

Abstract
BACKGROUND
Bone morphogenetic protein (BMP) receptor-II (BMPR2) heterozygous mutant (BMPR2+/−) mice have a similar genetic trait as certain patients with idiopathic pulmonary arterial hypertension (IPAH). To understand the role of the BMPR2 in the development of IPAH, we examined the phenotype of BMPR2+/− mice and its response to inflammatory stress.
METHODS AND RESULTS
BMPR2+/− mice were found to have the same life span, right ventricular systolic pressure (RVSP), and lung histology as those of wildtype mice under unstressed conditions. However, when treated with recombinant adenovirus expressing 5-lipoxygenase (Ad5LO), BMPR2+/− mice exhibited significantly higher RVSP than wildtype mice. The increase of RVSP occurred in the first two weeks after Ad5LO delivery. Modest but significant muscularization of distal pulmonary arterioles appeared in BMPR2+/− mice four weeks after Ad5LO treatment. Measurement of urinary metabolites of vasoactive molecules showed that cysteinyl leukotrienes, prostacyclin metabolites, and prostaglandin E2 were all increased to a similar degree in both BMPR2+/− and wildtype mice during 5LO transgene expression, while urinary endothelin-1 remained undetectable. Urinary thromboxane A2 metabolites, in contrast, were significantly higher in BMPR2+/− than wildtype mice and paralleled the increase in RVSP. Platelet activation markers, serotonin, and sP-selectin showed a trend toward higher concentrations in BMPR2+/− than wildtype mice. Cell culture studies found that BMP treatment reduced IL-1β-stimulated thromboxane A2 production in the pulmonary epithelial cell line A549.
CONCLUSIONS
BMPR2+/− mice do not develop pulmonary hypertension spontaneously; however, under inflammatory stress, they are more susceptible to an increase in RVSP, thromboxane A2 production, and vascular remodeling than wildtype mice.
Keywords: BMPR2, pulmonary hypertension, thromboxane
Introduction
Bone morphogenetic protein receptor-II (BMPR-II) transduces signals of bone morphogenetic proteins (BMPs), which are a family of peptides originally found to induce ectopic bone formation 1. BMPs were later found to be structurally related to TGF-βs, activins/inhibins, and Mullerian inhibiting substances 2–4; and to play critical roles in embryonic development, tissue morphogenesis, and cell differentiation 5.
Recent genetic linkage studies found that heterozygous germline mutations in the BMPR-II gene (BMPR2) are associated with idiopathic pulmonary arterial hypertension (IPAH) 6–8 (formally denoted as primary pulmonary hypertension 9), a disease characterized by a sustained increase of pulmonary artery pressure of unknown cause 10–12. Approximately 55% of the familial form and between 10–26% of the sporadic form of IPAH patients are found to have heterozygous BMPR2 mutations 8,13. Family members with BMPR2 mutations have only a 15-20% chance of developing (clinical) pulmonary hypertension 13. The onset of the disease varies widely both within families and among individuals carrying the same mutation 7,14. It has, thus, been suggested that additional genetic or environmental factors are required to develop pulmonary hypertension in individuals with heterozygous BMPR2 mutation. The nature of these additional factors is currently unknown.
Homozygous BMPR2 mutant (BMPR2−/−) mice have been shown to die in utero before mesoderm formation 15. Heterozygous BMPR2 mutant (BMPR2+/−) mice, by contrast, are reported to be morphologically normal and fertile 15. Transgenic mice expressing a dominant-negative BMPR-II gene have been recently examined 16. The study showed that postnatal expression of the mutant BMPR-II gene in smooth muscle leads to a significant increase of right ventricular systolic pressure (RVSP), and relatively modest pulmonary vascular remodeling in the mice.
In the present study, we examined the phenotype of BMPR2+/− mice with respect to: 1) their survival rate during prenatal development; 2) their RVSP and lung histology in the basal state; and 3) their responses to inflammatory stress resulting from adenovirus-mediated pulmonary overexpression of 5-lipoxygenase (5LO). Inflammation has been previously suggested to be one of the mechanisms involved in the development of pulmonary arterial hypertension (for reviews, see 17–20). 5LO catalyzes leukotriene formation and facilitates an inflammatory process 21. Increased 5LO expression or leukotriene production has been demonstrated in patients with PAH and an animal model of PAH 22–24. Thus, using this inflammatory mediator as a stimulus may yield useful information about the role of inflammation in the development of pulmonary hypertension in individuals with BMPR2 haploinsufficiency.
Methods
Animals
BMPR2 mutant mice were established by Beppu and colleagues via targeted gene disruption 15. As homozygous BMPR2 mutant mice are embryonic lethal, breeding of BMPR2+/− mice was carried out by crossing BMPR2+/− mice with wildtype mice (C57/BL6 mice). Genotyping of the offspring was performed after weaning. The mutated BMPR2 allele in the mice was identified by a polymerase chain reaction using primer pairs 5′-GCTAA AGCGC ATGCT CCAGA CTGCC TTG-3′ and 5′-AGGTT GGCCT GGAAC CTGAG GAAAT C-3′ 15. The amplification conditions were: 1 cycle of 120 sec at 95°C; 30 cycles of 30 sec at 95°C, 30 sec at 63°C and 90 sec at 72°C; and 5 min at 72°C.
The mice were maintained in Boston University Laboratory Animal Science Center, under stress-free conditions, i.e., unlimited food and water access, regular light cycles (12/12 hrs), no forced exercise, and generally pathogen-free. All animals received humane care. The study was approved by the Institutional Animal Care and Use Committee at Boston University.
Right heart catheterization
Mice underwent right heart catheterization at various time points during the study. Prior to the catheterization, mice were anesthetized with 90 mg/kg ketamine and 6 mg/kg xylazine. After dissection to expose the right jugular vein, a 1.4F Millar Mikro-Tip pressure catheter (Millar Instruments, Houston, TX) was inserted into the vein and advanced to the right ventricle. The catheter was connected to a transducer unit interfaced with a signal amplifier and recorder (Gould Instrument Systems, Valley View, OH), and the right ventricular systolic pressure (RVSP) was recorded.
Histology
Mice lungs were perfused with saline and inflated with 10% phosphate-buffered formalin at a pressure of 20 cm H2O. After fixation for 20 hr at 4°C, the lung tissue was processed and paraffin-embedded using a Hypercenter XP System and Embedding Center (Shandon Inc, Pittsburgh, PA), and cut into 5-μm sections. The tissue sections were then heat-dried on slides at 56°C for 1 hr. Deparaffinization and rehydration were carried out by immersing the slides in xylene (2 × 5min), 100% ethanol (2 × 1 min), 95% ethanol (2 × 1 min), and deionized water (5 min).
For hematoxylin and eosin staining, tissue sections were incubated in Gill-2 hematoxylin (Fisher, Fairlawn, NJ) for 2 min, rinsed with water for 2 min, dipped once in acid alcohol (70% ethanol and 1% concentrated HCl), rinsed with water for 1 min, dipped in 1% NH4OH for 15 sec, rinsed with water for 1 min, and dipped twice in 1% Eosin Y (Fisher, Fairlawn, NJ). The stained sections were dehydrated by incubation in 95% ethanol for 2 × 30 sec, 100% ethanol for 2 × 30 sec, and xylene for 2 × 2.5 min, then mounted with Cytoseal60.
For smooth muscle α-actin staining, the lung sections were incubated with a mouse monoclonal antibody against smooth muscle cell α-actin (Sigma, 1:800 dilution in 1% immunohistochemical grade BSA/PBS) at 4°C overnight. The sections were washed in PBS twice for 5 min and incubated with biotinylated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA), at a dilution of 1:500 in 1% BSA/PBS for 30 min at room temperature. The sections were then incubated with avidin DH and biotinylated-alkaline phosphatase H (provided in Vectastain ABC-AP kit, Vector Lab, Burlingame, CA) for 30 min, and then with alkaline phosphatase substrate (provided in Vector Red Substrate kit, Vector Lab) for 20 min. Two PBS washes were performed between these incubations, and the final washing was performed under running tap water for 5 min. The sections were counterstained with Harris Modified Hematoxylin for 10 sec, washed with water, dipped in acid alcohol, and rinsed with water.
Delivery of 5LO transgene
Recombinant replication-deficient adenovirus expressing human 5LO (Ad5LO) was prepared as previously described 25, and was delivered to the lungs of mice by intra-tracheal instillation. In this procedure, mice were anesthetized with ketamine (90 mg/kg)/xylazine (6 mg/kg), and given 1% lidocaine by local injection into the mid-line neck region. Surgical exposure of the trachea was then performed through a midline incision and subsequent blunt dissection to the level of the trachea. A 29-gauge needle bent at a 60-degree angle was inserted into the trachea, and 2 × 108 plaque forming units of Ad5LO were slowly instilled into the trachea. After instillation, the neck incision was closed with #4 silk surgical suture. Buprenorphine, 0.1 mg/kg sc, was given every 8–12 hours after the procedure for three doses, and animals were monitored until full recovery from anesthesia before returning them to regular cages.
RNA isolation and Northern blotting
Mice lungs were perfused with saline and homogenized immediately in 10 ml TRIzol Reagent (Invitrogen, Carlsbad, CA) followed by chloroform extraction for total RNA. Northern blotting was carried out by electrophoresing 20 μg RNA in 1.2 % agarose gels containing 4% formaldehyde. The RNA was transferred onto nitrocellulose membranes and hybridized with an [α-32P]-dCTP-labeled 5LO probe that encompasses nucleotides 45–290 of human 5LO cDNA (GenBank accession #J03600). Hybridization was carried out at 68°C overnight in MiracleHyb solution (Stratagene, La Jolla, CA) containing 10 μg/ml sheared salmon sperm DNA. The membrane was washed with 2 X SSC/0.1% SDS at room temperature for 15 min and then with 0.1 X SSC/0.1% SDS at 68°C for 20 min before exposure to X-ray film. The membrane was then washed in 0.1 X SSC/0.1% SDS at 100°C twice for 15 min to strip the bound 5LO probe, and hybridized with a [α-32P]-dCTP-labeled mouse GAPDH probe (Ambion, Austin, TX) to estimate total RNA loading.
Urine collection and measurement of urinary creatinine
Urine samples were collected from the mice two days before Ad5LO treatment (day –2) and on days 3, 7, 12, and 21 after the treatment. During the collection, mice were maintained in metabolic cages with water supplied but food withheld for 16 hr. Urine samples were collected over the 16-hr period and the mice were return to their regular cages after the collection. The urine samples were centrifuged at 20,000 × g for 5 min, and the supernatants were stored in aliquots at −80°C.
Urinary creatinine concentration was determined by the picric acid assay using creatinine assay reagents from Teco Diagnostics (Anaheim, CA). In this assay, 0.01 ml creatinine standards (1.25, 2.5, 5, and 10 mg/ml) or urine samples (diluted 5- to15-fold in water) were added to a 96-well plate, and mixed with 0.2 ml of Creatinine Picric Acid Reagent and Creatinine Buffer Reagent (mixed before assay at 1:1, v/v). The plate was incubated at 37°C for 40 min and read at 490 nm. The absorbance from this reading was registered as the total absorbance. Acetic Acid Reagent (0.01 ml) was then added to each well and the plate incubated at room temperature (22–25°C) for 15 min. The plate was read again at 490 nm and the absorbance from the second reading (non-specific absorbance) was subtracted from the first reading (total absorbance). The absorbance difference (specific absorbance) was used to construct the standard curve and calculate the sample urinary creatinine concentration.
Urinary vasoactive metabolites
The production of cysteinyl leukotrienes, prostacyclin, prostaglandin E2, endothelin-1, and thromboxane A2 in mice were determined by analyzing the urinary concentration of the products or their metabolites by ELISA. For cysteinyl leukotriene measurements, an ELISA kit detecting leukotrienes C4, D4, and E4 from Assay Designs (# 900-070, Ann Arbor, MI) was used. Urine samples were diluted a minimum 20-fold for the assay to avoid nonspecific binding. For urinary prostacyclin measurements, an ELISA kit detecting 6-keto-prostaglandin F1α and 2,3-dinor-6-keto-prostaglandin F1α, two major metabolites of prostacyclin in urine, was used (Assay Designs #901-025). Urine samples were diluted a minimum of 40-fold for these assays. For prostaglandin E2 measurements, a high sensitivity ELISA kit detecting prostaglandin E2 from Assay Designs (#930-001) was used. Urine samples were diluted a minimum of 20-fold for the assay. For endotheline-1 measurements, an ELISA kit detecting endothelin-1, -2, and -3 was used (#583151, Cayman Chemical, Ann Arbor, MI). Urine samples were diluted a minimum of 10-fold for the assay. For thromboxane A2 measurements, ELISA kits detecting thromboxane B2 (Assay Designs #901-002) and 11-dehydro-thromboxane B2 were used (Assay Designs #901-092), and urine samples were diluted a minimum of 50-fold and 30-fold for the assays using thromboxane B2 and 11-dehydro-thromboxane B2 kit, respectively. For urinary serotonin measurement, an ELISA kit from Labor Diagnostika Nord GmbH/Rocky Mountain Diagnostics (Colorado Spring, CO) was used. All samples were analyzed in duplicate and repeated at least twice.
Plasma soluble P-selectin measurement
Plasma soluble P-selectin (sP-selectin) was measured using a mouse sP-selectin Quantikine kit (#MPS00) from R&D Systems (Minneapolis, MN). Blood samples were collected from mice immediately after euthanasia, and mixed with EDTA as anticoagulant. The samples were centrifuged first at 2,000 × g for 20 minutes at room temperature and then at 10,000 × g for 10 minutes at 2 – 8° C. Aliquots of the supernatants were kept at −80°C before use.
A549 cell activity assay
The Pulmonary epithelial cell line A549 was obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained at 37ºC in RPMI 1640 medium containing 10% fetal bovine serum, 100 units penicillin, and 100 μg streptomycin. Treatment of the cells with IL-1β and/or BMPs was carried out in RPMI 1640 medium containing 1% fetal bovine serum. To determine the thromboxane A2 synthesis, the cells were washed with D-PBS and incubated with 0.01 mM arachidonic acid in D-PBS for 10 min at 37°C. The released thromboxane B2 in the assay medium was analyzed by ELISA as described above. Total cell protein concentration was determined using DC protein assay reagents from Bio-Rad (Hercules, CA).
Statistics
Data are presented as the mean ± SEM of the mean. Statistical analysis was performed by chi square analysis (Table 1), Student’s t-Test (Figs. 4, 10, and 11), or two-way ANOVA (Figs. 5, 7, 8, and 9) using the SigmaStat program. P<0.05 indicates statistical significance.
Table 1.
Genotypic ratio of offspring from BMPR2+/− and BMPR2+/+ breeding pairs*
| Offspring | Genotype | ||
|---|---|---|---|
| BMPR2+/− (B) | BMPR2+/+ (W) | B/W ratio | |
| Female (F) | 243 | 308 | 0.79 |
| Male (M) | 255 | 310 | 0.82 |
| Total | 498 | 618 | 0.81 |
| F/M ratio | 0.95 | 0.99 | |
The breeding was carried out by crossing BMPR2+/− male with wildtype (BMPR2+/+) female mice and the genotypes of the offspring were determined after weaning. The distribution of BMPR2+/− and BMPR2+/+ genotype in the offspring is significantly different, X2= 12.9, p< 0.001). No statistical difference exists between female and male distribution in either BMPR2+/− or BMPR2+/+ offspring.
Figure 4. RVSP in mice treated with Ad5LO.
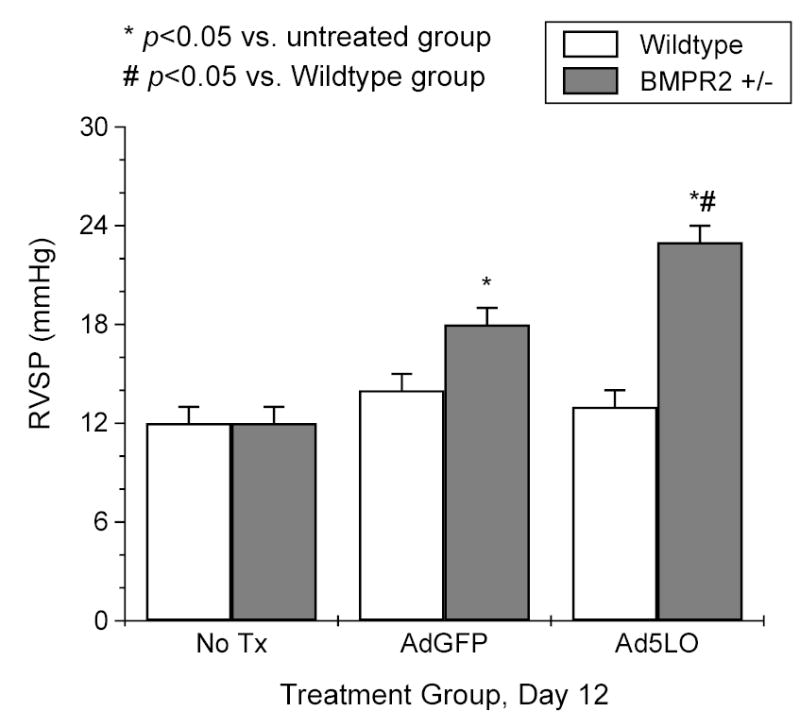
Wildtype (white column) and BMPR2+/− (gray column) mice were treated with nothing (No Tx), 2×108 pfu adenovirus expressing green fluorescence protein (AdGFP), or 2×108 pfu Ad5LO via intra-tracheal instillation at day 0. Right heart catheterization was performed on the mice at day 12 after the treatment. Data are presented as mean ± SEM; n = 6–10 per group. * indicates p<0.05 vs. no treatment group in the same type of mouse; # indicates p<0.05 BMPR2+/− vs. wildtype group with the same treatment. Statistical analysis was performed by Student’s t-Test.
Figure 10. Plasma soluble P-selectin (sP-selectin) and urinary serotonin concentrations.
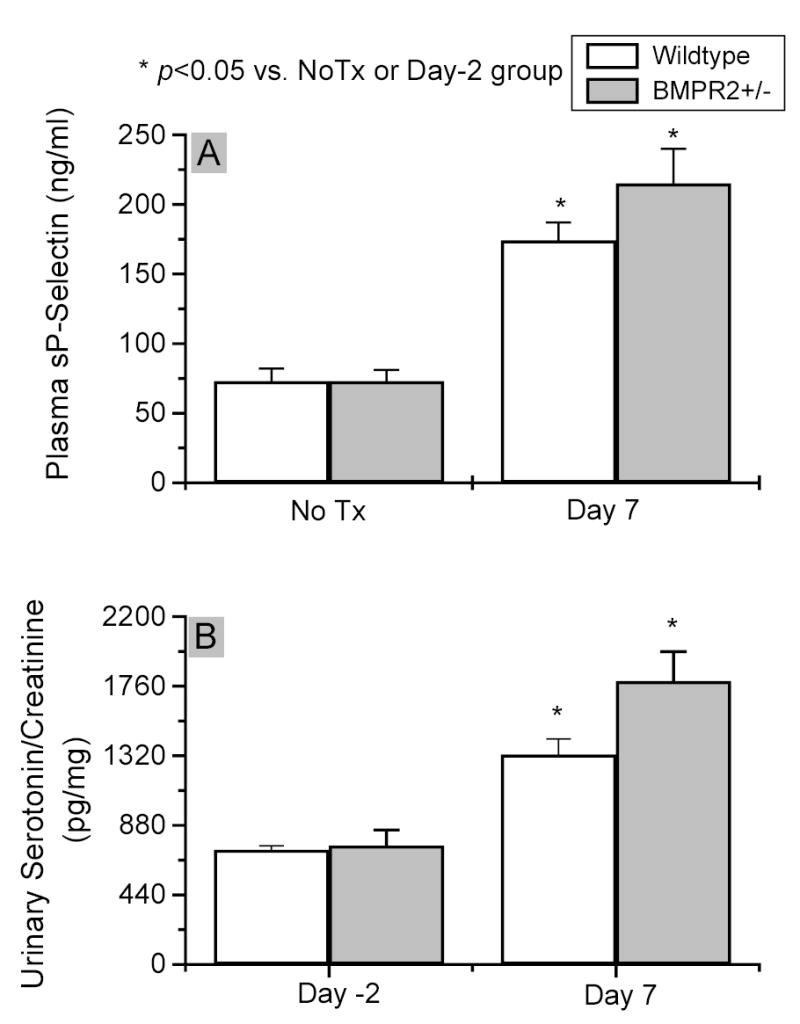
Plasma samples were prepared from wildtype (white column) and BMPR2+/− (gray column) mice with no treatment (No Tx) or treated with 2×108 pfu Ad5LO seven days previously (Day 7). sP-selectin was measured by ELISA (pane A). Urinary serotonin analysis was performed on the same samples as those used in Figure 7. * indicates p<0.05 vs. No Tx group (sP-selectin) or day -2 group (serotonin) in the same type of mice. The difference between BMPR2+/− and wildtype mice in both sP-selectin and serotonin measurement did not reach statistical significance. Data are presented as mean ± SEM; n = 6–10 per group. Statistical analysis was performed by Student’s t-Test.
Figure 11. Effect of BMPs and IL-1β on thromboxane B2 production in A549 cells.
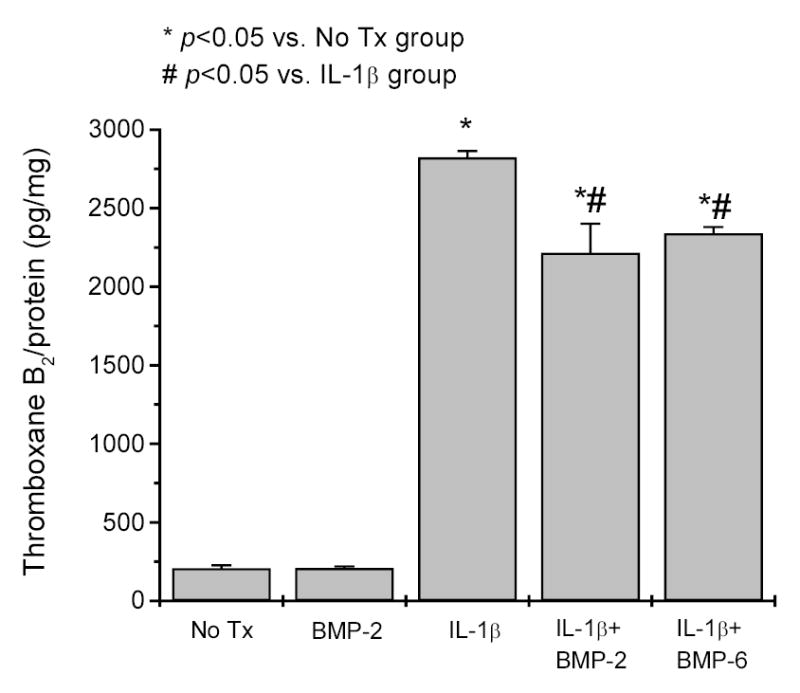
A549 cells were treated with 0.5 ng/ml IL-1β and/or 100 ng/ml BMP-2 or BMP-6 for 20 hrs. The cells were then washed and incubated with 0.01 mM arachidonic acid in D-PBS for 10 min. Thromboxane B2 released in the assay medium was analyzed by ELISA. * indicates p<0.05 vs. untreated cells; # indicates p<0.05 vs. the cells treated with IL-1β alone. Data are presented as mean ± SEM of three measurements each in duplicate. Statistical analysis was performed by Student’s t-Test.
Figure 5. Time course of right ventricular systolic pressure (RVSP) change in Ad5LO-treated mice.
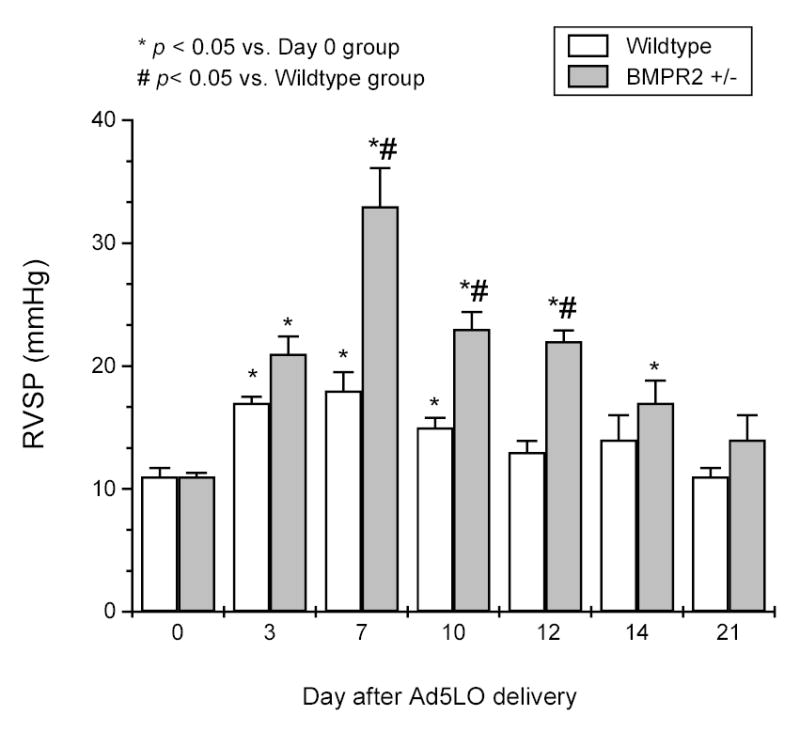
Wildtype (white column) and BMPR2+/− (gray column) mice were treated with 2×108 pfu Ad5LO via intra-tracheal instillation at day 0. RVSP was measured at days 0, 3, 7, 10, 12, 14, and 21 for each group. * indicates p<0.05 vs. day 0 group in the same type of mice; # indicates p<0.05 vs. wildtype group at the same time point. Data are presented as mean ± SEM; n = 6–10 per group. Statistical analysis was performed by two-way ANOVA.
Figure 7. Time course of cysteinyl leukotriene production in Ad5LO-treated mice.
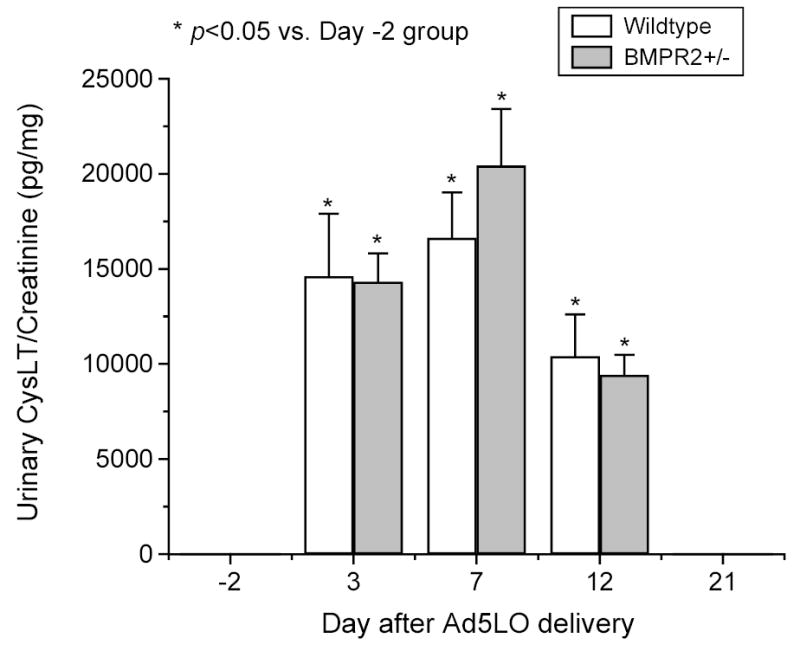
Wildtype (white column) and BMPR2+/− (gray column) mice were treated with 2×108 pfu Ad5LO via intra-tracheal instillation at day 0. Urine samples were collected from the mice 2 days before (day -2) and 3, 7, 12, and 21 days after the treatment. Urinary cysteinyl leukotrienes (CysLT) and creatinine were determined by ELISA and the picric acid assay, respectively. * indicates p<0.05 vs. day -2 group. Urinary CysLT concentration in either type of mice was undetectable at days –2 and 21; no statistical difference existed between BMPR2+/− and wildtype mice at any time point. Data are presented as mean ± SEM; n= 6–10 mice/group. Statistical analysis was performed by two-way ANOVA.
Figure 8. Prostacyclin (PGI2) and prostaglandin E2 (PGE2) production in Ad5LO-treated wildtype and BMPR2+/− mice.
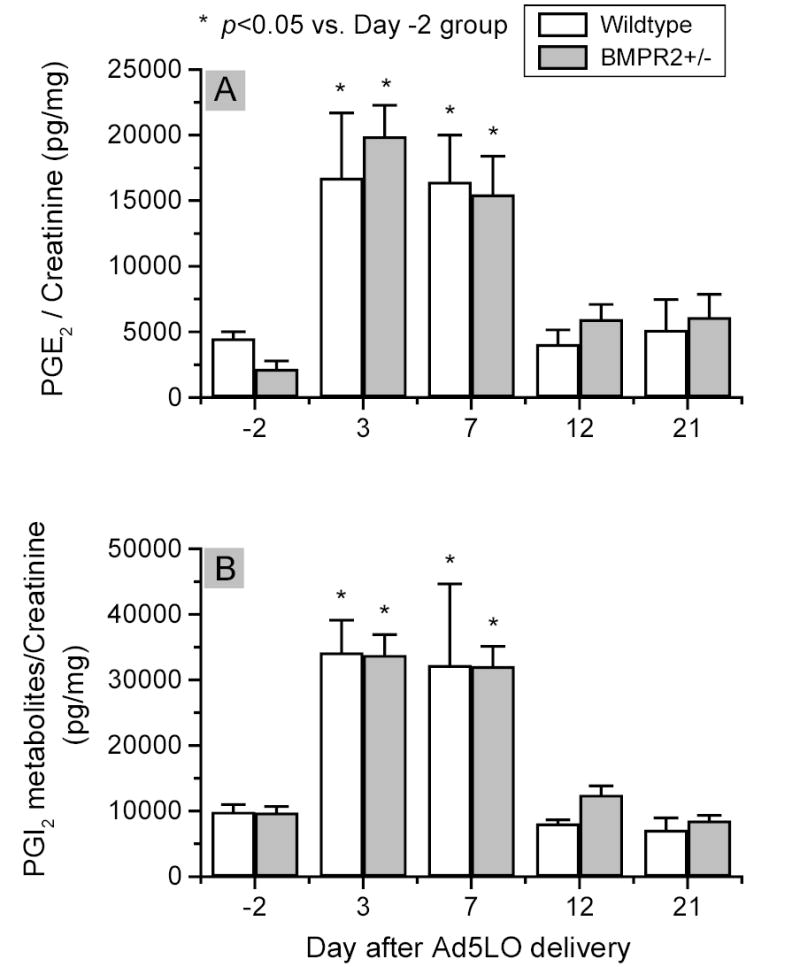
The analyses were performed on the same set of urine samples as that used in Figure 7. PGI2 production (Panel A) in the mice was determined by ELISA measuring urinary 6-keto-prostaglandin F1α and 2,3-dinor-6-keto-prostaglandin F1α concentration. * indicates p<0.05 vs. day -2 group. No statistical difference existed between BMPR2+/− and wildtype mice groups at any time point. Data are presented as mean ± SEM; n = 6–10 per group. Statistical analysis was performed by two-way ANOVA.
Figure 9. Thromboxane A2 production in Ad5LO-treated wildtype and BMPR2+/− mice.
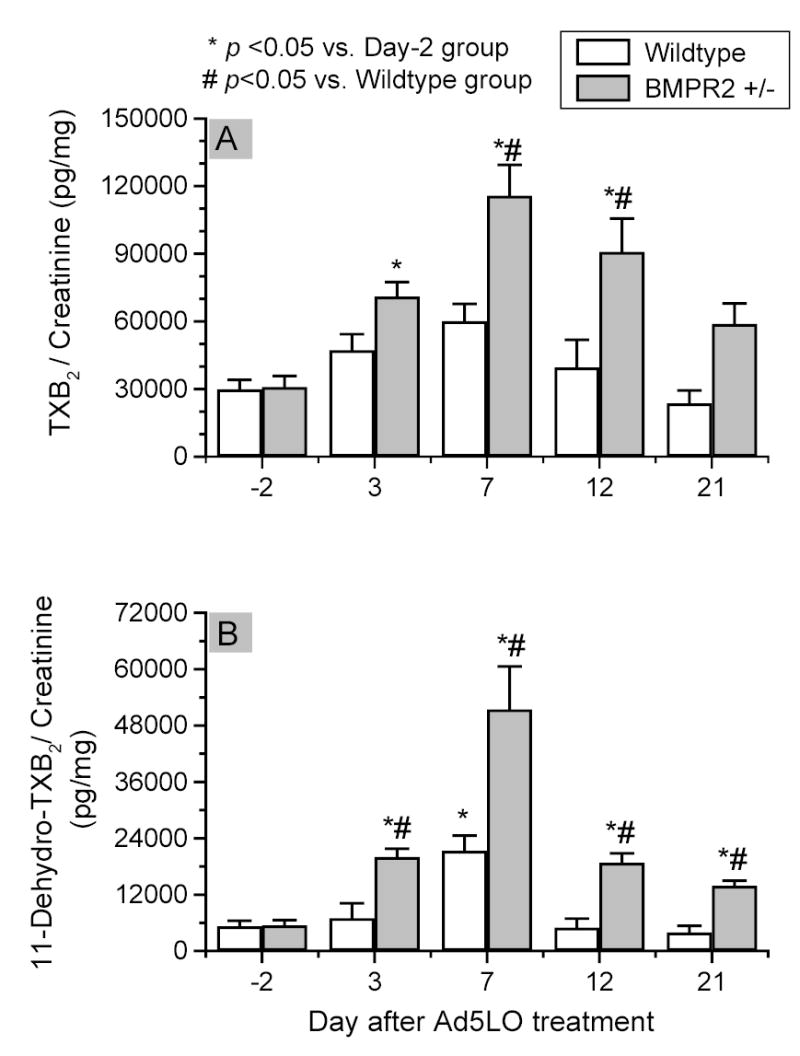
The analyses were performed on the same set of urine samples as that used in Figure 7. Urinary concentrations of thromboxane B2 (panel A) and 11-dehydro-thromboxane B2 (panel B) were determined by ELISA. * indicates p<0.05 vs. day -2 group; # indicates p<0.05 BMPR2+/− vs. wildtype group at the same time point. Data are presented as mean ± SEM; n = 6–10 per group. Statistical analysis was performed by two-way ANOVA.
Results
BMPR2+/− mice breeding
BMPR2+/− mice were bred by crossing BMPR2+/− with wildtype mice (C57/BL6 mice). The genotypes of the offspring were determined after weaning. Table 1 shows the summary of the numbers of BMPR2+/− and wildtype mice obtained from breeding, and the distribution of male and female mice in each population. As demonstrated, the numbers of BMPR2+/− and wildtype mice in the offspring are significantly different, 498 and 618, respectively (X2= 12.9, p< 0.001). The ratio of BMPR2+/− to wildtype mice was 81:100, indicating that approximately 20% of BMPR2+/− mice were lost in early development. The majority of the missing BMPR2+/− mice died in utero, although some may have died in the immediate postnatal stage as the litter number was not counted immediately after birth. The female/male distribution in either BMPR2+/− or wildtype offspring was not statistically different. The surviving BMPR2+/− mice were morphologically normal and had no apparent problems in the unstressed state. These mice also had a similar life span (~ 18 months) as wildtype mice.
Lung structure and right ventricular systolic pressure in BMPR2+/− mice
To assess lung development in the BMPR2+/− mice, lung tissue sections from BMPR2+/− and wildtype mice were prepared and stained with hematoxylin and eosin. As shown in Figure 1, no apparent gross abnormalities were observed in the sections from either mouse, although a minor increase in adhesion of leukocytes to the vessel wall was noted in the BMPR2+/− mice (Fig. 1B).
Figure 1. Lung histology of wildtype and BMPR2+/− mice.
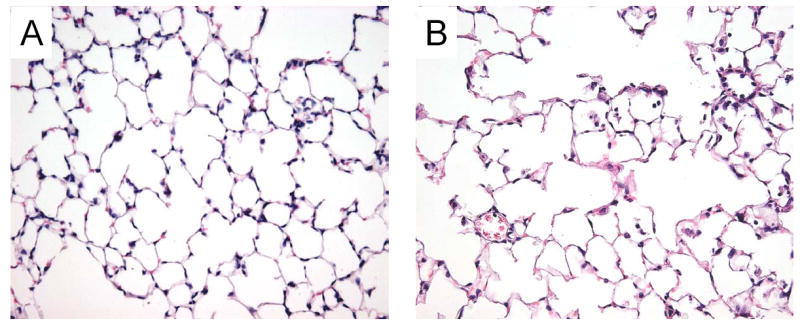
Paraffin-embedded lung tissues from a wildtype (A) and a BMPR2+/− (B) mouse were cut into 5-μm sections and stained with hematoxylin and eosin. Photomicrographs were taken at 400 × magnification.
The pulmonary artery pressure in the BMPR2+/− mice was assessed by measuring the right ventricular systolic pressure (RVSP) via right heart catheterization. In the absence of pulmonary valve disease, RVSP is equivalent to the pulmonary artery systolic pressure. As shown in Figure 2, the RVSP in the BMPR2+/− mice was similar to that of wildtype mice from 2–18 months of age, indicating that BMPR2+/− mice did not develop pulmonary hypertension spontaneously under unstressed conditions.
Figure 2. Right ventricular systolic pressure (RVSP).
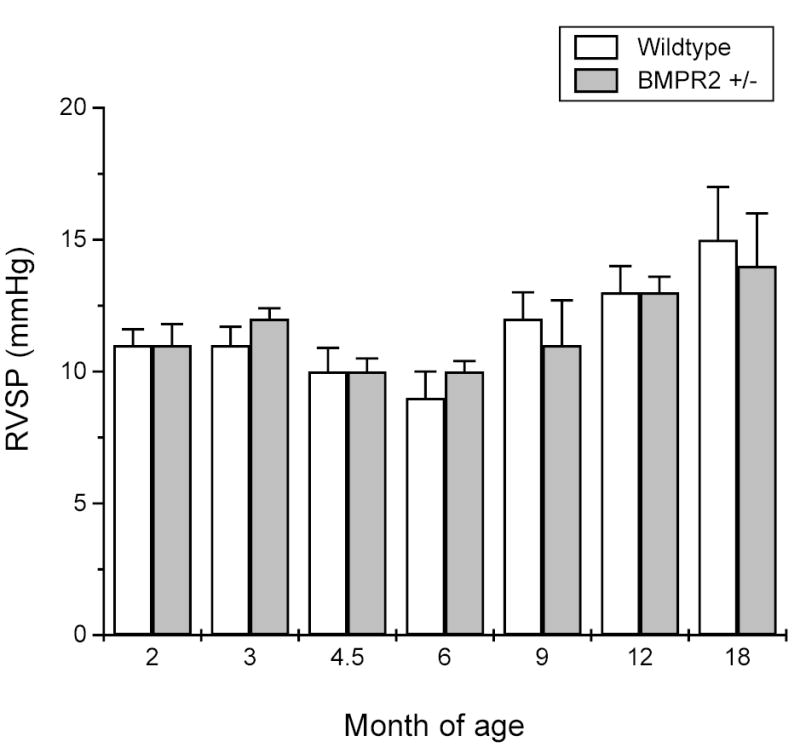
Right heart catheterization was performed on wildtype (white column) and BMPR2+/− (gray column) mice at 2, 3, 4.5, 6, 9, 12, or 18 months of age. No statistical differences were found between BMPR2+/− and wildtype mice groups and of any ages. Data are presented as mean ± SEM; n = 4–8 mice/group.
Expression of 5LO in lungs of mice
To examine the responses of BMPR2+/− mice to pulmonary inflammation, recombinant human 5LO was expressed in the lungs of the mice via adenovirus-mediated gene transfer. Approximately 2 × 108 plaque forming units of replication-deficient adenovirus expressing recombinant human 5LO (Ad5LO) was administered to the mice via intratracheal instillation. The time course of trangene expression was examined by Northern blotting. As shown in Figure 3, the expression of 5LO peaked at day 7 after the Ad5LO delivery and decreased significantly by day 21. No difference in transgene expression was found between wildtype and BMPR2+/− mice.
Figure 3. Northern blotting analysis of the time course of 5-lipoxygenase (5LO) transgene expression.
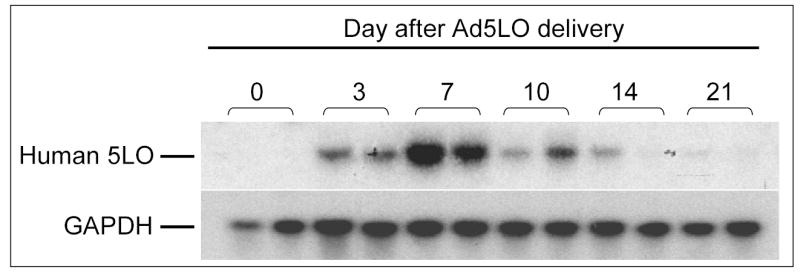
Wildtype mice were treated with 2×108 pfu Ad5LO via intra-tracheal instillation at day 0. Total RNA was isolated from the lung tissue of the mice at days 0, 3, 7, 10, 14, and 21. Northern blotting was carried out using an [α-32P]-dCTP-labeled human 5LO cDNA probe and a probe against mouse GAPDH. Two RNA samples from two individual mice were examined at each time point.
RVSP of mice after receiving Ad5LO
The RVSP of wildtype and BMPR2+/− mice was examined initially at day 12 after receiving Ad5LO, and compared to that of mice that received adenovirus expressing green fluorescence protein (AdGFP). The choice of the time point was based on a previous study, which showed that rats receiving Ad5LO and monocrotaline treatment developed pulmonary hypertension 10 days later 26. As shown in Figure 4, the RVSP was significantly increased in Ad5LO-treated BMPR2+/− mice compared to those with no treatment (23 ± 1 vs. 12 ± 1 mmHg, p < 0.05). The RVSP response of BMPR2+/− mice was not absolutely specific for exogenous 5LO expression, as the mice that received adenovirus expressing green fluorescent protein (AdGFP) had an elevated RVSP as well, although to a significantly milder degree (Fig. 4). Adenovirus-mediated gene transfer is known to cause inflammation. Thus, the increase in RVSP found in BMPR2+/− mice likely reflects a general response to inflammation, which is intensified by 5LO overexpression. Wildtype mice exhibited little increase in RVSP under either Ad5LO or AdGFP treatment, suggesting that BMPR2+/− mice are more sensitive to an inflammation-mediated RVSP increase than wildtype mice.
To investigate the time course of the change of RVSP in the mice, BMPR2+/− or wildtype mice were treated with Ad5LO and examined by right heart catheterization at days 0, 3, 7, 10, 12, 14, or 21 after Ad5LO delivery. As shown in Figure 5, the increase of RVSP in BMPR2+/− mice peaked at day 7 (33 ± 3 mmHg) and returned to near-normal by day 21 (14 ± 2 mmHg, compared to day 0, 12 ± 1 mmHg). The RVSP of wildtype mice increased mildly at day 7 (18 ± 2 mmHg) and became normal at day 12 (13 ± 1 mmHg). The increase of RVSP in the BMPR2+/− mice was consistent with the time course of transgene expression (shown in Fig. 3), and is significantly higher than that of wildtype mice.
To determine whether the change in RVSP was associated with pulmonary vascular remodeling, immunohistochemical staining for smooth muscle α-actin was carried out in lung tissue sections prepared from wildtype and BMPR2+/− mice after Ad5LO delivery. As shown in Figure 6, lung sections obtained at day 7 after Ad5LO delivery showed no sign of vascular remodeling in either type of mouse, although significant inflammatory cell infiltration was present. Increased muscularization of distal pulmonary arterioles was found in lung sections obtained at day 28 after Ad5LO treatment. Counting the α-actin-stained distal pulmonary arterioles (vessels that are located distal to terminal bronchioles, adjacent to the alveolar duct, non-collapsed, and with diameters less than 40 μm) showed that BMPR2+/− lungs had a significantly greater number of the muscularized vessels than lungs from wildtype mice (36 ± 3 vs. 7 ± 1) (Fig. 6, panel G). The muscularized distal arterioles accounted for approximately 10% of total distal arterioles in the BMPR2+/− lungs. The degree of muscularization (the thickness of the muscle layer), however, was mild. As this increase in muscularization occurred after the maximal increase in RVSP, the increased RVSP observed in BMPR2+/− mice in the first two weeks after Ad5LO delivery was not due to pulmonary vascular remodeling, but, rather, enhanced pulmonary vasoconstriction.
Figure 6. Muscularized distal pulmonary vessels in Ad5LO treated mice.
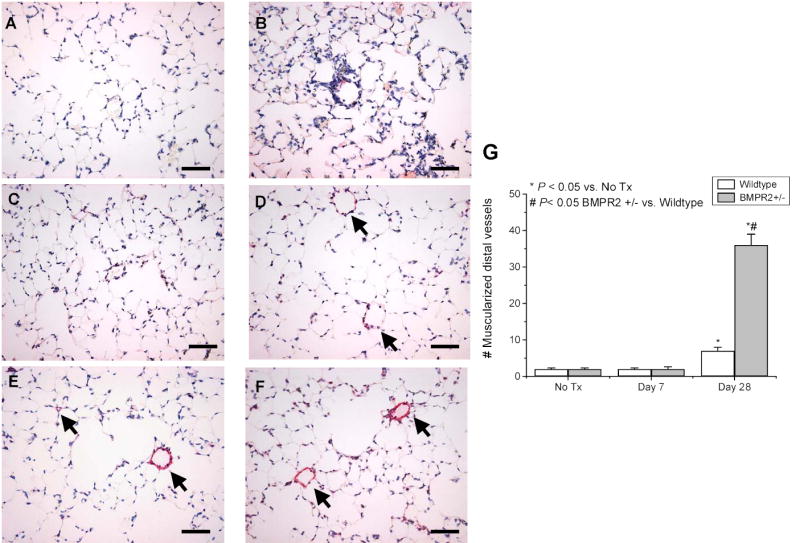
Mice were untreated (No Tx) or treated with 2×108 pfu Ad5LO at day 0, and lung tissues prepared at days 7 and 28. The lung sections were stained with anti-smooth muscle α-actin antibody (in red) and counterstained with hematoxylin (in blue). The number of muscularized pulmonary vessels with diameter less than 40 μm, located distal to terminal bronchioles, and adjacent to alveolar ducts was counted in 20 consecutive fields (200 x) per section. Panels A to E show photomicrographs (400x) of stained lung sections prepared at day 7 (panels A and B) and day 28 (panels C–E) from wildtype (panels A and C) and BMPR2+/− (panels B and D–F) mice. Panel G shows the average number of muscularized distal vessels in the lung sections obtained from 3–6 mice in each group. * indicates p<0.05 vs. No Tx group; # indicates p<0.05 BMPR2+/− vs. wildtype group at the same time point. Arrows indicate partially (panel D) and fully (panels E and F) muscularized distal pulmonary vessels. Bars = 50 μm.
Generation of vasoactive molecules during 5LO overexpression
To understand the molecular basis of the enhanced pulmonary vasoconstriction in BMPR2+/− mice during 5LO expression, urine samples were collected from the mice two days before (day -2) and 3, 7, 12, and 21 days after Ad5LO delivery. Urinary levels of several vasoactive molecules, including cysteinyl leukotrienes, prostacyclin metabolites, prostaglandin E2, thromboxane B2, and endothelin-1 were determined. As shown in Figure 7, the concentration of cysteinyl leukotrienes in urine was undetectable before Ad5LO delivery (day -2), but was markedly increased after 5LO expression. The production of cysteinyl leukotrienes peaked at day 7 (~20,000 pg/mg cysteinyl leukotrienes/urinary creatinine) and diminished by day 21. There was no difference between wildtype and BMPR2+/− mice in cysteinyl leukotriene production.
Urinary levels of prostacyclin metabolites, 6-keto-prostagaldin F1α and 2,3-dinor-6-keto-prostaglandin F1α, were next measured. As shown in Figure 8a, the basal level of urinary prostacyclin metabolites in BMPR2+/− and wildtype mice was similar, approximately 10,000 pg/mg (prostacyclin metabolites/urinary creatinine). The prostacyclin concentration was increased by 3–3.5 fold at days 3 and 7 after Ad5LO treatment and returned to the basal level by day 12. The increase was similar in BMPR2+/− and wildtype mice.
Urinary levels of prostaglandin E2, a pulmonary vasodilator, in BMPR2+/− and wildtype mice are shown in Figure 8b. The basal level of the compound in the two types of mice was 2,200–4,500 pg/mg (prostaglandin E2/urinary creatinine). During 5LO overexpression in the mice, the concentration increased to 16,000–20,000 pg/mg at days 3 and 7, and returned to the basal level by day 12. There was no significant difference in prostaglandin E2 production between the wildtype and BMPR2+/− mice. Interestingly, the pattern of prostaglandin E2 production in the mice was comparable to that of prostacyclin, which suggests that the increased production of these compounds was due to activation of an upstream enzyme(s) in prostaglandin synthesis, such as cyclooxygenase-2 27.
To determine whether the increased RVSP in the BMPR2+/− mice was due to over-production of vasoconstrictive molecules, the generation of endothelin-1 and thromboxane A2 in the mice during 5LO expression was examined. Results showed that urinary endothelin-1 was undetectable in these mice either before or after the Ad5LO delivery, indicating no marked increase in the production of this vasoactive peptide in these mice.
Thromboxane A2 production in the mice was assessed by measuring urinary concentrations of thromboxane B2 and 11-dehydro-thromboxane B2, metabolites of thromboxane A2. As shown in Figure 9, panel A, the basal level of urinary thromboxane B2 in both wildtype and BMPR2+/− mice was approximately 30,000 pg/mg (thromboxane B2/creatinine). During 5LO expression, thromboxane B2 concentration in the BMPR2+/− mice increased to 71,000, 116,000, 91,000, and 59,000 pg/mg at days 3, 7, 12, and 21, respectively. The change in thromboxane production in wildtype mice, by contrast, was less striking and did not reach statistical significance compared to pretreatment controls, with values of 47,000, 60,000, 39,000 and 24,000 pg/mg on days 3, 7, 12, and 21, respectively. Urinary 11-dehydro-thromboxane B2 in the Ad5LO treated mice showed a similar pattern of change as thromboxane B2 (Fig. 9, panel B). Thus, among the vasoactive molecules examined in this study, a difference between wildtype and BMPR2+/− mice was found only in thromboxane A2 production. The time course of the change in thromoboxane B2 level was comparable to that of the RVSP change in the mice.
Platelet activation
Platelet activation is the major source of thromboxane production in vivo 28, and urinary thromboxane metabolites have been used as markers for platelet activation. To determine whether or not the increased thromboxane A2 production in BMPR2+/− mice under inflammatory stress was due to augmented platelet activation, we examined two other platelet activation markers, plasma soluble P-selectin (sP-selectin) and urinary serotonin, in the mice. As shown in Figure 10, panel A, untreated wildtype and BMPR2+/− mice had similar plasma sP-selectin levels, 725 ± 26 and 751 ± 99 ng/ml, respectively. Ad5LO treatment increased the plasma sP-selectin concentration to 1,326 ± 100 and 1,791 ± 187 ng/ml in wildtype and BMPR2+/− mice, respectively. BMPR2+/− mice tended toward a higher sP-selectin level than wildtype mice, but the difference did not reach statistical significance. A similar trend was found in urinary serotonin concentrations in the two types of mice. As shown in Figure 10, panel B, before Ad5LO treatment, the urinary serotonin concentrations in wildtype and BMPR2+/− mice were similar, 73 ± 9 and 73 ± 8 pg/mg urinary creatinine, respectively. Seven days after Ad5LO treatment, urinary serotonin concentration increased to 173 ± 13 and 215 ± 25 pg/mg in wildtype and BMPR2+/− mice, respectively. The difference between wildtype and BMPR2+/− mice did not reach statistical significance. The relatively small difference in sP-selectin and serotonin levels found between the wildtype and BMPR2+/− mice after Ad5LO treatment suggested that platelet activation was not the primary source of the increased thromboxane A2 production in the Ad5LO-treated BMPR2+/− mice.
Effect of BMP on thromboxane A2 production in A549 cells
Pulmonary epithelial cells are another source of thromboxane A2 production in vivo 28,29. We, therefore, examined the effect of BMPs on thromboxane A2 production in a human pulmonary epithelial cell line, A549 cells. As shown in Figure 11, untreated A549 cells released a small amount of thromboxane B2 (207 ± 19 pg/mg total cell protein) in the basal state, and IL-1β stimulated thromboxane B2 production by more than 10-fold (2,822 ± 41 pg/mg). BMP-2 did not affect basal level of thromboxane B2 production (208 ± 11 pg/mg), but reduced IL-1β–stimulated production by ~ 20% (2,215 ± 186 pg/mg). BMP-6 had a similar effect (2,339 ± 41 pg/mg) as BMP-2 in the assay. These data indicated that BMP signaling did not directly affect thromboxane A2 production in A549 cells, but reduced the stimulating effect of IL-1β on the synthesis of this prostanoid.
Discussion
To understand the relationship between BMPR2 haploinsufficiency and pulmonary hypertension, this study examined BMPR2+/− mice and explored three issues: 1) whether BMPR2 haploinsufficiency affects the development of the mice; 2) whether haploinsufficiency alone leads to pulmonary hypertension; and 3) whether haploinsufficiency increases the susceptibility to pulmonary hypertension in the setting of inflammation. The results showed that BMPR2 heterozygosity caused approximately 20% fatality in mice during embryonic development, but did not lead to pulmonary hypertension under unstressed conditions. The study also showed that BMPR2+/− mice were more sensitive to inflammation-induced pulmonary hypertension than wildtype mice.
BMP signaling plays an important role in embryonic development, and is involved in gastrulation, mesoderm formation, neural patterning, skeletal development, and organogenesis (for reviews, see 5,30). Homozygous knockout mice deficient in BMP signaling proteins, such as BMP-2, BMP-4, BMPR2, BMPR-IA (Alk-3), ActR-I (Alk-2), Smad1, or Smad5, all die during embryonic development 30. By contrast, animals with heterozygous mutations in these genes are viable and reported to be grossly phenotypically normal. In this study, we found that approx. 20% of BMPR2+/− mice died in utero and/or during weaning, which suggests that haploinsufficiency of BMPR2 affects early development. Although we observed no apparent abnormality in the surviving BMPR2+/− mice under unstressed conditions in this study, some of these mice may have had minor developmental defects, and these defects could affect the phenotype of the mice under stressed conditions.
Inflammation has been suggested to be an important mechanism in the development of pulmonary arterial hypertension. Evidence supporting the hypothesis include: 1) inflammatory cells are found in the vicinity of remodeled pulmonary vessels with plexiform lesion; 2) proinflammatory cytokines and chemokines are increased in patients with pulmonary arterial hypertension; 3) pulmonary arterial hypertension is a common complication of autoimmune diseases involving systemic inflammation, such as scleroderma and systemic lupus erythematosus; and 4) in a rat model of pulmonary arterial hypertension, monocrotaline administration causes pulmonary endothelial injury and inflammation, which is followed by pulmonary vascular remodeling (for reviews, see 17–20). In the present study, we examined the effect of inflammation on the development of pulmonary hypertension in BMPR2+/− mice. Adenovirus-mediated overexpression of 5LO in the lung was used as the inflammatory stress because 5LO expression is increased in the human disease. The results showed that BMPR2+/− mice responded to the inflammation with an immediate marked increase of RVSP (peaking at day 7), and delayed muscularization of distal pulmonary arterioles (28 days after Ad5LO treatment). The early increase of RVSP is related to enhanced pulmonary vasoconstriction, due, at least in part, to increased thromboxane A2 production in the mice. The later muscularization of distal arterioles could be caused by the initial vasoconstriction, increased release of growth factors by inflammatory cells, and/or endothelial cell activation/injury caused by the inflammation. Further study is required to identify the specific mechanism. The overall degree of the muscularization found in the Ad5LO-treated BMPR2+/− mice is mild. This could be due to the transgene expression, as well, as the inflammation is transient. Further studies are required to demonstrate whether increased pulmonary vascular injury by sustained inflammation leads to extensive pulmonary vascular remodeling and persistent pulmonary hypertension in BMPR2+/− mice. BMPR2+/− mice were found to produce significantly higher amounts of thromboxane A2 than wildtype mice during 5LO overexpression. This observation is consistent with previous reports that thromboxane A2 production is significantly enhanced in patients with pulmonary arterial hypertension 31–33. Mechanistically, however, this finding raised the question of how BMPR2 deficiency leads to increased thromboxane A2 production. No report in the literature has linked BMP signaling to thromboxane A2 production. As the thromboxane A2 levels in the wildtype and BMPR2+/− mice were the same before Ad5LO delivery, the difference in thromboxane A2 production in these mice resides in the different responses to 5LO overexpression or inflammation. We, therefore, examined platelet activation in vivo and pulmonary epithelial cell activation in cell culture, as thromboxane synthase is most abundantly expressed in platelets (2,187 ng/mg protein) 34, and lung has the highest content of the enzyme, 765 ng/mg, among solid organ tissues 34. The cells in lung that express thromboxane synthase are mainly bronchial epithelial cells and alveolar macrophages in humans, and also small pulmonary artery smooth muscle cells in rats 29. Examining plasma sP-selectin and urinary serotonin, two platelet activation markers, showed that both of the markers tended to be higher in BMPR2+/− than wildtype mice, but neither difference reached statistical significance. Thus, platelet activation may not be the primary or the only source of the enhanced thromboxane A2 production in BMPR2+/− mice during 5LO expression. The pulmonary epithelial cell line, A549 cells, was found to produce a very small amount of thromboxane A2 under basal conditions, but the production of this prostanoid was stimulated markedly by IL-1β. BMP did not affect the basal level of thromboxane A2 production, but reduced IL-1β stimulation significantly by 20%. These effects suggest that BMP signaling does not regulate thromboxane A2 production directly, but interferes with IL-1β signaling.
IL-1β signaling activates two pathways, one leading to NFκB activation and the other, MAP kinase JNK and, subsequently, AP-1. A previous study has shown that BMP-7 inhibits IL-1β-induced JNK and AP-1 activation, but does not affect IL-1β-induced NFκB activation in human mesangial cells 35. If a similar selective inhibition occurs in pulmonary epithelial cells, it could explain the partial inhibitory effect of BMPs on the IL-1β-stimulated thromboxane A2 production in A549 cells. Further studies are required to understand the specific interaction between BMP and IL-1 signaling in these cells.
Thromboxane A2 is a potent vasoconstrictor and platelet activator 36,37. It also inhibits voltage-gated-potassium channels 38, and has synergistic effects with serotonin in causing vascular smooth muscle cell proliferation 39. These effects, when persistently produced by multiple inflammatory insults or stresses, could contribute to pulmonary vascular remodeling and sustained pulmonary hypertension. Understanding the relationship between BMPR2 haploinsufficiency and thromboxane A2 production under inflammatory stress could shed light on the mechanism of heterozygous BMPR2 mutation-mediated IPAH.
Acknowledgments
This study is supported by NIH grants HL58976, HL55993, and HL61795 (to JL); and grants from American Heart Association (0256282N) and Pfizer (Atorvastatin Research Award) (to YYZ).
References
- 1.Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–9. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- 2.Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–34. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 3.Massague J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 4.Kingsley DM. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994;8:133–46. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- 5.Hogan BL. Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580–94. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- 6.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 7.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, Higenbottam T, Gibbs JS, Egan J, Crozier A, Peacock A, Allcock R, Corris P, Loyd JE, Trembath RC, Nichols WC. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF- beta family. J Med Genet. 2000;37:741–5. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, Rich S, Fishman A. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 10.Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–7. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 11.Peacock AJ. Primary pulmonary hypertension. Thorax. 1999;54:1107–18. doi: 10.1136/thx.54.12.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet. 2003;361:1533–44. doi: 10.1016/S0140-6736(03)13167-4. [DOI] [PubMed] [Google Scholar]
- 13.Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, Coccolo F, Ventura C, Phillips JA, 3rd, Knowles JA, Janssen B, Eickelberg O, Eddahibi S, Herve P, Nichols WC, Elliott G. Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol. 2004;43:33S–39S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 14.Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, 3rd, Newman J, Williams D, Galie N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beppu H, Kawabata M, Hamamoto T, Chytil A, Minowa O, Noda T, Miyazono K. BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol. 2000;221:249–58. doi: 10.1006/dbio.2000.9670. [DOI] [PubMed] [Google Scholar]
- 16.West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94:1109–14. doi: 10.1161/01.RES.0000126047.82846.20. [DOI] [PubMed] [Google Scholar]
- 17.Voelkel NF, Cool C, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest. 1998;114:225S–230S. doi: 10.1378/chest.114.3_supplement.225s. [DOI] [PubMed] [Google Scholar]
- 18.Tuder RM, Voelkel NF. Pulmonary hypertension and inflammation. Journal of Laboratory & Clinical Medicine. 1998;132:16–24. doi: 10.1016/s0022-2143(98)90020-8. [DOI] [PubMed] [Google Scholar]
- 19.Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. 2002;45:173–202. doi: 10.1053/pcad.2002.130041. [DOI] [PubMed] [Google Scholar]
- 20.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–63. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 21.Samuelsson B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science. 1983;220:568–75. doi: 10.1126/science.6301011. [DOI] [PubMed] [Google Scholar]
- 22.Stenmark KR, James SL, Voelkel NF, Toews WH, Reeves JT, Murphy RC. Leukotriene C4 and D4 in neonates with hypoxemia and pulmonary hypertension. N Engl J Med. 1983;309:77–80. doi: 10.1056/NEJM198307143090204. [DOI] [PubMed] [Google Scholar]
- 23.Stenmark KR, Morganroth ML, Remigio LK, Voelkel NF, Murphy RC, Henson PM, Mathias MM, Reeves JT. Alveolar inflammation and arachidonate metabolism in monocrotaline- induced pulmonary hypertension. Am J Physiol. 1985;248:H859–66. doi: 10.1152/ajpheart.1985.248.6.H859. [DOI] [PubMed] [Google Scholar]
- 24.Wright L, Tuder RM, Wang J, Cool CD, Lepley RA, Voelkel NF. 5-Lipoxygenase and 5-lipoxygenase activating protein (FLAP) immunoreactivity in lungs from patients with primary pulmonary hypertension. Am J Respir Crit Care Med. 1998;157:219–29. doi: 10.1164/ajrccm.157.1.9704003. [DOI] [PubMed] [Google Scholar]
- 25.Zhang YY, Walker JL, Huang A, Keaney JF, Clish CB, Serhan CN, Loscalzo J. Expression of 5-lipoxygenase in pulmonary artery endothelial cells. Biochem J. 2002;361:267–276. doi: 10.1042/0264-6021:3610267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones JE, Walker JL, Song Y, Weiss N, Cardoso WV, Tuder RM, Loscalzo J, Zhang YY. Effect of 5-lipoxygenase on the development of pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol. 2004;286:H1775–84. doi: 10.1152/ajpheart.00281.2003. [DOI] [PubMed] [Google Scholar]
- 27.Brock TG, McNish RW, Peters-Golden M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J Biol Chem. 1999;274:11660–6. doi: 10.1074/jbc.274.17.11660. [DOI] [PubMed] [Google Scholar]
- 28.Nusing R, Ullrich V. Immunoquantitation of thromboxane synthase in human tissues. Eicosanoids. 1990;3:175–80. [PubMed] [Google Scholar]
- 29.Ermert L, Ermert M, Duncker HR, Grimminger F, Seeger W. In situ localization and regulation of thromboxane A(2) synthase in normal and LPS-primed lungs. Am J Physiol Lung Cell Mol Physiol. 2000;278:L744–53. doi: 10.1152/ajplung.2000.278.4.L744. [DOI] [PubMed] [Google Scholar]
- 30.Zhao GQ. Consequences of knocking out BMP signaling in the mouse. Genesis. 2003;35:43–56. doi: 10.1002/gene.10167. [DOI] [PubMed] [Google Scholar]
- 31.Barst RJ, Stalcup SA, Steeg CN, Hall JC, Frosolono MF, Cato AE, Mellins RB. Relation of arachidonate metabolites to abnormal control of the pulmonary circulation in a child. Am Rev Respir Dis. 1985;131:171–7. doi: 10.1164/arrd.1985.131.1.171. [DOI] [PubMed] [Google Scholar]
- 32.Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–5. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 33.Adatia I, Barrow SE, Stratton PD, Miall-Allen VM, Ritter JM, Haworth SG. Thromboxane A2 and prostacyclin biosynthesis in children and adolescents with pulmonary vascular disease. Circulation. 1993;88:2117–22. doi: 10.1161/01.cir.88.5.2117. [DOI] [PubMed] [Google Scholar]
- 34.Ullrich V, Nusing R. Thromboxane synthase. From isolation to function. Stroke. 1990;21:IV134–8. [PubMed] [Google Scholar]
- 35.Lee MJ, Yang CW, Jin DC, Chang YS, Bang BK, Kim YS. Bone morphogenetic protein-7 inhibits constitutive and interleukin-1 beta-induced monocyte chemoattractant protein-1 expression in human mesangial cells: role for JNK/AP-1 pathway. J Immunol. 2003;170:2557–63. doi: 10.4049/jimmunol.170.5.2557. [DOI] [PubMed] [Google Scholar]
- 36.Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci U S A. 1975;72:2994–8. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samuelsson B, Goldyne M, Granstrom E, Hamberg M, Hammarstrom S, Malmsten C. Prostaglandins and thromboxanes. Annu Rev Biochem. 1978;47:997–1029. doi: 10.1146/annurev.bi.47.070178.005025. [DOI] [PubMed] [Google Scholar]
- 38.Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ Res. 2003;93:656–63. doi: 10.1161/01.RES.0000095245.97945.FE. [DOI] [PubMed] [Google Scholar]
- 39.Pakala R, Willerson JT, Benedict CR. Effect of serotonin, thromboxane A2, and specific receptor antagonists on vascular smooth muscle cell proliferation. Circulation. 1997;96:2280–6. doi: 10.1161/01.cir.96.7.2280. [DOI] [PubMed] [Google Scholar]