

Abstract
Transcripts harboring 5′ upstream open reading frames (uORFs) are often found in genes controlling cell growth including receptors, oncogenes, or growth factors. uORFs can modulate translation or RNA stability and mediate inefficient translation of these potent proteins under normal conditions. In dysregulated cancer cells, where the gene product, for example Her-2 receptor, is overexpressed, post-transcriptional processes must exist that serve to override the inhibitory effects of the uORFs. The 5′ untranslated region (UTR) of Her-2 mRNA contains a short uORF that represses translation of the downstream coding region. We demonstrate that in Her-2 overexpressing breast cancer cells, the 3′ UTR of the Her-2 mRNA can override translational inhibition mediated by the Her-2 uORF. Within this 3′ UTR, a translational derepression element (TDE) that binds to a 38-kDa protein was identified. These results define a novel biological mechanism in which translational control of genes harboring a 5′ uORF can be modulated by elements in their 3′ UTRs.
Keywords: Her-2, uORF, 5′ and 3′ UTRs
Many studies have established a critical role for the Her-2 receptor in the biochemical pathways responsible for transduction of mitogenic signals from a variety of growth factor receptors (Olayioye et al. 2000; Yarden and Sliwkowski 2001). Overexpression of Her-2/neu protein is pro-oncogenic and has been implicated in ∼30% of solid tumors of the human breast, ovary, prostate, lung, bladder, stomach, esophagus, and pancreas (Hynes and Stern 1994; Holbro and Hynes 2004; Hynes and Lane 2005). Overexpression of Her-2 has been demonstrated to be the result of gene amplification as well as modulation of transcriptional or post-transcriptional control mechanisms (Ishii et al. 1987; Kraus et al. 1987; Bae et al. 2001; Vernimmen et al. 2003; Kuwada et al. 2005).
Her-2 mRNA and protein are expressed at basal levels in normal cells, but are increased to >50-fold in Her-2 positive tumor cells (Kraus et al. 1987; Vernimmen et al. 2003). The major 4.5 kb Her-2 transcript is translated into a protein of 185 kDa and is flanked by a 178-nucleotide (nt) 5′ untranslated region (UTR) (Ishii et al. 1987; Tal et al. 1987; Hollywood and Hurst 1993) and 612-nt 3′ UTR. The 5′ UTR is 78% GC rich and contains a six-codon upstream open reading frame (uORF) that precedes the Her-2 translation start codon by 5 nt and is highly conserved among mammalian species. Although the 612-nt 3′ UTR has no recognizable regulatory sequence elements, the 3′-UTR sequences have been reported to stabilize Her-2 mRNA in SKOV-3 ovarian cancer cells (Doherty et al. 1999).
Two distinct translational mechanisms have been reported to control Her-2 protein expression. One is a cell-type-independent mechanism that represses translation of Her-2 mRNA. This repression is mediated by the short uORF in the 5′ UTR of the Her-2 transcript. Mutagenesis experiments demonstrated that the translational repression was independent of the peptide coding sequence of the uORF, as well as the identity of the downstream cistron. This regulatory mechanism has been explored in detail and is believed to be the result of the inability of ribosomes to reinitiate at the downstream AUG codon due to short intercistronic spacing between the two ORFs (Child et al. 1999b).
The second post-transcriptional control mechanism that is not as well understood involves an increased cell-type-dependent Her-2 translational efficiency in transformed cells compared with that in primary cells. In primary human fibroblasts and human mammary epithelial cells, Her-2 mRNA is associated with monosomes and small polysome fractions (Child et al. 1999a). The Her-2 transcript cosediments with larger polysomes in Her-2 overexpressing cancer cells, indicating that it is translated with a higher efficiency in those cells. Importantly, no differences were observed in the sequence and size of the Her-2 mRNA in primary cells compared with that in transformed human cells.
In this report, we have tested the hypothesis that post-transcriptional processes mediate the efficiency of Her-2 translation in cancer cells through interactions between the 5′ and 3′ UTRs of Her-2 mRNA. We have identified a U-rich translational derepression element (TDE) in the 3′ UTR that can modulate the translational repression by the uORF in the Her-2 5′ UTR. The protein factors that directly bind to the TDE were identified and were shown to form complexes with the Her-2 mRNA in breast cancer cells. These results elucidate a novel post-transcriptional mechanism involved in the modulation of the translation efficiency of transcripts harboring uORFs in certain cancer cell types.
Results
Her-2 UTRs modulate luciferase expression in a cell-type-dependent manner
To investigate the role of the Her-2 UTRs in modulating translation efficiency, a series of gene constructs that contained the firefly luciferase (FLuc) reporter gene flanked by various combinations of either the Her-2 UTRs or control UTRs were made (Fig. 1A). Expression of the reporter gene constructs was studied by transfection into a panel of cell lines that express surface Her-2 protein at very low (MCF-10A, 293H), moderate (Hela, MCF-7), or very high (SKBR-3, BT-474, AU-565) levels. FLuc activity was normalized to the activity units from cotransfected β-galactosidase or Renilla luciferase reporter constructs. Transfection of each construct into various cell lines demonstrated that, compared with the vector-derived UTRs, the Her-2 5′ UTR represses translation of reporter transcripts in all of the cell lines studied (Fig. 1B). To compare the effects of the Her-2 5′ UTR on luciferase expression, we denoted all subsequent activity as fold increase over the repression caused by the 5′ Her-2 construct.
Figure 1.

Translational regulation of Luc mRNA by the Her-2 UTRs in a panel of cell lines. (A) Schematic of the firefly luciferase (Fluc) reporter constructs containing various combinations of 5′ and 3′ UTRs. (B) The reporter constructs were expressed via transient transfection in a number of cell lines. The activity was measured 72 h post-transfection and normalized to either Renilla luciferase or to β-galactosidase units. The results were expressed as fold increase over the 5′ UTR-Fluc units. (C) Reporter constructs containing 5′ Her-2 UTR in combination with 3′ UTR from either Her-2 or GAPDH were expressed transiently in SKBR-3 cells. Translational derepression specifically requires Her-2 3′ UTR since the Luc constructs containing GAPDH 3′ UTR do not derepress translational inhibition by the Her-2 5′ UTR in SKBR-3 cells.
We also monitored the luciferase activity of the reporter that harbored both the Her-2 5′ and 3′ UTRs. The results were striking since insertion of the 3′ Her-2 UTR into the construct above significantly abrogated the ability of the 5′ UTR to inhibit translation in a number of Her-2 expressing cell lines (Fig. 1B). The greatest modulation was observed in SKBR-3 (12-fold ± 3.5-fold) and BT-474 (10-fold ± 2.5-fold) cells. MCF-7 cells showed a significant (fourfold ± 1.5-fold) increase in reporter gene expression. These breast cancer cell lines also demonstrate the greatest overexpression of Her-2 protein. Conversely, reporter expression in 293H and MCF-10A cells was hardly affected (1.1-fold ± 0.86-fold) by the presence of Her-2 3′ UTR. The fact that these cells have low endogenous Her-2 expression suggests a possible role for cellular factors in modulating Her-2 expression in Her-2 overexpressing cells.
The translational derepression by the 3′ UTR was not due to changes in transcript stability or alternative 3′-end processing. As determined by Northern blot analysis, there were similar levels of transiently expressed RNAs in SKBR-3 cells (Fig. 3B, below). Sequencing of the 3′-RACE products of the transiently expressed mRNA confirmed that all transcripts used the vector encoded polyadenylation signal and mapped to the same 3′ end (data not shown).
Figure 3.

Identification of a U-rich TDE in the 3′ UTR of Her-2 mRNA. (A) The firefly luciferase activity of the constructs containing 3′-end deletions and internal deletions is shown. (B) Northern analysis of the transiently expressed firefly luciferase RNA and the endogenous GAPDH RNA shows stable accumulation of all transcripts 72 h post-transfection. The sequence alignment of the 73-nt U-rich TDE across mammalian species is shown in C.
Taken together, these results suggest that the translation efficiency of the Her-2 transcript in Her-2 positive cells is regulated by interactions between the 5′ and 3′ UTRs. These results are consistent with previous reports demonstrating an increased association of the Her-2 mRNA in larger polysome fractions of BT-474 breast cancer cells (Child et al. 1999a) and suggest a novel mechanism in which translational repression by the 5′ UTR can be modulated by the 3′ UTR of Her-2.
The 3′ UTR of Her-2 is required for translational derepression
We next asked whether the Her-2 3′ UTR was specifically required for the derepression. To test this, the 3′ UTR of the reporter gene harboring the Her-2 5′ and 3′ UTRs was replaced with GAPDH 3′ UTR. This reporter was transfected into SKBR-3 cells as described previously and luciferase activity was quantified. The results demonstrated that in the presence of Her-2 5′ UTR, the translation efficiency of the reporter harboring the 3′ UTR of GAPDH was greatly reduced compared with the reporter containing the Her-2 3′ UTR (Fig. 1C). No differences in the RNA levels of the constructs were detected by real-time PCR analysis (data not shown). Thus, the GAPDH 3′ UTR failed to overcome translation repression caused by the Her-2 5′ UTR. In the absence of the Her-2 5′ UTR, normal levels of translation were obtained with luciferase reporter under the control of the 3′ UTR of GAPDH (Fig. 1C), again demonstrating the inhibitory effect of the Her-2 5′ UTR on translation.
Translation of a uORF is required for the 3′ UTR of Her-2 to mediate translation derepression
We next examined whether the Her-2 uORF is utilized in cells overexpressing the Her-2 protein. The AUG start codon of the uORF is in an optimal Kozak sequence context and thus is expected to give rise to a six-amino-acid peptide upon initiation from this small ORF. Since the production of such a small peptide would be difficult to monitor, we mutated the uORF termination codon (UGA to GGA) and inserted one nucleotide so that the luciferase ORF would be in frame with the uORF in order to detect initiation at Her-2 uORF. Initiation at upstream AUG (uAUG) would result in the synthesis of an elongated luciferase protein. Transfection of this construct into SKBR-3 cells resulted in a 12-fold increase in activity in the reporter construct harboring the Her-2 uORF containing the GGA in-frame mutant compared with the reporter construct harboring the wild-type Her-2 uORF (Table 1). A further twofold increase (24 ± 3.5) was observed when the Her-2 3′ UTR was present (Table 1). These results suggest that the uAUG is used efficiently by the translating ribosomes and that removal of the upstream termination codon significantly reduced the 3′ UTR-mediated derepression. Mutation of the luciferase translation initiation AUG codon in the uORF in-frame construct to AAG did not have an appreciable effect on the expression of luciferase, supporting the conclusion that translation initiated efficiently at the uAUG (Table 1). To determine if the repression by the 5′ UTR of Her-2 is affected by the stop codon sequence in the uORF, the UGA in the uORF was mutated to UAA or UAG and luciferase expression was monitored. The repression of translation was found to be independent of the stop codon sequence (Table 1).
Table 1.
Effect of mutations in the 5′ Her-2–FLUC constructs

(WT) Wild type; (uORF) upstream open reading frame; (Luc) Luciferase cistron.
We next asked whether the interactions between the 5′ and 3′ UTRs required the translation of the uORF. To address this question, the translation initiation site of the uORF was removed by point mutagenesis (the AUG of the uORF to AAG) and the construct was transiently expressed in SKBR-3 cells. The translational repression was removed in the presence of a dysfunctional uORF. However, in presence of the mutant uORF and the wild-type Her-2 3′ UTR, there was no further synergistic increase in translational efficiency of the construct (Fig. 2A). This result suggests that the derepression of translation mediated by the 3′ UTR is dependent on the translation of the uORF. The mRNA levels of all of the transfected constructs reported in these studies were not significantly different as determined by real-time PCR (data not shown).
Figure 2.

(A) Derepression by the 3′ UTR requires translation of uORF in Her-2 5′ UTR. The constructs containing wild-type and mutated uORF (AUG to AAG) were transfected into SKBR-3 cells and the activity determined as described in Figure 1. Removal of the uAUG results in loss of the 3′-UTR-mediated derepression of translation. (B) Schematic depicting the location of uORF in SHIP-2 mRNA and the Fluc constructs containing chimeric Her-2 and SHIP-2 UTRs. (C) The 3′ Her-2 UTR derepresses heterologous Ship-2 uORF-mediated inhibition of translation of the reporter in SKBR-3 cells. (D) The table shows the relative levels of mRNA of transiently expressed constructs compared with that of 5′ Her-2 mRNA. The RT–PCR data show that the difference in the RNA levels of 5′ + 3′ SHIP-2 and 5′ SHIP-2 + 3′ Her-2 constructs did not contribute significantly to the derepression of SHIP-2 uORF.
We next asked if the increased translation efficiency observed was a consequence of internal translation initiation that was controlled by the Her-2 UTRs. Since the insertion of nucleotides predicted to result in strong secondary structures in the 5′ UTRs in mRNAs have previously been shown to inhibit cap-dependent translation (Kozak 1991a; Muhlrad et al. 1995), translational up-regulation by Her-2 UTRs in the presence of a secondary structure would suggest a UTR-mediated internal ribosome entry site (IRES) mechanism. To test this possibility a stem-loop (dG = −56.9 kcal/mol) was inserted close to the translation start site of the Her-2 5′ UTR and its effect on translation of the luciferase constructs was examined. The stem-loop inhibited the cap-dependent translation by >200-fold in the vector construct compared with that in the no-stem-loop control. The IRES from the 5′ UTR of HIF-1α served as a positive control and showed a 13-fold increase in translation in the presence of the stem-loop. However, there was no increase in translation of luciferase under the control of Her-2 5′ UTR alone or with the 3′ UTR, thus indicating that cap-dependent translation was required for the observed synergy (Table 2).
Table 2.
Effect of stem-loop on translation of the Her-2 UTR FLUC constructs
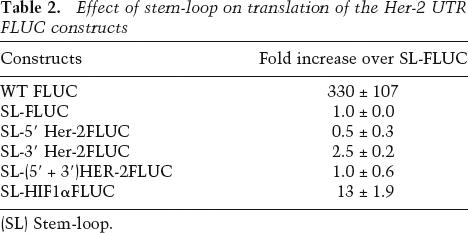
(SL) Stem-loop.
The 3′ UTR of Her-2 can promote translation derepression of other uORFs
We next determined whether translational derepression by the 3′ UTR of Her-2 specifically required the Her-2 uORF or whether it could promote derepression of other known uORFs. To test this, reporters in which the 5′ and 3′ Her-2 UTRs were replaced with the 5′ and 3′ UTRs of the SHIP-2 gene were constructed. The 5′ UTR of SHIP-2 RNA is similar in size to the Her-2 5′ UTR and harbors a 10-amino-acid uORF with similar spacing relative to the SHIP-2 protein coding region (Fig. 2B). The reporter constructs were transfected into SKBR-3 cells, and activity was monitored as described above. The results demonstrated that the 5′ UTR of SHIP-2 RNA also inhibited the translation of the reporter in SKBR-3 cells, and that this translational inhibition of the SHIP-2 uORF was not relieved when the SHIP-2 3′ UTR was inserted into the reporter gene (Fig. 2C). The presence of the Her-2 3′ UTR, however, demonstrated a 4.5-fold ± 1.1-fold increase in translation efficiency compared with that of the reporter containing the SHIP-2 5′ UTR. These results demonstrate that, although suboptimal compared with a reporter construct containing both the 5′ and 3′ UTRs from Her-2, the Her-2 3′ UTR could function in the context of a heterologus uORF sequence to derepress translation of the reporter. Quantitative real-time PCR analysis showed that both 5′ + 3′ SHIP-2 Fluc and 5′ SHIP-2 + 3′ Her-2 Fluc transcripts were expressed to similar levels and that the RNA levels did not account for the derepression of SHIP-2 uORF by Her-2 3′ UTR (Fig. 2D).
Identification of sequences required for translational derepression
We next defined more precisely the cis-acting elements in the 3′ UTR that are required for Her-2 uORF-mediated translational derepression. To accomplish this, deletions were made in the 612-nt 3′ UTR starting from the 3′ end of the reporter harboring the 5′ and 3′ UTRs of Her-2. Each resulting reporter construct was transfected in SKBR-3 cells and luciferase activity was determined. The results demonstrated that removal of the last 75 nt (Del 537–612) did not affect the ability of 3′ UTR in overcoming the inhibition by 5′ UTR (Fig. 4A, below). However, removal of an additional 73 nt (Del 465–612) completely abolished the derepression function of the 3′ UTR. These results demonstrated that a 73-nt sequence element (TDE) located between nucleotides 465 and 537 of the 3′ UTR is required to override the inhibition of Her-2 translation by the uORF. Northern blot analysis of the transiently expressed RNAs showed that the effect was independent of RNA levels in SKBR-3 cells (Fig. 3B).
Figure 4.

Derepression activity of TDE correlates with the UV-cross-linking of 38-kDa protein. (A) The luciferase activity of the Fluc constructs containing 5′ Her-2 UTR and internal and 3′-end deletions of Her-2 3′ UTR. (B) The 3′ Her-2 UTR UV-cross-linked to a 38-kDa protein from SKBR-3 extracts. Proteins UV-cross-linked to labeled RNA probes were resolved on 10%–14.5% SDS-PAGE as described in Materials and Methods. Molecular size markers are indicated on the right. (C) Competition of the UV-cross-linked band by 73-nt TDE but not by the sequences of the 5′ UTR of Her-2 mRNA. The T7 polymerase-transcribed 32P-labeled 3′ Her-2 UTR RNA was incubated with SKBR-3 cytoplasmic extracts in the absence of competitor (none) or in the presence of a 2.5-fold, fivefold, 20-fold, 50-fold, or 100-fold molar excess of unlabeled 465–537-nt RNA or with a 10-fold, 50-fold, 100-fold, or 500-fold molar excess of the 178-nt 5′ Her-2 UTR RNA.
To identify the nucleotides required for derepression, deletions were made within the 73-nt TDE. Deleting nucleotides 497–507 was found to decrease derepression activity of the 3′ UTR to 50% of the wild-type construct. A greater loss of derepression activity was obtained when a stretch of 22 nt was deleted from the Her-2 3′ UTR (485–507) or when almost all (Del 463–519) (Fig. 3A) or the entire 73-nt element (Del 465–537, Fig. 4A) was deleted. Sequence analysis of the 73-nt TDE showed the presence of uridine-rich stretches (Fig. 3C). Intriguingly, the TDE is 80% conserved at the nucleotide level across mammalian species.
To identify the minimal region required for derepression activity, deletions were made from the 5′ end of the 3′ UTR (data not shown). The removal of nucleotides 1–107, nucleotides 1–207, nucleotides 1–307, and nucleotides 1–465 from the 5′ end of the 3′ UTR completely abrogated derepression activity. In addition, removal of the first five nucleotides in a construct containing nucleotides 5–612 in the 3′ UTR resulted in loss of derepression activity, thus indicating that the regions in the 5′ end of the 3′ UTR are also required in addition to the TDE. Interestingly, the entire 3′ UTR was found to fold into a thermodynamically stable secondary structure with a ΔG of −228 kcal/mol (M-fold analysis; data not shown).
Identification of factors that interact with the TDE
We hypothesized that the TDE interacts with trans-acting factors to regulate translational derepression. To test this hypothesis, UV-cross-linking studies were performed to identify proteins that interact with the TDE. Several radiolabeled RNA transcripts were prepared that contained either the full-length Her-2 3′ UTR or a deletion construct lacking the TDE or the Her-2 5′ UTR. The RNAs were incubated with cytoplasmic extracts from the SKBR-3 cells and after 1 h, the protein–RNA complexes were cross-linked by UV irradiation. Following RNase A/T1 digestion, the radiolabeled RNA–protein complexes were resolved on SDS-PAGE and visualized by phosphorimager analysis. The results demonstrated that a predominant 38-kDa ± 4-kDa protein cross-linked to both the 612-nt Her-2 3′ UTR and to the 73-nt TDE, but did not cross-link to the 5′ UTR (Fig. 4B,C). The 38-kDa protein also did not cross-link to the RNA with 485–507-nt TDE internal deletion (Fig. 4B). The cross-linking activity largely correlated with the luciferase activity of the deletion constructs, indicating the possible involvement of the trans-acting factors in the derepression of translation (Fig. 4A,B).
Since the results demonstrated that a 38-kDa protein UV-cross-linked to the full-length 612-nt Her2 3′ UTR, competition experiments were performed to investigate the binding specificity of the 38-kDa protein (Fig. 4C). The cross-linking activity was effectively competed out by 50-fold molar excess of the cold-73-nt TDE (465–537) but not by a 500-fold molar excess of 178-nt Her-2 5′ UTR or by region 1–307 of Her-2 3′ UTR lacking the binding site (data not shown). These results show that the UV-cross-linking activity is specific to the 73-nt TDE in the Her-2 3′ UTR.
Identification of proteins binding to Her-2 3′ UTR in SKBR-3 cells
We hypothesized that the TDE in the Her-2 3′ UTR may function by recruiting protein factor(s) that facilitate interactions between the UTRs of Her-2 mRNA and the cellular translation machinery. To study the role of trans-acting factors in modulating interactions between the 5′ and 3′ UTRs, we utilized RNA affinity capture with in vitro biotinylated 3′ UTR transcripts to purify and identify the protein(s) that bound to the Her-2 3′ UTR TDE. SKBR-3 cytoplasmic extracts were precleared using RNA (1–407) lacking the TDE. Unbound proteins were subsequently incubated with the TDE-containing RNA (1–612) resin. After extensive washing, the bound proteins were eluted stepwise with 0.2 M, 0.5 M, and 1 M NaCl containing buffer A. Almost all of the UV-cross-linking activity was associated with the 0.5 M eluate. The proteins in the eluate were concentrated and resolved by SDS-PAGE. Protein bands were excised and their identity determined by mass-spectrometric analysis.
The mass-spectrometric analysis identified HuR and hnRNP A1 as Her-2 3′ UTR-binding proteins and suggested that multiple proteins may interact with 3′ UTR (data not shown). To determine which of the identified proteins directly bound to the TDE, in vitro binding affinity chromatography was again utilized with biotinylated RNA. The protein–RNA complexes were transferred to nitrocellulose, and the presence of the candidate proteins, HuR and hnRNP A1, as well as a number of other hnRNP proteins were examined by immunodetection with antibodies. The results demonstrated that hnRNP A1 bound not only to the full-length RNA (1–612) and the TDE (465–537) but also to RNA (1–407) lacking the TDE-binding site, indicating the presence of multiple binding sites in Her-2 3′ UTR (Fig. 5A). hnRNP C1/C2 and HuR bound specifically to the TDE and to the 1–612-nt RNA but not to the RNA (1–407) (Fig. 5A). Other hnRNP proteins (Q, K/J), transportin, and AUF (data not shown) were not detected. A very small proportion of hnRNP L (cf. the total input) was detected in the complex and could be the result of nonspecific binding under the conditions used. Overall, these results suggest that a large protein complex containing several known RNA-binding proteins assembles specifically on the Her-2 3′ UTR TDE.
Figure 5.

(A) A specific protein complex assembles on the 73-nt TDE. Western blot analysis of the protein complexes formed on biotinylated RNA encoding full-length 3′ UTR (1–612) or the 73-nt (465–537) or on RNA lacking the TDE (1–407). The complexes formed on RNA–streptavidin beads were resolved on SDS-PAGE and immunoblotted to nitrocellulose membrane. The blots were probed with antibodies to various proteins. (B) The UV-cross-linked proteins were immunoprecipitated by anti-HuR and anti-C1/C2 antibodies. The proteins cross-linked to the full-length 3′ UTR (1–612) or to the 73-nt binding site were incubated with various antibodies and the immunoprecipitated complexes resolved by SDS-PAGE and visualized by autoradiography.
We next determined which of the proteins detected in the Her-2 3′ UTR RNA complex were also present in the UV-cross-linked band. The protein extracts were UV-cross-linked to full-length 3′ UTR RNA (1–612) or the 73-nt TDE and after RNase treatment, the reactions were immunoprecipitated with various antibodies. Antibodies to the proteins HuR and C1/C2 immunoprecipitated the UV-cross-linked proteins whereas antibodies to hnRNP A1, L, Q, U, K/J, AUF, and transportin did not (Fig. 5B). These results confirm the presence of HuR and hnRNP C1/C2 as components of a RNP complex making direct contact with the Her-2 3′ UTR.
Characterization of the native complexes in SKBR-3 cells
To determine if HuR and C1/C2 exist as native complexes in vivo, extracts from SKBR-3 cells were subjected to immunoprecipitation by anti-HuR or by anti-C1/C2 antibodies. The immunoprecipitates were resolved on SDS-PAGE and, after transfer to nylon membranes, were probed with various antibodies. The hnRNP A1 and C1/C2 proteins were found to be present in the complex immunoprecipitated by anti-HuR antibody. In addition, hnRNP A1 and HuR were pulled down by antiC1/C2 antibody. However, the interaction between hnRNP C1/C2 and HuR was abrogated when the extract was treated with RNases before immunoprecipitation. This result suggests that RNA facilitates the interactions between factors and that there is no direct interaction between HuR and hnRNP C1/C2 (Fig. 6A). As seen with the in vitro assembled complexes, the in vivo complexes contained hnRNP A1 but did not contain AUF and other hnRNP proteins (L, Q, U, K/J, transportin, or A0). Thus, we conclude that HuR, hnRNP C1/C2, and A1 are present in the native complexes in SKBR-3 cells.
Figure 6.

(A) Identification of native complexes assembled in SKBR-3 cells. The SKBR-3 cytoplasmic extracts were left untreated or treated with RNase cocktail on ice for 30 min. The extracts were immunoprecipitated with anti-HuR or anti-C1/C2 antibodies and the complexes immobilized on nitrocellulose. The presence of PABP, AUF, HuR, C1/C2, A1, and other hnRNP proteins was examined by immunodetection with corresponding antibodies. (B) Association of the endogenous Her-2 mRNA with the HuR/C1/C2 complexes in SKBR-3 cells. The RNA–protein complexes in SKBR-3 cells were cross-linked by formaldehyde and the extracts were made as described in Materials and Methods. The extracts were immunoprecipitated with either anti-HuR or anti-C1/C2 antibodies and the RNA–protein cross-links were reversed by heat treatment. The RNA associated with the complexes was extracted with Trizol and used for first-strand cDNA synthesis. The presence of Her-2 RNA or nonspecific GAPDH RNA was detected by PCR using gene-specific primers.
Since the 3′ UTR of Her-2 mRNA was found to override the translational inhibition imposed by the uORF in the 5′ UTR, we were interested in determining whether there was association of the TDE complex with known translation factors. Thus, in addition to the proteins detected in the in vitro assembled complexes, the native immunoprecipitates were also examined for the presence of proteins known to mediate cap-dependent translation. While the poly(A)-binding protein (PABP) was detectable in the immunoprecipitates of both HuR and C1/C2 (Fig. 6A), other factors involved in mRNA translation, such as eIF4E, eIF4G, eIF3α, eIF5α, and eIF2α, were not detected (data not shown). The interaction of the PABP with the TDE complex was found to be RNA independent (Fig. 6A), indicating a direct association with both HuR and with hnRNP C1/C2.
HuR and hnRNP C1/C2 interact with the 3′ Her-2 UTR in cells
To study if the Her-2 mRNA interacts with the protein complexes in vivo, a ribonucleoprotein immunoprecipitation (RIP) assay was performed as described in Materials and Methods. Briefly, the in vivo SKBR3–RNP complexes were reversibly cross-linked with formaldehyde and the extracts immunoprecipitated with anti-HuR or anti-C1/C2-protein-G-Sepharose. Total RNA was isolated after the cross-links were reversed, DNase I treated, and the RNA was used for RT–PCR. As seen in Figure 6B, the Her-2 3′ UTR was detectable in the immunoprecipitates of anti-HuR and anti-C1/C2 antibodies. As expected, no product was detected when reverse transcriptase was omitted. The absence of a PCR product corresponding to GAPDH mRNA in the immunoprecipitates demonstrated the specificity of the RIP assay. These results indicate that the Her-2 mRNA is in complex with HuR and C1/C2 proteins in vivo in SKBR-3 cells.
Functional relevance of the protein complex in breast cancer cells
Since the proteins associating with the TDE complex (HuR, hnRNP C1/C2, PABP, and hnRNP A1) are ubiquitously expressed, we wanted to establish that the TDE–protein complex is functionally relevant to Her-2 translational derepression. Since the small interfering RNA (siRNA)-mediated knockdown of HuR and hnRNP C1/C2 resulted in significant cytotoxicity to SKBR3 cells, we used UV-cross-linking analysis to study if the cross-linked complex was detectable in the cell lines expressing Her-2 protein to different levels. The UV-cross-linking experiments with extracts made from a number of cell lines showed that the relative abundance of proteins that cross-link to the TDE correlates directly not only with the level of Her-2 expression but also with the fold-derepression activity of the Her-2 3′ UTR (Fig. 7A). The intensity of the UV-cross-linked complex was found to be stronger in cell lines that supported derepression by Her-2 3′ UTR and expressed high Her-2 protein levels (SKBR3, AU565, BT474, MCF-7) when compared with cell lines in which derepression by Her-2 3′ UTR is not observed and that have low Her-2 levels (293, HeLa). The results suggest that the expression levels of the protein complex may contribute to derepression activity and Her-2 expression in some cancer cells. Since these are cell extracts, however, we are unable to determine if the differences in cross-linking are directly due to relative abundance of the protein(s) or due to post-translational modifications of one or more of the cross-linked proteins that may influence affinity of interactions and/or intracellular distribution of the protein complex.
Figure 7.

Functional significance of the TDE–protein complex. UV-cross-linking studies were performed using extracts prepared from cell lines that express Her-2 to varying levels. A more intense cross-linked complex was detected in high Her-2 expressers (SKBR3, AU565, Calu3, MCF-7) compared with the low Her-2 expressers (293T, HeLa, HepG2). (B) Immunofluorescence microscopy of HuR in breast cancer cells. More cytoplasmic HuR was detected in SKBR-3 cells, a high Her-2 expresser compared with MCF-10A, a low Her-2 expressing cell line.
To study the intracellular distribution of one or more protein(s) in the TDE complex, we performed an in situ immunofluorescence experiment. As seen in Figure 7B, one of the proteins in the TDE complex, HuR, is differentially distributed between two breast cancer cell lines, SKBR3 and MCF10A. Although HuR is found predominantly in the nucleus of both cell lines, there is a significant portion of HuR within the cytoplasm of the high Her-2 expressing cell line SKBR3 compared with the primary breast cancer cell line, MCF10A. This observation is in accordance with the earlier reports on increased cytoplasmic HuR levels in higher grades of breast tumor samples (Denkert et al. 2004) and in colon cancer cells (Dixon et al. 2001).
Discussion
The 5′ UTRs of ∼5%–10% of eukaryotic mRNAs are known to harbor an uORF. Interestingly, a high percentage of RNAs containing uORFs encode oncogenes and growth factors (Kozak 1991b), and expression of these genes is highly regulated, as their protein products are important in cell growth and proliferation. Studies of a subset of RNAs harboring uORFs have shown that they can function by modulating translation efficiencies of downstream cistrons via a number of distinct mechanisms including translation termination and reinitiation as well as nascent-peptide-dependent ribosome stalling on the mRNA (for review, see Morris and Geballe 2000).
Overexpression of the Her-2 receptor is a critical factor in oncogenic transformation of certain breast cancers. In all mammals studied so far, the 5′ UTR of Her-2 mRNA contains an uORF that is highly conserved in its position, length, and sequence, suggesting an important role for the uORF in translational regulation. The Her-2 uORF is translated and strongly inhibits the expression of the downstream coding region in a peptide-sequence-independent manner (Child et al. 1999b). In the absence of the Her-2 3′ UTR, it has been reported that termination and reinitiation at the main Her-2 AUG occurs inefficiently due to short intercistronic spacing. Thus, post-transcriptional mechanisms must exist to repress the function of the uORF and allow for overexpression of Her-2 mRNA that contributes to oncogenic potential of tumor cells.
Tumor-specific Her-2 translational control requires the 3′ UTR
To investigate the potential role for translation control of the Her-2 mRNA, we focused on the function of the 5′ and 3′ UTRs. Utilizing reporter constructs where the Her-2 5′ and 3′ UTRs flank the firefly luciferase, we demonstrated that the uORF of Her-2 is derepressed in the presence of the 3′ Her-2 UTR in a tumor-cell-line-specific manner. Derepression of the uORF-mediated translation specifically required the presence of the Her-2 3′ UTR; a control reporter harboring the GAPDH 3′ UTR was unable to cause uORF derepression. In addition we demonstrated that derepression is independent of the nascent peptide, but requires translation of the uORF. We hypothesize that the translation of the uORF allows the ribosome to properly terminate at the uORF stop codon where derepression is facilitated by factors that interact with the Her-2 3′ UTR.
Deletion analysis of the 3′ UTR identified two important regions that are necessary to mediate derepression of the Her-2 uORF. The 5′-proximal portion of the 3′ UTR (1–465 nt) was required for translation derepression. This region has the potential to fold into thermodynamically stable strong secondary structures (M-fold) including IRES-like Y-type stem-loops (141–244 nt) (http://www.ba.itb.cnr.it/BIG/UTRScan; Le and Maizel 1997). A second sequence element, referred to as a TDE, located at 465–537 nt of the 3′ UTR was also shown to be necessary to mediate translation derepression of the uORF.
The Her-2 TDE binds a novel protein complex
Several RNA-binding proteins were identified that associated specifically with the TDE. Among the proteins identified, HuR and hnRNP C1/C2 proteins were found to bind specifically and directly to a short sequence (nucleotides 497–507) in the TDE (Fig. 4B). In addition, the hnRNP A1 protein was found to be associated with the immunoprecipitated complex formed on the TDE both in vitro and in vivo.
Both HuR and hnRNP C1/C2 bind to the U-rich sequences with high affinity (Gorlach et al. 1994; Lopez de Silanes et al. 2004). HuR is a highly conserved member of the Elav/Hu family of RNA-binding proteins involved in the modulation of RNA stability and translation (Good 1997). hnRNP C1/C2 proteins are nuclear proteins proposed to be involved in mRNA processing (Choi et al. 1986; Swanson et al. 1987). The hnRNP C1/C2 proteins have recently been implicated in translational control of the c-sis mRNA as well as poliovirus RNAs (Sella et al. 1999; Brunner et al. 2005). Post-translational modifications that mask the strong nuclear retention signal of hnRNP C1/C2 have been hypothesized to result in their retrafficking in the cytoplasm (Mayrand et al. 1993; Pinol-Roma and Dreyfuss 1993; Sella et al. 1999). Both HuR and hnRNP C1/C2 were shown to bind to a U-rich translational regulatory element in the 5′ UTR of p27kip RNA in a cell-cycle-dependent manner (Millard et al. 2000). The hnRNP A1 protein has been shown to be associated with pre-mRNA processing (Burd and Dreyfuss 1994) and export (Michael et al. 1995) of RNAs from the nucleus. hnRNP A1 binds to UAGGGU/A sequences in the RNA (Burd and Dreyfuss 1994), and one such consensus binding site is present in Her-2 mRNA (nucleotides 207–247).
In addition to proteins that directly cross-linked to the Her-2 TDE, the PABP was found to be associated with the native TDE complexes from SKBR-3 cells isolated by immunoprecipitation with anti-HuR and anti-C1/C2 antibodies. The PABP is normally bound to the poly(A) tract of the mRNA, although it has been implicated in binding to certain regions in 5′ (Yohn et al. 1998; Hornstein et al. 1999) and 3′ UTRs. PABP was shown to interact not only with components of translation initiation machinery (Sachs and Davis 1989; Tarun and Sachs 1996; Wells et al. 1998) but also with the translation termination release factor eRF3 (Hoshino et al. 1999). This has led to the notion that PABP most likely plays important roles in translation initation, termination, and recycling (Kahvejian et al. 2001, 2005; Wilkie et al. 2003; Kuhn and Wahle 2004). The observation that PABP interacts with TDE-binding proteins and key components of the translation initiation and termination process may suggest a role for PABP in regulating the translation derepression efficiency of the Her-2 transcript.
An important question is what determines the cell-type specificity that allows for translational derepression of the Her-2 transcript. HuR, hnRNP C1/C2, hnRNP A1 and PABP are ubiquitously and highly expressed proteins. It is conceivable that changes in localization or modification of these proteins, rather than absolute levels may be important. Post-translational modification of HuR (Li et al. 2002) and cell-environment-dependent nuclear–cytoplasmic shuttling of HuR (Yaman et al. 2002) has been reported earlier. Consistent with this idea, increased cytoplasmic HuR levels were observed in human breast carcinoma samples (Denkert et al. 2004). Increased cytoplasmic HuR levels were also shown to stabilize the COX-2 transcript in colon cancer cell lines (Dixon et al. 2001). Consistent with this observation, we found strikingly higher levels of HuR in the cytoplasm of SKBR-3 cells that overexpress Her-2 compared with MCF10A cells in which the derepression activity is not observed. This increase in cytoplasmic levels of HuR in cells that show high derepression activity, alone or in combination with post-translational modifications, altered affinities, and interactions of other proteins in the TDE complex, could account for the cell-type-specific derepression.
3′ UTRs can modulate translation efficiency
Derepression of the Her-2 uORF involves recruitment of a protein complex bound to the TDE of Her-2 3′ UTR to modulate translation termination/reinitiation efficiency of the Her-2 uORF, underscoring the importance of the termination/reinitiation process in gene regulation. This represents the first demonstration of how disregulation of this mechanism can lead to cancer.
The role of 3′ UTRs in regulating gene expression is well documented, and several lines of evidence indicate that the 3′ UTR can modulate the translation efficiency of an mRNA (Ostareck et al. 1997; Grskovic et al. 2003; Kuersten and Goodwin 2003; Hesketh 2004; Cho et al. 2005; de Moor et al. 2005; Espel 2005). The closed-loop model of eukaryotic mRNA structure is one conceptual framework that can explain how 3′ UTRs and their associated factors can modulate translation initiation efficiency (Tarun et al. 1997; Wells et al. 1998; Mangus et al. 2003). Interactions of the factors that bind to the 5′ cap, the 3′ poly(A) tract, and 5′ and 3′ UTRs promote contacts between the 5′ and 3′ ends of eukaryotic mRNAs that regulate translation initiation efficiency (Wilkie et al. 2003). The intriguing observation that PABP interacts with eRF3 and the factors at the 5′ cap suggests a further modification of the closed-loop model. This interaction suggests that terminating ribosomes may influence the RNP structure of the closed loop that affects subsequent rounds of translation initiation (Welch et al. 2000). Hence, logically the closed-loop model may include a more pretzel-like structure in which the 5′ and 3′ UTRs are interacting at the terminating ribosome to modulate translation reinitiation.
An important question to consider is how the Her-2 3′ UTR can modulate its translation initiation efficiency. At present there are only a few examples illustrating the role of uORFs in regulating the initiation of translation. Perhaps the best example of uORF regulation is the control of expression of GCN4 in the yeast Saccharomyces cerevisiae. The GCN4 mRNA harbors four uORFs and translation initiation/reinitiation efficiency of the uORFs governs the quantity of GCN4 protein produced (for review, see Hinnebusch 2000). The GCN4 uORF1 is always translated, and reinitiation following translation termination of uORF1 is very efficient. Translation reinitiation occurs either at the fourth uORF or at the AUG of the GCN4 protein-coding region depending on the nutrient supply. Under amino acid sufficient conditions, the ribosomes reinitiate at uORF4, but following translation termination, the GCN4 coding region is not translated. Under amino acid deficient conditions, the ribosomes bypass the fourth uORF and reinitiate further downstream at the GCN4 translation initiation site. Starvation generates uncharged tRNA that induces phosphorylation and subsequent inactivation of eIF2. When eIF2 levels fall, the ribosomes take longer to reacquire Met-tRNAi and do not translate the fourth uORF, but initiate translation at the GCN4 protein-coding region. Thus, regulation of GCN4 protein expression is an example of how modulation of translation reinitiation events of uORFs can control protein levels.
The 3′ UTR has also been shown to affect other aspects of the translation process. The translational recoding of UGA as selenocysteine is directed by a SECIS (selenocysteine insertion sequence) element in the 3′ UTRs of eukaryotic selenoprotein mRNAs (Copeland 2003). Proteins bound to SECIS elements found in the 3′ UTR alter translation termination efficiency at these stop codons so that selenocysteine amino acids are inserted. Clearly, there are a number of examples that can help to explain how the Her-2 3′ UTR modulates translation initiation efficiency.
Model of translational regulation by uORF and Her-2 3′ UTR
The results presented here suggest that the uORF in the Her-2 5′ UTR is a translational repressor that allows inefficient translation reinitiation at downstream cistrons. Factors bound to the Her-2 3′ UTR, however, can alter this process such that terminating ribosomes can reinitiate at the downstream Her-2 protein coding region (Fig. 8). Several lines of evidence are consistent with this hypothesis. In the primary human mammary epithelial cells, which express low levels of the Her-2 receptor, the majority of the Her-2 mRNA cosediments with small polysomes and suggests that 80S ribosomes may stall at the uUGA of the uORF. In contrast, the Her-2 mRNA is distributed throughout the polysome gradient in BT-474 breast cancer cells that overexpress Her-2 protein (Child et al. 1999a). This result suggests that the Her-2 transcript is efficiently translated and suggests that translation reinitiation occurs in the Her-2 protein-coding region.
Figure 8.

Model for translational derepression by Her-2 3′ UTR. (A) Based on earlier studies, the conceptual closed-loop model shows interactions of PABP with the cap complex at the 5′ end as well as its interaction with eRF3 at the stop codon near the 3′ end of the coding region. In the simplest scenario, the ribosomes translating the uORF remain stalled at the uUGA in cell lines where derepression is not significant. Under these conditions the translation of Her-2 protein is dependent on leaky scanning and/or inefficient reinitiation. (B) In cells overexpressing Her-2 protein, the highly folded 3′ UTR along with the protein complex docked on TDE facilitates rapid termination at the uUGA via the PABP–eRF3 contacts. The 40S ribsome that remains attached retains certain initiation factors, reacquires eIF2-Met-tRNAi-GTP, and is recycled to the main coding region with higher efficiency. The ribosomes terminating at the uUGA support formation of a pretzel-like mRNP structure as described in the Discussion. Such a structure would allow terminating ribosomes to recycle onto the same transcript through interactions with the 3′-UTR protein complex.
We hypothesize that the 80S ribosomes translating the short uORF can remain associated with the Her-2 mRNA (Fig. 8A). The interactions of the ribosomes with the Her-2 3′ UTR–protein complex may impact termination efficiency at the stop codon of uORF and facilitate efficient recharging of the ribsome to allow translation reinitiation at the Her-2 translation start site. The TDE-associated complex that includes the PABP may allow rapid dissociation of the 80S complex through its interactions with the translation termination release factor, eRF3, at the stop codon of the uORF so that the 40S subunit remains associated with the mRNA. The eRF3 complexes with the eRF1 and contributes to efficient termination at the stop codon (Stansfield et al. 1995a, b). Since the activity of the release factors can be modulated by other factors such as Upf1 (Czaplinski et al. 1998; Welch et al. 2000), we hypothesize that protein factors within the TDE protein complex may modulate activity of the release factors and subsequent recycling of the ribosomal subunits (Fig. 8B). Because of the small size of the uORF, the 40S subunit may retain certain translation initiation factors and rapidly reacquire eIF2/Met-tRNAi/GTP, so that they efficiently reinitiate at the Her-2 translation start site. It is also conceivable that the theoretical IRES-like structure within the Her-2 3′ UTR may mimic the 5′ cap and, in concert with the TDE-bound protein complex, may facilitate recruitment of translation factors for rapid reinitiation at the Her-2 translation start site.
The model described above is a modification of the closed-loop model of eukaryotic mRNA that describes how ribosomes are recycled following translation termination at the end of the protein-coding region (Fig. 8A; Kahvejian et al. 2001). In this scenario, terminating ribosomes proximal to the 3′ UTR can be recycled to the translation start site proximal to the 5′ UTR as a consequence of the interactions of factors associated with 5′ and 3′ UTRs, as well as factors associated with ribosomes. In the case of Her-2 uORF, we hypothesize that the factors associated with Her-2 3′ UTR can promote recycling of the terminating ribosomes at the uORF to facilitate their subsequent interaction at the Her-2 translation start site (“pretzel-loop”; Fig. 8B).
Interestingly, this model suggests that the single Her-2 uORF functions like a combination of the GCN4 uORF1 and uORF4 uORFs. In the absence of other elements and factors, the Her-2 uORF is analogous to the GCN4 uORF4 in that ribosomes that translate this uORF cannot efficiently translate downstream cistrons. Factors bound to the Her-2 3′ UTR, however, can alter the Her-2 uORF so that it functions more like the GCN4 uORF1, allowing efficient translation reinitiation to occur at downstream cistrons. The key difference is that, much like the SECIS elements that promote translation readthrough for synthesis of selenocyteine proteins, the Her-2 regulatory sequences that modulate this activity appear to be linearly distant from the site of action.
The work reported here describes a novel translation mechanism in which the translation efficiency of genes harboring uORFs in their 5′ UTRs can be governed by sequences in their 3′ UTRs. The detailed mechanism of how the 3′ UTR facilitates increased translation efficiency at the translation initiation site of the Her-2 protein-coding region requires further elucidation. Further dissection of the mechanism will help gain insight into the post-transcriptional mechanisms modulating translation of many regulatory genes and will facilitate the identification of new molecular targets for drug discovery to treat cancer.
Materials and methods
Generation of Her-2 constructs
The 178-nt 5′ UTR and the 612-nt 3′ UTR of Her-2 mRNA were amplified by PCR using SKBR-3 cDNA and the human genomic DNA (Promega), respectively, as template and then cloned into pcDNA3.1/FLUC vector, a pcDNA 3.1(+) (Invitrogen) plasmid containing a firefly luciferase (FLuc) expression cassette driven by a CMV promoter. Three different constructs were generated, including one in which the luciferase expression is controlled by the Her-2 5′ UTR alone (5′ Her-2), one controlled by only the Her-2 3′ UTR (3′ Her-2), and the one in which both the 5′ and the 3′ UTRs from Her-2 controlled Luc expression (5′ + 3′ Her-2). The GAPDH 3′ UTR was also PCR amplified and cloned into the above vectors to provide a control for specificity. The sequences of all constructs were confirmed by the dideoxysequencing method.
Cell culture and preparation of lysates
All cell lines were obtained from ATCC and all cell lines except SKBR-3 and MCF10A were cultured in Dulbecco’s Essential Modified Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Invitrogen) under humidified atmosphere containing 5% CO2. SKBR-3 cells were grown in McCoy’s medium containing 10% FBS, and MCF10A cells were grown in Mammary Epithelial Growth Medium (MEGM; Cambrex, Inc.) supplemented with 10% FBS and bovine pituitary extract. Whole-cell lysates for luciferase and β-galactosidase assays were prepared by lysing cells in passive lysis buffer (Promega) after two washes with PBS. The cytoplasmic extracts were prepared by swelling the cells in hypotonic buffer (10 mM HEPES at pH 7.4, 15 mM KCl, 1.5 mM magnesium acetate, 0.5 mM Pefabloc [Roche], 2 mM DTT) for 30 min followed by lysis by Dounce homogenizer. The lysates were clarified by centrifugation at 10,000 rpm for 10 min at 4°C. The total protein content was quantitated by the BCA microtiter protein assay reagents (Pierce).
Mutagenesis
Deletions in the 3′ UTR were made by amplifying regions of interest using polymerase chain reaction with appropriate primers. The primer sequences used for generating different deletions are listed in Table 3. Point mutations or internal deletions or insertions were made using the QuikChange mutagenesis kit (Stratagene) and all of the constructs were confirmed by dideoxy-chain termination DNA sequencing.
Table 3.
Sequences of oligonucleotides used for plasmid constructions

(S) Sense; (AS) antisense.
Transfection of cell-lines and reporter assays
Transient transfections were performed using Fugene (Roche Diagnostics) according to the manufacturer’s instructions. Briefly, SKBR-3 cells were seeded at a density of 5 × 105 cells per well of a 6-well plate and grown to 80%–90% confluency. Plasmid DNA (1.0 μg) and CMV-β-galactosidase DNA (0.5 μg) or Renilla luciferase DNA (0.125 μg) were incubated with 7 μL Fugene (Roche) in 100 μL serum-free media for 45 min and the DNA–Fugene complexes added drop-wise to 2 mL serum-containing media. Fresh media was added after 12 h and the cells grown for another 48 h. For steady-state experiments, cellular protein and/or total RNA was extracted 48 h post-transfection. The fold increase over the 5′ UTR was found to be a function of both the cell-passage number and the transfection efficiency. As a general guideline, most transfections were performed within passages 5–7 in culture. For most constructs, at least three independent transfection studies were performed. Firefly and Renilla luciferase reporter activity was determined using the Dual Luciferase Assay system (Promega) and the β-galactosidase activity was determined using the β-Gal assay kit (Invitrogen). Luciferase activity was normalized to β-Gal activity or to Renilla luciferase activity in each experiment.
RNA analysis
Total cellular RNA from transfected SKBR-3 cells was extracted using the RNAeasy extraction kit (Qiagen). The steady-state levels of RNA were determined by either Northern analysis or via real-time PCR. Northern blots were prepared and the blots hybridized using the Ultra-Hyb hybridization solution (Ambion). The gel-purified cDNA encoding firefly luciferase was labeled with [α-32P]-dCTP using Prime-it II random-primer labeling kit (Stratgene). The probes prepared from the GAPDH control were added along with luciferase for normalization. Radioactive signals were detected with a Storm PhosphorImager (Typhoon) and quantitated using the ImageQuant 2.2 software. Real-time PCR was performed using 5′ FAM–3′ TAMARA probes corresponding to the coding region of FLUC or 5′ VIC–3′ TAMARA probes for β-Actin (Applied Biosystems). The RT–PCR efficiency for both FLUC and actin was determined across a range of concentrations of input RNAs, and the Comparative CT analysis method (Jordan et al. 2004) was used to calculate the fold changes in expression of all constructs compared with that of the 5′ Her-2 Fluc RNA.
3′-end mapping
Total RNA from transfected and control SKBR-3 cells was isolated using RNAeasy kits (Qiagen). First-strand cDNA was synthesized from total RNA (1 μg) using the 3′-RACE Adaptor (Ambion) and MMLV RT. The cDNA (1 μL) was subjected to PCR using 3′-RACE primer and a gene-specific primer. The gene-specific primers were complementary to the coding regions of either luciferase (for transfected RNA) or the Her-2 receptor. The PCR products were analyzed by agarose gels, and the PCR products were submitted for dideoxy chain termination sequencing.
UV-cross-linking (UVXL) of RNA–protein complexes
RNA–protein binding reactions were performed using 3–5 μg of cytoplasmic extract, 10 fmol of 32P-UTP-labeled RNA, 2 μg tRNA, in a final volume of 20 μL using Binding Buffer A (20 mM HEPES-KOH at pH 7.5, 2.5 mM MgCl2, 100 mM KCl, 20% glycerol, 0.5 mM dithiothreitol, protease inhibitor tablets). Heparin was added to some reactions at a concentration of 0.25 μg/μL. Addition of heparin to 0.75 mg/mL did not have any significant influence on the pattern of UV-cross-linking. Reaction mixtures were incubated for 1 h at 37°C and UV irradiated at 254 nm for 10 min (StrataLinker 1800) on ice. The reaction mixtures were then treated with 4 μL of RNase cocktail (Ambion) for 30 min at 37°C. The samples were analyzed by 15% or 10%–14% Criterion gels (Bio-Rad) by SDS-PAGE and detected by phosphorimager analysis. For UVXL immunoprecipitation assays, UV-cross-linking was performed as described above, and the reactions were incubated for 2 h with specific antibodies at 4°C, followed by incubation with protein G beads (Amersham) for 1 h at 4°C. After washing with immunoprecipitation Buffer B (50 mM Tris at pH 7.5, 2 mM MgCl2, 1% NP40, 100 mM NaCl, Complete [EDTA-free] protease inhibitor tablets [Roche], Pefabloc [0.1 mM)], the RNA–protein complexes were resolved by SDS-PAGE and detected by PhosphorImager.
Biotin-RNA affinity purification
Biotinylated RNA was synthesized in vitro using a T7-Megascript RNA synthesis kit (Ambion, Inc.) in the presence of 14Biotin-UTP (Roche). The biotinylated RNA was bound to streptavidin-coated Dynabeads (Dynal, Inc.) according to the manufacturer’s recommendations. The cytoplasmic extracts from SKBR-3 were precleared by incubation with RNA-affinity resin containing RNA (1–407) in Binding Buffer A. The unbound fraction was incubated with RNA-affinity resin prepared with the RNA (1–612). After 2 h of incubation at 4°C, the unbound proteins were removed and after extensive washes with the binding buffer, the proteins were step-eluted with Binding Buffer A containing 0.25 mM or 0.5 mM NaCl. The proteins in 0.5 M eluate were resolved on SDS-PAGE and the protein bands (38–40 kDa) size were excised and submitted for mass-spectrometric analysis (City of Good Hope).
Native complex IP
The SKBR-3 cells grown to 80% confluency were washed twice with PBS and the cells scraped rapidly from the plates. The cells were suspended in IP-Buffer B containing 0.1% NP40 and 0.2 μM Pefabloc (Roche). Cytoplasmic extracts prepared from SKBR-3 cells were either left untreated or treated with RNase A/T1 cocktail (100 U) for 30 min on ice. The extracts were precleared on protein G sepharose for 1 h at 4°C. The unbound proteins incubated with anti-HuR or anti-hnRNP C1/C2 protein G sepharose for 2 h at 4°C. After 5 washes (10 min per wash, 4°C), the proteins bound to the resin were eluted into the SDS- PAGE buffer by heating to 100°C for 10 min and resolved on SDS-PAGE and transferred to the nitrocellulose membranes. The anti-HuR and C1/C2 immunoprecipitates were screened with the following antibodies: HuR (Invitrogen); hnRNPs C1/C2, L, Q, K, transportin, U, A1 (Sigma-Aldrich); eIF4E (Cell Signaling Technology); eIF2α, eIF5α, eIF3, PABP (SantaCruz); AUF-1 (Upstate Biotechnologies).
Ribonucleoprotein immunoprecipitation assay (RIA)
The RIA was performed according to Niranjanakumari et al. (2002), with some modifications. SKBR-3 cells (1.5 × 106) were washed twice in PBS after trypsinization and resuspended in 10 mL PBS containing 1% formaldehyde (16% stock; Polysciences, Inc.). After 40 min of cross-linking, the reaction was quenched with glycine (pH 7.0) to 0.25 M for 10 min. After two 50 mL washes with cold-PBS, the fixed cells were lysed by sonication and the insoluble material removed by centrifugation. The cell lysate was precleared on protein-G-sepharose for 1 h at 4°C, and the precleared lysate was incubated with Superase-in (Ambion) treated protein-G-sepharose coated with anti-HuR or anti-C1/C2 antibodies. After 3 h of incubation the beads were washed extensively in IP Buffer A and resuspended in 100 μL of 50 mM Tris.Cl (pH 7.0), 5 mM EDTA, 10 mM dithiothretol (DTT), and 1% SDS. Samples were incubated at 70°C for 45 min to reverse the cross-links. The total RNA bound to the beads was extracted with Trizol and precipitated with isopropanol. The samples were digested with DNase I (Ambion, Inc.) for 45 min at 37°C, followed by the removal with DNase-inactivating reagent (Ambion, Inc.). The RNA was used as template for cDNA synthesis, and the presence of specific targets was detected by PCR amplification using the gene-specific primers (Table 3).
Immunofluorescence microscopy
MCF-10A and SKBR-3 cells were grown on glass coverslips and were washed in PBS, fixed using 3.7% formaldehyde/PBS for 30 min, and permeablized with 0.5% Triton X-100/PBS for 5 min at room temperature. Fixed cells were blocked in 3% BSA/PBS for 1 h, and immunofluorescence staining was performed by incubating with anti-HuR antibody (Invitrogen) diluted in 3% BSA/PBS, followed by the anti-mouse IgG coupled to Alexafluor555 (Molecular Probes). Images were obtained using a Zeiss Axiovert 200 epi-fluorescence microscope and captured using IPLab version 3.6 for Windows software.
Acknowledgments
We thank Sergey Paushkin and Ellen Welch for helpful discussions, Charles Romfo for the SHIP-2 UTR and SL-Fluc constructs, and Joseph Colacino, Thomas Davis, and Pannayiota Triffilis for critical reading of the manuscript.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1388706
References
- Bae C.D., Juhnn Y.S., Park J.B. Post-transcriptional control of c-erb B-2 overexpression in stomach cancer cells. Exp. Mol. Med. 2001;33:15–19. doi: 10.1038/emm.2001.3. [DOI] [PubMed] [Google Scholar]
- Brunner J.E., Nguyen J.H., Roehl H.H., Ho T.V., Swiderek K.M., Semler B.L. Functional interaction of heterogeneous nuclear ribonucleoprotein C with poliovirus RNA synthesis initiation complexes. J. Virol. 2005;79:3254–3266. doi: 10.1128/JVI.79.6.3254-3266.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C.G., Dreyfuss G. RNA binding specificity of hnRNP A1: Significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 1994;13:1197–1204. doi: 10.1002/j.1460-2075.1994.tb06369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child S.J., Miller M.K., Geballe A.P. Cell type-dependent and -independent control of HER-2/neu translation. Int. J. Biochem. Cell Biol. 1999a;31:201–213. doi: 10.1016/s1357-2725(98)00068-5. [DOI] [PubMed] [Google Scholar]
- Child S.J., Miller M.K., Geballe A.P. Translational control by an upstream open reading frame in the HER-2/neu transcript. J. Biol. Chem. 1999b;274:24335–24341. doi: 10.1074/jbc.274.34.24335. [DOI] [PubMed] [Google Scholar]
- Cho P.F., Poulin F., Cho-Park Y.A., Cho-Park I.B., Chicoine J.D., Lasko P., Sonenberg N. A new paradigm for translational control: Inhibition via 5′–3′ mRNA tethering by Bicoid and the eIF4E cognate 4EHP. Cell. 2005;121:411–423. doi: 10.1016/j.cell.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Choi Y.D., Grabowski P.J., Sharp P.A., Dreyfuss G. Heterogeneous nuclear ribonucleoproteins: Role in RNA splicing. Science. 1986;231:1534–1539. doi: 10.1126/science.3952495. [DOI] [PubMed] [Google Scholar]
- Copeland P.R. Regulation of gene expression by stop codon recoding: Selenocysteine. Gene. 2003;312:17–25. doi: 10.1016/s0378-1119(03)00588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplinski K., Ruiz-Echevarria M.J., Paushkin S.V., Han X., Weng Y., Perlick H.A., Dietz H.C., Ter Avanesyan M.D., Peltz S.W. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes & Dev. 1998;12:1665–1677. doi: 10.1101/gad.12.11.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moor C.H., Meijer H., Lissenden S. Mechanisms of translational control by the 3′ UTR in development and differentiation. Semin. Cell Dev. Biol. 2005;16:49–58. doi: 10.1016/j.semcdb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Denkert C., Weichert W., Winzer K.J., Muller B.M., Noske A., Niesporek S., Kristiansen G., Guski H., Dietel M., Hauptmann S. Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin. Cancer Res. 2004;10:5580–5586. doi: 10.1158/1078-0432.CCR-04-0070. [DOI] [PubMed] [Google Scholar]
- Dixon D.A., Tolley N.D., King P.H., Nabors L.B., McIntyre T.M., Zimmerman G.A., Prescott S.M. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J.K., Bond C.T., Hua W., Adelman J.P., Clinton G.M. An alternative HER-2/neu transcript of 8 kb has an extended 3′ UTR and displays increased stability in SKOV-3 ovarian carcinoma cells. Gynecol. Oncol. 1999;74:408–415. doi: 10.1006/gyno.1999.5467. [DOI] [PubMed] [Google Scholar]
- Espel E. The role of the AU-rich elements of mRNAs in controlling translation. Semin. Cell Dev. Biol. 2005;16:59–67. doi: 10.1016/j.semcdb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Good P.J. The role of elav-like genes, a conserved family encoding RNA-binding proteins, in growth and development. Semin. Cell Dev. Biol. 1997;8:577–584. doi: 10.1006/scdb.1997.0183. [DOI] [PubMed] [Google Scholar]
- Gorlach M., Burd C.G., Dreyfuss G. The determinants of RNA-binding specificity of the heterogeneous nuclear ribonucleoprotein C proteins. J. Biol. Chem. 1994;269:23074–23078. [PubMed] [Google Scholar]
- Grskovic M., Hentze M.W., Gebauer F. A co-repressor assembly nucleated by Sex-lethal in the 3′ UTR mediates translational control of Drosophila msl-2 mRNA. EMBO J. 2003;22:5571–5581. doi: 10.1093/emboj/cdg539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh J., Hesketh J. 3′-Untranslated regions are important in mRNA localization and translation: Lessons from selenium and metallothionein. Biochem. Soc. Trans. 2004;32:990–993. doi: 10.1042/BST0320990. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A.G. Mechanism and regulation of initiator methionyl-tRNA binding to the ribosomes. In: Sonenberg N., et al., editors. Translational control of gene expression. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 185–243. [Google Scholar]
- Holbro T., Hynes N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- Hollywood D.P., Hurst H.C. A novel transcription factor, OB2-1, is required for overexpression of the proto-oncogene c-erbB-2 in mammary tumour lines. EMBO J. 1993;12:2369–2375. doi: 10.1002/j.1460-2075.1993.tb05891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstein E., Git A., Braunstein I., Avni D., Meyuhas O. The expression of poly(A)-binding protein gene is translationally regulated in a growth-dependent fashion through a 5′-terminal oligopyrimidine tract motif. J. Biol. Chem. 1999;274:1708–1714. doi: 10.1074/jbc.274.3.1708. [DOI] [PubMed] [Google Scholar]
- Hoshino S., Imai M., Kobayashi T., Uchida N., Katada T. The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3′-Poly(A) tail of mRNA. Direct association of erf3/GSPT with polyadenylate-binding protein. J. Biol. Chem. 1999;274:16677–16680. doi: 10.1074/jbc.274.24.16677. [DOI] [PubMed] [Google Scholar]
- Hynes N.E., Lane H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Hynes N.E., Stern D.F. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim. Biophys. Acta. 1994;1198:165–184. doi: 10.1016/0304-419x(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Ishii S., Imamoto F., Yamanashi Y., Toyoshima K., Yamamoto T. Characterization of the promoter region of the human c-erbB-2 protooncogene. Proc. Natl. Acad. Sci. 1987;84:4374–4378. doi: 10.1073/pnas.84.13.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R.C., Macabeo-Ong M., Shiboski C.H., Dekker N., Ginzinger D.G., Wong D.T., Schmidt B.L. Overexpression of matrix metalloproteinase-1 and -9 mRNA is associated with progression of oral dysplasia to cancer. Clin. Cancer Res. 2004;10:6460–6465. doi: 10.1158/1078-0432.CCR-04-0656. [DOI] [PubMed] [Google Scholar]
- Kahvejian A., Roy G., Sonenberg N. The mRNA closed-loop model: The function of PABP and PABP-interacting proteins in mRNA translation. Cold Spring Harb. Symp. Quant. Biol. 2001;66:293–300. doi: 10.1101/sqb.2001.66.293. [DOI] [PubMed] [Google Scholar]
- Kahvejian A., Svitkin Y.V., Sukarieh R., M’Boutchou M.N., Sonenberg N. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes & Dev. 2005;19:104–113. doi: 10.1101/gad.1262905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 1991a;266:19867–19870. [PubMed] [Google Scholar]
- Kozak M. An analysis of vertebrate mRNA sequences: Intimations of translational control. J. Cell Biol. 1991b;115:887–903. doi: 10.1083/jcb.115.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus M.H., Popescu N.C., Amsbaugh S.C., King C.R. Overexpression of the EGF receptor-related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J. 1987;6:605–610. doi: 10.1002/j.1460-2075.1987.tb04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuersten S., Goodwin E.B. The power of the 3′ UTR: Translational control and development. Nat. Rev. Genet. 2003;4:626–637. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- Kuhn U., Wahle E. Structure and function of poly(A) binding proteins. Biochim. Biophys. Acta. 2004;1678:67–84. doi: 10.1016/j.bbaexp.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Kuwada S.K., Kuang J., Li X. Integrin α5/β1 expression mediates HER-2 down-regulation in colon cancer cells. J. Biol. Chem. 2005;280:19027–19035. doi: 10.1074/jbc.M410540200. [DOI] [PubMed] [Google Scholar]
- Le S.Y., Maizel J.V., Jr. A common RNA structural motif involved in the internal initiation of translation of cellular mRNAs. Nucleic Acids Res. 1997;25:362–369. doi: 10.1093/nar/25.2.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Park S., Kilburn B., Jelinek M.A., Henschen-Edman A., Aswad D.W., Stallcup M.R., Laird-Offringa I.A. Lipopolysaccharide-induced methylation of HuR, an mRNA-stabilizing protein, by CARM1. Coactivator-associated arginine methyltransferase. J. Biol. Chem. 2002;277:44623–44630. doi: 10.1074/jbc.M206187200. [DOI] [PubMed] [Google Scholar]
- de Lopez Silanes I., Zhan M., Lal A., Yang X., Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangus D.A., Evans M.C., Jacobson A. Poly(A)-binding proteins: Multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 2003;4:223. doi: 10.1186/gb-2003-4-7-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrand S.H., Dwen P., Pederson T. Serine/threonine phosphorylation regulates binding of C hnRNP proteins to pre-mRNA. Proc. Natl. Acad. Sci. 1993;90:7764–7768. doi: 10.1073/pnas.90.16.7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael W.M., Choi M., Dreyfuss G. A nuclear export signal in hnRNP A1: A signal-mediated, temperature-dependent nuclear protein export pathway. Cell. 1995;83:415–422. doi: 10.1016/0092-8674(95)90119-1. [DOI] [PubMed] [Google Scholar]
- Millard S.S., Vidal A., Markus M., Koff A. A U-rich element in the 5′ untranslated region is necessary for the translation of p27 mRNA. Mol. Cell. Biol. 2000;20:5947–5959. doi: 10.1128/mcb.20.16.5947-5959.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris D.R., Geballe A.P. Upstream open reading frames as regulators of mRNA translation. Mol. Cell. Biol. 2000;20:8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D., Decker C.J., Parker R. Turnover mechanisms of the stable yeast PGK1 mRNA. Mol. Cell. Biol. 1995;15:2145–2156. doi: 10.1128/mcb.15.4.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjanakumari S., Lasda E., Brazas R., Garcia-Blanco M.A. Reversible cross-linking combined with immunoprecipitation to study RNA–protein interactions in vivo. Methods. 2002;26:182–190. doi: 10.1016/S1046-2023(02)00021-X. [DOI] [PubMed] [Google Scholar]
- Olayioye M.A., Neve R.M., Lane H.A., Hynes N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostareck D.H., Ostareck-Lederer A., Wilm M., Thiele B.J., Mann M., Hentze M.W. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell. 1997;89:597–606. doi: 10.1016/s0092-8674(00)80241-x. [DOI] [PubMed] [Google Scholar]
- Pinol-Roma S., Dreyfuss G. Cell cycle-regulated phosphorylation of the pre-mRNA-binding (heterogeneous nuclear ribonucleoprotein) C proteins. Mol. Cell. Biol. 1993;13:5762–5770. doi: 10.1128/mcb.13.9.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs A.B., Davis R.W. The poly(A) binding protein is required for poly(A) shortening and 60S ribosomal subunit-dependent translation initiation. Cell. 1989;58:857–867. doi: 10.1016/0092-8674(89)90938-0. [DOI] [PubMed] [Google Scholar]
- Sella O., Gerlitz G., Le S.Y., Elroy-Stein O. Differentiation-induced internal translation of c-sis mRNA: Analysis of the cis elements and their differentiation-linked binding to the hnRNP C protein. Mol. Cell. Biol. 1999;19:5429–5440. doi: 10.1128/mcb.19.8.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfield I., Jones K.M., Kushnirov V.V., Dagkesamanskaya A.R., Poznyakovski A.I., Paushkin S.V., Nierras C.R., Cox B.S., Ter Avanesyan M.D., Tuite M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995a;14:4365–4373. doi: 10.1002/j.1460-2075.1995.tb00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfield I., Jones K.M., Tuite M.F. The end in sight: Terminating translation in eukaryotes. Trends Biochem. Sci. 1995b;20:489–491. doi: 10.1016/s0968-0004(00)89113-6. [DOI] [PubMed] [Google Scholar]
- Swanson M.S., Nakagawa T.Y., LeVan K., Dreyfuss G. Primary structure of human nuclear ribonucleoprotein particle C proteins: Conservation of sequence and domain structures in heterogeneous nuclear RNA, mRNA, and pre-rRNA-binding proteins. Mol. Cell. Biol. 1987;7:1731–1739. doi: 10.1128/mcb.7.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal M., King C.R., Kraus M.H., Ullrich A., Schlessinger J., Givol D. Human HER2 (neu) promoter: Evidence for multiple mechanisms for transcriptional initiation. Mol. Cell. Biol. 1987;7:2597–2601. doi: 10.1128/mcb.7.7.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarun S.Z., Jr., Sachs A.B. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J. 1996;15:7168–7177. [PMC free article] [PubMed] [Google Scholar]
- Tarun S.Z., Jr., Wells S.E., Deardorff J.A., Sachs A.B. Translation initiation factor eIF4G mediates in vitro poly(A) tail-dependent translation. Proc. Natl. Acad. Sci. 1997;94:9046–9051. doi: 10.1073/pnas.94.17.9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernimmen D., Gueders M., Pisvin S., Delvenne P., Winkler R. Different mechanisms are implicated in ERBB2 gene overexpression in breast and in other cancers. Br. J. Cancer. 2003;89:899–906. doi: 10.1038/sj.bjc.6601200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch E.M., Wong W., Peltz S.W. Translation termination: It’s not the end of the story. In: Sonenberg N., et al., editors. Translational control of gene expression. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 467–485. [Google Scholar]
- Wells S.E., Hillner P.E., Vale R.D., Sachs A.B. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell. 1998;2:135–140. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- Wilkie G.S., Dickson K.S., Gray N.K. Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem. Sci. 2003;28:182–188. doi: 10.1016/S0968-0004(03)00051-3. [DOI] [PubMed] [Google Scholar]
- Yaman I., Fernandez J., Sarkar B., Schneider R.J., Snider M.D., Nagy L.E., Hatzoglou M. Nutritional control of mRNA stability is mediated by a conserved AU-rich element that binds the cytoplasmic shuttling protein HuR. J. Biol. Chem. 2002;277:41539–41546. doi: 10.1074/jbc.M204850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y., Sliwkowski M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Yohn C.B., Cohen A., Danon A., Mayfield S.P. A poly(A) binding protein functions in the chloroplast as a message-specific translation factor. Proc. Natl. Acad. Sci. 1998;95:2238–2243. doi: 10.1073/pnas.95.5.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]