. 2003;98:95–104. doi: 10.1016/j.virusres.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 113.Disbrow GL, Sunitha I, Baker CC, Hanover J, Schlegel R. Codon optimization of the HPV-16 E5 gene enhances protein expression. Virology. 2003;311:105–114. doi: 10.1016/s0042-6822(03)00129-6. [DOI] [PubMed] [Google Scholar]
- 114.Liu WJ, Gao F, Zhao KN, Zhao W, Fernando GJ, Thomas R, Frazer IH. Codon modified human papillomavirus type 16 E7 DNA vaccine enhances cytotoxic T-lymphocyte induction and anti-tumour activity. Virology. 2002;301:43–52. doi: 10.1006/viro.2002.1584. [DOI] [PubMed] [Google Scholar]
- 115.Cid-Arregui A, Juarez V, zur HH. A synthetic E7 gene of human papillomavirus type that yields enhanced expression of the protein in mammalian cells and is useful for DNA immunization studies. J.Virol. 2003;77:4928–4937. doi: 10.1128/JVI.77.8.4928-4937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leder C, Kleinschmidt JA, Wiethe C, Muller M. Enhancement of capsid gene expression: preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J.Virol. 2001;75:9201–9209. doi: 10.1128/JVI.75.19.9201-9209.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Baud D, Ponci F, Bobst M, De Grandi P, Nardelli-Haefliger D. Improved efficiency of a Salmonella-based vaccine against human papillomavirus type 16 virus-like particles achieved by using a codon-optimized version of L1. J.Virol. 2004;78:12901–12909. doi: 10.1128/JVI.78.23.12901-12909.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mossadegh N, Gissmann L, Muller M, Zentgraf H, Alonso A, Tomakidi P. Codon optimization of the human papillomavirus 11 (HPV 11) L1 gene leads to increased gene expression and formation of virus-like particles in mammalian epithelial cells. Virology. 2004;326:57–66. doi: 10.1016/j.virol.2004.04.050. [DOI] [PubMed] [Google Scholar]
- 119.Stacey SN, Jordan D, Williamson AJ, Brown M, Coote JH, Arrand JR. Leaky scanning is the predominant mechanism for translation of human papillomavirus type 16 E7 oncoprotein from E6/E7 bicistronic mRNA. J.Virol. 2000;74:7284–7297. doi: 10.1128/jvi.74.16.7284-7297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Remm M, Remm A, Ustav M. Human papillomavirus type 18 E1 protein is translated from polycistronic mRNA by a discontinuous scanning mechanism. J.Virol. 1999;73:3062–3070. doi: 10.1128/jvi.73.4.3062-3070.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kanduc D. Translational regulation of human papillomavirus type 16 E7 mRNA by the peptide SEQIKA, shared by rabbit alpha(1)-globin and human cytokeratin 7. J.Virol. 2002;76:7040–7048. doi: 10.1128/JVI.76.14.7040-7048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.von Knebel DM, Oltersdorf T, Schwarz E, Gissmann L. Correlation of modified human papilloma virus early gene expression with altered growth properties in C4-1 cervical carcinoma cells. Cancer Res. 1988;48:3780–3786. [PubMed] [Google Scholar]
- 123.von Knebel DM, Rittmuller C, zur HH, Durst M. Inhibition of tumorigenicity of cervical cancer cells in nude mice by HPV E6-E7 anti-sense RNA. Int.J.Cancer. 1992;51:831–834. doi: 10.1002/ijc.2910510527. [DOI] [PubMed] [Google Scholar]
- 124.Steele C, Cowsert LM, Shillitoe EJ. Effects of human papillomavirus type 18-specific antisense oligonucleotides on the transformed phenotype of human carcinoma cell lines. Cancer Res. 1993;53:2330–2337. [PubMed] [Google Scholar]
- 125.Tan TM, Ting RC. In vitro and in vivo inhibition of human papillomavirus type 16 E6 and E7 genes. Cancer Res. 1995;55:4599–4605. [PubMed] [Google Scholar]
- 126.Alam S, Bromberg-White J, McLaughlin-Drubin M, Sen E, Bodily JM, Meyers C. Activity and therapeutic potential of ORI-1001 antisense oligonucleotide on human papillomavirus replication utilizing a model of dysplastic human epithelium. Anticancer Res. 2005;25:765–777. [PubMed] [Google Scholar]
- 127.Clawson GA, Miranda GQ, Sivarajah A, Xin P, Pan W, Thiboutot D, Christensen ND. Inhibition of papilloma progression by antisense oligonucleotides targeted to HPV-11HPV-11 E6/E7 RNA. Gene Ther. 2004;11:1331–1341. doi: 10.1038/sj.gt.3302303. [DOI] [PubMed] [Google Scholar]
- 128.He Y, Huang L. Growth inhibition of human papillomavirus 16 DNA-positive mouse tumor by antisense RNA transcribed from U6 promoter. Cancer Res. 1997;57:3993–3999. [PubMed] [Google Scholar]
- 129.Vaish NK, Kore AR, Eckstein F. Recent developments in the hammerhead ribozyme field. Nucleic Acids Res. 1998;26:5237–5242. doi: 10.1093/nar/26.23.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wisotzkey JD, Krizenoskas A, DiAngelo S, Kreider JW. Cleavage of cottontail rabbit papillomavirus E7 RNA with an anti-E7 ribozyme. Biochem. Biophys. Res. Commun. 1993;192:833–839. doi: 10.1006/bbrc.1993.1490. [DOI] [PubMed] [Google Scholar]
- 131.He YK, Lu CD, Qi GR. In vitro cleavage of HPV-16 E6 and E7 RNA fragments by synthetic ribozymes and transcribed ribozymes from RNA-trimming plasmids. FEBS Lett. 1993;322:21–24. doi: 10.1016/0014-5793(93)81102-6. [DOI] [PubMed] [Google Scholar]
- 132.Lu D, Chatterjee S, Brar D, Wong KK., Jr. Ribozyme-mediated in vitro cleavage of transcripts arising from the major transforming genes of human papillomavirus type 16. Cancer Gene Ther. 1994;1:267–277. [PubMed] [Google Scholar]
- 133.Chen Z, Kamath P, Zhang S, Weil MM, Shillitoe EJ. Effectiveness of three ribozymes for cleavage of an RNA transcript from human papillomavirus type 18. Cancer Gene Ther. 1995;2:263–271. [PubMed] [Google Scholar]
- 134.Fedor MJ. The catalytic mechanism of the hairpin ribozyme. Biochem.Soc.Trans. 2002;30:1109–1115. doi: 10.1042/bst0301109. [DOI] [PubMed] [Google Scholar]
- 135.Lian Y, De Young MB, Siwkowski A, Hampel A, Rappaport J. The sCYMV1 hairpin ribozyme: targeting rules and cleavage of heterologous RNA. Gene Ther. 1999;6:1114–1119. doi: 10.1038/sj.gt.3300920. [DOI] [PubMed] [Google Scholar]
- 136.Alvarez-Salas LM, Cullinan AE, Siwkowski A, Hampel A, DiPaolo JA. Inhibition of HPV-16 E6/E7 immortalization of normal keratinocytes by hairpin ribozymes. Proc. Natl. Acad. Sci.; U.S.A. 1998. pp. 1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 138.Jiang M, Milner J. Selective silencing of viral gene expression in HPV-positive human cervical carcinoma cells treated with siRNA, a primer of RNA interference. Oncogene. 2002;21:6041–6048. doi: 10.1038/sj.onc.1205878. [DOI] [PubMed] [Google Scholar]
- 139.Butz K, Ristriani T, Hengstermann A, Denk C, Scheffner M, Hoppe-Seyler F. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene. 2003;22:5938–5945. doi: 10.1038/sj.onc.1206894. [DOI] [PubMed] [Google Scholar]
- 140.Tang S, Tao M, McCoy JP, Jr., Zheng ZM. Short-term induction and long-term suppression of HPV-16 oncogene silencing by RNA interference in cervical cancer cells. 2005 December 12; doi: 10.1038/sj.onc.1209244. Doi:10.1038/sj.onc.1209244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hall AH, Alexander KA. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J.Virol. 2003;77:6066–6069. doi: 10.1128/JVI.77.10.6066-6069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yoshinouchi M, Yamada T, Kizaki M, Fen J, Koseki T, Ikeda Y, Nishihara T, Yamato K. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol.Ther. 2003;8:762–768. doi: 10.1016/j.ymthe.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 143.Mitchell LG, McGarrity GJ. Gene therapy progress and prospects: reprograming gene expression by trans-splicing. Gene Ther. 2005;12:1477–1485. doi: 10.1038/sj.gt.3302596. [DOI] [PubMed] [Google Scholar]
- 144.Patrick M, Puttaraju M, DiPasquale J, Quintero J, Yang Y, Garcia-Blanco MA, Mitchell LG, Baker CC. Factors that influence spliceosome mediated RNA trans-splicing efficacy. Molecular Therapy. Mol.Ther. 2005 in press. [Google Scholar]
- 145.Harper DM, Franco EL, Wheeler C, Ferris DG, Jenkins D, Schuind A, Zahaf T, Innis B, Naud P, De Carvalho NS, Roteli-Martins CM, Teixeira J, Blatter MM, Korn AP, Quint W, Dubin G. Efficacy of a bivalent L1 virus-like particle vaccine in prevention of infection with human papillomavirus types 16 and 18 in young women: a randomised controlled trial. Lancet. 2004;364:1757–1765. doi: 10.1016/S0140-6736(04)17398-4. [DOI] [PubMed] [Google Scholar]
- 146.Villa LL, Costa RL, Petta CA, Andrade RP, Ault KA, Giuliano AR, Wheeler CM, Koutsky LA, Malm C, Lehtinen M, Skjeldestad FE, Olsson SE, Steinwall M, Brown DR, Kurman RJ, Ronnett BM, Stoler MH, Ferenczy A, Harper DM, Tamms GM, Yu J, Lupinacci L, Railkar R, Taddeo FJ, Jansen KU, Esser MT, Sings HL, Saah AJ, Barr E. Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in young women: a randomised double-blind placebo-controlled multicentre phase II efficacy trial. Lancet Oncol. 2005;6:271–278. doi: 10.1016/S1470-2045(05)70101-7. [DOI] [PubMed] [Google Scholar]
- 147.Koutsky LA, Ault KA, Wheeler CM, Brown DR, Barr E, Alvarez FB, Chiacchierini LM, Jansen KU. A controlled trial of a human papillomavirus type 16 vaccine. N.Engl.J.Med. 2002;347:1645–1651. doi: 10.1056/NEJMoa020586. [DOI] [PubMed] [Google Scholar]
- 148.Lai MC, Teh BH, Tarn WY. A human papillomavirus E2 transcriptional activator. The interactions with cellular splicing factors and potential function in pre-mRNA processing. J.Biol.Chem. 1999;274:11832–11841. doi: 10.1074/jbc.274.17.11832. [DOI] [PubMed] [Google Scholar]
- 149.Zamore PD, Haley B. Ribo-gnome: the big world of small RNAs. Science. 2005;309:1519–1524. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
- 150.Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–4662. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- 151.Middleton K, Peh W, Southern S, Griffin H, Sotlar K, Nakahara T, El Sherif A, Morris L, Seth R, Hibma M, Jenkins D, Lambert P, Coleman N, Doorbar J. Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J.Virol. 2003;77:10186–10201. doi: 10.1128/JVI.77.19.10186-10201.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu X, Mayeda A, Tao M, Zheng ZM. Exonic splicing enhancer-dependent selection of the bovine papillomavirus type 1 nucleotide 3225 3′ splice site can be rescued in a cell lacking splicing factor ASF/SF2 through activation of the phosphatidylinositol 3-kinase/Akt pathway. J.Virol. 2003;77:2105–2115. doi: 10.1128/JVI.77.3.2105-2115.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]