

Abstract
Cellular glutathione peroxidase is a key intracellular antioxidant enzyme that contains a selenocysteine residue at its active site. Selenium, a selenocysteine-incorporation sequence in the 3′- untranslated region of the glutathione peroxidase mRNA, and other translational cofactors are necessary for “read-through” of a UGA-stop codon that specifies selenocysteine incorporation. Aminoglycoside antibiotics facilitate read-through of premature stop codons in prokayotes and eukaryotes. We studied the effects of G418, an aminoglycoside, on cellular glutathione peroxidase expression and function in mammalian cells. Insertion of a selenocysteine-incorporation element along with a UGA-codon into a reporter construct allows for read-through only in the presence of selenium. G418 increased read-through in selenium-replete cells as well as in the absence of selenium. G418 treatment increased immunodetectable endogenous or recombinant glutathione peroxidase, but reduced the specific activity of the enzyme. Tandem mass spectrometry experiments indicated that G418 caused a substitution of L-arginine for selenocysteine. These data show that G418 can affect the biosynthesis of this key antioxidant enzyme by promoting substitution at the UGA-codon.
Cellular glutathione peroxidase (GPx-1) is one of several mammalian glutathione peroxidases that reduce hydrogen and lipid peroxides using glutathione (GSH) as a reductant (1). GPx-1 is ubiquitously expressed and is localized mostly to the cytosol. Each monomer of the tetrameric enzyme contains a selenocysteine (SEC) residue at the active site of the protein (2,3). This selenium-containing amino acid is incorporated during translation through the recognition of a UGA-codon as a site for SEC-incorporation rather than as a site for termination of translation.
Unique mechanisms are involved in the biosynthesis of SEC as well as in the insertion of this selenium-containing amino acid into the nascent polypeptide chain [reviewed in (4,5)]. A special tRNASEC that can recognizes the UGA codon is first charged with a serine that is enzymatically converted to a SEC (6). The specificity of SEC incorporation at a UGA codon in selenoprotein transcripts, rather than at UGA-stop codons, is ensured by the presence of additional cis-sequences within the transcripts (7–9). Most widely studied is the SEC insertion sequence, or SECIS element, which forms a stem-loop structure in the 3′ UTR of the selenoprotein transcripts. Prior studies have shown that insertion of a UGA within the protein coding region of heterologous transcripts can be properly decoded as a selenocysteine codon provided that the SECIS-element and surrounding sequences are present in the 3′ UTR of the transcript (10,11). Recognition of the SEC codon also requires the presence of unique translational cofactors (12), such as the efSEC, and specific RNA binding proteins, such as SBP2 (12,13), which bind to the SECIS element and promote SEC insertion.
Overall, SEC incorporation appears to be an inefficient process. It can be affected by selenium concentration, the sequence context of the UGA-codon, and the distance between the UGA and the SECIS element. (9,14–18). Substitution of a SEC codon with other codons has been shown to increase protein production but drastically reduce the activities of selenoenzymes (19–22).
Aminoglycoside antibiotics (AMG) are known for their ability to disrupt translational fidelity in bacterial and mammalian cell systems (23–26). Recently, these properties have been exploited to overcome nonsense mutations in mammalian proteins associated with human disease (27–31). It has long been known that AMG antibiotics can also promote oxidative stress in susceptible cells (32–34). Owing to the specialized mechanism of translation required for recoding a UGA as a site for selenocysteine incorporation, we tested whether G418, an AMG, could alter SEC incorporation, protein expression, and enzyme activity of the antioxidant enzyme GPx-1 in mammalian cells.
METHODS
Cell lines
COS7 cells were passaged in Dulbecco’s modified eagle medium with high glucose (DMEM) (Gibco BRL) and 10% fetal calf serum (FCS). Bovine aortic endothelial cells (BAECs) were obtained from Clonetics and passaged in Dulbecco’s modified eagle medium with high glucose (DMEM) (Gibco BRL) and 10% fetal calf serum (FCS).
Vectors for read-through translation
The pGL2 UGA vector was provided by Donna Driscoll (11). Briefly, this vector contains a TGA codon substituted for the glycine 38 codon of the luciferase protein and a PvuII site added to the 3′ untranslated region (UTR) of the luciferase gene; an SV40 promoter and enhancer drives transcription in this vector. To study the influence of the human GPx-1 SECIS element on SEC-dependent translation, the GPx-1 SECIS element was isolated from a human GPx-1 cDNA construct (35) and constructs with the SECIS element in the sense (hSECIS/S) and antisense (hSECIS/AS) orientation were made as described (36). A GPx1 promoter fragment was cloned in place of the SV40 promoter. These constructs were denoted GPx1/UGA (construct with no SECIS element), GPx1/hSECIS/S, and GPx1/hSECIS/AS.
Transfections and read-through assays
COS7 cells were seeded at 50,000 cells/well in 12-well tissue culture plates in DMEM supplemented with 0.1% fetal calf serum (FCS), in the presence or absence of 10 ng/ml sodium selenite. After 5 d of growth, each well was transfected with 2 μg of one of the luciferase constructs, 20 ng CMV renilla construct, and 5 μl Superfect (Qiagen, Valencia, CA) for 2 h as described (36). All luciferase activities were corrected for renilla luciferase activities before any other comparisons were made.
GPx-1 Enzyme Assay
Cell lysates were prepared by sonication in 50 mM Tris, pH 7.5, 5 mM EDTA, and 1 mM DTT. Activity was determined by an indirect assay that links GPx1-mediated oxidation of glutathione with the recycled reduction of GSSG to GSH by glutathione reductase using NADPH as a reductant according to the commercially available Calbiochem assay kit.
Western Blot
Lysates were prepared as for the GPx1 assay. Samples (50–100 μg) were separated by electrophoresis in 12% polyacrylamide SDS gels. After transfer to nitrocellulose membranes (Hybond, Amersham, Piscataway, NJ.), GPx-1 or actin was detected using a mouse anti-human GPx-1 monoclonal antibody (MBL, Woburn, MA) or a polyclonal rabbit anti-β-actin antibody (Sigma, St. Louis, MO), respectively, followed by HRP-conjugated secondary antibodies (Sigma, St. Louis, MO., or Cell Signaling, Beverly, MA) and enhanced chemiluminescence (ECL, Amersham, Piscataway, NJ.). To detect recombinant His-tagged GPx-1, anti-HisG antibody (Invitrogen, Carlsbad, CA) was also used as a primary antibody.
Recombinant His-tagged Gpx-1
Human GPx-1 cDNA was amplified by PCR using upstream (GGTAAGGATCCAATGTGTGCTGCTCGGCT A) and downstream (AGGCTCGAGCGATAAGCTTTGCTGACACC) primers to add BamHI and XhoI cloning sites that were used to ligate the cDNA in frame with the protein coding cassette of the pcDNA/HisB plasmid (Invitrogen, Carlsbad, CA). A total of 36 amino acids, including a “Histag” near the N-terminus, was added to the native 202 amino acid GPx-1 protein. COS7 cells were transfected with the recombinant expression vector and permanent transfectants were selected with 100 μg/ml Zeocin™ (Invitrogen, Carlsbad, CA), a member of the bleomycin/phleomycin family of antibiotics.
To isolate the recombinant GPx-1 (rGPx-1), cells were harvested from confluent 150 mm dishes and lysed in 50 mM NaH2PO4, 500 mM NaCl, 30 mM imidazole, 2 % Tween 20, and 10 % glycerol, pH 7.9, on ice. Nuclei and cellular debris were pelleted in a cold microcentrifuge, and supernatants were mixed with Ni-NTA magnetic beads (Qiagen) and rotated for 2.5 hr at 4° C. Beads were collected in a magnetic tube stand and washed several times with cold 50 mM NaH2PO4, 500 mM NaCl, 75 mM imidazole, and 2 % Tween 20, pH 7.9. Beads from several plates were pooled and eluted in 50 mM NaH2PO4, 500 mM NaCl, 300 mM imidazole, and 2 % Tween 20, pH 7.9. Micro BioSpin6 Chromatography Columns (Biorad, Hercules, CA) were used to reduce the concentration of salts in the protein preparation, and partially purified rGPx-1 was stored at −80° C.
In-gel GPx-1 assay
In-gel GPx-1 assays were used to assess rGPx-1 enzyme activity according to the procedures described by Lin and colleagues (37). Briefly, samples were separated on 10% Tris-HCl/SDS gels without prior heat denaturation. After rinsing the gels in 25% isopropanol,10 mM Tris-HCl, pH 7.9, gels were equilibrated in 50 mM Tris-HCl, pH 7.9 for 15 min. The gel was then exposed for 15 min to freshly prepared substrate solution containing 13 mM GSH and 0.004% H2O2 in 50 mM Tris-HCl, pH 7.9. After a brief rinse in distilled water, the gel was developed with freshly prepared 1.2 mM thiazolyl blue tetrazolium bromide and 1.6 mM phenazine methosulfate for 15 minutes. After several washes in distilled water, GPx-1 activity can be visualized as clearing in the otherwise purple-stained gel. Various amounts of bovine GPx-1 were included on gels as activity standards, and the approximate activity was based on calculating the density of the reversed image using the Biorad Versadoc scanner and QuantityOne Software.
Characterization by mass spectrometry
rGPx-1 preparations were separated on SDS-PAGE gels and stained with Coomasie Blue. Gel bands were excised and trypsin digested, and tryptic fragments were analyzed by LC-MS/MS at the Dana Farber Proteomics Center (Boston, MA). Sequence analysis was performed using an ESI-ion trap. Samples were analyzed by nanoflow HPLC microelectrospray ionization on a Finnigan LCQ ion trap (Thermo-Finnigan San Jose, CA). A gradient was applied from 95% A to 80% A for 30 min then to 75% A in 15 min and finally to 10% A in 60 min (where A= 0.1% formic acid in water, B= 0.1% formic acid in acetonitrile) at 200 nl/min over a self-packed, flame-pulled C-18 integrated column and electrospray emitter into the LCQ Deca. The 100 um ID × 8 cm column fused silica (Polymicro Technologies, Phoenix AZ) was slurry packed with Magic C18AQ 5u 200 Å, (Michrom BioResources Auburn, CA). Spectra were acquired in a data-dependent mode throughout the gradient. CID fragmentation was achieved by collision energy of 35%. The capillary temperature was 150° C, and the electrospray voltage was 1.9 kV. Data were analyzed using the Sequest algorithm by searching the updated nonredundant database from NCBI.
RESULTS
G418 induces selenium independent read-through
A series of reporter gene vectors was used to study the components necessary for selenium-dependent translation. We started with the pGL2 UGA vector (11), which has a TGA mutation in the protein coding region of the luciferase gene, and modified it by adding a human GPx-1 promoter as well as the human GPx-1 SECIS element to the 3′ UTR of the luciferase gene (Fig. 1A). Normal read-through of a UGA stop codon only occurs infrequently, at a level of 10−4 in normal intact cells (26). As expected, in constructs lacking a SECIS element, mutation of the luciferase gene to introduce a UGA codon in the protein coding region reduced luciferase activity to levels 10−5 that of a wildtype luciferase with the same promoter (data not shown). Addition of a SECIS element in the sense orientation within the 3′ UTR of the luciferase gene allowed for a low level of read-through dependent on the levels of selenium supplementation (Fig. 1B); by contrast, the presence of a SECIS element in the antisense orientation did not allow for an increase in luciferase activity. These data confirm that read-through of the UGA codon requires selenium and a SECIS element.
Fig. 1. G418 increases read-through of selenocysteine (SEC) codons.
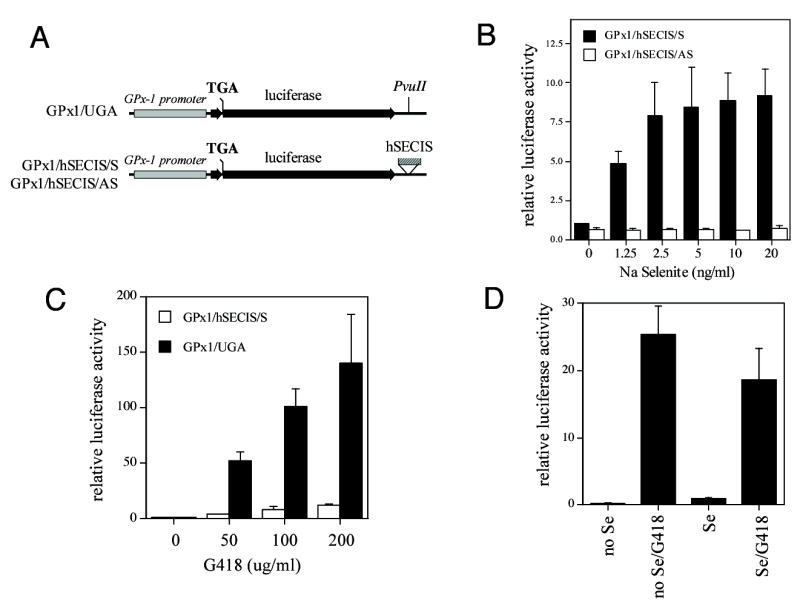
Luciferase constructs with UGA codons in the protein coding region were cotransfected with a renilla luciferase construct into COS7 cells cultured in the presence and absence of selenium. After 48 hr treatment, luciferase and renilla luciferase activity were measured in cell extracts. Shown are luciferase activities ± standard deviations. (A) Map of the luciferase constructs. The GPx-1 promoter (grey bar) was inserted upstream of the luciferase protein coding region (black bar), which has a TGA mutation. The vector GPx1/UGA has a PvuII site in the 3′ UTR of the luciferase gene whereas the GPx1/hSECIS/S and GPx1/hSECIS/AS have the human GPx-1 SECIS element (hatched bar) inserted at the PvuII site in the sense or antisense orientation, respectively. (B) Read-through of SEC codons requires selenium and a SECIS element in the sense orientation. COS7 cells were cultured in selenium-deficient media (media with 0.1% FCS) for 5 days prior to transfection. After transfection, selenium-deficient media was supplemented with various concentrations of sodium selenite. (C) G418 increased read-through in a dose-dependent manner, in the absence or presence of a SECIS element. COS7 cells were grown in the presence of 10% FCS and treated with various doses of G418 after transfection. (D) G418 stimulates read-through in the absence of selenium. COS7 cells were grown in the absence of selenium as in (A). G418 (200 μg/ml) and selenium (Se, 10 ng/ml) were added after transfection.
To test the effects of G418 on read-through translation, cells grown in media with 10% FCS were transfected with the GPx1/hSECIS/S and GPx1/UGA constructs and treated with G418. The addition of G418 led to a striking dose-dependent increase in read-through to a maximum of 12-fold in cells transfected with the GPx1/hSECIS/S construct and treated with 200 μg/ml G418 in the presence of selenium (Fig. 1C). By contrast, the increase in read-through was 140-fold at the highest level of G418 tested (200 μg/ml) with the GPx1/UGA construct that lacks a SECIS element. These data show that unlike selenium-dependent UGA read-through, G418-induced read-through is also increased in the absence of a SECIS element. G418-mediated read-through is also enhanced by the absence of selenium (Fig. 1D): the expression of the GPx1/hSECIS/S was increased 28-fold by G418 in the absence of selenium.
AMG can increase endogenous GPx-1 expression in selenium depleted cells
In cell culture systems, selenium restriction is difficult to ensure owing to the requirement of serum for cellular proliferation and the presence of inorganic selenium in serum as well as in the form of SEC in serum proteins. We found that COS7 cells will survive in media with only 0.1% FCS. In this media, selenium levels are below the limits of detection by atomic absorption spectrometry (data not shown). After 10 days of selenium restriction, immunodetectable GPx-1 protein expression was decreased by approximately 95% (Fig. 2A). The addition of G418 significantly increased immunodetectable protein 5.3-fold in selenium-deficient cells but had no effect on GPx-1 enzyme activity (Fig. 2B). Protein and enzyme activity levels were not significantly altered by G418 treatment in cells cultured in parallel with 10 ng/ml sodium selenite. The relative enzyme level/protein band density (as an estimate of specific enzyme activity) was decreased with G418 treatment in both the selenium-depleted and -replete cells. These data show that GPx-1 protein expression and activity can both be modified by the presence of G418. It is well established that SEC is part of redox-active core of the GPx-1 enzyme and that substitution of other amino acids for SEC in other redox-active selenoproteins greatly reduces their activity (38,39). Thus, the enhanced read-through caused by G418 most likely involves an amino acid substitution at the SEC insertion site that renders the enzyme less active than wild type. Our data also suggest that high levels of selenium may be protective against the effects of G418 on selenium-mediated expression of GPx-1, as the significant effects of G418 on GPx-1 protein expression were only detected in selenium-deficient cells.
Fig. 2. Endogenous GPx-1 protein is increased by G418 treatment.
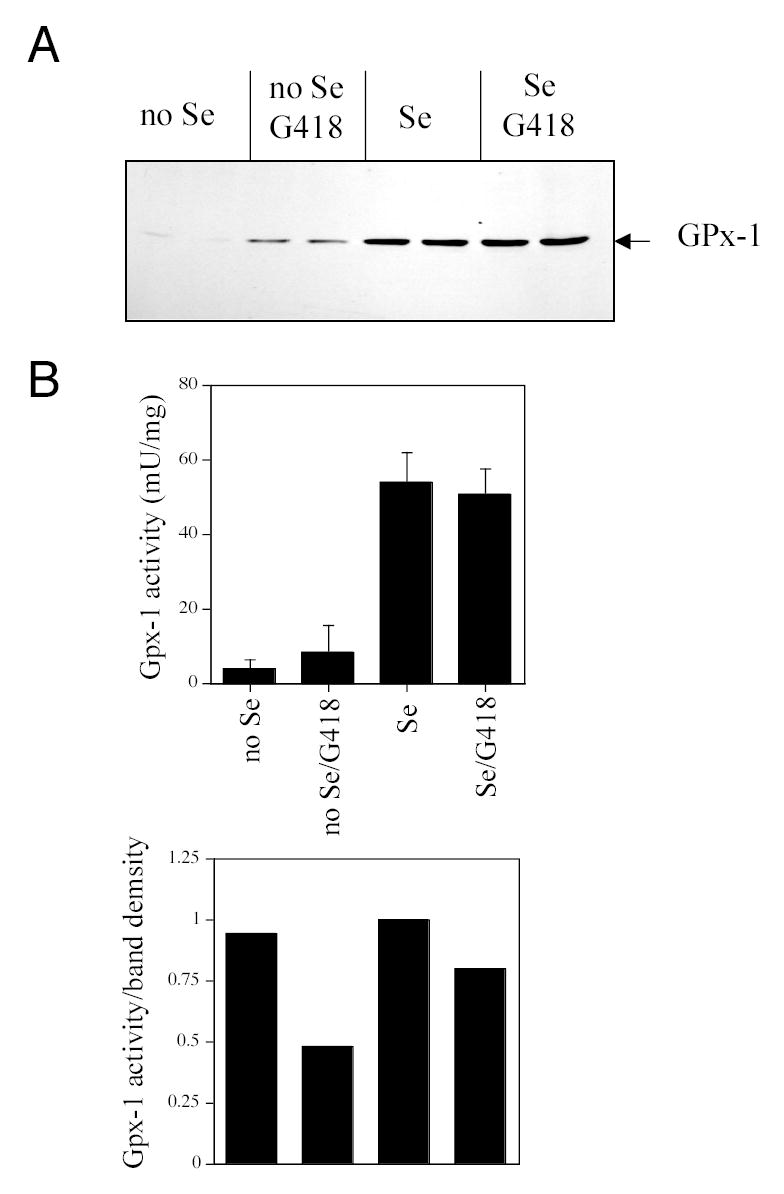
COS7 cells were cultured in selenium-deficient media (no Se) or selenium-replete media (Se, plus 10 ng/ml sodium selenite). After 7 days, treatments were continued for 3 additional days with the addition of G418 (200 μg/ml). (A) Western blot with a mouse anti-human GPx-1 antibody. (B) GPx-1 enzyme activity was measured in extracts; upper plot shows mU/mg ± standard deviations; lower plot shows GPX-1 activity normalized for band density, relative to the Se condition which was arbitrarily set to 1.
We next examined whether G418 could interfere with the production of GPx-1 in COS7 cells that were first selenium starved and then resupplied with selenium. For these experiments, COS7 cells were first cultured in selenium-deficient media for 7 days and then selenium was added at 10 ng/ml for 3 days to assess whether G418 would interfere with the new synthesis of GPx-1 in these cells. Under these conditions, G418 treatment caused a detectable decrease in GPx-1 activity (Fig. 3A); by six days of treatment these differences were abolished (data not shown). In contrast, BAECs cultured continually in the presence of adequate selenium showed a higher level of basal GPx-1 activity and a greater significant reduction in GPx-1 activity after G418 treatment (Fig. 3B). These data suggest that BAECs are more sensitive to G418 than COS7 cells.
Fig. 3. G418 treatment reduces GPx-1 activity in COS7 and bovine aortic endothelial cells (BAEC).
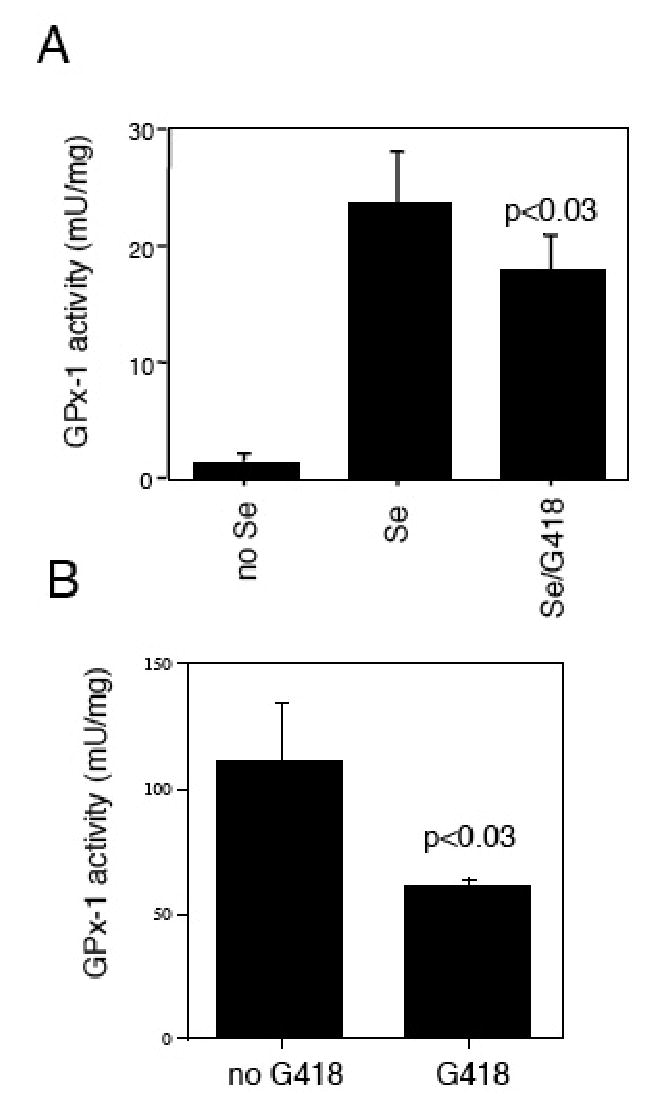
Shown are GPx-1 enzyme activity levels (mU/mg) + standard deviations. (A) G418 interferes with GPx-1 production after selenium replacement. COS7 cells were first selenium starved for 7 days, followed by a 3 day treatment with no selenium or with added selenium (10 ng/ml) or G418 in the presence of selenium (10 ng/ml). (B) BAEC were cultured continually in the presence of media with 10% FCS and 10 ng/ml selenium plus G418.
G418 modifies the expression and activity of rGPx-1
To study the effects of selenium and G418 on GPx-1 enzyme function, a His-tagged recombinant GPx-1 (rGPx1) was isolated from permanently transfected COS7. Endogenous GPx-1 protein is difficult to study in cells grown with less than adequate levels of selenium because of the low levels of expression. Similarly, the His-tagged protein levels are substantially reduced by low selenium growth conditions; however, unlike the endogenous protein, the rGPx-1 is readily purified on Ni-NTA magnetic beads or on Ni-NTA columns, allowing for in vitro analysis. To provide sufficient quantities of cells for the isolation of the rGPx-1, we used media with 2% FCS, which would significantly decrease available selenium while allowing for rapid proliferation of COS7. Under these low selenium conditions, addition of 200 μg/ml G418 reproducibly increased the production of rGPx-1 (Fig. 4A).
Fig. 4. G418 increases recombinant GPx-1 protein (rGPx-1) but decreases rGPx-1 activity.
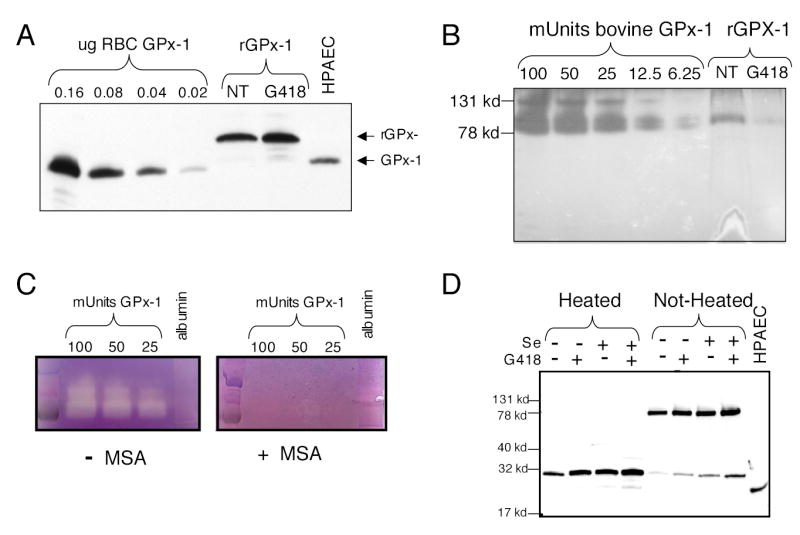
Recombinant protein was isolated from permanently transfected cells grown in media with 2% FCS with the addition of selenium (10ng/ml) and/or G418 (200 μg/ml). His-tagged rGPx-1 was isolated using Ni-magnetic beads. Samples were analyzed by Western blot with the mouse anti-human GPx-1 antibody and by in-gel enzyme activity assays. (A) Western blot of rGPx-1 isolated from cells treated with G418 and those not treated (NT). Various amounts of commercially available human GPx-1 were used as controls on the gel. HPAEC indicates samples from human pulmonary arterial endothelial cells. (B) Reversed image of an in-gel GPx-1 assay using the same rGPx-1 samples used for panel (A). Commercially available bovine GPx-1 was used as standards for the in-gel assay. Samples were run on SDS/PAGE gel without heat denaturation to preserve the active form of the enzyme. (C) The in-gel assay is specific for GPx-1 activity. MSA, a GPx-1 inhibitor, blocks the formation of the achromatic bands. Albumin (1 μg) was used as a non-specific protein. (D) Western blot or rGPx-1 from cells treated with G418 or selenium. Heat-denaturation reduces the rGPx-1 to the monomeric form. Most of the rGPx-1 is in the active tetrameric form (non-heated sample).
In order to test the activity of the rGPx-1, we established an in-gel-assay using methods described previously (37). Although the active GPx-1 enzyme has been described as a tetramer (estimated molecular weight 92 kD), higher molecular mass forms were sometimes detectable in the in-gel assay, in both the commercially available bovine GPx-1 used as standards and in samples of the rGPx-1 (Fig. 4B). In stained activity gels, the gel appears purple with achromatic bands representing sites of enzyme activity. Commercially available GPx-1 was used to show that enzyme activity is proportional to the size of the band. Figure 4B shows an inverse image of an in-gel assay: less than 6.25 mU of enzyme was detected in this system and rGPx1 isolated from the untreated cells had more activity than that isolated from the G418 treated cells, even though there was more immunodetectable protein in the G418-treated sample than in the untreated sample (Fig. 4A). Addition of MSA, a GPx-1 inhibitor, blocked specifically the detectable enzyme activity (Fig. 4C). Heat denaturation resolved rGPx-1 into the monomeric form (Fig. 4D), but most of the isolated rGPx-1 existed in the tetrameric or active form of the enzyme (Fig. 4D, not heated). Addition of selenium or G418 increased the levels of rGPx-1 produced in the transfectants. To illustrate the dose-dependent effect of G418, transfectants were treated with 0, 50, 100, and 200 μg/ml G418. Western blot analysis was used to normalize samples according to rGPx-1 levels, and then samples were used in the in-gel assay. Enzyme activity was reduced as the levels of G418 increased, indicating that the apparent specific activity of the protein was lowered in a dose-dependent manner by G418 (Fig. 5B): 200 μg/ml G418 lowered the relative enzyme activity by approximately 85%, as determined by estimating intensities from the in-gel assay corrected for intensities from the Western blot analysis.
Fig. 5. G418 causes a dose-dependent decrease in rGPx-1 activity.
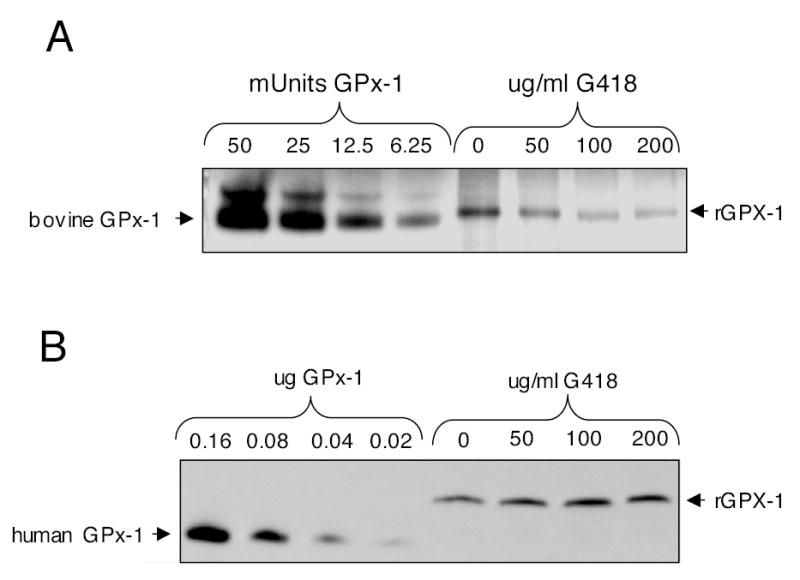
COS7 cells were grown in 2% FCS in the presence of various doses of G418. The rGPx-1 was isolated on Ni-magnetic beads as described. After initial Western blots to normalize protein levels, samples were used for in-gel and Western blot analysis. (A) In-gel assay. (B) Western blot showing relative loading of rGPx-1 protein in the various samples.
G418 modifies the sequence of rGPx-1.
To determine the nature of the read-through of the UGA-codon caused by G418, mass spectrometry was used to characterize the purified rGPx-1. LC-MS/MS analysis of tryptic peptides from rGPx-1 isolated from G418-treated cells showed a unique peptide ion attributable to an arginine substitution in place of the UGA-encoded SEC (Fig. 6). This substitution was only found in preparations from treated cells. Notably, arginine is a site for trypsin digestion, resulting in a shorter peptide in the substituted protein (VLLIENVASLR) than in the control protein (VLLIENVASLSecGTTVR). No other substituted peptides were identified in the LC-MS/MS analysis.
Fig. 6. Characterization of the rGPx-1 by mass spectrometry.
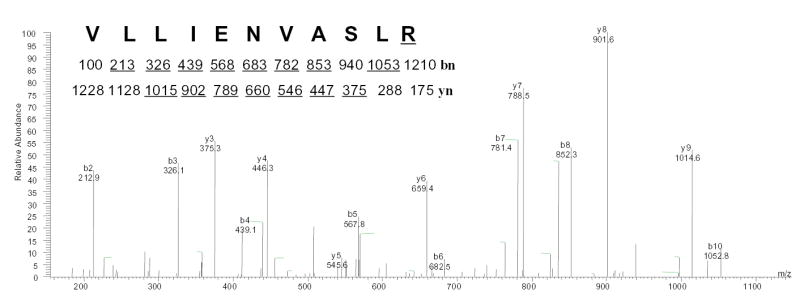
rGPx-1 purified from G418 treated COS7 cells was subjected to mass spectrometry analysis to determine the mass of the peptide, followed by amino acid sequencing by tandem mass spectrometry. The experimentally determined mass for the peptide, [M+2H]2+ =614.01, matched the predicted mass for the Sec-containing peptide (74–89) of rGPx-1 after conversion of the selenocysteine (residue 84) to arginine (underlined). This double-charged precursor ion was completely fragmented and was not detectable. yn and bn are the ions in which amino acid residues are sequentially cleaved from the NH2- terminal and COOH-terminal, respectively. The predicted molecular masses for fragment ions of types b and y are show under the sequence, and ions observed in the spectrum are underlined
DISCUSSION
These findings suggest that GPx-1 translation is highly sensitive to G418 in COS7 cells grown under conditions of selenium restriction. In the reporter gene experiments, this effect manifests as additional luciferase activity, possibly because the AMG-induced misincorporation of amino acids is more efficient than SEC incorporation, especially under selenium-depleted conditions or when constructs without SECIS elements were assayed. The increase in luciferase activity was readily detected because the luciferase enzyme does not rely on SEC for its activity. In the rGPx-1 and the endogenous GPx-1 experiments, enzyme activity relies on SEC incorporation at the redox-active catalytic site. Thus, in the case of recombinant or endogenous GPx-1 proteins, AMG-induced amino acid misincorporation at the UGA codon manifests as increased production of protein that has a lower specific activity.
AMG are thought to alter translation by altering proofreading at the ribosome, resulting in amino acid misincorporation. The ability of AMG to read through stop codons in bacterial and mammalian cells has long been studied (23–26,40,41), but no one has analyzed their effects on SEC incorporation. Recent studies suggest the utility of AMG treatment to overcome premature stop codons in crucial cellular proteins where premature termination of protein synthesis is responsible for certain genetic forms of diseases, such as cystic fibrosis (27,29,30), Hurler syndrome (28), Duchenne’s and Becker’s muscular dystrophy (31,42), and nephrogenic diabetes insipidus (43). AMG, however, have been known to cause other complications, such as deafness and renal dysfunction, primarily via mechanisms that involve oxidative stress (32,44,45). In some studies, the levels of antioxidant enzyme activity, including GPx-1 activity, were found to be decreased in AMG-induced nephrotoxicity (33,46,47), suggesting a cause for the increased oxidative stress in this model. Other studies have found insignificant decreases in GPx-1 activity in AMG nephrotoxicity (48,49).
Our findings provide the first example of specific amino acid recoding caused by AMG-induced stop codon read-through in mammalian cells. Although arginine was the only substitution found, there may be other low-level amino acid substitutions that occur below the levels of detection in our system. Insertion of arginine, however, may not be a random event: of six possible arginine codons two, CGA and AGA, have only a single base difference with the UGA codon. Alternatively, as arginine is a site for trypsin digestion, it is possible that the insertion of an arginine may have created a shorter peptide that was more readily detected in the LC-MS/MS experiment. Nonetheless, any amino acid substitution at the selenocysteine site will decrease enzyme activity, as SEC is essential for normal active site function in the protein.
Different factors appear to be involved in recognition of the UGA as a site for SEC incorporation and recognition of UGA as a stop codon in mammalian cells (5,8,50). Translation termination appears to be highly influenced by the bases surrounding the stop codon (14,51). Factors, such as eRF1 and eRF3, are necessary for the recognition of stop codons during translation and the release of the polypeptide chain (52). Recoding of UGA as a site for SEC incorporation involves a different set of cofactors (12,13,53–55), and eRF factors apparently have no influence on the level of SEC incorporation at the UGA-codon (9).
Transcripts that contain UGA codons in the middle of a protein coding region, such as those encoding selenoproteins, may be subject to nonsense-mediated decay, a process whereby mRNAs encoding truncated proteins are eliminated (15,56). This process occurs in the nucleus in parallel with RNA splicing, and not in the cytoplasm during translation (57). GPx-1 mRNA is apparently subject to nonsense-mediated decay only under conditions of selenium restriction (15); selenium repletion appears to increase GPx-1 mRNA levels in cultured cells (58). GPx-1 mRNA has also been shown to be decreased by selenium restriction in vivo; however, not all selenoprotein transcripts are tightly regulated by selenium levels, and some selenoprotein transcripts increase with selenium restriction (17,59).
Our reporter gene system utilizes a luciferase construct that lacks an intron in the protein coding region, as does the recombinant GPx-1 construct; thus, these constructs would not be subject to nonsense-mediated decay. We have found, however, that G418 increased the translation of the reporter construct, the recombinant construct, and the endogenous GPx-1 protein in cells cultured under low levels of selenium.
Several lines of evidence suggest that the translational efficiency of GPx-1 mRNA is lower than that of other selenoproteins (11, 17). Other evidence suggests that substitution of a SEC-codon with another amino acid codon increases protein expression (10, 19–22). Thus, recoding of a UGA-codon in the mRNA to allow for SEC incorporation is an inefficient process.
Based on in vitro studies of prokaryotic translation using polyribonucleotides (23), stop codon substitutions are not the only mutations caused by AMG. In mammalian translation, however, AMG has only been studied as an agent to suppress stop codons. We suggest that owing to the inefficient process of SEC incorporation at UGA-codons, AMG-mediated substitution at a SEC-codon may be more frequent than at other amino acid codons. Our data indicate that selenium levels and, therefore, the abundance of SEC-incorporation factors may modulate the effects of AMG. Thus, tissue susceptibilities to AMG-induced downregulation of GPx-1 activity may reflect, in part, cell-specific differences in selenium status that contribute to differential expression of various translational cofactors involved in SEC-incorporation.
Acknowledgments
We wish to thank the members of the Loscalzo laboratory for their helpful comments; Earl Gillespie and James Lee of the Protein Analysis Group Molecular Biology Core Facilities at Dana Farber Cancer Institute for their assistance with mass spectrometry; and Mark McComb and David H. Perlman for advice on sample preparation and handling. We also thank Stephanie Tribuna for her excellent assistance preparing this manuscript. This research was funded by R01 HL59876, P01 HL55993, and N01 HV28178.
References
- 1.Behne D, Kyriakopoulos A. Annu Rev Nutr. 2001;21:453–473. doi: 10.1146/annurev.nutr.21.1.453. [DOI] [PubMed] [Google Scholar]
- 2.Ursini F, Maiorino M, Brigelius-Flohe R, Aumann KD, Roveri A, Schomburg D, Flohe L. Methods Enzymol. 1995;252:38–53. doi: 10.1016/0076-6879(95)52007-4. [DOI] [PubMed] [Google Scholar]
- 3.Stadtman TC. Ann N Y Acad Sci. 2000;899:399–402. doi: 10.1111/j.1749-6632.2000.tb06203.x. [DOI] [PubMed] [Google Scholar]
- 4.Driscoll DM, Copeland PR. Annu Rev Nutr. 2003;23:17–40. doi: 10.1146/annurev.nutr.23.011702.073318. [DOI] [PubMed] [Google Scholar]
- 5.Copeland PR. Gene. 2003;312:17–25. doi: 10.1016/s0378-1119(03)00588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leinfelder W, Zehelein E, Mandrand-Berthelot MA, Bock A. Nature. 1988;331(6158):723–725. doi: 10.1038/331723a0. [DOI] [PubMed] [Google Scholar]
- 7.Berry MJ, Banu L, Harney JW, Larsen PR. Embo J. 1993;12(8):3315–3322. doi: 10.1002/j.1460-2075.1993.tb06001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berry MJ, Harney JW, Ohama T, Hatfield DL. Nucleic Acids Res. 1994;22(18):3753–3759. doi: 10.1093/nar/22.18.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grundner-Culemann E, Martin GW, 3rd, Tujebajeva R, Harney JW, Berry MJ. J Mol Biol. 2001;310(4):699–707. doi: 10.1006/jmbi.2001.4809. [DOI] [PubMed] [Google Scholar]
- 10.Leonard JL, Leonard DM, Shen Q, Farwell AP, Newburger PE. J Cell Biochem. 1996;61(3):410–419. doi: 10.1002/(sici)1097-4644(19960601)61:3<410::aid-jcb8>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 11.Lesoon A, Mehta A, Singh R, Chisolm GM, Driscoll DM. Mol Cell Biol. 1997;17(4):1977–1985. doi: 10.1128/mcb.17.4.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen Q, Wu R, Leonard JL, Newburger PE. J Biol Chem. 1998;273(10):5443–5446. doi: 10.1074/jbc.273.10.5443. [DOI] [PubMed] [Google Scholar]
- 13.Copeland PR, Driscoll DM. J Biol Chem. 1999;274(36):25447–25454. doi: 10.1074/jbc.274.36.25447. [DOI] [PubMed] [Google Scholar]
- 14.McCaughan KK, Brown CM, Dalphin ME, Berry MJ, Tate WP. Proc Natl Acad Sci U S A. 1995;92(12):5431–5435. doi: 10.1073/pnas.92.12.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moriarty PM, Reddy CC, Maquat LE. Mol Cell Biol. 1998;18(5):2932–2939. doi: 10.1128/mcb.18.5.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fletcher JE, Copeland PR, Driscoll DM, Krol A. RNA. 2001;7(10):1442–1453. [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss Sachdev S, Sunde RA. Biochem J. 2001;357(Pt 3):851–858. doi: 10.1042/0264-6021:3570851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehta A, Rebsch CM, Kinzy SA, Fletcher JE, Copeland PR. J Biol Chem. 2004;279(36):37852–37859. doi: 10.1074/jbc.M404639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berry MJ, Maia AL, Kieffer JD, Harney JW, Larsen PR. Endocrinology. 1992;131(4):1848–1852. doi: 10.1210/endo.131.4.1396330. [DOI] [PubMed] [Google Scholar]
- 20.Lee SR, Bar-Noy S, Kwon J, Levine RL, Stadtman TC, Rhee SG. Proc Natl Acad Sci U S A. 2000;97(6):2521–2526. doi: 10.1073/pnas.050579797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong L, Holmgren A. J Biol Chem. 2000;275(24):18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- 22.Kuiper GG, Klootwijk W, Visser TJ. Endocrinology. 2003;144(6):2505–2513. doi: 10.1210/en.2003-0084. [DOI] [PubMed] [Google Scholar]
- 23.Davies J, Gorini L, Davis BD. Mol Pharmacol. 1965;1(1):93–106. [PubMed] [Google Scholar]
- 24.Martin R, Mogg AE, Heywood LA, Nitschke L, Burke JF. Mol Gen Genet. 1989;217(2–3):411–418. doi: 10.1007/BF02464911. [DOI] [PubMed] [Google Scholar]
- 25.Kotra LP, Haddad J, Mobashery S. Antimicrob Agents Chemother. 2000;44(12):3249–3256. doi: 10.1128/aac.44.12.3249-3256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manuvakhova M, Keeling K, Bedwell DM. Rna. 2000;6(7):1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howard M, Frizzell RA, Bedwell DM. Nat Med. 1996;2(4):467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 28.Keeling KM, Brooks DA, Hopwood JJ, Li P, Thompson JN, Bedwell DM. Hum Mol Genet. 2001;10(3):291–299. doi: 10.1093/hmg/10.3.291. [DOI] [PubMed] [Google Scholar]
- 29.Zsembery A, Jessner W, Sitter G, Spirli C, Strazzabosco M, Graf J. Hepatology. 2002;35(1):95–104. doi: 10.1053/jhep.2002.30423. [DOI] [PubMed] [Google Scholar]
- 30.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E. N Engl J Med. 2003;349(15):1433–1441. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 31.Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, Atkins JF, Flanigan KM. Ann Neurol. 2004;55(3):422–426. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- 32.Hoppe G, Chai YC, Sears J. Arch Biochem Biophys. 2003;414(1):19–23. doi: 10.1016/s0003-9861(03)00144-9. [DOI] [PubMed] [Google Scholar]
- 33.Pedraza-Chaverri J, Maldonado PD, Medina-Campos ON, Olivares-Corichi IM, Granados-Silvestre MA, Hernandez-Pando R, Ibarra-Rubio ME. Free Radic Biol Med. 2000;29(7):602–611. doi: 10.1016/s0891-5849(00)00354-3. [DOI] [PubMed] [Google Scholar]
- 34.Wu WJ, Sha SH, Schacht J. Audiol Neurootol. 2002;7(3):171–174. doi: 10.1159/000058305. [DOI] [PubMed] [Google Scholar]
- 35.Weiss N, Zhang YY, Heydrick S, Bierl C, Loscalzo J. Proc Natl Acad Sci U S A. 2001;98(22):12503–12508. doi: 10.1073/pnas.231428998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Handy DE, Zhang Y, Loscalzo J. J Biol Chem. 2005;280(16):15518–15525. doi: 10.1074/jbc.M501452200. [DOI] [PubMed] [Google Scholar]
- 37.Lin CL, Chen HJ, Hou WC. Electrophoresis. 2002;23(4):513–516. doi: 10.1002/1522-2683(200202)23:4<513::AID-ELPS513>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 38.Gasdaska JR, Harney JW, Gasdaska PY, Powis G, Berry MJ. J Biol Chem. 1999;274(36):25379–25385. doi: 10.1074/jbc.274.36.25379. [DOI] [PubMed] [Google Scholar]
- 39.Maiorino M, Aumann KD, Brigelius-Flohe R, Doria D, van den Heuvel J, McCarthy J, Roveri A, Ursini F, Flohe L. Z Ernahrungswiss 37 Suppl. 1998;1:118–121. [PubMed] [Google Scholar]
- 40.Gorini L, Kataja E. Proc Natl Acad Sci U S A. 1964;51:995–1001. doi: 10.1073/pnas.51.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh A, Ursic D, Davies J. Nature. 1979;277(5692):146–148. doi: 10.1038/277146a0. [DOI] [PubMed] [Google Scholar]
- 42.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. J Clin Invest. 1999;104(4):375–381. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulz A, Sangkuhl K, Lennert T, Wigger M, Price DA, Nuuja A, Gruters A, Schultz G, Schoneberg T. J Clin Endocrinol Metab. 2002;87(11):5247–5257. doi: 10.1210/jc.2002-020286. [DOI] [PubMed] [Google Scholar]
- 44.Cuzzocrea S, Mazzon E, Dugo L, Serraino I, Di Paola R, Britti D, De Sarro A, Pierpaoli S, Caputi A, Masini E, Salvemini D. Eur J Pharmacol. 2002;450(1):67–76. doi: 10.1016/s0014-2999(02)01749-1. [DOI] [PubMed] [Google Scholar]
- 45.Ali BH. Food Chem Toxicol. 2003;41(11):1447–1452. doi: 10.1016/s0278-6915(03)00186-8. [DOI] [PubMed] [Google Scholar]
- 46.Soejima A, Ishizuka S, Suzuki M, Miyake N, Fukuoka K, Nagasawa T. Nephron. 1998;80(3):331–339. doi: 10.1159/000045194. [DOI] [PubMed] [Google Scholar]
- 47.Erdem A, Gundogan NU, Usubutun A, Kilinc K, Erdem SR, Kara A, Bozkurt A. Nephrol Dial Transplant. 2000;15(8):1175–1182. doi: 10.1093/ndt/15.8.1175. [DOI] [PubMed] [Google Scholar]
- 48.Yazar E, Elmas M, Altunok V, Sivrikaya A, Oztekin E, Birdane YO. Can J Vet Res. 2003;67(3):239–240. [PMC free article] [PubMed] [Google Scholar]
- 49.Atessahin A, Karahan I, Yilmaz S, Ceribasi AO, Princci I. Pharmacol Res. 2003;48(6):637–642. doi: 10.1016/s1043-6618(03)00227-5. [DOI] [PubMed] [Google Scholar]
- 50.Namy O, Rousset JP, Napthine S, Brierley I. Mol Cell. 2004;13(2):157–168. doi: 10.1016/s1097-2765(04)00031-0. [DOI] [PubMed] [Google Scholar]
- 51.Ozawa Y, Hanaoka S, Saito R, Washio T, Nakano S, Shinagawa A, Itoh M, Shibata K, Carninci P, Konno H, Kawai J, Hayashizaki Y, Tomita M. Gene. 2002;300(1–2):79–87. doi: 10.1016/s0378-1119(02)01042-9. [DOI] [PubMed] [Google Scholar]
- 52.Kisselev L, Ehrenberg M, Frolova L. Embo J. 2003;22(2):175–182. doi: 10.1093/emboj/cdg017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zavacki AM, Mansell JB, Chung M, Klimovitsky B, Harney JW, Berry MJ. Mol Cell. 2003;11(3):773–781. doi: 10.1016/s1097-2765(03)00064-9. [DOI] [PubMed] [Google Scholar]
- 54.Low SC, Harney JW, Berry MJ. J Biol Chem. 1995;270(37):21659–21664. doi: 10.1074/jbc.270.37.21659. [DOI] [PubMed] [Google Scholar]
- 55.Tujebajeva RM, Copeland PR, Xu XM, Carlson BA, Harney JW, Driscoll DM, Hatfield DL, Berry MJ. EMBO Rep. 2000;1(2):158–163. doi: 10.1093/embo-reports/kvd033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun X, Moriarty PM, Maquat LE. Embo J. 2000;19(17):4734–4744. doi: 10.1093/emboj/19.17.4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferraiuolo MA, Lee CS, Ler LW, Hsu JL, Costa-Mattioli M, Luo MJ, Reed R, Sonenberg N. Proc Natl Acad Sci U S A. 2004;101(12):4118–4123. doi: 10.1073/pnas.0400933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chada S, Whitney C, Newburger PE. Blood. 1989;74(7):2535–2541. [PubMed] [Google Scholar]
- 59.Wingler K, Bocher M, Flohe L, Kollmus H, Brigelius-Flohe R. Eur J Biochem. 1999;259(1–2):149–157. doi: 10.1046/j.1432-1327.1999.00012.x. [DOI] [PubMed] [Google Scholar]