

Abstract
Transcription regulation is a fundamental biological process, and extensive efforts have been made to dissect its mechanisms through direct biological experiments and regulation modeling based on physical–chemical principles and mathematical formulations. Despite these efforts, transcription regulation is yet not well understood because of its complexity and limitations in biological experiments. Recent advances in high throughput technologies have provided substantial amounts and diverse types of genomic data that reveal valuable information on transcription regulation, including DNA sequence data, protein–DNA binding data, microarray gene expression data, and others. In this article, we propose a Bayesian error analysis model to integrate protein–DNA binding data and gene expression data to reconstruct transcriptional regulatory networks. There are two unique aspects to this proposed model. First, transcription is modeled as a set of biochemical reactions, and a linear system model with clear biological interpretation is developed. Second, measurement errors in both protein–DNA binding data and gene expression data are explicitly considered in a Bayesian hierarchical model framework. Model parameters are inferred through Markov chain Monte Carlo. The usefulness of this approach is demonstrated through its application to infer transcriptional regulatory networks in the yeast cell cycle.
Keywords: gene expression, Markov chain Monte Carlo, misclassification
Transcription regulation involves synthesizing and/or degrading mRNA in response to the need of a biological system. Understanding its mechanisms has been a central topic in biology for many decades because of its importance as a fundamental biological process. Recently, large amounts of diverse types of genomic data, e.g., DNA sequence data (1–4), microarray gene expression data (5–7), and protein–DNA binding data (8–13) have been collected, and they yield valuable information on different aspects of transcription regulation. Motivated by the availability of these data, many computational approaches have been developed to model transcription regulation, where it is often formulated as an interaction network between sets of transcription factors (TFs) and genes being regulated (14–18). The potentially large numbers of nodes (TFs or genes), high connectivity, and transient behaviors of the network results in high complexity in such models. Because different types of genomic data reveal different aspects of the underlying transcriptional regulatory network (TRN), inference of TRNs based on information integration from different data types is expected to provide a more thorough understanding than that based on each data type alone. However, information integration is nontrivial because different data types are related to each other through a complex biological system, and they are generally collected with much noise.
In this article, we focus on transcription regulation in yeast as many diverse data types are available and much knowledge has been accumulated in the literature on this organism. Several research groups have attempted to dissect yeast transcription regulation by using multiple types of genomic data. For example, sequence data and gene expression data were considered jointly to infer regulatory elements (i.e., binding motif) (7, 18–20), and gene expression data and protein–DNA binding data were considered jointly to infer TRNs (12, 16). In the latter approach, the focus was on reconstructing large-scale TRNs for many yeast TFs (e.g., 113 TFs in ref. 10). However, information from different data types was not jointly modeled in a coherent framework in the existing approaches, and the associated measurement errors were not explicitly considered.
To address the limitations in the existing methods, we introduce a Bayesian model for inferring TRNs from two valuable data sources, microarray gene expression data and protein–DNA binding data, in this article. The advantages of our approach are that the model parameters have clear biological interpretations and the measurement errors for both data types are explicitly modeled in a coherent fashion. We also introduce Markov chain Monte Carlo (MCMC) methods for the inference of TRNs and other model parameters. The usefulness of this Bayesian error analysis model (BEAM) is illustrated through its application for studying yeast cell-cycle transcription regulation.
Results
In BEAM, there are three submodels: a system model, a misclassification model, and an exposure model. The system model relates gene expression and true TRN through chemical reaction models, the misclassification model connects true binary TRN with the observed protein–DNA binding data, and the exposure model specifies priors for transcription regulations in TRN. We have developed an MCMC algorithm to infer model parameters {βt, σt2; t = 1, …, T}, R, p, q, πR} from gene expression data {Yt; t = 1, …, T} and protein–DNA binding data [binding intensity matrix B and binary binding network (BN) W with threshold of P ≤ 0.001]. The notation can be found in Methods and Table 3, which is published as supporting information on the PNAS web site. We selected eight cell-cycle regulators, Fkh1, Fkh2, Swi4, Mcm1, Ace2, Ndd1, Mbp1, and Swi5 (see Data Sources). For 786 cell-cycle genes, we used the binding data of these eight cell-cycle regulators for these genes and their α-arrest cell-cycle gene expression data at 18 time points as inputs for BEAM. The details of BEAM and statistical inference are described in Methods. Our goal is to infer the underlying true TRN (R) of these eight regulators for cell-cycle genes, while we also gain understanding on the system model parameters, {βt, σt2; t = 1, …, T}, and the network parameters, {p, q, πR}. Our MCMC algorithm converged quickly (see Fig. 6, which is published as supporting information on the PNAS web site), and we chose a burn-in of 1,000 iterations followed with 4,000 iterations in our analysis.
Sensitivity Analysis.
We studied the sensitivity to prior specifications of model parameters. We specified noninformative priors Beta(1,1) for {p, q, πR}. The posterior means of the model parameters {βt, σt2} from either improper or proper priors are plotted across 18 time points in Fig. 1a. These estimates are robust with respect to prior specifications. The Pearson correlation coefficients between the posterior probabilities for TRN of each TF using different priors are generally high (above 0.9 except 0.78 for Fkh1). Using 0.5 as a threshold for inferring TRN (i.e., all TF gene pairs whose posterior probability is >0.5 are considered regulatory targets), the binary TRN was obtained for each gene. A Venn diagram showing the overlaps among BEAM results with improper prior, proper prior, and the observed BN is shown in Fig. 1b. The posterior TRN assuming an improper prior is consistent with that using a proper prior, and they both differ from BN. However, the overlap between the inferred TRN and the observed BN is still high. The posterior means and 90% credible intervals of parameters p, q, and πR are summarized in Table 1. Note that the inferred value of p is low, suggesting high false-negative rates of the observed protein–DNA binding data. One possible explanation is that for those genes whose expression profiles cannot be well explained by the observed protein–DNA binding data, the inclusion of other relevant TFs may improve model inference on their TRNs. Overall, model results are robust to prior specification. To avoid redundancy, we only present the results using improper priors in the following discussion.
Fig. 1.

Sensitivity to prior specification. (a) The posterior mean profiles of relative TF activities or σt2 versus 18 time points based on either improper (red triangle line) or proper (green cross line) prior distributions. (b) Venn diagram showing the overlaps of the inferred regulatory targets based on BEAM using improper and proper priors and those with the observed protein–DNA binding data.
Table 1.
Comparisons of BEAM results with either improper or proper prior distributions
| Parameters | Proper | Improper |
|---|---|---|
| p | 0.067 (0.056, 0.079) | 0.069 (0.057, 0.081) |
| q | 0.947 (0.941, 0.952) | 0.947 (0.941, 0.952) |
| πP | 0.290 (0.264, 0.315) | 0.264 (0.237, 0.291) |
We investigated the sensitivity of model parameter inference to different TF choices. When the system model included the eight cell-cycle regulators and different informative priors were assumed for {p, q, πR}, the posterior means of the relative TF activities were rather stable, except that Fkh1, Fkh2, and Ndd1 showed small variations (Fig. 2a). When we replaced Fkh2 and Ace2 with two TFs unspecific to cell-cycle regulation, Dal82 and Yap3, the posterior means based on different prior specifications are plotted in Fig. 2b, where a rather large variation of the inferred protein activities can be observed for Dal82 and Yap3. The results for Fkh1 became more unstable, but Ndd1 and other cell-cycle regulators stayed quite stable. This observation may be caused by the low quality of the binding data for Fkh1, marginally acceptable quality for Fkh2, but good quality for Ndd1 and other cell-cycle TFs (Table 2). Hence, these results suggest that the inference results are rather robust across multiple runs for the cell-cycle regulators whose protein–DNA binding data have good quality. Furthermore, robustness under different prior specifications may also serve as a diagnostic tool for the relevance of a certain TF in the system model.
Fig. 2.

Sensitivity analysis: the posterior means of the inferred relative activities of TFs versus 18 time points. (a) The system model included eight cell-cycle regulators. (b) The system model included six cell-cycle regulators and two TFs not specific to cell-cycle genes. The values for (p, q, πR) used are: (0.4, 0.8, 0.2), (0.2, 0.8, 0.5), (0.1, 0.8, 0.2), and (0.2, 0.9, 0.2).
Table 2.
Known DNA binding motifs and the reported qualities of the protein–DNA binding data
| TF | Binding site | Source | Binding data quality |
|---|---|---|---|
| Fkh2 | GTMAACAA | 13 | Acceptable (300) |
| Fkh1 | GTMAACAA | 13 | Poor (376) |
| Swi4 | CGCSAAA | TRANSFAC, ref. 13 | Good (289) |
| Mcm1 | CCNNNWWRGG | TRANSFAC, SCPD | Good (225) |
| Ace2 | GCTGGT | TRANSFAC, SCPD | Good (179) |
| Ndd1 | CCNRWWNNGG | 13 | Good (176) |
| Mbp1 | WCGCGW | TRANSFAC, ref. 13 | Good (223) |
| Swi5 | KGCTGR | TRANSFAC, SCPD, ref. 13 | Good (279) |
TRANSFAC, transcription factor database (www.gene-regulation.com/pub/databases.htm). SCPD, promoter database of Saccharomyces cerevisiae (http://rulai.cshl.edu/SCPD).
Correlation with Binding Motif in Sequence Data.
In the absence of complete knowledge of the true underlying TRN to assess the validity of our results, we evaluated our BEAM results indirectly through binding motifs. If our model performs well in terms of better identifying the regulatory targets of a TF, there should be enrichment for the binding motif for this TF in the upstream sequences of the genes inferred to be regulated by this TF.
According to the literature (Table 2), we identified binding motifs for the eight cell-cycle TFs and used these motifs to scan the upstream sequences of the cell-cycle genes. For a set of genes and a given TF, the enrichment of the binding motif for this TF is defined as the proportion of genes with at least one such motif in their upstream sequences. The gene set can be selected from the inferred TRN (R) or the observed BN (W). We calculated the enrichment score as a function of the number of inferred regulated genes per TF, and the results are shown in Fig. 3. Fig. 3 has one plot for each of the eight TFs and two curves within a plot, with one representing the motif enrichment score from the observed BN (marked with squares) and the other corresponding to that from the inferred TRN through BEAM (marked with up-triangles). It is easy to see that the motifs are more enriched in the inferred TRNs for Ace2, Ndd1, Mbp1, and Swi5 compared with the observed BN, similar between both networks for Swi4, but less enriched for Fkh1, Fkh2, and Mcm1.
Fig. 3.

The motif enrichment score versus the number of inferred regulatory genes for each of the eight cell-cycle TFs. The red dashed line with triangle symbols represents the BEAM results, and the black dashed line with square symbols represents results based on protein–DNA binding data.
The relatively poor performance for Fkh1 and Fkh2 may be explained by the lower qualities of the protein–DNA binding data on these two TFs (Table 2), coupled with the fact the binding motifs used in scanning were derived from the binding data itself. Sensitivity analysis results of these two TFs (Fig. 2) also revealed the relatively larger variations in their inferred activities. Different from Fkh1 and Fkh2, the binding data quality of Mcm1 was good and the binding motif was consistent across several databases, yet the binding motif was not enriched as much in the inferred TRN. Based on the literature, Mcm1 interacts with many coactivators or corepressors to regulate gene transcription (21–25). If Mcm1 barely functions as a regulator alone, its posterior activities at different time points may be influenced by its cofactors. The nonlinear effect of various protein complexes between Mcm1 and its cofactors may lead to the poor performance of BEAM in inferring its regulatory targets. However, the BEAM results for the other five TFs are promising. Overall, the results suggest that BEAM can effectively integrate protein–DNA binding data and gene expression data to better infer regulatory targets of TFs provided the protein–DNA binding data have good quality. We selected 0.5 as the threshold for inference of regulatory targets from the posterior network because at this level the dichotomized posterior TRN has similar motif enrichment as that in the observed BN with a rather stringent threshold of P ≤ 0.001. This inferred TRN for yeast cell cycle is discussed in the following section.
Inferred TRN for Yeast Cell Cycle.
A Venn diagram based on the inferred TRN and the observed BN is plotted in Fig. 4a, and the difference between the two networks can be clearly seen. With similar motif enrichment for each TF, 200 regulatory targets were inferred by BEAM but not in the observed BN (W) defined with threshold P ≤ 0.001. A total of 94 genes with weak binding evidence (P > 0.05) were inferred to be regulatory targets by BEAM, whereas 67 genes with strong binding evidence (P ≤ 0.001) were not inferred as regulatory targets. In general, we found that genes in the former group tended to have strong gene expression variations and clear cell-cycle patterns (Fig. 4b), and genes in the latter group tended to show weak expression changes over all of the time points (Fig. 4c). The above results indicate that the inferred TRN does effectively integrate gene expression data to explain strong in vivo gene regulation in a real biological process. However, because the exact TRN in the cell cycle of the α-arrest experiment is still unknown, our model results can only serve as an exploratory tool to guide the reconstruction of in vivo time-independent TRN.
Fig. 4.

Comparisons between the inferred TRN from BEAM and the observed protein–DNA binding data. (a) Venn diagram for comparisons. All cell-cycle genes (786 total) are in the black circle. The inferred regulated genes based on BEAM (343 genes) are in the sky-blue circle, the inferred regulated genes based on the observed BN with a P ≤ 0.001 cut-off (210 genes) are in the red circle, and the inferred regulated genes based on the observed BN with a P ≤ 0.05 cut-off (400 genes) are in the orange circle. (b and c) Focus on Mbp1, gene expression profiles versus 18 time points are plotted for 57 genes that were inferred to be regulatory targets by BEAM but had weak binding evidence (b) and 29 genes with strong binding evidence but were not inferred to be regulated by Mbp1 (c).
Conclusions
In this article, we have proposed a Bayesian model, BEAM, to integrate protein–DNA binding data with gene expression data with explicit consideration of the measurement errors in these genomic data for the inference of TRNs. MCMC was implemented for statistical inference, and this procedure yielded robust results when relevant TFs were studied in the model. When applied to dissect TRNs in the yeast cell cycle, we observed that the inferred regulatory targets for those TFs with good-quality binding data and robust regulatory role were more enriched for known binding motifs in their upstream sequences compared with those inferred based on protein–DNA binding data alone.
We have focused on this set of TRNs, both because the yeast cell cycle is a well studied biological process, and more importantly, because we assumed that the underlying TRNs for this process are time-independent. This assumption may be valid for some core biological processes, but is likely invalid when experimental conditions change (13) or nonessential processes are studied. If we think of the whole TRN as an organized network according to its biological functions, TRN in the yeast cell cycle is only one module of the full network. It is unclear how to model transient behavior of the currently incomplete network to account for the variations of observed gene expression or protein–DNA binding data. Hence, modeling at a smaller scale like BEAM may be one reason for us to obtain some insights on TRNs in yeast cell cycle.
Although we have focused on small-scale networks, there lacks ground truth even for this system to evaluate computational methods for TRN reconstruction. Simulations have been used to study and demonstrate the good performance of a similar method (N.S. and H.Z., unpublished results); however, they are rather limited by the models used to simulate data. In this article, we used DNA sequence data to evaluate the model results by examining the enrichment of known binding motif of a TF in the upstream sequences of the inferred regulatory targets. Although some researchers (16) evaluated their results by using Gene Ontology (www.geneontology.org) information, such information is rather incomplete and nonspecific, making it less useful compared with motif enrichment.
The motif enrichment results are encouraging. A number of modifications of BEAM may further improve TRN inference. Note that in BEAM the constant error structures are assumed for both types of data. These simplifying assumptions on errors may lead to the failed TRN inference for Fkh1 and Fkh2 because different measurement errors are embedded in their binding data. A more sophisticated error structure such as TF-specific error may be required to account for varying data qualities. In addition, the measurement errors in the binding intensities (B) may also need to be incorporated. In the system model, the equilibrium assumptions of transcription initiation and the quasi steady-state assumption of mRNA synthesis and degradation may not hold. Incorporation of kinetics in the system model may be crucial to improve BEAM. In summary, we note that BEAM provides a flexible framework for incorporating the above suggested extensions through modifying submodels, integrating more data types, and adjusting error assumptions.
Methods
BEAM has three submodels: a system model, a misclassification model, and an exposure model. The three submodels amalgamate into a hierarchical structure (Fig. 5). The details of each submodel are described in the following.
Fig. 5.

The hierarchical structure of BEAM. The unknown parameters are in ovals, and the observations are in rectangles.
System Model.
The system model describes the relationship between measured gene expression levels, TRN, and TF activities. A TRN is represented by a matrix with each row corresponding to the specific TRN of each gene and each column corresponding to the regulation targets of each TF. The (i, j)th entry in the matrix describes the role of the jth TF in regulating gene i. In our analysis, the transcription activity of each gene is measured through gene expression data.
We model mRNA regulation as a closed reacting system, which involves proteins, chromosomes, nucleotide bases, mRNA, and intermediate species. We assume that specific bindings of TFs or interacting TFs to immobilize partial TFs from cell solution onto DNA sequences and the reaction for the bound TFs to further recruit RNA polymerase II complex (Pol II) onto promoter region of DNA are accomplished by a set of reversible reactions. For this given set of chemical species, there may exist multiple sets of independent reactions to relate the species as reactants and products. However, the minimization of free (or Gibbs) energy determines that a specific set of reversible reactions actually happens. Here we assume that all reversible reactions reach equilibrium. The amounts of atomic species are conserved in this closed system, so that an overall chemical reaction stoichiometry of transcription initiation is given in the chemical equation (CE):
 |
where there are a total of J TFs as regulators of gene i, the stoichiometric coefficient aij represents the effective abundance of bound TFj involved in the regulation of gene i, and DNAi is the sequence of gene i. The product of transcription initiation is an immobilized compound denoted by [DNAi(TF1)αi1 … (TFJ)αiJPol II]. Although we cannot obtain the set of reversible reactions and write the formulae for their equilibrium constants, we can obtain an apparent equilibrium constant as the product of all equilibrium constants. The activities of intermediate species cancel out in this formula based on stoichiometry analysis (CE). This apparent equilibrium constant can be expressed as
 |
The combinatorial effect among TFs is included in the selected set of reactions constrained by minimum Gibbs energy. Therefore, the apparent equilibrium constant reflects this combinatorial effect. In this article, we assume that TRN is time-independent, so the combinatorial effect on the equilibrium constant is time-invariant. This assumption should generally hold for the TRN in the yeast cell cycle. Therefore, if we assume that reaction equilibrium is reached at each time point, the apparent equilibrium constant (Keq) does not change for the TRN of a given gene. We further assume that there are sufficient RNA Pol II complexes in cells so that [Pol II] = 1 and [DNAi] remains constant. Stoichiometry analysis on this process then leads to Eq. 2:
 |
The complex in Eq. 2 is modeled to catalyze the mRNA synthesis. Assume that nucleotide bases are sufficient, the mRNA synthesis rate reaches maximum value, which is proportional to the activity of [DNAi(TF1)ai1…(TFJ)aiJPol II]. By a quasi steady-state assumption for mRNA synthesis and degradation, we have
By combining Eqs. 2 and 3 and performing log transformation, we have
 |
where y′it = [mRNAi]t, y′i0 = [mRNAi]0, β′jt = [TFj]t, β′j0 = [TFj]0, and t = 0 refers to a reference sample, e.g., asynchronized cell sample, which may be considered as an average quantity over multiple time points. Let βjt = log2(β′jt/β′j0) and Yit = log2(Y′it/Y′i0), where Yit represents the relative gene expression level at time t, and βjt is the unknown relative activity of TFj at time t and needs to be estimated.
The above model describes the expected gene expression levels. However, microarray data are noisy and the biological system is intrinsically stochastic. In BEAM, we assume that the observed gene expression data differ from the expected level described in Eq. 4 by an error term, and we further assume that the errors for all of the genes at the same time point have the same distribution, while allowing the errors at different time points to have different distributions. This leads to the following system model for all genes in vector notation,
where Yt = (y1t, …, yNt)T, βt = (β1t, …, βJt)T, A = {aij; i = 1, …, N, j = 1, …, J}, et = (e1t, …, eNt)T, eit ∼iid N(0, σ t2), N is the number of genes, and J is the number of TFs. In our following discussion, we consider different prior distributions for β and σ 2 in BEAM (26), a noninformative improper prior (Eq. 6a) and a proper prior (Eq. 6b)
where Σβ0 is a diagonal matrix with diagonal elements being σβj02, IG stands for inverse Gamma distribution, βj0 ∼ U(−2,2), σβj02 ∼ U(0.001, 1.0), n0 = 5, and σ 02 = 2. This system model links relative mRNA abundance of gene i with relative activities of TFs through TRN. The elements of TRN are stoichiometric coefficients for transcription initiation. Because aij is specific between TFj and gene i, the same TF may have different impacts on different genes, and different TFs may have different regulation roles on the same gene. We use the measured relative protein-binding intensity of a TF to approximate the value of aij if TFj regulates gene i. Therefore, we consider TRN as consisting of two components: a binary network (rij), indicating the involvement of TFj in the transcriptional regulation of gene i; and if rij is 1, we use the relative binding intensity (bij) to approximate the stoichiometric coefficient aij for TFj and gene i. So the element (aij) in TRN can be expressed as the product of bij and rij.
Therefore, complete knowledge of R = {rij} and B = {bij} determines the TRN. Here we use a threshold level, e.g., 0.001, to define the observed binary protein–DNA BN, W = {wij}. We consider W as the surrogate of true binary TRN (R). Note that because of nonequivalence between physical binding and regulation, W and R can differ. In the following, we describe a misclassification model to account for the discrepancy between W and R.
Misclassification Model.
Recall that rij denotes the underlying regulation relationship between TFj and gene i, wij denotes the observed physical binding data dichotomized through a threshold, we introduce p and q to denote the sensitivity and specificity of W with respect to R defined in the following:
where we assume that p and q are constants for any pair of TF and gene. In BEAM, we assume that the prior distributions for p and q are Beta(b1, c1) and Beta(b2, c2), respectively, where different specifications of b1, c1, b2, and c2 represent different prior beliefs on p and q.
Exposure Model.
For this submodel, we specify the prior distribution of the binary TRN R, which describes the probability of rij being 1. We assume that the rij are independent and have the same Bernoulli distribution with parameter πR. We assume a Beta prior distribution Beta(b3, c3) for πR.
MCMC Algorithm for Statistical Inference.
In our model set-up, a large number of unknown parameters {{βt, σt2; t = 1, …, T}, R, p, q, πR} need to be inferred based on the observed gene expression data {Yt; t = 1, …, T} and protein–DNA binding data B = {bij} and W = {wij}. Let A = {bijrij}. We propose to use the following MCMC algorithm iterated between the following two steps for statistical inference: (i) sample {{βt, σt2; t = 1, …, T}, p, q, πR} conditional on the updated estimates of R; and (ii) sample R conditional on the updated estimates of {{βt, σt2; t = 1, …, T }, p, q, πR}. These two steps are described in detail in the following.
In the first step, given the current estimates of the components in the regulatory matrix R, the system model reduces to a standard linear regression model. The parameters {βt, σt2; t = 1, …, T} can be sampled as follows:
- For the improper prior (Eq. 6a), the conditional posteriors of βt and σt2 are (26):
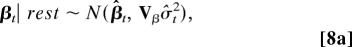
where Vβ = (ÂTÂ)−1, each element in TRN  is âij and âij = bijr̂ij, β̂t = VβÂTYt, st is the sample standard deviation for the errors associated with gene expression data at time point t, and R̂ is the current estimate for the TRN.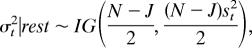
The parameters related to TRN, i.e., parameters in the Beta distributions of {p, q, πR}, are sampled based on the comparisons between the estimated regulatory matrix R̂ and the observed binding matrix W. Recall that we assumed Beta distributions Beta(b1, c1), Beta(b2, c2), and Beta(b3, c3) for these three parameters. Therefore, it is easy to see that the posterior distributions for these three parameters are
where the estimated rij is compared with wij, and n̂kl represents the number of TF gene pairs whose estimated true regulation is k (r̂ij = k) and the observed one is l (wij= l) with k and l being either 0 or 1; n̂0 (n̂1) is the number of elements with r̂ij = 0 (r̂ij = 1) in R. Therefore we have n̂1 = n̂10 + n̂11, and n̂0 = n̂00 + n̂01.
In the second step, given the sampled parameter estimates of {{βt, σt2; t = 1, …, T}, p, q, πR}, we sample the TRN R by sampling each row in R (which corresponds to the regulation pattern for each gene) one at a time from K = 2J possible patterns as follows,
 |
where Lik is the log-likelihood for each pattern k, and Lik = LikR + LikY + constant. LikY is the log-likelihood contribution from gene expression data, which is −Σt=1T((Yit − Ŷikt)2/2σ̂t2), and LikR = n̂1 logπ̂R + n̂11 log p̂ + n̂10 log(1 − p̂) + n̂0 log(1 − π̂R) + n̂01log (1 − q̂) + n̂00 logq̂, and it represents the log-likelihood contribution from protein–DNA binding data. We repeat this for each of the N genes to obtain the updated R̂ for the next iteration.
Based on the sampled parameter values, we can derive the posterior distributions for all of the unknown parameters in the model. For example, we can obtain the marginal posterior distribution of the regulation between TFj and gene i by using the proportion of sampled rij that is 1. These posterior probabilities can then be used to infer the presence or absence of regulation of TFj on gene i through specifying a threshold, e.g., the posterior mean of πR, such that all of the entries below this cut-off are inferred to have no regulation effect, whereas all of the entries having values above this cutoff are inferred to have a regulation role.
Data Sources.
For gene expression data, we used the yeast α-arrest gene expression data reported by Spellman et al. (7). There were 18 time points covering two cell cycles. Spellman et al. identified 800 cell cycle genes For protein–DNA binding data, we used the data in Lee et al. (10), where protein–DNA binding data for 113 TFs on 6,270 genes were reported. Among the 800 cell-cycle genes, 794 genes had protein–DNA binding data. We further removed eight genes because all of their expression levels were missing in the α-arrest experiment, resulting in a total of 786 cell-cycle genes being studied here.
From protein–DNA binding data of each TF, a threshold (e.g., 0.05) can be set for the observed statistical evidence for binding to separate the 786 genes into two classes: those genes with P ≤ 0.05 are considered to be bound by this TF and those with P > 0.05 are considered not bound by this TF. For each time point, we calculated the t statistics to test the null hypothesis that these two groups of genes have the same average gene expression level at the given time point and selected the maximum t statistic across all 18 time points to represent the correlations between the binding data and gene expression data of this specific TF. Then we randomly permuted the binding data of this given TF with 786 genes 10,000 times. A total of 20 TFs were selected at family wise error rate < 0.07 whose binding patterns were associated with gene expression variations during the cell cycle (Table 4, which is published as supporting information on the PNAS web site).
Then we evaluated the binding specificity of these 20 TFs to 786 cell-cycle genes versus all 6,270 yeast genes. For each TF, we constructed a 2 × 2 table with rows representing 786 cell-cycle genes or other yeast genes and columns representing the number of genes being bound or unbound by the given TF. A hypergeometric distribution was used to estimate the statistical significance of the binding specificity of the given TF to the cell cycle genes. We ranked the specificity of 113 TFs. The top 11 cell-cycle-specific binding TFs are among the chosen 20 TFs (Fig. 7 and Table 4, which are published as supporting information on the PNAS web site).
We applied hierarchical clustering analysis on the protein–DNA binding intensities of the 20 TFs and 786 cell-cycle genes, and the results are shown in Fig. 7. Nine of these 20 TFs in a tight cluster are well known cell-cycle regulators. We removed one cofactor, Swi6, from the list and focused on eight other cell-cycle regulators (Fkh1, Fkh2, Swi4, Mcm1, Ace2, Ndd1, Mbp1, and Swi5) in our following analysis. These eight TFs, except Ace2 (which has a rank of 37 with P being 0.007), have top cell-cycle specificity ranks among all 113 TFs. These results are consistent with literature (Saccharomyces Genome Database, www.yeastgenome.org).
Our objective is to infer TRN between the eight TFs and 786 cell-cycle genes. The missing entries in gene expression data and protein–DNA binding data were assumed to be missing at random and the k-nearest neighbor method was used to infer their values, where k was set to 5. The Euclidean distance was used to determine the nearest neighbors for a given gene (27).
Supplementary Material
Acknowledgments
We thank two reviewers for their constructive comments. This work was supported in part by National Science Foundation Grant DMS-0241160, National Cancer Institute Grant CA-57030, and the Texas A&M Center for Environmental and Rural Health via National Institute of Environmental Health Sciences Grant P30-ES09106.
Abbreviations
- TF
transcription factor
- TRN
transcriptional regulatory network
- MCMC
Markov chain Monte Carlo
- BEAM
Bayesian error analysis model
- BN
binding network
- Pol II
polymerase II complex
- CE
chemical equation.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bassett D. E., Jr., Basrai M. A., Connelly C., Hyland K. M., Kitagawa K., Mayer M. L., Morrow D. M., Page A. M., Resto V. A., Skibbens R. V., et al. Curr. Opin. Genet. Dev. 1996;6:763–766. doi: 10.1016/s0959-437x(96)80033-5. [DOI] [PubMed] [Google Scholar]
- 2.Goffeau A., Barrell B. G., Bussey H., Davis R. W., Dujon B., Feldmann H., Galibert F., Hoheisel J. D., Jacq C., Johnston M., et al. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 3.Mewes H. W., Hani J., Pfeiffer F., Frishman D. Nucleic Acids Res. 1998;26:33–37. doi: 10.1093/nar/26.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kellis M., Patterson N., Endrizzi M., Birren B., Lander E. S. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- 5.Cho R. J., Campbell M. J., Winzeler E. A., Steinmetz L., Conway A., Wodicka L., Wolfsberg T. G., Gabrielian A. E., Landsman D., Lockhart D. J., et al. Mol. Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 6.Chu S., DeRisi J., Eisen M., Mulholland J., Botstein D., Brown P. O., Herskowitz I. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 7.Spellman P. T., Sherlock G., Zhang M. Q., Iyer V. R., Anders K., Eisen M. B., Brown P. O., Botstein D., Futcher B. Mol. Biol. Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren B., Robert F., Wyrick J. J., Aparicio O., Jennings E. G., Simon I., Zeitlinger J., Schreiber J., Hannett N., Kanin E., et al. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 9.Iyer V. R., Horak C. E., Scafe C. S., Botstein D., Snyder M., Brown P. O. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 10.Lee T. I., Rinaldi N. J., Robert F., Odom D. T., Bar-Joseph Z., Gerber G. K., Hannett N. M., Harbison C. T., Thompson C. M., Simon I., et al. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 11.Horak C. E., Snyder M. Methods Enzymol. 2002;350:469–483. doi: 10.1016/s0076-6879(02)50979-4. [DOI] [PubMed] [Google Scholar]
- 12.Bar-Joseph Z., Gerber G. K., Lee T. I., Rinaldi N. J., Yoo J. Y., Robert F., Gordon D. B., Fraenkel E., Jaakkola T. S., Young R. A., et al. Nat. Biotechnol. 2003;21:1337–1342. doi: 10.1038/nbt890. [DOI] [PubMed] [Google Scholar]
- 13.Harbison C. T., Gordon D. B., Lee T. I., Rinaldi N. J., Macisaac K. D., Danford T. W., Hannett N. M., Tagne J. B., Reynolds D. B., Yoo J., et al. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W., Cherry J. M., Botstein D., Li H. Proc. Natl. Acad. Sci. USA. 2002;99:16893–16898. doi: 10.1073/pnas.252638199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segal E., Yelensky R., Koller D. Bioinformatics. 2003;19:i273–i882. doi: 10.1093/bioinformatics/btg1038. [DOI] [PubMed] [Google Scholar]
- 16.Gao F., Foat B. C., Bussemaker H. J. BMC Bioinformatics. 2004;5:31. doi: 10.1186/1471-2105-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beer M. A., Tavazoie S. Cell. 2004;117:185–198. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- 18.Hughes J. D., Estep P. W., Tavazoie S., Church G. M. J. Mol. Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- 19.Liu X. S., Brutlag D. L., Liu J. S. Nat. Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- 20.Roven C., Bussemaker H. J. Nucleic Acids Res. 2003;31:3487–3490. doi: 10.1093/nar/gkg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lydall D., Ammerer G., Nasmyth K. Genes Dev. 1991;5:2405–2419. doi: 10.1101/gad.5.12b.2405. [DOI] [PubMed] [Google Scholar]
- 22.Ercan S., Reese J. C., Workman J. L., Simpson R. T. Mol. Cell Biol. 2005;25:7976–7987. doi: 10.1128/MCB.25.18.7976-7987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carr E. A., Mead J., Vershon A. K. Nucleic Acids Res. 2004;32:2298–2305. doi: 10.1093/nar/gkh560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pramila T., Miles S., GuhaThakurta D., Jemiolo D., Breeden L. L. Genes Dev. 2002;16:3034–3045. doi: 10.1101/gad.1034302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mead J., Bruning A. R., Gill M. K., Steiner A. M., Acton T. B., Vershon A. K. Mol. Cell Biol. 2002;22:4607–4621. doi: 10.1128/MCB.22.13.4607-4621.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelman A., Carlin J. B., Stern H. S., Rubin D. B. Bayesian Data Analysis. London: Chapman & Hall; 2003. [Google Scholar]
- 27.Troyanskaya O., Cantor M., Sherlock G., Brown P., Hastie T., Tibshirani R., Botstein D., Altman R. B. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.