

Abstract
Bacteria form biofilms by adhering to biotic or abiotic surfaces. This phenomenon causes several problems, including a reduction in the transport of mass and heat, an increase in resistance to antibiotics, and a shortening of the lifetimes of modules in bioindustrial fermentors. To overcome these difficulties, we created a biofilm production-deficient Escherichia coli strain, BD123, by deleting genes involved in curli biosynthesis and assembly, Δ(csgG-csgC); colanic acid biosynthesis and assembly, Δ(wcaL-wza); and type I pilus biosynthesis, Δ(fimB-fimH). E. coli BD123 remained mostly in the form of planktonic cells under the conditions tested and became more sensitive to the antibiotics streptomycin and rifampin than the wild-type E. coli MG1655: the growth of BD123 was inhibited by one-fourth of the concentrations needed to inhibit MG1655. In addition, the transformation efficiency of BD123 was about 20 times higher than that of MG1655, and the production and secretion of recombinant proteins were ∼16% and ∼25% greater, respectively, with BD123 than with MG1655. These results indicate that the newly created biofilm production-deficient strain of E. coli displays several key properties that substantially enhance its utility in the biotechnology arena.
In natural environments, most bacteria tend to be associated with solid surfaces instead of existing as free-swimming entities. The attached bacterial cells form a monolayer on a solid surface and then organize into a three-dimensional (3-D) biofilm (4). The biofilm protects the cells from being exposed to hostile environments, such as biocides, bacteriophages, and protozoa (3). Biofilm formation, however, is disadvantageous for industrial fermentation for several reasons. First, the production of cell surface components for biofilm formation may cost the bacteria energy that could be better used for driving processes that promote cell growth. The polymeric substances used in the building of 3-D biofilm structures are known to retard the diffusion of nutrients. The resulting nutrient-limited growth conditions cause the cells in a biofilm to exist in a slow-growing or starved state (5, 8, 24). Biofilms also result in increased resistance to antibiotics relative to freely growing bacterial cells. Because some antibiotics bind to exopolysaccharides on the surfaces of cells, bacterial populations produce a subset of cells known as “persisters,” which are hibernating cells that neither grow nor die in the presence of antibiotics (12). The phenotypic changes caused by growth on biofilm surfaces also protect cells from the effects of antibiotics (5). Finally, biological and electrochemical processes mediated by biofilm cells cause biocorrosion and biofouling of the inner surfaces of industrial machinery, which shorten the lifes of fermentor modules (23).
Two major approaches have been used for the inhibition of biofilm formation in bioindustry (23). One approach is the addition of biofilm-inhibiting agents to the growth media, and the other is the development of new materials to which bacteria cannot adhere. The limitations of these approaches are the production of environmental pollution and the high cost of materials. Therefore, new methods are needed for the elimination of biofilms from bacterial populations. One effective method for halting the formation of biofilms might be the development of a biofilm production-deficient bacterial strain by genetic manipulation.
In Escherichia coli, adhesion to inert surfaces and the development of multilayered cell clusters require the synthesis of curli, which allow cells to bind to various kinds of surfaces and to each other (18, 19). Colanic acid is also a major component of the biofilm architecture in E. coli, and this viscous capsular polysaccharide allows the formation of voluminous biofilms (6, 18). In addition to curli and colanic acid, type I pili are needed for the initial attachment of bacteria to inert surfaces and to other cells. Pili are proteinaceous appendages on the surfaces of bacteria, and these structures accomplish adhesion by overcoming electrostatic repulsive forces between substratum surfaces and the bacterial envelopes (17, 21). These observations suggest that, to make a strain of E. coli that is biofilm production deficient, these bacterial extracellular components must be eliminated.
In this study, E. coli K-12 strain MG1655 was genetically engineered to eliminate biofilm formation by the deletion of genes involved in the biosynthesis and assembly of curli and colanic acid and type I pilus biosynthesis. The resulting biofilm production-deficient strain, BD123, remained mostly planktonic under the conditions tested. Also, BD123 exhibited several advantageous properties that make it a prime host strain for use in bioindustry. These include an increased susceptibility to antibiotics, a higher transformation efficiency, and better production and secretion of recombinant proteins than MG1655.
MATERIALS AND METHODS
Bacterial strains, plasmids, enzymes, and chemicals.
The bacterial strains used in this work are listed in Table 1. Plasmids pKD3 and pKD46 were obtained from B. L. Wanner (7), pST76-K and pST76-ASceP from G. Posfai (16), pKO3 from G. M. Church (14), and pACYC-ABC and pHOPE from J. S. Rhee (1). pCSI was constructed by cloning the cat gene of pKD3 and the sacB gene of pKO3 into the KpnI and BamHI sites of pST76-K, respectively. pEndo was constructed by cloning xynA of pJHKJ4 (11) into the EcoRI and HindIII sites of pKK223-3 (Amersham Pharmacia, Piscataway, NJ). All enzymes were purchased from New England Biolabs (Beverly, MA), except LA Taq polymerase (Takara, Japan), and all antibiotics and chemicals were from Sigma-Aldrich (St. Louis, MO). Ampicillin and chloramphenicol were used at concentrations of 50 and 17 μg/ml, respectively.
TABLE 1.
Strains and primer sequences used in this study
| Strain or primer | Description or sequence | Source or reference |
|---|---|---|
| E. coli K-12 strains | ||
| MG1655 | λ− F−ilvG rfb-50 rph-1 | 2 |
| BD1 | MG1655 Δ(csgG-csgC)a | This study |
| BD2 | MG1655 Δ(wacL-wza)b | This study |
| BD3 | MG1655 Δ(fimB-fiimH)c | This study |
| BD12 | MG1655 Δ(csgG-csgC) Δ(wacL-wza) | This study |
| BD13 | MG1655 Δ(csgG-csgC) Δ(fimB-fimH) | This study |
| BD23 | MG1655 Δ(wacL-wza) Δ(fimB-fimH) | This study |
| BD123 | MG1655 Δ(csgG-csgC) Δ(wacL-wza) Δ(fimB-fimH) | This study |
| BD123ccp | BD123 containing pBACccp; Cmr | This study |
| Primersd | ||
| Cur-r | CTTCTCTCTATTGCAATCAC | |
| Cur-a1 | TGGGGATGGGTGGCTGTTTCC | |
| Cur-a2 | CGATATTTCAACAAATTAAGCAGATAAAAAAGCCGGAATTATC | |
| Cur-b1 | GATCAGCTAGCCCATGGGTATGGGACGGTTAAACAGTTCGGTGG | |
| Cur-b2 | ACTTTTCTGAAGAGGGCGGCCA | |
| Cur-c1 | CTTAATTTGTTGAAATATCG | |
| Cur-c2 | GCCGATCAACGTCTCATTTTCGCCTTAAGTGTACAGCAGGCTTTTTGG | |
| Cur-f | CGCGTTTTCATAACGCCTCC | |
| Cur-cr | GAAGATCTGCTAGCAGTAAAATGCCGGATGATAATTCCG | |
| Cur-cf | CGGGATCCATGATTAGTCATCCTTGAGGGTTGTG | |
| Col-r | GCTTCCCGCAAGGCCTATGC | |
| Col-a1 | GTTATCGATGATCAGGTTGCGC | |
| Col-a2 | CTGCCAAAGTGATAAATAAACACGCCAGCTTGCTGCAGGCTTTATAG | |
| Col-b1 | GATCAGCTAGCCCATGGGTATGGGCGGCGATATTAACGTCCAC | |
| Col-b2 | ATGATGAAATCCAAAATGAAATTG | |
| Col-c1 | TGTTTATTTATCACTTTGGCAG | |
| Col-c2 | GCCGATCAACGTCTCATTTTCGCGTAATAACCTCACATTATCCCTG | |
| Col-f | ACCCGTGCGCGGTCACGCTGC | |
| Col-cr | GACGCTAGCCGAGCTTGCCGTCAGGAACGTGCGTCGG | |
| Col-cf | CTAGCTAGCTTACCGAATTGTTATCTTGCCTGCTATTCC | |
| Pil-r | GGAAGGTTTAAGTCGTAGTGACC | |
| Pil-a1 | GAAAAACGATGAAGAATAAGGCTG | |
| Pil-a2 | ACCAGCATTAGCAATGTCCTGTGACGATGCCCAAGATAATCCTGG | |
| Pil-b1 | GATCAGCTAGCCCATGGGTATGGGGCGGGGTGGCGATTAAAGC | |
| Pil-b2 | TTATTGATAAACAAAAGTCACGCCA | |
| Pil-c1 | TCACAGGACATTGCTAATGCTGGT | |
| Pil-c2 | GCCGATCAACGTCTCATTTTCGCCGATGACCGCCGCCGGGATTATC | |
| Pil-f | GGCAATCTTGAGCGCCCAACG | |
| Pil-cr | GAAGATCTGGTCACTTCTGAAGTCGATCTGGAGAGG | |
| Pil-cf | GAAGATCTGAACCAGGGTAGTCCGGCAGAGTAACG |
Deletion size, 4.4 kb.
Deletion size, 21.3 kb.
Deletion size, 8.3 kb.
Cur-, Col-, and Pil- indicate gene clusters involved in curli, colanic acid, and type I pilus synthesis and assembly, respectively. The primer target sites are indicated by lowercase letters (r, a1, a2, b1, b2, c1, c2, and f) in Fig. 1A.
Construction of a biofilm production-deficient E. coli strain.
To construct the biofilm production-deficient E. coli BD123 strain, the genes related to biofilm formation, csgG-csgC, wcaL-wza, and fimB-fimH, were sequentially deleted from the MG1655 genome by λ-Red-mediated markerless deletion, as illustrated in Fig. 1A.
FIG. 1.

Construction of deletion mutants. (A) The target regions of the MG1655 chromosome were replaced with a DNA cassette that contained a positive selectable marker (cat), a negative selectable marker (sacB), an I-SceI cleavage site, and homology arms generated by recombinant PCR. The selection markers in the recombinant genome were removed by intramolecular homologous recombination mediated by I-SceI cleavage, and markerless deletion mutants were selected on LB plates with 5% sucrose. A, B, and C represent chosen DNA segments (homology arms) for the deletion of a target region (between A and C). Primers for recombinant PCR are labeled by lowercase letters (a1, a2, b1, b2, c1, and c2) and arrows. I indicates an I-SceI recognition site. (B) Multiplex PCRs were conducted using locus-specific primers, r and f, that flanked the endpoints of each target region (Table 1). The successful construction of BD1, BD2, and BD3 produced PCR products of 1.0, 1.5, and 2.1 kb (b), respectively, compared to the PCR products of 5.5, 22.8, and 9.8 kb (a) of the wild-type MG1655.
To delete a chromosomal target region (between boxes A and C [Fig. 1]), a linear DNA cassette that carried three homology arms (boxes A, B, and C), a positive selectable marker (cat), a negative selectable marker (sacB), and an I-SceI meganuclease recognition site was generated by recombinant PCR using plasmid pCSI as a template. The cassette was electrotransformed into MG1655 harboring pKD46, which was needed to replace the chromosomal target region by λ-Red recombination. The deletion recombinants were selected on LB plates containing chloramphenicol, and the helper plasmid pKD46 was cured by growing the recombinants at 42°C.
The remaining selectable markers were removed from the resulting recombinant strain by intramolecular homologous recombination mediated by the I-SceI meganuclease expressed from pST76-ASceP (16). The markerless deletion strains were selected on LB plates with 5% sucrose, generating E. coli BD1, BD2, and BD3 (Fig. 1A).
BD12, BD13, BD23, and BD123 were then constructed by a series of sequential and combinatorial deletions of genes involved in biofilm formation, as described above. At each step, the deletion of each target region was verified by multiplex PCR using pairs of primers that flanked the endpoints of each target region.
For a complementation assay, the genes related to biofilm formation, csgG-csgC, wcaL-wza, and fimB-fimH, were amplified from the MG1655 genomic DNA using primers Cur-cr/Cur-cf, Col-cr/Col-cf, and Pil-cr/Pil-cf, respectively (Table 1), and the amplified biofilm-related genes were cloned into pCC1BAC (Epicenter, Wisconsin), producing pBACccp.
Quantification of biofilm formation.
Biofilm cells were quantified as described by Vianney et al. (22). For biofilm cell culture, bacterial cells were grown at 30°C in a 24-well polystyrene plate with M9 medium containing 0.2% glucose (2 ml per well). After 24 h of growth, the cell supernatant was removed from the plate and was referred to as planktonic cells. The surface-attached cells of the wells were recovered in M9 glucose medium (1 ml) by scraping and pipetting up and down. To determine the adherence percentage for each bacterial strain, the biomasses of the planktonic and surface-attached cells were estimated by measuring the optical density at 600 nm. A minimum of three independent assays were performed, and mean optical densities were calculated.
Transmission electron microscopy.
Biofilm formation was observed visually by transmission electron microscopy (TEM) as described by Prigent-Combaret et al. (18). Cells were grown at 30°C in 24-well polystyrene plates with M9 glucose medium (2 ml per well) for 48 h. For negative staining, the cells were carefully recovered and allowed to adhere to carbon-coated 300 mesh grids. After being stained with a 2% aqueous solution of uranyl acetate, the grids were examined with a Carl Zeiss EM 912 Omega energy-filtering TEM.
Assays to assess antibiotic susceptibility and transformation efficiency.
Either MG1655 or BD123 was grown at 30°C in two 96-well plates with M9 glucose medium (0.2 ml per well). After 48 h of precultivation for biofilm formation, the initial supernatant (0.2 ml) from one of two 96-well plates was transferred to a new 96-well plate and used as planktonic cells. The cells in the other 96-well plate were used as total cells. In order to avoid the inoculum effect, the cell masses for both samples were normalized by measuring the optical density at 600 nm. To each well of the plates with either planktonic or total cells, serial dilutions of streptomycin or rifampin were added. After an additional 10 h of cultivation, the cells were recovered by scraping and pipetting up and down. The total number of viable cells in each well was estimated by the standard plate-counting method (13). These cells were diluted 105- to 107-fold with M9 glucose medium and plated on LB agar plates. Colonies were counted after incubation overnight at 37°C. Cell survival was expressed as numbers of CFU per well for total cells and numbers of CFU per ml for planktonic cells. For all sample types, CFU measurements were taken in triplicate for each of three experiments, and mean CFU values were calculated.
Transformation efficiencies of MG1655 and BD123 were measured with pUC19 by the standard CaCl2 heat shock transformation method (20). Three independent assays were performed, and mean values were calculated.
Endoxylanase activity assay.
Endoxylanase activity was determined by measuring the increase in the number of reducing ends released from hydrolysis of xylan as a substrate (11). The cells that harbored pEndo containing a Bacillus subtilis xynA gene that did not have the signal sequence necessary for protein secretion were grown at 30°C in a 24-well plate with LB medium (2 ml per well). Expression of the endoxylanase gene in the transformants was induced by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After 24 h of cultivation, the cells were harvested by centrifugation at 10,000 × g for 10 min at 4°C. The resulting pellets were resuspended in 1 ml of Tris-EDTA buffer (pH 8.0), and the solution was sonicated thoroughly to disrupt the cells. The cytoplasmic fraction of the transformants (0.5 ml) and 1% (wt/vol) xylan in 50 mM potassium phosphate buffer (pH 6.5) (0.5 ml) were mixed together and incubated at 60°C for 10 min, after which the DNS reagent (3 ml; 3′5′-dinitrosalicylic acid, 1.87 g; NaOH, 3.5 g; Rochelle salt, 54 g; and phenol, 1.6 ml in 250 ml of distilled water) was added. After the mixture was boiled for 10 min, endoxylanase activity in the samples was estimated by measuring the absorbance at 550 nm. One unit of enzyme activity was defined as the amount of enzyme that liberated 1 μmol of reducing sugar per min. The experiments were performed three times, and mean values were calculated.
Lipase activity assay.
The plasmid pHOPE, which contained a gene from Pseudomonas fluorescens SIK W1 that encoded a lipase, or pACYC-ABC, which contained a cassette encoding a lipase secretion system (1), was electroporated into MG1655 and BD123 cells. The cells that carried the plasmids were selected on LB agar plates supplemented with chloramphenicol or ampicillin. Colonies were selected, and the cells were grown at 25°C in a 24-well plate with LB medium (2 ml per well). Expression and secretion of the lipase in the transformants were induced by the addition of 1 mM IPTG and 15 μg/ml chlortetracycline, respectively. After 24 h of cultivation, the culture supernatant was collected by centrifugation at 10,000 × g for 10 min at 4°C. The resulting pellets were resuspended in 0.5 ml of Tris-EDTA buffer (pH 8.0), and the suspension was sonicated thoroughly to disrupt the cells.
Lipase activity was assayed quantitatively by spectrophotometry with p-nitrophenyl palmitate (1). The p-nitrophenyl palmitate was dissolved in acetonitrile to a concentration of 10 mM. Ethanol and 50 mM Tris · HCl buffer (pH 8.5) were subsequently added to yield a solution with a final composition of 1:4:95 of acetonitrile/ethanol/buffer. The cytoplasmic fraction (0.2 ml), which contained the intracellular lipase, or the culture supernatant (0.2 ml), which contained the secreted lipase, was added to 0.8 ml of the prepared solution that contained p-nitrophenyl palmitate, and the mixture was incubated at 45°C for 10 min. Lipase activity in the samples was detected by measuring the absorbance at 405 nm. One unit of enzyme activity was defined as the amount of enzyme necessary to release 1 μmol of fatty acids produced per min. Three independent assays were performed, and mean values were calculated.
RESULTS
Construction of a biofilm production-deficient E. coli strain by the deletion of genes that specify curli, colanic acid, and type I pili.
To construct a biofilm production-deficient E. coli strain, genes involved in biofilm formation (csgG-csgC, wcaL-wza, and fimB-fimH) were sequentially and combinatorially deleted from the MG1655 genome. First, genes involved in curli biosynthesis and assembly (csgG-csgC), colanic acid biosynthesis and assembly (wcaL-wza), or type I pili biosynthesis (fimB-fimH) were deleted, producing strains BD1, BD2, and BD3, respectively (Fig. 1). Strains BD12, BD13, and BD23 were then constructed by the combinatorial deletion of one of the two remaining sets of genes involved in biofilm production. Finally, a strain, BD123, which lacked the ability to form a biofilm, was successfully constructed by the complete deletion of csgG-csgC, wcaL-wza, and fimB-fimH (Table 1 and Fig. 1). After each round of gene removal, the deletion of the target regions was confirmed by PCR using a pair of primers, r and f, that flanked the endpoints of each segment to be deleted (Fig. 1). The successful deletion of csgG-csgC, wcaL-wza, and fimB-fimH produced PCR products of 1.0, 1.5, and 2.1 kb, respectively, as confirmed on a 1.0% agarose gel (Fig. 1B). Deletion of the biofilm-related genes did not have any negative effects on the growth of the deletion strains under the conditions tested (data not shown).
The ability of MG1655 and its deletion mutants to carry out biofilm formation was assessed by comparing the biomasses of surface-attached and planktonic cells, as estimated by measuring the optical density at 600 nm. The adherence abilities of cells were determined by calculating an adherence percentage, which was expressed as the ratio of the biomass of attached cells to the biomass of total cells times 100. The adherence percentages of the deletion mutant strains BD1, BD2, and BD3 were about 14.9 ± 1.6, 17.3 ± 2.8, and 16.2 ± 3.6, respectively, whereas that of MG1655 was 30.0 ± 1.1 (Fig. 2). Among the gene clusters involved in biofilm formation, the deletion of curli-related genes (csgG-csgC) was the most effective treatment in terms of its ability to decrease bacterial adherence (∼50% reduction), whereas the deletion of the colanic acid-related genes (wcaL-wza) was the least effective (∼42% reduction).
FIG. 2.

Assessment of biofilm formation of E. coli mutants. Bacteria were grown for 24 h at 30°C in 24-well polystyrene plates with M9 medium containing 0.2% glucose. The adherence percentage is expressed as the ratio of the biomass of attached cells to the biomass of total cells times 100 (refer to Materials and Methods): (adherence percentage) = (biomass of attached cells)/(biomass of total cells) × 100. The data shown are the means and standard deviations for three independent experiments.
The introduction of the second deletion into the above-mentioned individual-deletion strains decreased the adherence percentage a bit more, but to a lesser extent than did the first deletion (Fig. 2). There was, however, a substantial decrease in the adherence percentage of the BD123 strain, which contained deletions in all three regions [Δ(csgG-csgC) Δ(wcaL-wza) Δ(fimB-fimH)] on the same chromosome. The deficiency in biofilm formation of BD123 was further examined by TEM, which revealed that no extracellular components or biofilm matrices were present, even though these structures were clearly visible with MG1655 (Fig. 3). The BD123 cells remained planktonic and displayed clear, smooth, biofilm-free surfaces.
FIG. 3.

TEM analysis of biofilm formation. Negative staining of biofilms was performed on wild-type MG1655 (A), BD123 (B), and BD123(pBACccp) (C) using TEM. Fibrillar structures are not observed in the biofilm production-deficient BD123 strain.
To further confirm that the biofilm deficiency in BD123 was the result of the deletion of genes related to biofilm formation, a complementation experiment was performed. The plasmid pBACccp, which carried the curli-, colanic acid-, and type I pilus-related gene clusters (csgG-csgC, wcaL-wza, and fimB-fimH), was constructed. When pBACccp was transformed into BD123, the biofilm production-deficient BD123 strain regained the ability to form biofilms and adherence ability (Fig. 2 and 3).
Antibiotic susceptibility and DNA transformation efficiency of the biofilm production-deficient BD123 strain.
The effects of biofilms on antibiotic susceptibility were examined with both MG1655 and BD123. A transcription inhibitor, rifampin, and a translation inhibitor, streptomycin, were used for the antibiotic susceptibility experiments. The antibiotic susceptibilities of BD123 were 15- and 20-fold greater for streptomycin and rifampin, respectively, than those of MG1655. Specifically, the number of MG1655 cells that survived at 64 μg/ml of streptomycin or 256 μg/ml of rifampin was similar to the number of BD123 cells that survived at 16 μg/ml of streptomycin or 64 μg/ml of rifampin (Fig. 4).
FIG. 4.

Antibiotic susceptibilities of the biofilm production-deficient E. coli BD123. Antibiotic sensitivities to streptomycin (A) and rifampin (B) were examined. The cells were grown for 48 h to induce biofilm formation before being treated with serial dilutions of the antibiotics (refer to Materials and Methods). Cell survival was expressed as CFU per well for total cells and CFU per ml for planktonic cells.
In addition to antibiotic susceptibility, the transformation efficiencies of both BD123 and MG1655 were assessed by using pUC19. BD123 exhibited a transformation efficiency that was ∼20 times higher than that of MG1655. Specifically, an average of 7.9 × 106 CFU/μg of pUC19 was obtained with BD123 compared to an average of 3.4 × 105 CFU/μg of pUC19 with MG1655 (Table 2).
TABLE 2.
Transformation efficiencies of the biofilm-deficient E. coli BD123a
| Amt of pUC19 transformed (ng) | Transformation efficiency (CFU/μg)
|
|
|---|---|---|
| MG1655 | BD123 | |
| 1 | 3.3 (±0.7) × 105 | 7.4 (±1.0) × 106 |
| 10 | 3.4 (±1.2) × 105 | 8.1 (±0.8) × 106 |
| 100 | 3.6 (±0.8) × 105 | 8.1 (±1.4) × 106 |
The transformation efficiencies of the biofilm-deficient BD123 and MG1655 were compared by the standard CaCl2 heat shock transformation method.
Protein expression and secretion properties of the biofilm production-deficient BD123.
The effects of biofilm formation on the production of recombinant proteins in both BD123 and MG1655 were studied. The genes tested were those that encoded an endoxylanase from B. subtilis and a lipase from P. fluorescens SIK W1. For the estimation of the amount of each protein expressed in the cytoplasmic space, lysates of the cultured BD123 and MG1655 cells that harbored pEndo or pHOPE were used to measure the endoxylanase and lipase activities, respectively. The amounts of the two enzymes present in the cell cytoplasm were ∼16% higher in the BD123 cells than in the MG1655 cells (Fig. 5). Specifically, the amounts of endoxylanase activity in the cytoplasm of the BD123 and MG1655 cells were 7.3 ± 0.5 U/ml and 6.4 ± 0.4 U/ml, respectively. Furthermore, the BD123 cells contained a lipase activity of 5.2 ± 0.1 U/ml compared to the lipase activity of 4.5 ± 0.1 U/ml in the MG1655 cells (Fig. 5).
FIG. 5.
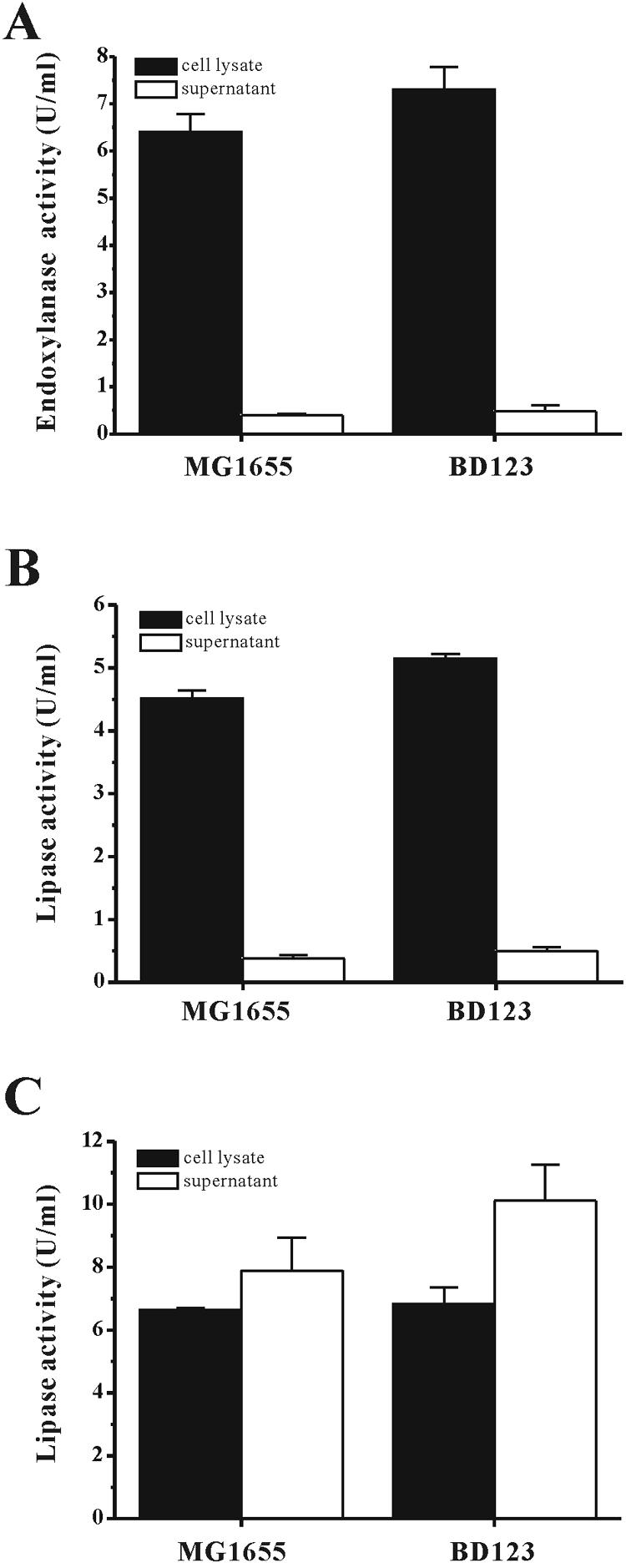
Production of recombinant proteins in the biofilm production-deficient strain E. coli BD123. MG1655 and BD123 carrying pEndo (A), pHOPE (B), and both pHOPE and pABC-ACYC (C) were grown for 24 h in 24-well plates. The amounts of lipase and endoxylanase activities in the cytoplasmic space and the supernatant were determined by spectrophotometry (refer to Materials and Methods).
For estimation of the yields of lipase secreted by the BD123 and MG1655 cells harboring pHOPE and pABC-ACYC, a spectrophotometric method that measured the p-nitrophenyl palmitate hydrolysis activity of the lipase was used. The amount of lipase secreted into the medium by the BD123 cells was ∼25% higher than that secreted by the MG1655 cells. For both BD123 and MG1655 cells, secretion of the lipase into the culture medium peaked at 24 h after induction with IPTG and chlortetracycline (data not shown), at which time the extracellular lipase activity in the BD123 medium was 10.0 ± 1.1 U/ml compared to 7.9 ± 1.0 U/ml for the MG1655 medium. The amount of lipase activity that remained inside the cells was similar for both bacterial strains under the growth conditions used for this series of experiments (Fig. 5).
DISCUSSION
This study focused on the construction and characterization of a biofilm production-deficient strain of E. coli, BD123, that was constructed by the deletion of the curli-, colanic acid-, and type I pilus-related gene clusters [Δ(csgG-csgC) Δ(wcaL-wza) Δ(fimB-fimH)] from the genome of E. coli MG1655.
Although deletion of either the curli-, colanic acid-, or type I pilus-related genes or the combined deletion of two of these three gene clusters was effective in decreasing the adherence ability of E. coli MG655 to some extent (42 to 60%), none of these manipulations completely eliminated the ability of MG655 to form biofilms. However, when all three gene clusters were deleted from the MG1655 genome, biofilm formation was almost completely abolished, and planktonic cells were produced. The conclusion that the deficiency in biofilm formation of BD123 was due to the deletion of the genes csgG-csgC, wcaL-wza, and fimB-fimH was further verified by a complementation experiment with these genes. When these biofilm-related genes were reintroduced into the biofilm production-deficient BD123 cells using a low-copy-number plasmid, pBACccp, the transformed BD123 cells regained the ability to form biofilms and adherence ability (Fig. 2 and 3). It has been reported that curli are involved in cell-to-cell and cell-to-surface attachment, that colanic acid contributes to the 3-D structure of biofilms, and that type I pili are required for the initial attachment of cells to surfaces (4, 6, 17, 19). It appears that together, these three components orchestrate the formation of multilayered biofilms. Therefore, if one wishes to block the ability of E. coli to generate biofilms, all three gene clusters must be deleted from the bacteria.
When inspected with TEM, BD123 cells were shown to exhibit smoother surfaces, probably because there were no curli, colanic acid, and type I pili on the surfaces of the cells, and the cells could not form 3-D biofilm structures (Fig. 3). Even though BD123 was not capable of forming substantial biofilms, some fibers were occasionally apparent on the cell surfaces. Whether these fibrillar structures could contribute to biofilm formation is unknown. Elimination of the biofilm-related genes did not affect the growth rate of the cells (data not shown).
The elimination of biofilm formation resulted in increased antibiotic susceptibility. Inhibition of the growth of BD123 required only one-fourth the amount of antibiotics needed to inhibit the growth of MG1655 to the same degree. However, the growth of planktonic MG1655 cells was inhibited at concentrations of antibiotics that were similar to those needed for the inhibition of BD123 growth, suggesting that antibiotic resistance is higher in MG1655 populations due to the presence of bacterial cells expressing the biofilm phenotype. Antibiotic pressures commonly used in laboratories and bioindustry increase the development of antibiotic-resistant microorganisms. Therefore, the availability of a commonly used bacterial host that displays a substantial increase in antibiotic sensitivity would obviate the overuse of antibiotics and thus be of great value to biologists involved in basic and applied research.
The transformation efficiency with plasmid DNA was increased more than 20-fold in BD123 compared to that in MG1655. It is possible that the elimination of surface appendages augments a cell's ability to take up DNA. Several procedures developed thus far have been shown to increase the transformation efficiency of E. coli 10- to 20-fold (10, 15). These manipulations involve the mutation of genes whose products participate in the synthesis of cell wall components or in the degradation of DNA. In addition, treatment with various chemicals has also been shown to enhance transformation efficiency (10, 15). It may be beneficial to use these approaches with BD123 to further magnify transformation efficiency.
The elimination of biofilm formation in BD123 also led to an ∼16% increase in the expression of recombinant proteins relative to MG1655. Even though the effect in terms of overexpression is marginal for the biofilm production-deficient strain BD123, it is still valuable from the practical and economical point of view for biotechnology companies because it gives an additional advantage to the industrial strains that have been improved through other genetic manipulations. It is likely that the production of numerous extracellular proteins and the assembly of multicomponent cell surface structures are expensive for bacterial cells in terms of energy use. If this is the case, then one would predict that the removal of unnecessary extracellular components from bacterial cells would spare energy that can then be used to produce desired materials, such as recombinant proteins. In addition, colanic acid matrices retard solute diffusion and entrap proteins (4, 5). The entrapment of proteins in the matrices could be minimized by using our biofilm production-deficient E. coli strain, as shown by an ∼25% increase in lipase secretion into the medium by BD123 relative to the amount secreted by MG1655.
Although biofilm formation was almost completely inhibited in BD123, the deletion of its remaining surface determinants might be needed if one wished to completely abolish the biofilm formation potential of E. coli. This might be accomplished by the elimination of other cell surface structures and components, such as flagella, cellulose, and polygalacturonic acid polysaccharide (the pgaABCD locus), all of which are involved in one or more of the developmental stages of biofilm formation in E. coli (9).
Acknowledgments
This work was supported by grants from the 21C Frontier Program of Microbial Genomics and Applications (MG05-0204-1-0), the National Research Laboratory Program (M1-0204-00-0102), and the Molecular and Cellular BioDiscovery Research Program (M1-0106-00-0200) of the Ministry of Science and Technology of Korea and by grants from the Basic Research Program of the Korea Science and Engineering Foundation (R01-2005-000-11010-0) and the Korea Research Foundation (KRF-2004-042-D00072).
REFERENCES
- 1.Ahn, J. H., J. G. Pan, and J. S. Rhee. 1999. Identification of the tliDEF ABC transporter specific for lipase in Pseudomonas fluorescens SIK W1. J. Bacteriol. 181:1847-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collando-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 3.Brown, M. R. W., and P. Gilbert. 1993. Sensitivity of biofilms to antimicrobial agents. J. Appl. Bacteriol. 74:87S-97S. [DOI] [PubMed] [Google Scholar]
- 4.Costerton, J. W., Z. Lewandowski, D. E. Caldwell, D. R. Korber, and H. M. Lappin-Scott. 1995. Microbial biofilms. Annu. Rev. Microbiol. 49:711-745. [DOI] [PubMed] [Google Scholar]
- 5.Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318-1322. [DOI] [PubMed] [Google Scholar]
- 6.Danese, P. N., L. A. Pratt, and R. Kolter. 2000. Exopolysaccharide production is required for development of Escherichia coli K-12 biofilm architecture. J. Bacteriol. 182:3593-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert, P., P. J. Collier, and M. R. Brown. 1990. Influence of growth rate on sensitivity to antimicrobial agents: biofilms, cell cycle, dormancy, and stringent response. Antimicrob. Agents Chemother. 34:1865-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houdt, R. V., and C. W. Michiels. 2005. Role of bacterial cell surface structure in Escherichia coli biofilm formation. Res. Microbiol. 156:626-633. [DOI] [PubMed] [Google Scholar]
- 10.Inoue, H., H. Nojima, and H. Okayama. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23-28. [DOI] [PubMed] [Google Scholar]
- 11.Jeong, K. J., I. Y. Park, M. S. Kim, and S. C. Kim. 1998. High-level expression of an endoxylanase gene from Bacillus sp. in Bacillus subtilis DB104 for the production of xylobiose from xylan. Appl. Microbiol. Biotechnol. 50:113-118. [DOI] [PubMed] [Google Scholar]
- 12.Keren, I., N. Kaldalu, A. Spoering, Y. Wang, and L. Kim. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230:13-18. [DOI] [PubMed] [Google Scholar]
- 13.Knowles, D. J., and B. J. Weston. 1984. A simple rapid technique to measure neutrophile or serum bactericidal activity. J. Immunol. Methods 72:411-420. [DOI] [PubMed] [Google Scholar]
- 14.Link, A. J., D. Phillips, and G. M. Church. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 179:6228-6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molchanova, E. S., N. A. Likhacheva, A. A. Kim, and B. N. Il'iashenko. 1983. Escherichia coli K-12 mutants with an increased efficiency of plasmid transformation. Genetika 19:375-380. [PubMed] [Google Scholar]
- 16.Posfai, G., V. Kolisnychenko, Z. Bereczki, and F. R. Blattner. 1999. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 27:4409-4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pratt, L. A., and R. Kolter. 1998. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotoxis and type I pili. Mol. Microbiol. 30:285-293. [DOI] [PubMed] [Google Scholar]
- 18.Prigent-Combaret, C., G. Prensier, T. T. L. Thi, O. Vidal, P. Lejeune, and C. Dorel. 2000. Developmental pathway for biofilm formation in curli-producing Escherichia coli strains: role of flagella, curli, and colonic acid. Environ. Microbiol. 2:450-464. [DOI] [PubMed] [Google Scholar]
- 19.Römling, U., Z. Bian, M. Hammar, W. D. Sierralta, and S. Normark. 1998. Curli fibers are highly conserved between Salmonella typhimurium and Escherichia coli with respect to operon structure and regulation. J. Bacteriol. 180:722-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, New York, N.Y.
- 21.van Loosdrecht, M. C., J. Lyklema, W. Norde, and A. J. Zehnder. 1990. Influence of interfaces on microbial activity. Microbiol. Rev. 54:75-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vianney, A., G. Jubelin, S. Renault, C. Dorel, P. Lejeune, and J. C. Lazzaroni. 2005. Escherichia coli tol and rcs genes participate in the complex network affecting curli synthesis. Microbiology 151:2487-2497. [DOI] [PubMed] [Google Scholar]
- 23.Videla, H. A. 1994. Biocorrosion of nonferrous metal surfaces, p. 231-241. In G. G. Geesey, Z. Lewandowski, and H. Flemming (ed.), Biofouling and biocorrosion in industrial water systems. CRC Press, Boca Raton, Fla.
- 24.Williams, I., W. A. Venebles, D. Lloyd, F. Paul, and I. Critchley. 1997. The effects of adherence to silicone surfaces on antibiotic sensitivity in Staphyloccus aureus. Microbiology 143:2407-2413. [DOI] [PubMed] [Google Scholar]