

Abstract
Short peptides of 11 residues were synthesized and tested against the economically important plant pathogenic bacteria Erwinia amylovora, Pseudomonas syringae, and Xanthomonas vesicatoria and compared to the previously described peptide Pep3 (WKLFKKILKVL-NH2). The antimicrobial activity of Pep3 and 22 analogues was evaluated in terms of the MIC and the 50% effective dose (ED50) for growth. Peptide cytotoxicity against human red blood cells and peptide stability toward protease degradation were also determined. Pep3 and several analogues inhibited growth of the three pathogens and had a bactericidal effect at low micromolar concentrations (ED50 of 1.3 to 7.3 μM). One of the analogues consisting of a replacement of both Trp and Val with Lys and Phe, respectively, resulted in a peptide with improved bactericidal activity and minimized cytotoxicity and susceptibility to protease degradation compared to Pep3. The best analogues can be considered as potential lead compounds for the development of new antimicrobial agents for use in plant protection either as components of pesticides or expressed in transgenic plants.
Phytopathogenic bacteria are responsible for great losses in economically important crops such as vegetables and fruit (1). Although a great amount of research has been undertaken to overcome the damage caused by this type of bacteria, a major difficulty encountered is the lack of effective control against some severe diseases (37). Plant protection against these pathogens is mainly based on copper derivatives and antibiotics. However, these compounds are regarded as environmental contaminants, and resistant strains of plant-pathogenic bacteria have been reported in several crops (34, 48).
In recent years, much attention has been paid to searching for new classes of antibiotics to which bacteria cannot develop resistance. Endogenous antimicrobial peptides have emerged as good candidates (26, 27, 51). These peptides have been found in a variety of sources, including mammals, amphibians, insects, and plants, and they are known to play important roles in the host defense system and innate immunity (12-14, 21, 22, 43).
Most antimicrobial peptides are cationic and have the ability to adopt an amphipathic conformation in which positively charged and hydrophobic groups segregate onto opposing faces of an α-helix, a β-sheet, or other tertiary structures (7, 9, 11, 25, 29, 46, 49, 51). Natural antimicrobial peptides exhibit a broad spectrum of activity against bacteria, fungi, enveloped viruses, parasites, and tumor cells. They are lytic, have a synergistic activity with conventional antibiotics, neutralize endotoxins, and have very few reported side effects (7, 9, 11, 25, 29, 46, 49). Their mechanism of action has been studied mainly in bacteria. Amphiphilicity is thought to be a key requisite for these peptides to interact with their primary target, the cell membrane. They do not interact with a specific receptor but, rather, disturb bilayer integrity, either by disruption or pore formation, probably by direct peptide-lipid interactions (7, 9, 11, 25, 29, 46, 49). This unique mode of action makes the induction of resistance difficult, because it requires dramatic changes in phospholipid membrane composition and/or organization (51).
Cecropins are some of the best studied cationic antimicrobial peptides, representing a family of highly basic α-helical peptides first found in the hemolymph of the giant silk moth Hyalophora cecropia as a response to a bacterial infection (32, 47). In particular, cecropin A is a 37-amino-acid linear peptide, which consists of a strongly basic N-terminal amphipathic α-helical domain connected to a hydrophobic C-terminal α-helix by a flexible hinge. Cecropin A displays powerful lytic activity against gram-positive and gram-negative bacteria but has no cytotoxic effects against human erythrocytes and other eukaryotic cells (6, 32, 47). The basis of this selectivity has been attributed to the absence of acidic phospholipids and presence of sterols in eukaryotic cells (18). However, major concerns about the use of cecropin A as a pesticide in plant protection are the high production cost of such a long peptide and its sensitivity to protease degradation. Searches for shorter, more potent, nontoxic, and more stable peptides have led to the identification of synthetic peptides with broader and higher activity than their natural counterparts (3, 5, 6, 8, 16, 17, 19, 23, 33, 41, 50). In particular, the 11-residue sequence WKLFKKILKVL-NH2 (Pep3), derived from the well-known cecropin A(1-7)-melittin(2-9) hybrid peptide (8, 17, 23), is sufficient for antifungal and antibacterial activities (4, 16).
Reports on the activity of short synthetic cecropin A-melittin hybrid peptides concerning plant-pathogenic microorganisms have been focused mainly on fungal plant pathogens (4, 16). Studies involving plant-pathogenic bacteria are practically limited to Erwinia carotovora (4). Our current research is oriented to develop new control methods against economically important plant pathogenic bacteria, such as Erwinia amylovora, the causal agent of fire blight of rosaceous plants; Xanthomonas vesicatoria, the cause of bacterial spot of tomato and pepper; and Pseudomonas syringae, the cause of several blight diseases (15, 38, 40). To date, apart from the antibiotics streptomycin and tetracycline registered in certain countries, no effective method to treat these plant diseases has been described.
The production of antimicrobial peptides by self-defending genetically improved plants constitutes an effective means for improving crop protection against bacterial diseases (42). However, the use of genetically modified crops is governed by several restrictions in some countries. Therefore, peptide synthesis has been regarded as a useful alternative, but a major concern is the high production cost associated with the preparation of large peptides. Within this context, we were interested in the identification of short synthetic peptides (≤11 residues) with specific activity against E. amylovora, X. vesicatoria, and P. syringae. In particular, our study was centered on the sequence of Pep3, which shows interesting antibacterial activities, but it has not been tested against the above plant- pathogenic bacteria.
In the present study we report the activity of Pep3 and 22 new analogues against the phytopathogenic bacteria E. amylovora, X. vesicatoria, and P. syringae. Analogues were designed based on the α-helical wheel diagram of Pep3 and synthesized by the solid-phase method. Circular dichroism (CD) spectroscopy was employed to investigate the secondary structure of the peptides. The main objective was to obtain new peptides with higher bactericidal activity and lower cytotoxic effects and sensitivity to protease degradation than Pep3.
MATERIALS AND METHODS
Peptide synthesis.
All peptides listed in Table 1 were synthesized by the solid-phase method using 9-fluorenylmethoxycarbonyl (Fmoc)-type chemistry, tert-butyloxycarbonyl side chain protection for Lys and Trp, and tert-butyl for Tyr. All the Fmoc acid derivatives, reagents, and solvents were obtained from Senn Chemicals International (Gentilly, France). Fmoc-Rink-4-methylbenzhydrylamine resin (0.64 mmol/g; Novabiochem, Darmstadt, Germany) was used as solid support to obtain C-terminal peptide amides and 4-hydroxymethylphenoxypropionic acid polyethylene glycol-polystyrene resin (0.23 mmol/g; Perspective Biosystems, Framingham, Mass.) to synthesize C-terminal peptide acids BP01, BP05, and BP07. Peptides were obtained with >90% purity by high-performance liquid chromatography (HPLC). Electrospray ionization mass spectrometry was used to confirm peptide identity.
TABLE 1.
Sequences and α-helicity of synthetic peptides
| Peptide | Sequencea | [θ]222b | α-Helix (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pep3 | W | K | L | F | K | K | I | L | K | V | L-NH2 | −9,048 | 23.5 |
| BP01 | W | K | L | F | K | K | I | L | K | V | L-OH | NDc | ND |
| BP02 | K | L | F | K | K | I | L | K | K | L-NH2 | ND | ND | |
| BP03 | K | L | F | K | K | I | L | K-NH2 | ND | ND | |||
| BP04 | W | K | L | F | K-NH2 | ND | ND | ||||||
| BP05 | W | K | L | F | K-OH | ND | ND | ||||||
| BP06 | K | I | L | K | V | L-NH2 | ND | ND | |||||
| BP07 | K | I | L | K | V | L-OH | ND | ND | |||||
| BP08 | Ac-W | K | L | F | K | K | I | L | K | V | L-NH2 | −10,851 | 29.5 |
| BP09 | Ts-W | K | L | F | K | K | I | L | K | V | L-NH2 | −13,341 | 37.8 |
| BP10 | Bz-W | K | L | F | K | K | I | L | K | V | L-NH2 | −13,119 | 37.1 |
| BP11 | Bn-W | K | L | F | K | K | I | L | K | V | L-NH2 | −10,665 | 28.9 |
| BP12 | Pam-W | K | L | F | K | K | I | L | K | V | L-NH2 | ND | ND |
| BP13 | F | K | L | F | K | K | I | L | K | V | L-NH2 | ND | ND |
| BP14 | Y | K | L | F | K | K | I | L | K | V | L-NH2 | ND | ND |
| BP33 | L | K | L | F | K | K | I | L | K | V | L-NH2 | −8,918 | 23.1 |
| BP15 | K | K | L | F | K | K | I | L | K | V | L-NH2 | −9,892 | 26.3 |
| BP16 | K | K | L | F | K | K | I | L | K | K | L-NH2 | ND | ND |
| BP76 | K | K | L | F | K | K | I | L | K | F | L-NH2 | −9,188 | 24.0 |
| BP17 | W | K | L | F | K | K | I | L | K | K | L-NH2 | ND | ND |
| BP18 | W | K | L | F | K | K | I | L | K | W | L-NH2 | −8,922 | 23.1 |
| BP19 | W | K | L | F | K | K | I | L | K | F | L-NH2 | −9,950 | 26.5 |
| BP20 | W | K | L | F | K | K | I | L | K | Y | L-NH2 | −10,316 | 27.7 |
Boldfaced letters indicate the modifications introduced into the Pep3 sequence. Ac, acetyl; Ts, tosyl; Bz, benzoyl; Bn, benzyl; Pam, palmitoyl.
Determined in 50% trifluoroethanol in 10 mM sodium phosphate buffer (pH 7.4).
ND, not determined.
Bacterial strains and growth medium.
The following plant-pathogenic bacterial strains were used: E. amylovora PMV6076 (Institut National de la Recherche Agronomique, Angers, France), P. syringae pv. syringae EPS94 (Institut de Tecnologia Agroalimentària, Universitat de Girona, Spain) and X. vesicatoria 2133-2 (Instituto Valenciano de Investigaciones Agrarias, Valencia, Spain). All bacteria were stored in liquid LB medium supplemented with glycerol (20%) and maintained at −80°C. E. amylovora PMV6076 and P. syringae pv. syringae EPS94 were scraped from LB agar after growth for 24 h and X. vesicatoria 2133-2 was scraped after 48 h at 25°C. The cell material was suspended in sterile water to obtain a suspension of 108 CFU ml−1.
Antimicrobial activity.
Lyophilized peptides were solubilized in sterile Milli-Q water to a final concentration of 1,000 μM and filter sterilized through a 0.22-μm-pore-size filter. For MIC assessment, dilutions of the synthetic peptides were made to obtain final concentrations of 750, 500, 250, 200, 150, 125, 100, 75, 50, and 25 μM. For cecropin A, the concentrations were 250, 200, 150, 125, 100, 75, 50, 25, 12.5, 6.25, and 3.12 μM. Twenty microliters of each dilution was mixed in each well of a microtiter plate with 20 μl of the corresponding suspension of the bacterial indicator, 160 μl of tryptic soy broth (BioMèrieux, France), to a total volume of 200 μl. Two replicates for each strain, peptide, and concentration were used. Positive controls contained water instead of peptide, and negative controls contained peptides without bacterial suspension. Microbial growth was automatically determined by optical density measurement at 600 nm (Bioscreen C; Labsystem, Helsinki, Finland). Microplates were incubated at 25°C with 20 s of shaking before hourly absorbance measurements for 48 h. The experiment was repeated twice.
The MIC was defined as the lowest peptide concentration with no growth at the end of the experiment. Since it may be that certain compounds have the same MIC but different inhibition-dose relationships, an additional parameter, the 50% effective dose (ED50), was determined for the measurement of activity. Growth was measured as the area under the curve. Inhibition of growth (I) was calculated as a percentage of the positive control using the following equation: I = 100 × [(AC − AS)/AC], where AC is the area under the curve of the control, and AS is the area under the curve of a given peptide concentration.
The ED50 is the concentration required to obtain 50% inhibition of growth and was calculated from the inhibition-dose data for each peptide. Inhibition data were fitted to the probit-dose model (20, 36). The equation of the probit-dose model is as follows: y = ø{[log10(x) − λ]/τ}, where y is the proportion of inhibition, x is the peptide concentration, ø denotes the cumulative distribution function for the standard normal, λ is a parameter equivalent to ED50, and τ is the peptide efficiency.
Regression and parameter estimation were performed by a nonlinear least-squares method using the NLIN procedure of the PC-Statistical Analysis System, version 8.2 (SAS Institute Inc., Cary, NC).
Analysis of bactericidal activity.
The bactericidal activity of peptides was determined for the reference peptide Pep3 and the analogues BP11, BP15, BP33, and BP76. LB broth-grown cultures of E. amylovora, P. syringae, and X. vesicatoria inoculated at 4 × 106 CFU ml−1 were incubated in a 5 μM concentration of the corresponding peptide. Aliquots of 500 μl were removed at 30-min intervals during 3 h and diluted 10-fold, and the dilutions were plated on LB agar plates. The CFU were counted after a 48-h incubation at 25°C. Values were expressed as percentages of survival from the start of the experiment.
Hemolytic activity.
The hemolytic activity of the peptides was evaluated by determining hemoglobin release from erythrocyte suspensions of fresh human blood (5%, vol/vol). Blood was aseptically collected using a BD Vacutainer K2E system with EDTA (Belliver Industrial State, Plymouth, United Kingdom) and stored for less than 2 h at 4°C. Blood was centrifuged at 6,000 × g for 5 min, washed three times with Tris buffer (10 mM Tris, 150 mM NaCl, pH 7.2) and diluted with Tris buffer. Peptides were solubilized in Tris buffer to final concentrations of 800, 400, 200, 100, 50, 25, and 12.5 μM. Fifty microliters of human red blood cells was mixed with 50 μl of the peptide solution and incubated under continuous shaking for 1 h at 37°C. Then, the tubes were centrifuged at 3,500 × g for 10 min. Eighty-microliter aliquots of the supernatant were transferred to 100-well microplates (Bioscreen) and diluted with 80 μl of Milli-Q water. Hemolysis was measured as the absorbance at 540 nm with a Bioscreen plate reader. Complete hemolysis was determined in TRIS buffer plus melittin (Sigma-Aldrich Corporation, Madrid, Spain) as a positive control. The percentage of hemolysis (H) was calculated using the following equation: H = 100 × [(Op − Ob)/(Om − Ob)], where Op is the density for a given peptide concentration, Ob is the density for the buffer, and Om is the density for the melittin positive control.
To analyze dose-response relationships in hemolysis, the 50% hemolytic dose (HD50) was determined following the procedure previously described for the analysis of antibacterial activity.
Susceptibility to protease degradation.
Digestion of Pep3, BP08, BP09, BP10, BP15, BP20, BP33, and BP76 by proteinase K (Sigma-Aldrich Corp., Madrid, Spain) was carried out by treating 50 μg/ml peptide with 1 μg/ml proteinase K in 100 mM Tris buffer, pH 7.6, at room temperature. The peptide cleavage after 5, 10, 15, 30, and 45 min was monitored by HPLC using a Kromasil (4.6- by 40-mm column; 3.5-μm particle size) C18 reverse-phase column. Linear gradients of 0.1% aqueous trifluoroacetic acid and 0.1% trifluoroacetic acid in CH3CN were run from 0.98:0.02 to 0:1 over 7 min with UV detection at 220 nm. Digestion was estimated as the percentage of degraded peptide calculated from the decrease of the HPLC peak area of the native peptide.
CD spectroscopy.
CD measurements were obtained using a Jasco spectropolarimeter (J-810; Easton, MD) at 25°C. Spectra were obtained in a fused quartz cell with 1-mm path length over a wavelength range of 190 to 250 nm at 0.1-nm intervals, 50 nm/min speed, 0.5-s response time, and 1-nm bandwidth. Peptides were dissolved to a 100 μM concentration in 50% (vol/vol) trifluoroethanol in 10 mM sodium phosphate buffer at pH 7.4. A baseline correction was made with only solvent in the cell. Data were expressed in terms of mean residue ellipticity [θ] (degrees × cm2 dmol−1), calculated per mol of total amide groups present in the different peptides. The percent helicity of the peptide was calculated as follows: α-helix (%) = 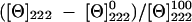 , where [Θ]222 is the experimentally observed absolute mean residue ellipticity at 222 nm. Values for [Θ]2220 and [Θ]222100, corresponding to 0 and 100% helix content at 222 nm, were estimated to be −2,000 and −30,000 (degrees × cm2 dmol−1), respectively (33).
, where [Θ]222 is the experimentally observed absolute mean residue ellipticity at 222 nm. Values for [Θ]2220 and [Θ]222100, corresponding to 0 and 100% helix content at 222 nm, were estimated to be −2,000 and −30,000 (degrees × cm2 dmol−1), respectively (33).
RESULTS
Design of peptides.
Primary structures of Pep3 analogues reported here are shown in Table 1. All peptides were prepared as C-terminal amides except BP01, BP05, and BP07. Analogues BP08 to BP12 were blocked at the N termini with an acetyl, tosyl, benzoyl, benzyl, or palmitoyl group, respectively. To examine whether the entire sequence of Pep3 is necessary for its full antibacterial activity, N- and C-terminal deletion analogues (BP02 to BP07) were synthesized. Analogues BP13 to BP20, BP33, and BP76 were designed based on the ideal α-helical wheel diagram of Pep3 (Fig. 1). We investigated the effect of individually replacing tryptophan and valine residues with amino acids possessing various degrees of hydrophobicity and hydrophilicity. Thus, tryptophan was replaced with Phe (BP13), Tyr (BP14), Lys (BP15), or Leu (BP33). Valine was replaced with Lys (BP17), Trp (BP18), Phe (BP19), or Tyr (BP20). Tryptophan and valine were replaced with Lys and Phe, respectively (BP76). The hydrophilic surface area of Pep3 was further increased by replacing both tryptophan and valine residues with Lys (BP16).
FIG. 1.

Edmunson wheel projection of the 11-mer peptides that were synthesized. Black background, hydrophilic amino acids (Lys); white background, hydrophobic amino acids; gray background, residues that can be either hydrophilic (Lys) or hydrophobic (Leu, Trp, Tyr, and Phe), depending on the sequence as shown in Table 1.
Antibacterial activity.
The peptides synthesized were tested for antibacterial activity against E. amylovora, P. syringae, and X. vesicatoria. Results obtained are shown in Table 2. Pep3, which has not been previously tested against these bacteria, inhibited the growth of all three pathogens. The ED50 values of the peptide were in the range of 3.6 to 5.5 μM. The C-terminal peptide acid derivative BP01 was significantly less active than Pep3, and the N- and C-terminal deletion analogues (BP02 to BP07) were inactive against these pathogens (data not shown).
TABLE 2.
Antimicrobial activity against three plant-pathogenic bacteria and cytotoxicity of selected peptides
| Peptide | MIC (μM)
|
ED50 (μM)
|
HD50 (μM) | ||||
|---|---|---|---|---|---|---|---|
| E. amylovora | P. syringae | X. vesicatoria | E. amylovora | P. syringae | X. vesicatoria | ||
| Pep3 | 7-10 | 7-10 | 7-10 | 5.5 | 5.5 | 3.6 | 104 |
| BP08 | 10-12 | 7-10 | 2-5 | 9.0 | 4.3 | 2.0 | 17 |
| BP09 | 12-15 | 12-15 | <2 | 11.2 | 6.9 | NDa | 10 |
| BP10 | 15-20 | 15-20 | <2 | 15.1 | 9.5 | ND | 11 |
| BP11 | 7-10 | 5-7 | 2-5 | 5.0 | 3.8 | 2.5 | 30 |
| BP12 | 50-100 | 50-100 | 25-50 | 60.0 | 56.8 | 14.7 | 6 |
| BP15 | 5-7 | 2-5 | 12-15 | 4.3 | 1.6 | 7.3 | 334 |
| BP18 | 5-7 | 5-7 | <2 | 3.0 | 2.5 | ND | 26 |
| BP19 | 5-7 | 5-7 | <2 | 1.3 | 1.9 | ND | 32 |
| BP20 | 2-5 | 2-5 | 2-5 | 3.2 | 2.7 | 2.3 | 42 |
| BP33 | 5-7 | 5-7 | 10-12 | 4.3 | 3.2 | 4.1 | 190 |
| BP76 | 2-5 | 2-5 | 2-5 | 2.5 | 2.1 | 1.9 | 203 |
| Cecropin Ab | <1 | <1 | <1 | <0.3c | <0.3 | <0.3 | ND |
ND, not determined.
Cecropin A was included for comparison purposes.
Estimated visually from graphs; lowest concentration tested.
Except for the N-palmitoylated peptide BP12, derivatization of the N terminus of Pep3 produced peptides (BP08 to BP11) with significant biological activity, with ED50 values ranging from 2 μM to 15.1 μM. The N-benzylated analogue BP11 was the most inhibitory peptide within this group and slightly better than Pep3 (ED50 of 2.5 to 5.0 μM).
Replacement of Trp in Pep3 with Lys (BP15) or Leu (BP33) induced a slight increase in the overall activity (ED50 values of 1.6 to 7.3 μM and 3.2 to 4.3 μM, respectively). In contrast, replacement of Trp with Phe (BP13) or Tyr (BP14) resulted in analogues with poor antibacterial activity (data not shown).
Analogues BP18, BP19 and BP20, which have Trp, Phe, or Tyr instead of Val, were considerably more active than the parent peptide Pep3 (ED50 of 1.3 to 3.2 μM). In contrast, BP17, which has Lys instead of Val, did not show antibacterial activity (data not shown).
Double replacement of Trp and Val with Lys and Phe (BP76), respectively, also led to a significant increase of the antibacterial activity compared to Pep3 (ED50 of 1.9 to 2.5 mM). In contrast, BP16, which possesses two Lys residues instead of Trp and Val residues, was not active (data not shown).
MICs of these cecropin A-derived peptides were also determined (Table 2) and were in good agreement with the ED50 values. In general, X. vesicatoria was more sensitive to each of the peptides than were the other two bacteria. Complete inhibition of X. vesicatoria was observed at 2 μM for peptides BP09, BP10, BP18, and BP19. On the basis of MICs, BP15, BP20, and BP76 were the most potent peptides against P. syringae (MIC of 2 to 5 μM), and the MICs of BP20 and BP76 were the lowest against E. amylovora (MIC of 2 to 5 μM).
Since some of the analogues synthesized showed similar antibacterial activity against the bacterial strains tested, we evaluated their bactericidal activity by comparing the time course to kill mid-logarithmic-phase culture suspensions of E. amylovora, P. syringae, and X. vesicatoria. As shown in Fig. 2, at the concentration tested, all the peptides had a slower bactericidal effect against P. syringae than against the other two pathogens, except for BP11, which had no effect on E. amylovora. BP15 showed higher bactericidal activity against E. amylovora than Pep3 and similar bactericidal activity against P. syringae and X. vesicatoria. Notably, BP76 was the peptide with the highest bactericidal effect against E. amylovora and X. vesicatoria. Its activity against X. vesicatoria was nearly complete after a 90-min exposure period at a concentration of 5 μM.
FIG. 2.

Kinetics of survival of P. syringae, X. vesicatoria, and E. amylovora in the presence of Pep3 (○) or selected analogues. Bacterial suspensions were untreated (▪) or treated with 5 μM concentrations of BP11 (▵), BP15 (⋄), BP33 (⧫), or BP76 (•), and viable cells were determined at different time intervals.
Hemolytic activity.
The toxicity of the peptides to eukaryotic cells was tested by their ability to lyse human red blood cells, and results were compared to melittin as a standard. Results are shown in Table 2. The HD50 values of the peptides ranged from 6 to 334 μM. Pep3 showed moderate hemolytic activity (HD50 of 104 μM). Derivatization of the N terminus of Pep3 (BP08 to BP12) caused a significant increase in hemolytic activity (HD50 of 6 to 30 μM). The N-palmitoylated analogue, BP12, was the most hemolytic peptide (HD50 of 6 μM). BP33, which has a Leu instead of a Trp residue, was twofold less hemolytic than Pep3 (HD50 of 190 μM), and BP15, containing a Lys instead of a Trp residue, was more than threefold less hemolytic than Pep3 (HD50 of 334 μM). The replacement of Val in Pep3 with Trp, Phe, or Tyr resulted in peptides (BP18 to BP20) with a high level of hemolytic activity (HD50 of 26 to 42 μM). Replacement of Trp and Val with Lys and Phe (BP76), respectively, led to a twofold decrease of hemolytic activity (HD50 of 203 μM).
Susceptibility to proteolysis.
The susceptibility of some peptides to proteolysis was studied by exposure to proteinase K and monitoring the degradation by reverse-phase HPLC over time (Fig. 3). Pep3, BP15, BP33, and the N-terminal derivatized peptides BP08 to BP10 showed similar stabilities. Interestingly, BP76 turned out to be twofold more stable than Pep3. While Pep3 was completely degraded after 45 min of incubation with the enzyme, BP76 underwent only a 50% degradation.
FIG. 3.

Kinetics of digestion of Pep3 (○) and selected analogues BP08 (—), BP20 (✻), BP33 (⧫), and BP76 (•) by proteinase K.
α-Helical structures determined by CD.
We investigated the secondary structures of the 11-residue peptides which showed the highest biological activity by analyzing their CD spectra in 50% (vol/vol) trifluoroethanol in 10 mM sodium phosphate buffer at pH 7.4. The α-helical contents obtained are listed in Table 1. The spectra of all peptides were characteristic of a typical α-helical conformation with two negative minimum bands at 208 and 222 nm. Results showed that Pep3 and its analogues have a moderate α-helical content (23.1 to 37.8%).
DISCUSSION
Despite the existence of natural potent antimicrobial peptides (e.g., magainins and cecropins), a search for more potent and shorter peptides with a broader spectrum of activity has led to the identification of highly potent synthetic peptides. Several design strategies have been devised in order to develop new compounds with maximum antimicrobial activity and minimum cytotoxicity. For instance, juxtaposition of the N-terminal sections of cecropin A and melittin has resulted in hybrids with better antimicrobial spectra than the parent peptide cecropin A and less hemolytic activity than melittin. In particular, Pep3, an 11-mer peptide corresponding to cecropin A(2-8)-melittin(6-9), is effective as a bactericidal and fungicidal (4, 16) agent.
In the present work, we have evaluated the biological activity of Pep3 against E. amylovora, X. vesicatoria, and P. syringae. Although antimicrobial peptides have been tested against these pathogens (2, 39), to the best of our knowledge, inhibition of these bacteria by Pep3 or short synthetic peptides (≤11 residues) has not been previously reported. Our results showed that Pep3 inhibits growth of these plant-pathogenic bacteria at low micromolar concentrations (MIC of <10 μM).
Using Pep3 as a template, we have synthesized new analogues with improved biological properties. Inhibitory concentrations of peptides for the different bacteria varied significantly, with X. vesicatoria being the most susceptible pathogen. These results are in agreement with previous reports, which showed that X. vesicatoria was more susceptible than P. syringae and E. carotovora to peptides such as MII, MSI-99, and cecropin B (2). These different levels of susceptibility of bacteria to peptides with different amino acid sequences have been attributed to variation in the components of the plasma membranes of the target microorganism, e.g., charge and lipid composition, which would influence rates of binding of cationic peptides to the membranes (31).
The mechanism of action of antimicrobial peptides against gram-negative bacteria is thought to involve first the so-called “self-promoted uptake” pathway across the outer membrane (45). Then, cationic peptides interact with the negatively charged phospholipids of the inner membrane, followed by either channel formation or simple membrane disruption (7, 9, 11, 25, 29, 46, 49). Since the 11-residue hybrid peptide Pep3 is not long enough to span the entire width of the membrane, a “carpet-like” mechanism seems more plausible. In such a mechanism, peptides first lie parallel to the surface of the phospholipid bilayer with their hydrophobic sides facing the membranes and their cationic sides facing outside, and this is followed by membrane permeation/disintegration once a threshold concentration is reached (46). Therefore, it appears that the ability to adopt an amphipathic structure is important for activity of this type of peptide. Other parameters that modulate the activity are the degree of structure formation, the net positive charge, and the overall hydrophobicity. It has been shown that the accomplishment of these structural parameters is not stringently necessary for activity against several gram-negative bacteria, but rather a good balance between them is required. In the design of short peptides, a fine-tuning of charge, helix-forming propensity, an adequate hydrophobicity, and, in particular, the number of aliphatic residues, is even more important (24).
The design of the peptides in the present work was based on the above-mentioned rationale. The amphipathic character of Pep3 becomes evident when it is represented as an ideal α-helix by means of an Edmunson wheel plot. Accordingly, analogues BP13 to BP20, BP33, and BP76 were designed based on this ideal α-helical wheel diagram. Replacement of amino acids located at the interface, e.g., Trp and Val, with residues with various degrees of hydrophilicity and hydrophobicity, resulted in great changes in antibacterial activity. Replacement of Trp in Pep3 with Phe or Tyr and acetylation, benzoylation, or tosylation of the N terminus of Pep3 decreased antibacterial activity, whereas replacement of Trp with Leu or Lys or benzylation induced a slight increase in the overall activity. Replacement of Val with hydrophobic aromatic residues afforded peptides considerably more active than Pep3 (BP18 to BP20), but when a Lys was incorporated, no activity was observed (BP17). Double replacement of both Trp and Val with Lys and Phe (BP76), respectively, induced a significant increase of antibacterial activity, whereas an increase in the size of the hydrophilic face by incorporation of two Lys residues (BP16) resulted in complete loss of activity. Consequently, structural features that seem to be important for the antibacterial activity are a basic N terminus and a hydrophobic C terminus. In contrast to antibacterial activity that was dramatically influenced by single- or double-residue replacement, the α-helical content of peptides was not significantly affected.
As previously described with other cationic peptides (25, 28) and unlike conventional antibiotics that in many cases are bacteriostatic, Pep3, BP11, BP15, BP33, and BP76 showed a bactericidal effect against the three bacteria tested at concentrations around the MIC.
Peptide cytotoxicity was also strongly influenced by the nature of the amino acid replacement in Pep3. Replacement of Trp in Pep3 with a cationic residue such as Lys produced the less hemolytic peptides BP15 and BP76. In contrast, derivatization of the N terminus of Pep3 or replacement of Val with more hydrophobic residues resulted in peptides with higher cytotoxicity. This result is in accordance with previous studies on antimicrobial peptides reporting that an increase in the peptide hydrophobicity is shown to be related to an increase in cytotoxicity (41). This selectivity has been attributed to the differences in membrane lipid composition between bacteria and mammalian cells. The absence of acidic phospholipids and presence of sterols reduce the susceptibility of eukaryotic cells to lytic peptides (35). Moreover, replacement of Trp with an aliphatic amino acid such as Leu (BP33) also led to a decrease in hemolytic activity. Peptides containing tryptophan residues have been previously reported to display high hemolytic activity, which has been attributed to the ability of tryptophan to assume a defined orientation when binding to cholesterol (10).
The peptides reported here are comparable in terms of activity to antibiotics, such as streptomycin, used in agriculture for bacterial disease control with an in vitro activity of 2 to 9 μM and operational doses for field treatment of around 100 μM. In the case of the best peptides described here, these concentrations are not expected to present toxic effects in regard to the HD50.
Protease digestion stability is a desired property in antimicrobial peptides to assure a reasonable half-life of the molecules in the plant environment. Proteases from epiphytic microorganisms or intrinsic to the plant in internal tissues may degrade antimicrobial peptides (4, 16). Again, certain replacements of amino acids in Pep3 had a strong influence in susceptibility to protease digestion. N-terminal derivatization or single residue replacement did not improve the stability of Pep3. In contrast, double replacement of both Trp and Val with Lys and Phe, respectively, was associated with an important increase in peptide stability. The double replacement for natural amino acids were equivalent to methods commonly used to increase peptide stability such as replacement of a natural amino acid with a d-amino acid or peptide cyclization (30, 44).
In conclusion, we have obtained peptides effective against E. amylovora, X. vesicatoria, and P. syringae. Moreover, we have improved the bactericidal activity and minimized cytotoxic effects and sensitivity to protease degradation of the previously described antimicrobial peptide Pep3. Therefore, these peptides might be considered as potential lead compounds for the development of antimicrobial agents for use in plant protection either as pesticide ingredients or as agents expressed in transgenic plants. Optimization of their antimicrobial properties by combinatorial chemistry and ex vivo and in vivo studies are currently in progress.
Acknowledgments
Rafael Ferre is the recipient of a predoctoral fellowship from the Ministry of Education and Science of Spain (MEC). This work was supported by grants AGL2001-2354, AGL2001-2349-C03-01, AGL2003-03354, and AGL2004-07799-C03-01 from MEC of Spain and CIRIT from the Catalonian Government.
REFERENCES
- 1.Agrios, G. N. 1998. Plant pathology, 4th ed. Academic Press, San Diego, Calif.
- 2.Alan, A. R., and E. Earle. 2002. Sensitivity of bacterial and fungal plant pathogens to the lytic peptides, MSI-99, magainin II, and cecropin B. Mol. Plant-Microbe Interact. 15:701-708. [DOI] [PubMed] [Google Scholar]
- 3.Alberola, J., A. Rodríguez, O. Francino, X. Roura, L. Rivas, and D. Andreu. 2004. Safety and efficacy of antimicrobial peptides against naturally acquired leishmaniasis. Antimicrob. Agents Chemother. 48:641-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali, G. S., and A. S. N. Reddy. 2000. Inhibition of fungal and bacterial plant pathogens by synthetic peptides: in vitro growth inhibition, interaction between peptides, and inhibition of disease progression. Mol. Plant-Microbe Interact. 13:847-859. [DOI] [PubMed] [Google Scholar]
- 5.Andreu, D., and R. B. Merrifield. 1985. N-terminal analogues of cecropin A: synthesis, antibacterial activity, and conformational properties. Biochemistry 24:1683-1688. [DOI] [PubMed] [Google Scholar]
- 6.Andreu, D., R. B. Merrifield, H. Steiner, and H. G. Boman. 1983. Solid-phase synthesis of cecropin A and related peptides. Proc. Natl. Acad. Sci. USA 80:6475-6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreu, D., and L. Rivas. 1998. Animal antimicrobial peptides: an overview. Biopolymers 47:415-433. [DOI] [PubMed] [Google Scholar]
- 8.Andreu, D., J. Ubach, A. Boman, B. Wåhlin, D. Wade, R. B. Merrifield, and H. G. Boman. 1992. Shortened cecropin A-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett. 296:190-194. [DOI] [PubMed] [Google Scholar]
- 9.Bechinger, B. 2004. Structure and function of membrane-lytic peptides. Crit. Rev. Plant Sci. 23:271-292. [Google Scholar]
- 10.Blondelle, S. E., and K. Lohner. 2000. Combinatorial libraries: a tool to design antimicrobial and antifungal peptide analogues having lytic specificities for structure-activity relationship studies. Biopolymers 55:74-87. [DOI] [PubMed] [Google Scholar]
- 11.Boman, H. G. 2003. Antibacterial peptides: basic facts and emerging concepts. J. Intern. Med. 254:197-215. [DOI] [PubMed] [Google Scholar]
- 12.Brodgen, K. A., M. Ackermann, P. B. McCray, Jr., and B. F. Tack. 2003. Antimicrobial peptides in animals and their role in host defences. Int. J. Antimicrob. Agents 22:465-478. [DOI] [PubMed] [Google Scholar]
- 13.Broekaert, W. F., B. P. A. Cammue, M. F. C. DeBolle, K. Thevissen, G. W. De Samblanx, and R. W. Osborn. 1997. Antimicrobial peptides from plants. Crit. Rev. Plant Sci. 16:297-323. [Google Scholar]
- 14.Bulet, P., R. Stöcklin, and L. Menin. 2004. Antimicrobial peptides: from invertebrates to vertebrates. Immunol. Rev. 198:169-184. [DOI] [PubMed] [Google Scholar]
- 15.Cabrefiga, J., and E. Montesinos. 2005. Analysis of aggressiveness of Erwinia amylovora using disease-dose and time relationships. Phytopathology 95:1430-1437. [DOI] [PubMed] [Google Scholar]
- 16.Cavallarin, L., D. Andreu, and B. San Segundo. 1998. Cecropin A-derived peptides are potent inhibitors of fungal plant pathogens. Mol. Plant-Microbe Interact. 11:218-227. [DOI] [PubMed] [Google Scholar]
- 17.Chicharro, C., C. Granata, R. Lozano, D. Andreu, and L. Rivas. 2001. N-terminal fatty acid substitution increases the leishmanicidal activity of CA(1-7)M(2-9), a cecropin-melittin hybrid peptide. Antimicrob. Agents Chemother. 45:2441-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen, B., J. Fink, R. B. Merrifield, and D. Mauzerall. 1988. Channel-forming peptides of cecropins and related model compounds incorporated into planar lipid membranes. Proc. Natl. Acad. Sci. USA 85:5072-5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fink, J., R. B. Merrifield, A. Boman, and H. G. Boman. 1989. The chemical synthesis of cecropin D and an analogue with enhanced antibacterial activity. J. Biol. Chem. 264:6260-6267. [PubMed] [Google Scholar]
- 20.Finney, D. J. 1971. Probit analysis, 3rd ed. Cambridge University Press, Cambridge, United Kingdom.
- 21.Ganz, T., and R. I. Lehrer. 1998. Antimicrobial peptides of vertebrates. Curr. Opin. Immunol. 10:41-44. [DOI] [PubMed] [Google Scholar]
- 22.García-Olmedo, F., A. Molina, J. M. Alamillo, and P. Rodríguez-Palenzuela. 1998. Plant defense peptides. Biopolymers 47:479-491. [DOI] [PubMed] [Google Scholar]
- 23.Giacometti, A., O. Cirioni, W. Kamysz, G. D'Amato, C. Silvestri, M. S. Del Prete, J. Lukasiak, and G. Scalise. 2004. In vitro activity and killing effect of the synthetic hybrid cecropin A-melittin peptide CA(1-7)M(2-9)NH2 on methicillin-resistant nosocomial isolates of Staphylococcus aureus and interactions with clinically used antibiotics. Diagn. Microbiol. Infect. Dis. 49:197-200. [DOI] [PubMed] [Google Scholar]
- 24.Giangaspero, A., L. Sandri, and A. Tossi. 2001. Amphipathic α helical antimicrobial peptides: a systematic study of the effects of structural and physical properties on biological activity. Eur. J. Biochem. 268:5589-5600. [DOI] [PubMed] [Google Scholar]
- 25.Hancock, R. E. W. 2001. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 1:156-164. [DOI] [PubMed] [Google Scholar]
- 26.Hancock, R. E. W., and R. Lehrer. 1998. Cationic peptides: a new source of antibiotics. Trends Biotechnol. 16:82-88. [DOI] [PubMed] [Google Scholar]
- 27.Hancock, R. E. W., and A. Patrzykat. 2002. Clinical development of cationic antimicrobial peptides: from natural to novel antibiotics. Curr. Drug Targets Infect. Disord. 2:79-83. [DOI] [PubMed] [Google Scholar]
- 28.Hancock, R. E. W., and A. Rozek. 2002. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol. Lett. 206:143-149. [DOI] [PubMed] [Google Scholar]
- 29.Hancock, R. E. W., and M. G. Scott. 2000. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. USA 97:8856-8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong, S. Y., J. E. Oh, and K.-H. Lee. 1999. Effect of d-amino acid substitution on the stability, the secondary structure and the activity of membrane-active peptide. Biochem. Pharmacol. 58:1775-1780. [DOI] [PubMed] [Google Scholar]
- 31.Huang, H. W. 2000. Action of antimicrobial peptides: two-state model. Biochemistry 39:8347-8352. [DOI] [PubMed] [Google Scholar]
- 32.Hultmark, D., A. Engstrom, H. Bennich, R. Kapur, and H. G. Boman. 1982. Insect immunity: isolation and structure of cecropin D and four minor antibacterial components from Cecropia pupae. Eur. J. Biochem. 127:207-217. [DOI] [PubMed] [Google Scholar]
- 33.Lee, D. G., Y. Park, I. Jin, K.-S. Hahm, H.-H. Lee, Y.-H. Moon, and E.-R. Woo. 2004. Structure-antiviral activity relationships of cecropin A-magainin 2 hybrid peptide and its analogues. J. Pept. Sci. 10:298-303. [DOI] [PubMed] [Google Scholar]
- 34.Loper, J. E., M. D. Henkels, R. G. Roberts, G. G. Grove, M. J. Willet, and T. J. Smith. 1991. Evaluation of streptomycin, oxytetracycline and copper resistance of Erwinia amylovora isolated from pear orchards in Washington state. Plant Dis. 75:287-290. [Google Scholar]
- 35.Matsuzaki, K., K. Sugishita, N. Fujii, and K. Miyajima. 1995. Molecular basis for membrane selectivity of antimicrobial peptide, magainin 2. Biochemistry 34:3421-3429. [DOI] [PubMed] [Google Scholar]
- 36.Montesinos, E., and A. Bonaterra. 1996. Dose-response models in biological control of plant pathogens: an empirical verification. Phytopathology 86:856-863. [Google Scholar]
- 37.Montesinos, E., P. Melgarejo, M. A. Cambra, and J. Pinochet (ed.). 2000. Enfermedades de los frutales de pepita y hueso. Ediciones Mundi Prensa, Barcelona, Spain.
- 38.Montesinos, E., and P. Vilardell. 2001. Effect of bactericides, phosphonates and nutrient amendments on blast of dormant flower buds of pear: a field evaluation for disease control. Eur. J. Plant Pathol. 107:787-794. [Google Scholar]
- 39.Mourgues, F., M. Brisset, and E. Chevreau. 1998. Activity of different antibacterial peptides on Erwinia amylovora growth, and evaluation of the phytotoxicity and stability of cecropins. Plant Sci. 139:83-91. [Google Scholar]
- 40.Ninot, A., N. Aletà, C. Moragrega, and E. Montesinos. 2002. Evaluation of a reduced copper spraying program to control bacterial blight of walnut. Plant Dis. 86:583-587. [DOI] [PubMed] [Google Scholar]
- 41.Oh, D., S. Y. Shin, S. Lee, J. H. Kang, S. D. Kim, P. D. Ryu, K.-S. Hahm, and Y. Kim. 2000. Role of the hinge region and the tryptophan residue in the synthetic antimicrobial peptides, cecropin A(1-8)-magainin 2(1-12) and its analogues, on their antibiotic activities and structures. Biochemistry 39:11855-11864. [DOI] [PubMed] [Google Scholar]
- 42.Osuky, M., G. Zhou, L. Osuska, R. E. Hancock, W. W. Kay, and S. Misra. 2000. Transgenic plants expressing cationic peptide chimeras exhibit broad-spectrum resistance to phytopathogens. Nat. Biotechnol. 18:1162-1166. [DOI] [PubMed] [Google Scholar]
- 43.Otvos, L., Jr. 2000. Antibacterial peptides isolated from insects. J. Pept. Sci. 6:497-511. [DOI] [PubMed] [Google Scholar]
- 44.Rozek, A., J.-P. S. Powers, C. L. Friedrich, and R. E. W. Hancock. 2003. Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 42:14130-14138. [DOI] [PubMed] [Google Scholar]
- 45.Sawyer, J. G., N. L. Martin, and R. E. W. Hancock. 1988. Interaction of macrophage cationic proteins with the outer membrane of Pseudomonas aeruginosa. Infect. Immun. 56:693-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shai, Y. 2002. Mode of action of membrane active antimicrobial peptides. Biopolymers 66:236-248. [DOI] [PubMed] [Google Scholar]
- 47.Steiner, H., D. Hultmark, A. Engstrom, H. Bennich, and H. G. Boman. 1981. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 292:246-248.7019715 [Google Scholar]
- 48.Sundin, G. W., and C. L. Bender. 1993. Ecological and genetic analysis of copper and streptomycin resistance in Pseudomonas syringae pv. syringae. Appl. Environ. Microbiol. 59:1018-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tossi, A., L. Sandri, and A. Giangaspero. 2000. Amphipathic, α-helical antimicrobial peptides. Biopolymers 55:4-30. [DOI] [PubMed] [Google Scholar]
- 50.Wade, D., D. Andreu, S. A. Mitchell, A. M. V. Silveira, A. Boman, H. G. Boman, and R. B. Merrifield. 1992. Antibacterial peptides designed as analogues or hybrids of cecropins and melittin. Int. J. Pept. Protein Res. 40:429-436. [DOI] [PubMed] [Google Scholar]
- 51.Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389-395. [DOI] [PubMed] [Google Scholar]