

Abstract
Enteroviruses can be easily transmitted through the fecal-oral route and cause a diverse array of clinical manifestations. Recent outbreaks associated with enteroviral contamination in aquatic environments have called for the development of a more efficient and accurate virus monitoring system. To develop a simple, rapid, and direct method for identifying enteroviral infections, we generated a fluorescent reporter system in which genetically engineered cells express a hybrid fluorescent indicator composed of a linker peptide, which is exclusively cleaved by the 2A protease (2Apro), flanked with a cyan fluorescent protein (CFP) and a yellow fluorescent protein undergoing fluorescence resonance energy transfer. The covalent linkage between two fluorophores is disrupted due to 2Apro activity upon viral infection, which results in an increase in CFP intensity. This allows the rapid (within 7.5 h) detection of very low numbers (10 PFU or fewer) of infectious enteroviruses.
Enteroviruses, including coxsackievirus, echovirus, poliovirus (PV), and the numbered enteroviruses, are the second most common viral infectious agents in the world (41). These viruses are transmitted through the fecal-oral route and can cause an array of clinical manifestations, ranging from mild febrile illness to potentially fatal syndromes including paralytic poliomyelitis, aseptic meningitis, and encephalitis (27, 28, 40). Enteroviruses pose a public health threat because they are stable in aquatic environments and have low infectious doses (13, 14, 50).
The importance of water as a vehicle for the transmission of enteroviruses, coupled with the very low infectious dose for many of these viruses, has resulted in the need for methods that can reliably and rapidly detect small numbers (e.g., 1 PFU per 1,000 liters of water) of infectious virus particles in environmental samples. Conventional methods for the detection and quantification of human enteroviruses rely on the production of cytopathic effects in mammalian cell cultures; however, traditional infectivity assays are too time-consuming and labor-intensive for routine applications. Studies have been directed toward applying molecular methods, such as reverse transcriptase PCR, for more sensitive, specific, and rapid detection of viral genomes. However, PCR-based methods cannot indicate the capability of the viruses to cause infections, as the presence of the viral genome does not correlate with infectivity.
Viral replication inside the host cells is known to be a virus- and host-specific event and has been used to identify infectious viruses. Some virus-inducible reporter systems have been established for viral detection based on transcription from viral promoters that are specific for virus-infected cells (35). These transgenic cell lines provide a high level of sensitivity and specificity to facilitate the detection process. However, no research has been conducted beyond developing the concept of using a viral promoter-dependent reporting system. This strategy is not applicable to some RNA viruses, such as enteroviruses and hepatitis A virus (HAV), which exhibit no defined viral promoter region. However, most positive-strand RNA viruses, including enteroviruses, encode virus-specific proteases that cleave the primary translation product into mature proteins (46). Viral proteases represent logical targets for the detection of infectious viruses, because the cleavage event proceeds in a defined manner and is ubiquitous within various virus families.
Enteroviruses, members of the Picornaviridae, are nonenveloped viruses with a positive-strand RNA genome (31). Upon infection, the viral RNA genome is translated immediately into a single polypeptide, which is subsequently cleaved by two viral proteases, 2A (2Apro) and 3C (3Cpro), to produce mature viral proteins (31, 46, 52). The 2Apro of enteroviruses is a cysteine protease with a unique chymotrypsin-like property. During virus replication, 2Apro carries out the primary cleavage between its own N terminus and the C terminus of the preceding capsid precursor, VP1 (5). Subsequently, the 2A protease performs an additional intermolecular cleavage in the VP3 region to release mature viral polymerase (25). The cleavage mediated by 2Apro is highly specific and dependent on the primary sequences. This proteolytic processing occurs with 100% efficiency and high specificity (1). Furthermore, 2Apro is a diffusible protein and can carry out intermolecular cleavage of its substrate in the enterovirus-infected cells. 2Apro serves as a good candidate for virus detection, because the enzyme is highly expressed at an early stage of infection and the proteolysis of 2Apro is extremely efficient and selective.
In this study, we report a novel fluorescence-based detection method using fluorescence resonance energy transfer (FRET) between a donor (enhanced cyan fluorescent protein [ECFP]) and an acceptor (enhanced yellow fluorescent protein [EYFP]) fluorophore joined by a linker peptide containing the proteolytic site of 2Apro (Fig. 1). FRET is a phenomenon in which fluorescence energy is transferred from an excited fluorophore to a light-absorbing molecule, located in close proximity. On protease cleavage, the in vivo separation of the fluorescent protein pair can be monitored using fluorescence microscopy, which allows for the determination of active viral replication within the host cells. Poliovirus (PV) was used as a model virus to demonstrate the utility of the method for rapid, in vivo detection of infectious enteroviruses. By taking advantage of this unique feature of the viral proteases, this method may be applicable to other viruses portraying similar characteristics.
FIG. 1.

Schematic representation of fluorescent indicator for monitoring 2Apro-mediated proteolytic processing. (A) In the presence of 2Apro, the linker peptide is cleaved via the protease and results in the increased ECFP signal. (B) The amino acid sequence of the linker peptide is shown, and the cleavage site between two amino acids is shown in boldface.
MATERIALS AND METHODS
Cell culture and viruses.
Buffalo green monkey kidney (BGMK) cells were cultured in 4.0% (wt/vol) autoclavable Eagle's minimal essential medium-modified Earle's salts (AMEM; Irvine Scientific, Santa Ana, CA), 8 mM HEPES, 0.0075% (wt/vol) NaHCO3, 80 mM l-glutamine, 10 mM minimal essential medium nonessential amino acids (Gibco BBL, Grand Island, NY), 1,000 U/ml penicillin, 1,000 U/ml streptomycin, 2,000 U/ml nystatin, and 2 mg/ml kanamycin supplemented with 8% (wt/vol) fetal bovine serum (HyClone, Logan, UT) at 37°C in 5% (vol/vol) CO2. All virus strains used in this study were obtained from the American Type Culture Collection. Coxsackievirus B6 (CVB6), echovirus 11 (EV11), and poliovirus type 1 (strain LSc) (PV1) were cultivated in BGMK cells and purified as previously described (42). The cell line was also used to determine the virus concentration in the stock solution. HAV was used for the specificity test as a negative control. HAV concentration was determined by plaque assay using FRhK-4 cells as the host.
The viruses were quantified using plaque-forming assays as described previously (11). Briefly, 10-fold serial dilutions of virus stocks were distributed on confluent cell monolayers, and samples were incubated for 30 min at room temperature. A maintenance medium containing 1.5% (wt/vol) agarose in normal growth medium without fetal bovine serum was applied to the cells, which were then incubated at 37°C for 48 h. Plaques were visualized after overlay removal, cell fixation, and crystal violet staining.
Cloning, expression, and purification of 2Apro.
Plasmids pMal-c.CV2A, pMal-c.EV2A, and pMal-c.PV2A, which encode 2Apro of CVB6, EV11, and PV1, respectively, were constructed as described previously (39). Briefly, the 2Apro gene of CVB6, EV11, and PV1 was amplified by reverse transcriptase PCR using the viral RNA genome as template and primers with internal restriction sites. Primers used for this study include the following: CVB6 forward, 5′CCGGAATTCATGGGAGCCTTTGGGCAACAATC; CVB6 reverse, 5′CCCCAAGCTTCTATTACTGTTCCATGGCATCGTCCT; EV11 forward, 5′CGCGGATCCATGGGAGCTTTTGGATACCAATC; EV11 reverse, 5′CCCCAAGCTTCTATTACTGCTCCATCGCATCGTCCT; PV1 forward, 5′CGCGGATCCATGGGATTCGGACACCAAAAC; and PV1 reverse, 5′CCCCAAGCTTCTATTATTGTTCCATGGCTTCTTCTT. The PCR products were then digested with designed restriction enzymes and ligated into the pMal-c.2x vector with the same enzymatic digestion. Plasmids pMal-c.CV2A, pMal-c.EV2A, and pMal-c.PV2A were transformed into Escherichia coli (strain JM109) for the expression of 2Apro with maltose binding protein (MBP) fusion.
CVB6 2Apro, EV11 2Apro, and PV1 2Apro were expressed as MBP fusion proteins (MBP-2Apro) in order to enhance protein solubility while maintaining proteolytic activity (34). The MBP fusion proteins were constructed in order to enhance the protein solubility in E. coli. As reported previously, the proteolytic activity of the 2Apro fusion protein remains the same even in the presence of extra MBP sequences at the N terminus (34). Therefore, the MBP-2Apro fusion protein was used for the following study.
MBP-2Apro was expressed and purified as described previously, with some modifications (34). Briefly, E. coli cells transformed with either pMal-c.CV2A, pMal-c.EV2A, or pMal-c.PV2A were grown in Luria-Bertani (LB) medium containing 0.2% (wt/vol) glucose and 100 μg/ml ampicillin until the optical density reached 0.5, and then isopropyl-β-d-thiogalactopyranoside was added to 0.5 mM final concentration. Cells were grown at 30°C for an additional 5 h before harvesting. Cells were sonicated, and the crude extracts were collected for amylose affinity chromatography to purify the MBP fusion protein. The purified 2Apro was analyzed by sodium dodecyl sulfate-12.5% polyacrylamide gel electrophoresis, concentrated by Centricon Plus-20 (Millipore, Billerica, MA), and stored in a pH 7.4 column buffer containing 100 mM sodium phosphate, 100 mM NaCl, 1 mM EGTA, and 1 mM dithiothreitol.
Cloning, expression, and purification of 1D2A-FRET substrate.
A multiple cloning strategy was used to create plasmid pUC19.1D2A-FRET containing a fusion protein of ECFP and EYFP linked by the PV 2Apro cleavage sequences (peptide 1D2A, genome nucleotides 3359 to 3410). The gene for EYFP (32) was amplified by PCR (forward primer, 5′GAGCTCATGGTGAGCAAGGGCGAGGAG; reverse primer, 5′GAATTCTTACTTGTACAGCTCGTCCAT) and ligated to the pGEM-T vector. The resulting plasmid pGEM-EYFP was doubly digested by SacI and EcoRI endonucleases, and the 728-bp fragment was inserted into the same restriction sites of the pUC19 plasmid (New England Biolabs, Ipswich, MA) to generate pUC19.EYFP. The coding sequence of ECFP was obtained from the YELLOWcam3.1 plasmid (53) digested with HindIII and SphI, and the 728-bp fragment corresponding to ECFP was ligated to pUC19.EYFP to create pUC19.FRET. A DNA fragment encoding the PV 2Apro targeted polypeptide linker region (amino acid sequence, STKDLTTYGFGHQNKA) was inserted into the SphI and SacI restriction site between ECFP and EYFP. The resulting construct, pUC19.1D2A-FRET, was then digested with BamHI and XhoI, and the fragment corresponding to 1D2A-FRET was ligated to the pET-14b vector (Novagen, San Diego, CA) digested with the same enzymes to generate the construct pET.1D2A-FRET. The resulting plasmid expressed 1D2A-FRET substrate as a fusion product containing an N-terminal polyhistidine tag and was transformed into E. coli (strain BL21).
A mammalian expression vector encoding the 1D2A-FRET substrate was also constructed. Plasmid pUC19.1D2A-FRET was digested with HindIII and EcoRI, and the excised fragment was then cloned into the mammalian expression vector pcDNA3 (Invitrogen, Carlsbad, California) containing a neomycin resistance gene as a selectable marker. The resulting plasmid, pcDNA3.1D2A-FRET, was used to transfect BGMK cells for the establishment of the engineered cell line.
The fluorogenic substrate 1D2A-FRET was expressed as a polyhistidine fusion protein in order to facilitate the purification process through a Ni-nitrilotriacetic column. The 1D2A-FRET substrate was expressed and purified as described by the manufacturer (QIAGEN, Valencia, CA). E. coli cells transformed with pET.1D2A-FRET were grown in LB medium containing 50 μg/ml ampicillin at 30°C and harvested at an optical density of 1.6. The bacteria were lysed in a French pressure cell, and the crude extract was prepared for affinity chromatography over a Ni column (QIAGEN) for purification.
In vitro cleavage assay and Western blots.
An in vitro cleavage assay using purified MBP-2Apro and its fluorogenic substrate, 1D2A-FRET, was carried out at 30°C for 120 min in phosphate-buffered saline with 5 mM MgCl2 and 5% glycerol. Samples were taken after incubation, and the fluorescence intensity of ECFP and EYFP was determined by a 96-well microplate reader with designed filter sets (POLARstar Optima; BMG Labtechnologies, Inc., Durham, NC). The fluorescence intensity of the fluorophores was measured by using the fluorimeter at excitation and emission wavelengths of 430 ± 10 nm and 480 ± 10 nm for ECFP and 480 ± 10 nm and 520 ± 10 nm for EYFP. Western blotting against GFP variant-specific antibodies (Clontech, Mountain View, CA) was conducted to verify the fluorimeter results.
Transfection of pcDNA.1D2A-FRET in BGMK cells.
Parental BGMK cells were transfected with pcDNA3.1D2A-FRET, and clones resistant to G418 were obtained. BGMK cells at 95% confluence in 6-well plates were washed with serum-free, antibiotic-free AMEM medium prior to transfection. Vector pcDNA3.1D2A-FRET was introduced into BGMK cells by lipofection (39) using LIPOFECTAMINE PLUS Reagent (Invitrogen) following the manufacturer's instructions. The transfected cells were selected for neomycin resistance in regular growth medium consisting of 4 mg/ml G418 (Sigma-Aldrich Corp., St. Louis, MO). A prescreening procedure for the clones with high EYFP intensity was carried out in a 96-well microplate reader. A total of 12 clones were obtained and analyzed via flow cytometer-activated cell sorting. The clone with the most uniform and strongest EYFP expression level was selected for the subsequent study. The established engineered cells were cultivated using normal growth medium containing 4 mg/ml G418.
Acquisition of fluorescent images of the reporter cell line.
The stably transformed reporter cells expressing the FRET fusion protein were cultured in 16-well slide chambers to full confluence 16 h prior to infection. Infection was conducted as described previously. The reporter cells without viruses were used as the negative control for each experiment. The reporter cells were also challenged with HAV (100 or 1,000 PFU) to test the substrate specificity of the 2A protease. Samples were mounted 7.5 h postinfection and examined using a confocal microscope (Leica TCS SP2/UV). To observe the in vivo FRET phenomenon, the cells were visualized by fluorescent microscopy at excitation and emission of 440 nm and 480 nm for ECFP and 480 nm and 520 nm for EYFP.
RESULTS
Fluorogenic 2Apro substrate and in vitro cleavage assay.
To demonstrate the utility of the FRET cleavage assay for detecting viral replication, a fluorogenic indicator containing an ECFP-EYFP pair joined by a 16-amino-acid linker containing the consensus 1D2A peptide sequence from poliovirus was constructed, and it was used as a substrate for 2Apro to test the trans-cleavage ability (Fig. 1). Different amounts of MBP-2Apro were added to purified 1D2A-FRET, and the changes in ECFP fluorescence intensity were measured to illustrate the direct cleavage event mediated by 2Apro (Fig. 2). As expected, cleavage of the 1D2A site resulted in the separation of ECFP and EYFP and a dose-dependent increase in the ECFP intensity. The result was also confirmed by Western blotting analysis (Fig. 2). No detectable cleavage products were observed in the reaction without 2Apro, while reactions with MBP-2Apro exhibited extensive proteolysis, resulting in the production of cleaved ECFP and EYFP fragments (Fig. 2). The amount of cleavage products was in agreement with the increase in ECFP intensity, indicating that PV 2Apro directly cleaved the fluorogenic substrate in vitro.
FIG. 2.
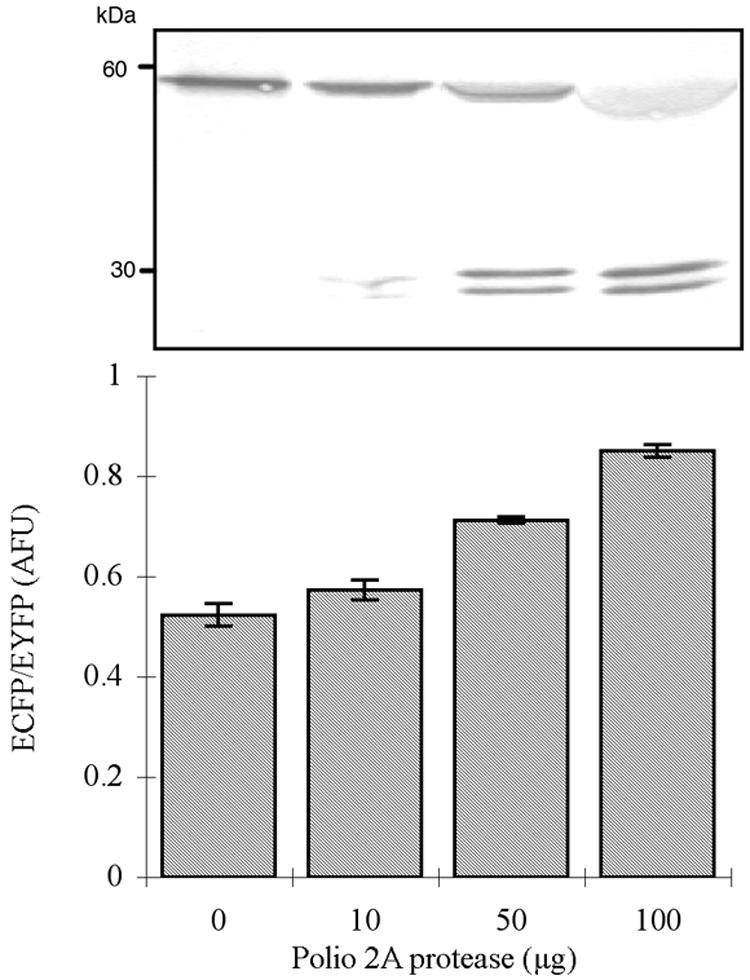
The fluorimetric assay of the FRET substrate with PV 2Apro in vitro. Each experiment was performed in triplicate. Western blot analysis of GFP variants shows that the upper band represents the conjugated FRET substrate with a molecular mass of approximately 60 kDa, and the lower band represents the proteolytic products with a molecular mass of approximately 30 kDa. AFU, artificial fluorescent unit.
Measurement of the kinetic parameters Km and kcat was achieved by measuring the proteolytic process of the fluorogenic substrate at various concentrations. The calculated Km and kcat values were 0.38 g liter−1 and 0.015 min−1, respectively. The fluorogenic substrate exhibited kinetics similar to those of the 1D2A peptide, indicating that the flanking fluorescent proteins would not interfere with the 2Apro-mediated cleavage efficiency (6).
Establishment of a stably transformed cell line.
A potential issue with all transgenic cell lines is cell toxicity due to the high expression level of the target protein. Based on preliminary observations, the high expression level of the fluorogenic substrate had no effect on the genetically modified BGMK cells: the morphologies and the growth rate (approximately 24 h/cell cycle) of the bioreporter cells and their parental ones are indistinguishable. The engineered cell line also showed no difference from its parental cell line in terms of the susceptibility to the viruses used in this study, as similar cytopathic effects were observed when infected with the same viral doses (see the supplemental material). The expression level of the fluorescent indicator protein remained stable for up to 40 passages.
Expression of the fluorescent substrates was verified by examining the bioreporter cells under a confocal microscope using a 440-nm/480-nm filter set for ECFP (Fig. 3A) and a 480-nm/530-nm filter set for EYFP (Fig. 3B), which was monitored as an internal control. When the images in Fig. 3A and B were superimposed, the cells with intact fluorogenic substrate could be seen in yellow (Fig. 3C). A baseline level of ECFP signal was observed due to the partial quenching of ECFP by EYFP. The cell lysate of the parental BGMK cells exhibited a background level of ECFP signal and was negative for ECFP and EYFP antibodies, while the genetically modified cells showed a high expression level of FRET substrate and were positive for ECFP and EYFP antibodies (data not shown).
FIG. 3.

Fluorescence microscopy analysis of the reporter cells. Fluorescence intensity of ECFP was monitored via a 440-nm/480-nm filter set (A) and a 480-nm/530-nm filter set for EYFP (B), and the superimposed image is shown in panel C.
In vivo detection of enteroviral infection.
The reporter cells infected with PV1 synthesize 2Apro, which cleaves the fluorogenic substrate and results in an increased ECFP intensity. The reporter assay was tested by challenging the reporter cells with various concentrations of PV1, and the samples were examined by fluorescent microscopy as described above. The uninfected reporter cells or cells infected with HAV were used as negative controls and are shown in yellow (Fig. 4A), while cells that allowed active viral replication are shown in green (Fig. 4B and C). Figure 4B and C show a dose-dependent increase in ECFP intensity after infection of the reporter cells with different concentrations of PV1.
FIG. 4.

Fluorescence microscopy analysis of the reporter cells challenged with two different concentrations of PV. Control cells (A) show no sign of increased ECFP signal, while cells challenged with either 100 PFU (B) or 1,000 PFU (C) of virus exhibit increased ECFP intensity.
Monolayers containing approximately 105 cells were infected with 1, 10, 100, or 1,000 PFU in total. Infected cells with increased ECFP signal were detected as soon as 7.5 h postinfection, and the number of cells with increased ECFP intensity correlated to the amount of virus added. A detection limit of less than 10 PFU was achieved (Table 1). A parallel experiment using a plaque assay was also performed with the same sample set to verify the microscopy results. As expected, the results from in vivo cleavage assay were in agreement with the plaque assay. The 2Apro-mediated cleavage of the fluorescent substrates was also confirmed by Western blotting of extracts from the reporter cells infected with various concentrations of PV1 (data not shown).
TABLE 1.
Qualitative examination of infectious viruses
| Virus | Detection at the following virus titer (PFU)a
|
|||
|---|---|---|---|---|
| 1 | 10 | 100 | 1,000 | |
| PV1 | +/− | + | ++ | +++ |
| EV11 | +/− | + | ++ | +++ |
| CVB6 | − | +/− | ++ | +++ |
The sensitivity of the fluorogenic detection system was determined to be undetectable (−), detectable in some assays (+/−), or positive (+ through +++). +, few cells (<10) showed in green color under fluorescent microscope; ++, moderate number of cells showed in green (between 10 to 100); and +++, numerous green cells (more than 100) in a given experimental unit. Each experiment was replicated 10 times.
To demonstrate the potential utility of the reporter assay for other enteroviruses, the reporter cells were challenged with two other enteroviruses, CVB6 and EV11, to determine the specificity of the fluorogenic substrate. Both CVB6 and EV11 induced an increased ECFP intensity at a level similar to those induced by PV1. The numbers of cells with higher ECFP signal than the control were also in proportion to the virus concentration in a given sample. Parallel experiments using plaque assay and Western blotting analysis of the cell extracts from the reporter cells infected with CVB6, EV11, and PV1 again confirmed the microscopic observations (data not shown).
Direct cleavage assays of CVB6 and EV11 2Apro.
In vitro cleavage assays of CVB6 and EV11 2Apro and the fluorogenic substrate were performed to demonstrate that the proteolytic event was mediated directly via these two enzymes. Both CVB6 and EV11 2Apro were expressed as MBP fusion proteins and purified via amylase affinity chromatography as mentioned previously. The same fluorimeter assay described before was applied by incubating the fluorogenic substrate with PV1, CVB6, or EV11 2Apro. Figure 5 shows that both CVB6 and EV11 2Apro are able to mediate the proteolytic processing of the FRET substrate in vitro. The purified CVB6 MBP-2Apro fusion protein has enzymatic kinetics similar to those of PV1 2Apro, with Km and kcat values of 0.43 g liter−1 and 0.025 min−1, respectively. However, the EV11 MBP-2Apro fusion protein exhibits much higher catalytic efficiency, with a Km of 2.92 g liter−1 and a kcat of 2.08 min−1.
FIG. 5.
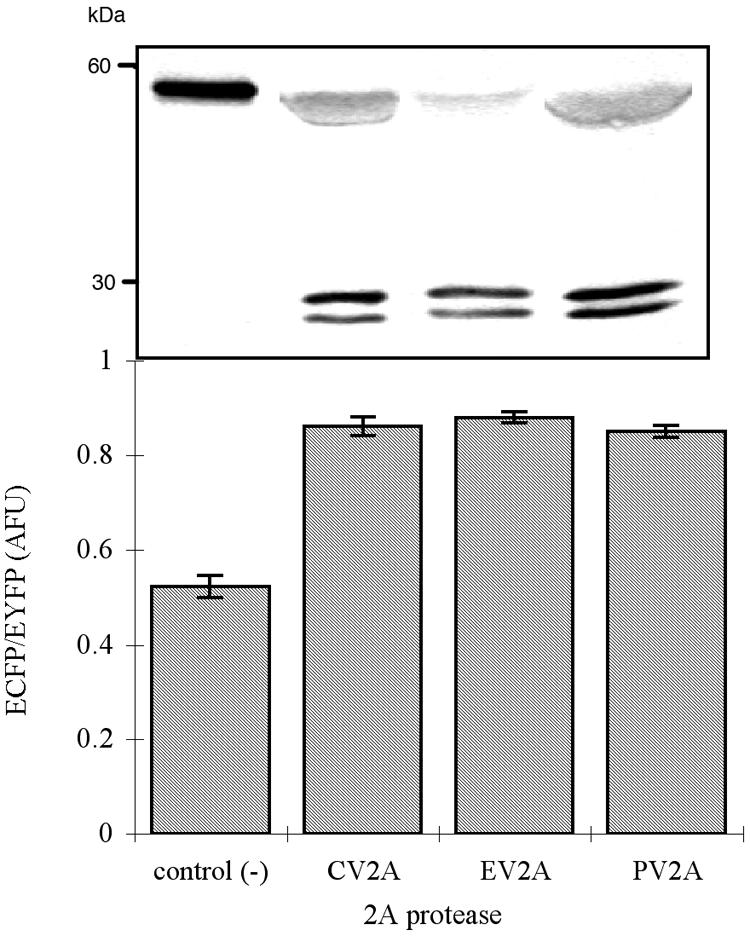
The fluorimetric assay of the FRET substrate with various 2A proteases in vitro. Each experiment was performed in triplicate, and 100 μg of each protease was added to the reaction. Western blotting of total cell lysate from the bioreporter cells using anti-GFP variant monoclonal antibody shows the same result as the in vitro assay, in which the upper band indicates the intact FRET substrate and the lower bands are the cleaved products. AFU, artificial fluorescent unit.
DISCUSSION
Protection against exposure to viral pathogens in water presents a particular challenge, because viral occurrence does not correlate well with the occurrence of indicator organisms (4). Conventional culture-based assay is the only method that detects infectious viruses, but it is too time-consuming for routine water quality surveillance. Engineered reporter cell lines, which can be designed to exhibit specific characteristics upon infection, have the potential to provide the means for the rapid detection of infectious viruses. The method developed in this study provided rapid (7.5 h postinfection) and sensitive (≤10 PFU) detection of infectious viral particles by taking advantage of the viral replication process and specifically engineered cells.
For many viruses, viral proteases play a crucial role in the replication cycle. Substrate specificity and the efficiency of the 2A protease make it a perfect target for a diagnostic marker and antiviral drug development (6, 17, 23). In this study, we demonstrated that a reporter cell line expressing fluorogenic protease substrate responds well to viral infection, and the infected cells could be detected easily through fluorescent microscopy. The reporter cells responded to all three enteroviruses tested with different sensitivities and did not respond to HAV, which is also a member of Picornaviridae, but it does not encode protease 2A (37). The reporter cells achieved the same detection limit for PV1 and EV11 (≤10 PFU) but had a higher detection limit for CVB6 (≤100 PFU) (Table 1).
The 2Apro-mediated cleavage of the 1D2A junction is the first proteolytic event upon enteroviral infection; therefore, 2Apro is an excellent candidate for the rapid detection of infectious enteroviruses. Previous reports have shown that the critical amino acid residues in the 1D2A junction between VP1 and VP2 in enterovirus polyproteins determine the 2Apro-mediated proteolytic efficiency (6, 17, 42). It has been shown that a 16-amino-acid peptide corresponding to the 1D2A junction derived from the poliovirus polyprotein, amino acids 874 to 889 (Fig. 1B), can serve as a good in vitro substrate for poliovirus 2Apro (6). In addition, it has also been shown that a linker peptide less than 20 amino acids in length is sufficient for efficient fluorescence energy transfer between the donor and acceptor pair (33).
The reporter cells are sensitive to all three enteroviruses tested, suggesting that the cell line established in this study has the potential to allow the detection of a variety of enteroviruses, if they have protease cleavage sites similar to that of PV. This method may also serve as a model system for the development of bioreporters for viruses with unique proteases. Despite the fact that research regarding tissue engineering has been extensively conducted, there are limited studies involving the application of engineered cell lines for the detection of infectious viruses. The results presented in this study demonstrate that genetically engineered cell lines can be applied to monitor active infection events in cell culture. However, the current approach is only semiquantitative. Using suspended reporter cells coupled with a flow cytometer as an end point analytical tool might provide more quantitative information.
In addition, the developed approach may also be adapted for anti-protease drug development. Due to their unique protein structures and essential roles in viral replication, viral proteases appear to be ideal targets for antiviral intervention (36). Numerous compounds with potent in vitro activity against viral proteases, including the human immunodeficiency virus type 1 protease, human rhinovirus protease 3C, and coxsackievirus protease 2A, have been reported, but only some of them have been marketed at this point due to the lack of clinical efficacy (23, 36, 50). The fluorescence resonance energy transfer-based protein substrates of viral proteases will allow rapid kinetic assays ideal for searching for protease inhibitors.
Supplementary Material
Acknowledgments
Plasmid SCAT3 carrying the EYFP gene was a kind gift from Atsushi Miyawaki, Advanced Technology Development Group, Brain Science Institute, Saitama, Japan, and plasmid YC3.3 carrying the ECFP gene was a generous gift from Roger Tsien, Howard Hughes Medical Institute, University of California, San Diego. We also thank Gabriela Jenikova and Cindy Wu for their help with molecular cloning.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org.
REFERENCES
- 1.Alvey, J., E. Wyckoff, S. Yu, R. Lloyd, and E. Ehrenfeld. 1991. cis- and trans-cleavage activities of poliovirus 2A protease expressed in Escherichia coli. J. Virol. 65:6077-6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angres, B., and G. Green. April. 1999, posting date. Dual labeling using ECFP and EYFP in standard fluorescence microscopy. CLONTECHniques [Online.] http://www.clontech.com/clontech/archive/APR99UPD/dual.shtml.
- 3.Ashbolt, N. 2004. Microbial contamination of drinking water and disease outcomes in developing regions. Toxicology 198:229-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banks, W., C. Klohe, and D. Battigelli. 2001. Occurrence and distribution of enteric viruses in shallow ground water and factors affecting well vulnerability to microbiological contamination in Worcester and Wicomico Counties, Maryland. U.S. Geological Survey water-resources investigations report 01-4147, p. 23. U.S. Geological Survey, Baltimore, Md.
- 5.Blom, N., J. Hansen, D. Blaas, and S. Brunak. 1996. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci. 5:2203-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bovee, M. L., B. J. Lamphear, R. E. Rhoads, and R. E. Lloyd. 1998. Direct cleavage of eIF4G by poliovirus 2A protease is inefficient in vitro. Virology 245:241-249. [DOI] [PubMed] [Google Scholar]
- 7.Cuthbert, J. 2001. Hepatitis A: old and new. Clin. Microbiol. Rev. 14:38-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahling, D. 1991. Detection and enumeration of enteric viruses in cell culture. Crit. Rev. Environ. Control 21:237-263. [Google Scholar]
- 9.Fout, G., B. Martinson, M. Moyer, and D. Dahling. 2003. A multiplex reverse transcription-PCR method for detection of human enteric viruses in groundwater. Appl. Environ. Microbiol. 69:3158-3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujimoto, T., M. Chikaira, S. Yoshida, H. Ebira, A. Hasegawa, A. Totsuka, and O. Nishio. 2002. Outbreaks of central nervous system disease associated with hand, foot, and mouth disease in Japan during the summer of 2000: detection and molecular epidemiology of enterovirus 71. Microbiol. Immunol. 46:621-627. [DOI] [PubMed] [Google Scholar]
- 11.Glass, J. S., R. Van Sluis, and W. Yanko. 1978. Practical method for detecting poliovirus in anaerobic digester sludge. Appl. Environ. Microbiol. 35:983-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon, G., G. Berry, X. Liang, B. Levine, and B. Herman. 1998. Quantitative fluorescence resonance energy transfer measurements using fluorescence microbiology. Biophys. J. 74:2702-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haas, C. N., J. B. Rose, C. P. Gerba, and R. Regli. 1993. Risk assessment of viruses in drinking water. Risk Anal. 13:545-552. [DOI] [PubMed] [Google Scholar]
- 14.Hafliger, D., P. Hubner, and L. Luthy. 2000. Outbreaks of viral gastroenteritis due to sewage-contaminated drinking water. Int. J. Food Microbiol. 54:123-126. [DOI] [PubMed] [Google Scholar]
- 15.Harpur, A., F. Wouters, and P. Bastiaens. 2001. Imaging FRET between spectrally similar GFP molecules in single cells. Nat. Biotechnol. 19:167-169. [DOI] [PubMed] [Google Scholar]
- 16.He, L., D. Olson, X. Wu, T. Karpova, J. McNally, and P. Lipsky. 2003. A flow cytometric method to detect protein-protein interaction in living cells by directly visualizing donor fluorophore quenching during CFP-YFP fluorescence resonance energy transfer (FRET). Cytometry 55A:71-85. [DOI] [PubMed] [Google Scholar]
- 17.Hellen, C. U. T., C. K. Lee, and E. Wimmer. 1992. Determinants of substrate recognition by poliovirus-2A proteinase. J. Virol. 66:3330-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, Y. T., P. Yam, H. Yan, and Y. Sun. 2002. Engineered BGMK cells for sensitive and rapid detection of enteroviruses. J. Clin. Microbiol. 40:366-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jares-Erijman, E., and T. Jovin. 2003. FRET imaging. Nat. Biotechnol. 21:1387-1395. [DOI] [PubMed] [Google Scholar]
- 20.Kindhauser, M. (ed.). 2003. Global defense against the infectious disease threat. World Health Organization, Geneva, Switzerland.
- 21.Kohi, T., K. Heinze, R. Kuhlemann, A. Koltermann, and P. Schwille. 2002. A protease assay for two-photon crosscorrelation and FRET analysis based solely on fluorescent proteins. Proc. Natl. Acad. Sci. USA 99:12161-12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kung, S., Y. Wang, C. Lin, R. Kuo, and W. Liu. 2000. Rapid diagnosis and quantification of herpes simplex virus with a green fluorescent protein reporter system. J. Virol. Methods 90:205-212. [DOI] [PubMed] [Google Scholar]
- 23.Lalezari, J. P., E. DeJesus, D. W. Northfelt, G. Richmond, P. Wolfe, R. Haubrich, D. Henry, W. Powderly, S. Becker, M. Thompson, F. Valentine, D. Wright, M. Carlson, S. Riddler, F. F. Haas, R. DeMasi, P. R. Sista, M. Salgo, and J. Delehanty. 2003. A controlled phase II trial assessing three doses of enfuvirtide (T-20) in combination with abacavir, amprenavir, ritonavir and efavirenz in non-nucleoside reverse transcriptase inhibitor-naive HIV-infected adults. Antivir. Ther. 8:279-287. [PubMed] [Google Scholar]
- 24.Lamphear, B. J., and R. E. Rhoads. 1996. A single amino acid change in protein synthesis initiation factor 4G renders cap-dependent translation resistant to picornaviral 2A proteases. Biochemistry 35:15726-15733. [DOI] [PubMed] [Google Scholar]
- 25.Lee, C., and E. Wimmer. 1988. Proteolytic processing of poliovirus polyprotein: elimination of 2Apro-mediated, alternative cleavage of polypeptide 3CD by in vitro mutagenesis. Virology 166:405-414. [DOI] [PubMed] [Google Scholar]
- 26.Liu, W., J. Sun, C. Lin, R. Kuo, and S. Kung. 2001. An indicator cell assay for detection of human cytomegalovirus based on enhanced green fluorescent protein. J. Virol. Methods 96:85-92. [DOI] [PubMed] [Google Scholar]
- 27.Lukashev, A., V. Lashkevich, G. Koroleva, J. Ilonen, G. Karganova, V. Reznik, and A. Hinkkanen. 2003. Molecular epidemiology of enteroviruses causing uveitis and multisystem hemorrhagic disease of infants. Virology 307:45-53. [DOI] [PubMed] [Google Scholar]
- 28.McIntyre, J. O., B. Fingleton, K. S. Wells, D. W. Piston, C. C. Lynch, S. Gautam, and L. M. Matrisian. 2004. Development of a novel fluorogenic proteolytic beacon for in vivo detection and imaging of tumour-associated matrix metalloproteinase-7 activity. Biochem. J. 377:617-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMinn, P. 2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol. Rev. 26:91-107. [DOI] [PubMed] [Google Scholar]
- 30.Mead, P., L. Slutsker, V. Dietz, L. McCaig, J. Bresee, C. Shapiro, P. Griffin, and R. Tauxe. 1999. Food-related illness and death in the United States. Emerg. Infect. Dis. 5:607-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melnick, J. 1996. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, p. 609-712. In B. N. Fields and D. M. Knipe (ed.), Fields virology, 3rd ed. Lippincott-Raven, Philadelphia, Pa.
- 32.Nagai, T., and A. Miyawaki. 2004. A high-throughput method for development of FRET-based indicators for proteolysis. Biochem. Biophys. Res. Commun. 319:72-77. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen, A., and P. Daugherty. 2005. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 23:355-360. [DOI] [PubMed] [Google Scholar]
- 34.Novoa, I., F. Martinez-Abarca, P. Fortes, J. Ortin, and L. Carrasco. 1997. Cleavage of p220 by purified 2Apro in cell-free systems: effects on translation of capped and uncapped mRNAs. Biochemistry 36:7802-7809. [DOI] [PubMed] [Google Scholar]
- 35.Olivo, P. 1996. Transgenic cell lines for detection of animal viruses. Clin. Microbiol. Rev. 9:321-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patick, A., and K. Potts. 1998. Protease inhibitors as antiviral agents. Clin. Microbiol. Rev. 11:614-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pintó, R. M., S. Guix, J. F., González-Dankaarta, S. Caballero, G. Sánchez, K. Guob, E. Ribes, and A. Bosch. 2002. Hepatitis A virus polyprotein processing by Escherichia coli proteases. J. Gen. Virol. 83:359-368. [DOI] [PubMed] [Google Scholar]
- 38.Rider, T., M. Petrovick, F. Nargi, J. Harper, E. Schwoebel, R. Mathews, D. Blanchard, L. Bortolin, A. Young, J. Chen, and M. Hollis. 2003. A B cell-based sensor for rapid identification of pathogens. Science 301:213-215. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook, J., and D. W. Russell. 2001. Molecular cloning, 3rd ed, p. 16.7-16.13. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 40.Sato, M., M. Hosoya, K. Honzumi, M. Watanabe, N. Ninomiya, S. Shigeta, and H. Suzuki. 2003. Cytokine and cellular inflammatory sequence in enteroviral meningitis. Pediatrics 112:1103-1107. [DOI] [PubMed] [Google Scholar]
- 41.Sawyer, M. 2002. Enterovirus infections: diagnosis and treatment. Semin. Pediatr. Infect. Dis. 13:40-47. [DOI] [PubMed] [Google Scholar]
- 42.Sobsey, M. D., and J. S. Glass. 1980. Poliovirus concentration from tap water with electropositive adsorbent filters. Appl. Environ. Microbiol. 40:201-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sommergruber, W., H. Ahorn, H. Klump, J. Seopelt, A. Zoephel, F. Fessl, E. Krystek, D. Blaas, E. Kuechler, H. D. Liebig, and T. Skern. 1994. 2A proteinase of coxsackie- and rhinovirus cleave peptides derived from eIF-4γ via a common recognition motif. Virology 198:741-745. [DOI] [PubMed] [Google Scholar]
- 44.Stellrecht, K., I. Harding, A. Woron, M. Lepow, and R. Venezia. 2002. The impact of an enteroviral RT-PCR assay on the diagnosis of aseptic meningitis and patient management. J. Clin. Virol. 25(Suppl.1):S19-S26. [DOI] [PubMed] [Google Scholar]
- 45.Strauss, J. 1990. Viral proteinases. Semin. Virol. 1:307-384. [Google Scholar]
- 46.Strauss, J., and E. Strauss. 2002. Viruses and human disease, p. 58-70. Academic Press, New York, N.Y.
- 47.U.S. Environmental Protection Agency. 1984. Manual of methods for virology. U.S. Environmental Protection Agency publication EPA 600-4-84-013. Government Printing Office, Cincinnati, Ohio.
- 48.U.S. Environmental Protection Agency. 1995. Virus monitoring protocol for the information collection requirements rules. U.S. Environmental Protection Agency publication EPA 814-B-95-002. Government Printing Office, Cincinnati, Ohio.
- 49.Walter, J., and D. Mitchell. 2003. Astrovirus infection in children. Curr. Opin. Infect. Dis. 16:247-253. [DOI] [PubMed] [Google Scholar]
- 50.Wang, Q. M., R. B. Johnson, L. N. Jungheim, J. D. Cohen, and E. C. Villareal. 1998. Dual inhibition of human rhinovirus 2A and 3C proteases by homophthalimides. Antimicrob. Agent. Chemother. 42:916-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilhelmi, I., E. Roman, and A. Sanchez-Fauquier. 2003. Viruses causing gastroenteritis. Clin. Microbiol. Infect. 9:247-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wimmer, E., C. Hellen, and X. Cao. 1993. Genetics of poliovirus. Annu. Rev. Genet. 27:353-436. [DOI] [PubMed] [Google Scholar]
- 53.Xia, Z., and Y. Liu. 2001. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscope. Biophys. J. 81:2395-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, J., Y. Ma, S. Taylor, and R. Tsien. 2001. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA 98:14997-15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.