

Abstract
Amyloid fibrils are associated with >20 fatal human disorders, including Alzheimer's, Parkinson's, and prion diseases. Knowledge of how soluble proteins assemble into amyloid fibrils remains elusive despite its potential usefulness for developing diagnostics and therapeutics. In at least some fibrils, runaway domain swapping has been proposed as a possible mechanism for fibril formation. In runaway domain swapping, each protein molecule swaps a domain into the complementary domain of the adjacent molecule along the fibril. Here we show that T7 endonuclease I, a naturally domain-swapped dimeric protein, can form amyloid-like fibrils. Using protein engineering, we designed a double-cysteine mutant that forms amyloid-like fibrils in which molecules of T7 endonuclease I are linked by intermolecular disulfide bonds. Because the disulfide bonds are designed to form only at the domain-swapped dimer interface, the resulting covalently linked fibrils show that T7 endonuclease I forms fibrils by a runaway domain swap. In addition, we show that the disulfide mutant exists in two conformations, only one of which is able to form fibrils. We also find that domain-swapped dimers, if locked in a close-ended dimeric form, are unable to form fibrils. Our study provides strong evidence for runaway domain swapping in the formation of an amyloid-like fibril and, consequently, a molecular explanation for specificity and stability of fibrils. In addition, our results suggest that inhibition of fibril formation for domain-swapped proteins may be achieved by stabilizing domain-swapped dimers.
Keywords: disulfide bond, fibrillization, protein aggregation, protein design
The association of amyloid fibril formation with >20 fatal human diseases, including Alzheimer's, Parkinson's, and prion diseases, has stimulated much work on the molecular basis of fibril formation (1–3). Although the proteins involved in different amyloid diseases lack similarities in sequence and structure, the fibrils do share similar elongated morphologies (4), dye-binding properties (5, 6), and a characteristic “cross-β” diffraction pattern (7). These similarities suggest that amyloid-like fibrils share similar underlying features of molecular structure and implicate that there may exist some general organizing principles for amyloid fibril formation. Recently the structure of a cross-β spine has been determined, revealing a pair of β-sheets tightly intermeshed by a dry “steric zipper” (8). Cross-β diffraction pattern shared by amyloids suggest that a similar cross-β spine is at the center of many or all amyloid-like fibrils (9). But the cross-β spine is formed from only a short segment (≈6 residues of each molecule), so the question remains how the full-length protein rearrange its structure to assemble from a soluble form into amyloid fibrils.
One proposed mechanism for fibril formation is called runaway domain swapping. Domain swapping refers to an oligomeric assembly in which two or more protein molecules exchange a small domain to form intertwined oligomers (10). All of the domain-swapped oligomers characterized so far are close-ended oligomers (11), which are unable to further oligomerize into higher-order oligomers. To form amyloid-like fibrils, domain swapping must take place in an open-ended fashion in which each protein molecule swaps a domain into the complementary domain of the adjacent molecule along the fibrils, permitting addition of molecules at both ends to elongate fibrils (12). This open-ended domain swapping is named runaway domain swapping. Runaway domain swapping can account for some important features of amyloid fibrils. For example, the well established species barrier in the prion diseases indicates that fibril formation has high sequence specificity. Transmission between animals of different species is much less efficient than between animals of the same species (13). In vitro studies show that cross-seeding, i.e., by using the fibrils of one protein to seed another protein, strongly depends on sequence similarity (14). Presence of inhomogeneous species of protein molecules, even with only one residue difference, often results in inhibition of fibril formation (15–17). Because runaway domain swapping uses the same native interactions that specify a unique protein fold, a runaway domain swapping mechanism for fibril formation would unsurprisingly confer high sequence specificity to amyloid fibrils.
Despite being theoretically plausible, the runaway characteristic of domain swapping in fibril formation lacks substantial experimental evidence. The role of domain swapping in amyloid fibril formation has been studied in several proteins (18–28). For example, a strong correlation between the ability to domain swap and the ability to form fibrils has been shown in the studies of cystatin C. The cystatin C L68Q variant found in the amyloid plaques of hereditary cystatin C amyloid angiopathy patients exists mostly in the dimer form in vitro at body temperature, whereas the wild-type protein stays as a monomer (18). The crystal structures of cystatin C show that the dimers are formed by the mechanism of domain swapping (19, 20). Stabilization of the monomeric form by either intramolecular disulfide bonds, or binding to its antibody or substrate, inhibits fibril formation (21). But no evidence was given as to whether a runaway type of domain swap is required for the fibril formation of cystatin C. Recently, studies of complementary active site mutants of ribonuclease A showed that the amyloid-like fibrils formed by the ribonuclease A mutants retain enzymatic activity, which, by experimental design, can be achieved only by a domain swap (28). However, the results of ribonuclease A work can be explained by a simple stack of domain-swapped dimers, leaving the question of whether the domain swap is of a runaway type unanswered.
Here we assess the role of domain swapping in amyloid fibril formation by using a model system, the enzyme T7 endonuclease I (T7EI). T7EI is a DNA junction resolving enzyme from bacteriophage T7 (29). T7EI exists naturally as a domain-swapped dimer as shown in the crystal structure (30). Strictly speaking, T7EI should be classified as a candidate for 3D domain swapping, because no monomeric structures of T7EI have been solved. The swapped domain includes the N-terminal 44 residues that form a long α-helix and two β-strands, the hinge loop comprises residues 45–48, and the core domain is formed from residues 49–149. Here we show that T7EI forms amyloid-like fibrils under native conditions. By engineering two disulfide bonds at the domain-swapped dimer interface, the process of fibril formation was followed by gel electrophoresis. We find that the fibrils formed by the disulfide mutant are connected by intermolecular disulfide bonds, a linkage that is achieved only when the protein adopts a runaway type of domain swap in the fibrils, thus providing definitive evidence for runaway domain swapping in the fibril formation of T7EI.
Results
T7EI Forms Amyloid-Like Fibrils.
T7EI forms large-molecular-mass aggregates upon incubation at 37°C for 3 to 7 days at neural pH at concentrations >200 μM. These aggregates do not enter the stacking gel during native PAGE (Fig. 1a). The sample at 4°C remains mostly as soluble dimers, with a minority component of a small oligomer (Fig. 1a). Electron microscopy reveals that T7EI forms curvilinear fibrils with a diameter of ≈10 nm (Fig. 1b). X-ray crystallography studies of orientated fibrils show strong meridional reflections at ≈4.7 Å, characteristic of cross-β structure (Fig. 1c). A weak reflection at 10.5 Å is also present, indicative of β-sheets. The T7EI fibrils bind Congo red (CR), resulting in a red shift of the absorption spectrum (Fig. 1d). The fibrils also bind thioflavin T (ThT), giving rise to characteristic fluorescence emission peak at 482 nm (Fig. 1e). Thus, T7EI fibrils have amyloid properties.
Fig. 1.

Amyloid-like properties of T7EI fibrils. (a) Native PAGE of the T7EI protein incubated at 4°C and 37°C. Elongated fibrils are formed only at 37°C and stay on the surface of the stacking gel (open arrowhead). The soluble dimer is indicated by the filled arrowhead. (b) Electron micrograph of the T7EI fibrils. (Scale bar: 100 nm.) (c) X-ray diffraction pattern of oriented T7EI fibrils. A sharp meridional reflection at ≈4.7 Å is a characteristic of cross-β structure. A weak 10.5-Å reflection is indicative of β-sheets. (d) The absorbance of CR solution alone and with T7EI protein incubated at 37°C. Binding of the fibrils results in a red shift of the absorption spectrum, characteristic of amyloid formation. (e) Fluorescence emission spectrum of thioflavin T (ThT) alone and with T7EI fibrils. Binding of amyloid-specific ThT to the fibrils gives an emission peak at 482 nm.
Design of a Double-Cysteine Mutant to Study the Organization of T7EI Fibrils.
To study the molecular organization of T7EI fibrils, we took a protein engineering approach by designing a double-cysteine mutant, L19C/S95C. The two cysteines are located on different domains at the domain-swapped, dimer interface, so the formation of disulfide bonds serves as an indicator of a domain swap (Fig. 2a). The rationale is that if the protein forms fibrils by a runaway type of domain swap, the fibrils formed by the double-cysteine mutant would consist of long oligomers connected by intermolecular disulfide bonds. The distance between the Cα atoms of Leu-19 of one molecule and Ser-95 of the other molecule in dimeric T7EI is 6.7 Å, within the range of Cα-Cα distances of natural disulfides (31). As expected, in solution, L19C/S95C exists as a doubly disulfide-linked dimer in the absence of the reducing agent DTT and is reduced to monomers in the presence of DTT (Fig. 2b). The free thiol content of L19C/S95C was found to be <5% by using a 5,5′-dithiobis(2-nitrobenzoic acid) assay. That virtually all cysteines in L19C/S95C are involved in disulfide bond formation also is confirmed by a fluorescence-staining assay (see below).
Fig. 2.

Design of the double-cysteine mutant L19C/S95C of T7EI. (a) Ribbon diagram of T7EI with Leu-19 and Ser-95 shown in space-filling models. (b) SDS/PAGE showing that T7EI L19C/S95C is locked as a disulfide-bridged dimer in the absence of disulfide-breaking agent DTT (lane 1) and is released into monomers in the presence of DTT (lane 2).
Introduction of replacements L19C and S95C does not disrupt the native structure of T7EI, as demonstrated by a gel-shift assay showing that the disulfide-linked dimer L19C/S95C is capable of binding junction 3, a substrate for T7EI (ref. 32; Fig. 3, lanes 1 and 2). In a control experiment, the engineered protein, like the wild-type protein (33), does not bind to duplex DNA, indicating that binding to junction 3 is specific (Fig. 3, lanes 3 and 4). Because the junction binds to the T7EI surface formed by the dimer (34), the gel-shift assay confirms that the domain swap and normal function is preserved in the doubly disulfide-linked L19C/S95C dimer.
Fig. 3.
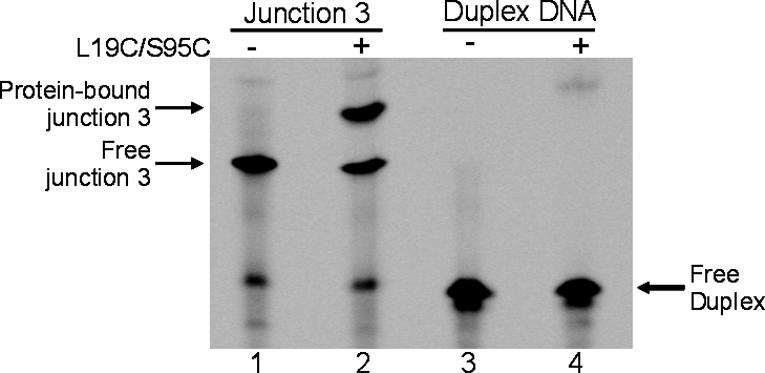
Demonstration of wild-type activity in the designed mutant T7EI L19C/S59C by a gel-shift assay. Fluorescently labeled DNA junction 3, a synthetic substrate for T7EI, was loaded either alone (lane 1) or with L19C/S95C protein (lane 2). The presence of L19C/S95C resulted in a more slowly migrating band that corresponds to the protein-bound junction 3 (lane 2). As a control, a duplex DNA that comprises the fluorescently labeled strand of the junction 3 and its complementary strand was loaded either alone (lane 3) or with L19C/S95C protein (lane 4). No binding of duplex DNA was detected in the presence of L19C/S95C protein.
T7EI Forms Fibrils via a Fibrillization-Competent Conformation.
Pilot experiments (data not shown) showed that incubation of the disulfide-linked dimers of T7EI L19C/S95C did not lead to fibril formation. Adding sulfhydryl reductants such as DTT or Tris(2-carboxyethyl)phosphine (TCEP) resulted in the conversion of soluble dimers into fibrils, indicating that conformational changes that involve the opening of the dimer interface may be required. To investigate the conformational changes during fibril formation, the effect of reducing agents was studied by incubating the disulfide-linked dimer with various amounts of DTT. We find that a second dimeric form of L19C/S95C, termed dimer II, is created by treating the doubly disulfide-linked dimer, termed dimer I, with small amounts of DTT. Fig. 4a shows that the proportion of dimer II increases at the expense of dimer I with increasing concentration of DTT. Dimer II runs more slowly on both native and denaturing gels (Fig. 4 a and b), suggesting that dimer II is less compact than dimer I. There is also a higher oligomer band in the presence of high concentrations of DTT, suggesting that this higher oligomer species is assembled from dimer II (Fig. 4a, lanes 4 and 5). SDS/PAGE indicates that dimer II contains at least one intact disulfide bond, because dimer II runs as a dimer in the presence of SDS (Fig. 4b, lanes 2–4). The conversion between dimer I and dimer II is reversible in solution, because dimer I is present even with excess DTT (Fig. 4a, lane 5). After dialyzing away DTT, dimer II is converted back to dimer I (data not shown). This result indicates that dimer II is not a kinetically trapped conformer. We also performed a gel-shift assay to check whether dimer II is capable of binding junction 3. We find that T7EI binds to junction 3 in the presence of DTT (data not shown), suggesting that dimer II may be able to bind to junction 3. However, the result also could be explained by binding to dimer I, because dimer I and dimer II are in equilibrium.
Fig. 4.

Fibril formation of T7EI L19C/S95C requires a fibrillization-competent conformation. (a) Two dimers of the double-cysteine mutant L19C/S95C revealed by native PAGE in various concentrations of DTT. A new dimer (dimer II) appears in the presence of DTT, as shown by increasing amount of dimer II with increasing concentrations of DTT (lanes 2–5). The doubly disulfide-linked dimer in the absence of DTT is dimer I. DTT also results in the formation of a higher oligomer (lanes 4 and 5). (b) Dimer II is disulfide-linked, as shown by SDS/PAGE of L19C/S95C. Both dimer I and dimer II are seen in DTT concentrations up to 100 μM (lanes 2–4). This result indicates intermolecular disulfide bridges in both dimer I and dimer II. In the presence of 1 mM DTT (lane 5), both dimers are converted to monomers. There are also two forms of monomers in low concentrations of DTT on SDS gel (lanes 2–4) that correspond to the intramolecularly disulfide-bonded monomer (lower band) and nondisulfide-bonded monomer (upper band). (c) Dimer II but not dimer I forms fibrils, as shown by native PAGE of L19C/S95C after incubation at 37°C for 18 h. The 4°C samples (19 hours old) are loaded for comparison. In the presence of 25 μM and 50 μM DTT (lanes 4 and 6), dimer II disappears and fibrils form as shown by the high-molecular-mass band on the surface of the stacking gel and at the boundary between the stacking gel and the running gel. Dimer I does not disappear at these DTT concentrations. However, in the presence of 100 μM and 1 mM DTT (lanes 8 and 10), all dimer I disappears, indicating that the equilibrium between dimer I and dimer II is shifted toward dimer II that subsequently forms fibrils at 37°C.
By a fluorescence-staining assay, we confirmed that dimer I is doubly intermolecularly disulfide-bonded but that dimer II has one intermolecular disulfide bond and two free cysteines. The T7EI L19C/S95C protein in the presence of various concentrations of TCEP was run on a native PAGE. Then the gel was stained with a thiol-specific fluorescent label. After fluorescence staining, the same gel was stained with Coomassie blue to reveal the protein bands. The results show that, after TCEP is removed by running the gel, all of the cysteines in dimer I participate in disulfide bond formation, but the cysteines in dimer II remain freely available to react with fluorescent labels (Fig. 5). Taken together, these results suggest that one of the disulfide bonds in dimer II is broken, unlatching its subunits and reducing its mobility, whereas half of the dimer remains closed with its disulfide bond intact. And the conformation of dimer II is distinct from that of dimer I, because when the cysteines in dimer I form disulfide bonds on the native gel, the newly freed cysteines in dimer II retain the reduced state.
Fig. 5.

Dimer II but not dimer I of T7EI L19C/S95C contains free cysteines. T7EI L19C/S95C in the presence of increasing concentrations of the reducing agent TCEP was run on a native PAGE. (Left) The gel was stained with a thiol-specific fluorescent label and scanned for fluorescence. (Right) T7EI L19C/S95C on the native gel was stained with Coomassie blue after fluorescence scanning.
By treating dimers with varying concentrations of DTT, we find that dimer I does not form fibrils but dimer II does. As shown in Fig. 4c, at moderate concentrations of DTT, fibrils grow at the expense of dimer II but not dimer I (lanes 4 and 6). But at concentrations of DTT that are sufficient to completely reduce the dimers, even dimer I is converted to fibrils (Fig. 4c, lanes 8 and 10). This result again indicates that dimer I and dimer II are in equilibrium. The inability of dimer I to form fibrils suggests that the simple close-ended, dimeric domain swap is not sufficient for fibril formation. The fact that only dimer II is able to form fibrils suggest that the fibrillization of T7EI requires a fibrillization-competent conformation, in which one domain of each dimer is free to swap into its complementary domain of the next molecule along the fibril.
T7EI Forms Amyloid-Like Fibrils by a Runaway Domain-Swapping Mechanism.
A runaway-type linkage in the T7EI fibrils is revealed by disulfide-linked oligomers. To check whether the T7EI L19C/S95C fibrils contain disulfide-linked oligomers, the fibrils were solubilized in 2% SDS in the presence or absence of excess DTT and were run on a SDS/PAGE to reveal their oligomeric status. Disulfide-linked oligomers would be broken down by the reducing agent DTT. In Fig. 6, T7EI L19C/S95C is initially incubated at 37°C for either 18 h or 6 days, before being run on SDS/PAGE. For the samples incubated for 18 h, small oligomers are found (Fig. 6a, lanes 3, 5, and 7), but are largely broken down by the addition of DTT (Fig. 6a, lanes 4, 6, and 8). This result shows that the oligomers are held together by disulfide bonds. For samples incubated for 6 days, much larger fibrils now are found to be trapped in the stacking gel or at the junction of the running and stacking gels (Fig. 6b, lanes 5, 7, and 9). These fibrils are also linked by disulfide bonds because they are mostly converted into monomers by DTT (Fig. 6b, lanes 6, 8, and 10). Because each T7EI L19C/S95C dimer contains only four cysteine residues, two must hold the monomers of the dimer together and each of the remaining two must be disulfide-linked to other dimers, these disulfide-linked small oligomers and large fibrils can be accounted for only by a runaway domain swap. This type of linkage is shown schematically in Fig. 7.
Fig. 6.

Monomers of T7EI L19C/S95C are cross-linked into small oligomers and fibrils by disulfide bonds, demonstrating an organization based on a runaway domain swap. (a) SDS/PAGE of T7EI L19C/S95C incubated at 37°C for 18 h. This gel shows that the fibrils contain higher oligomers (lanes 3, 5, and 7), and these oligomers are disulfide-bridged because they are reduced to monomers by DTT (lanes 4, 6, and 8). The sample with 1,000 μM DTT does not show a lot of higher oligomers (lane 9) because excess DTT is present in the sample. (b) SDS/PAGE of L19C/S95C incubated at 37°C for 6 days. After 6 days at 37°C, the SDS-solubilized fibrils show even higher oligomers that remain at the boundary of the stacking gel and running gel (lanes 5, 7, and 9, filled arrowheads) or on the surface of the stacking gel (lane 9, open arrowhead), and most of these fibrils are converted into monomers by adding DTT in the gel-loading buffer (lanes 6, 8, and 10).
Fig. 7.

Schematic model for the fibril formation of T7EI L19C/S95C. Each subunit is colored either in red or blue. Black dots represent disulfide bonds, and SH represents free cysteine. In dimer I, both disulfide bonds are intact and the protein is locked in close-ended dimers and unable to form fibrils. In dimer II, half of the domain-swapped molecule is unlatched by the reduction of one of the two disulfide bonds. Upon incubation at 37°C, dimer II changes to an open form (denoted as dimer II*), in which half of the dimer opens up, exposing the interface that remains protected in dimer I. Open-ended dimer II* readily fibrillizes via runaway domain swapping. The hinge-loop region of the domain-swapped protein forms a zipper spine in the fibrils.
Discussion
Although high-resolution structures of amyloid fibrils formed by a full-length protein are still beyond reach, protein engineering reveals the essential mechanism of amyloid fibril formation of T7EI. Previous studies by using engineered disulfide bonds to prevent the monomeric form of cystatin C from domain swapping showed that the monomeric cystatin C forms fewer fibrils, implying that domain swapping may be required for the fibril formation of cystatin C (21). Mutagenesis of the hinge loop region of another domain-swapped protein, suc1, showed that there is excellent correlation between domain swapping and aggregation, suggesting that they may share a common mechanism (35). In this work, by designing disulfide bonds at the domain-swapped dimer interface of T7EI, we find that, if locked in a dimer form by disulfide bonds, T7EI is unable to form amyloid-like fibrils (Fig. 4c, lanes 1 and 2). Our results suggest that stacking of domain-swapped dimers is not the mechanism of fibril formation. Rather, fibrillization requires the opening of one of the two domain-swapped dimer interfaces.
The process of fibril formation was followed by using gel electrophoresis by incubating the disulfide mutant of T7EI with the disulfide-breaking agent DTT. We find that when the conformational changes are no longer restrained by disulfide bonds, T7EI can adopt two conformations: dimer I and dimer II. Judging by gel electrophoresis, dimer I has the same conformation before and after incubating with DTT (Fig. 4 a and b). When DTT is removed from the system, the disulfide bonds reforms readily in dimer I (Fig. 5), providing strong support that dimer I, even when disulfide bonds are broken, has the same conformation as the disulfide-linked dimer. On the other hand, dimer II has a distinct conformation from dimer I. Dimer II runs more slowly than dimer I on both native and SDS gels, indicating that dimer II is less compact than dimer I (Fig. 4 a and b). When DTT is removed, dimer II still contains free cysteines (Fig. 5), suggesting that cysteines 19 and 95, which are close enough to form disulfide bonds in dimer I, are far apart in dimer II. SDS/PAGE of dimer II shows that dimer II also contains disulfide bonds (Fig. 4b). Therefore, we conclude that dimer II is a partially open-ended dimer, in which half of the domain-swapped molecule is unlatched and the other half remains closed. A model of dimer II is depicted in Fig. 7.
Upon incubation at 37°C, dimer II forms fibrils, whereas dimer I remains as a soluble dimer (Fig. 4c). Because incubation of dimer II at 4°C did not lead to fibril formation (Fig. 4c, lanes 3, 5, 7, and 9), we speculate that, at 37°C, there are further conformational changes in which half of the dimer II completely opens up to expose the dimer interface to become “activated dimer II” (denoted as dimer II* in Fig. 7). And dimer II* is competent for fibrillization via a runaway domain swap. Exactly how dimer II converts to dimer II* is not clear and requires additional investigation.
The finding of two dimer forms of T7EI is reminiscent of a fundamental principle of amyloid research: Amyloid proteins exist in both normal forms that carry out cellular function and aberrant forms that readily form amyloid fibrils. For example, the prion protein has been shown to exist in two conformations: a normal form, PrPC, and a disease form, PrPSc (36). PrPSc forms amyloid fibrils that may be associated with prion disease and can serve as a template to convert PrPC to the disease form. Other amyloid-forming proteins are also thought to exist in amyloidogenic conformation under certain conditions (37–39). However, unlike other proteins, whose amyloidogenic conformations are transiently populated during fibrillization, dimer II of T7EI is a stable, dominant conformation (Fig. 4). Further characterization of the structure of dimer II may offer insights on the structural switches underlying fibril formation.
The rationale behind the design of the double-cysteine mutant of T7EI is that the amyloid-like fibrils of T7EI would consist of disulfide-linked long oligomers if the fibrils were formed by runaway domain swapping as depicted in Fig. 7. By solubilizing the fibrils in 2% SDS with or without DTT, we show that, indeed, the T7EI fibrils formed by the double-cysteine mutant are composed of long oligomers connected by intermolecular disulfide bonds (Fig. 6). Since the terminology of domain swapping was introduced in 1994 (10) and implication of runaway domain swapping as a mechanism for protein aggregation was proposed in 1995 (12), experimental evidence for domain swapping in amyloid fibril formation has been largely speculative (18–27). Previous studies are limited to just offer correlations between the ability to domain swap and the ability to form fibrils, making the role of domain swapping in fibril formation an open question. Recently, domain swapping was found to exist in the amyloid-like fibrils of ribonuclease A (28), but whether the domain swap is a runaway type was not answered. Our finding provides definitive evidence on the role of runaway domain swapping in amyloid-like fibril formation of T7EI. Runaway domain swapping may represent a mechanism for fibril formation in at least some proteins such as cystatin C and ribonuclease A.
Runaway domain swapping in T7EI also highlights the importance of native-like interactions in amyloid fibril formation. Although there may be conformational changes in the conversion of dimer II to dimer II* upon incubation at 37°C, T7EI most likely retains native geometry, because the fibrils contain intermolecular disulfide bonds that are designed to form only when the domain-swapped dimer interface is reestablished. The role of native structure in amyloid formation has also been suggested in other proteins such as β2 microglobulin (40). Runaway domain swapping also is consistent with the general concept of the zipper-spine model of amyloid fibrils (22), in which a small segment forms the cross-β spine of the fibril consisting a steric zipper (8), and the rest of the protein forms a runaway domain swap around the central spine. X-ray diffractions of the oriented fibrils of T7EI show characteristic cross-β diffraction pattern (Fig. 1c), indicting the existence of a zipper-spine.
In summary, we provide definitive evidence that T7EI forms amyloid-like fibrils by a runaway, domain-swapping mechanism. Using a protein engineering approach, we find that the double-cysteine mutant of T7EI exists in two conformations: a native conformation that is unable to form fibrils and a second conformation that readily forms fibrils. In the second conformation, half of the dimer is unlatched and exposing the domain-swapped dimer interface, which is necessary for domain swapping. Gel electrophoresis analysis of the fibrils formed by the double-cysteine mutant revealed that the fibrils are connected by intermolecular disulfide bonds, providing strong evidence that the fibrils are formed via a runaway domain-swapping mechanism. We also find that the doubly disulfide-linked domain-swapped dimers are unable to form fibrils, suggesting that stabilization of the dimers may be a strategy for inhibiting amyloid fibril formation of domain-swapped proteins.
Materials and Methods
T7EI Mutants.
All T7EI clones in this work contain an E65K mutation, which makes the enzyme inactive (41) and, thus, allows high level expression in Escherichia coli. The mutant L19C/S95C contains mutations of E65K, C113A, L19C, and S95C. Mutagenesis was performed by using the QuikChange site-directed mutagenesis kit (Stratagene). Full-length T7EI construct contains 172 residues, including an N-terminal His-tag sequence of 23 amino acids (33).
Protein Expression and Purification.
Expression of T7EI proteins in E. coli BL21(DE3)pLysS cells (Novagen) was induced with 1 mM isopropyl β-d-thiogalactoside when cells grew to an absorbance of A600 ≈ 0.6 at 37°C. The induction was allowed to proceed at 25°C for 4 h. The cells were harvested by centrifugation and resuspended in buffer A (50 mM sodium phosphate, pH 8.0/1 M NaCl). The cells were then sonicated, and the cell debris was pelleted by centrifugation. The supernatant was filtered by using 0.22 μm Steriflip filter units (Millipore) and loaded onto a 5-ml HisTrap column (Amersham Pharmacia) equilibrated with buffer A. Proteins were eluted with a linear imidazole gradient (50–500 mM) in 10-column volumes. Protein concentration was determined by UV absorption at 280 nm by using an extinction coefficient of 24.75 × 103 M−1·cm−1 for E65K (41), and 24.88 × 103 M−1·cm−1 for L19C/S95C after adjustment for cysteines by using an extinction coefficient of 125 M−1·cm−1 for cysteines (42).
Fibril Formation.
Fibrils form upon incubation of T7EI proteins in buffer B (50 mM Tris, pH 7.5/40 mM NaCl), or buffer C (50 mM Mops, pH 6.8/40 mM NaCl) at 37°C without agitation.
Fibril Characterization.
For information on fibril characterization, see Supporting Materials and Methods, which is published as supporting information on PNAS web site.
Gel Electrophoresis.
Gel electrophoresis was performed by using PhastSystem (Amersham Pharmacia). For native PAGE, native buffer strips for basic proteins (2% agarose/4.4% β-alanine/4% acetic acid) were prepared, and 4–15% gradient PhastGel were used. The proteins were loaded directly on the gel. For SDS/PAGE, SDS buffer strips and 8–25% gradient PhastGel were used. The proteins were mixed with gel-loading buffer (125 μM Tris, pH 6.8/4% SDS/20% glycerol/2 mg/ml bromophenol blue) and boiled for 10 min before being loaded on the gel.
5,5′-Dithiobis(2-Nitrobenzoic Acid) Assay.
The 100 μM 5,5′-dithio-bis(2-nitrobenzoic acid) in buffer C was mixed with T7EI L19C/S95C and incubated at room temperature for 5 min before measuring absorbance at 412 nm. An extinction coefficient of 14,150 M−1·cm−1 for 2-nitro-5-thiobenzoic acid was used to calculate the concentration of free thiols (43).
Gel-Shift Assay.
Fluorescently labeled junction 3 was prepared by annealing stoichiometric quantities of b, h, r, and x strands of 34-nt oligos (GenScript). The fluorescent label, FAM, was covalently linked to the 5′ end of the b strand. The duplex DNA was prepared by hybridization of fluorescently labeled b strand and unlabeled b′-strand. For gel-shift assay, 1 μM T7EI L19C/S95C in buffer B was mixed with either 0.5 μM junction 3 or duplex DNA and incubated at room temperature for 1 h. The mixture was run on a 4–15% gradient PhastGel under native conditions. The gel was scanned for fluorescence by using Molecular Imager FX (Bio-Rad) equipped with an external laser. The synthesized oligos have the following sequences, all written 5′ to 3′: b strand, CCTCCGTCCTAGCAAGGGGCTGCTACCGGAAGGG; h strand, CCCTTCCGGTAGCAGCCTGAGCGGTGGTTGAAGG; r strand, CCTTCAACCACCGCTCAACTCAACTGCAGTCTGG; x strand, CCAGACTGCAGTTGAGTCCTTGCTAGGACGGAGG; b′ strand, CCCTTCCGGTAGCAGCCCCTTGCTAGGACGGAGG.
Fluorescence Staining.
T7EI proteins in the presence of various concentrations of reducing agent, TCEP, were run on a 4–15% gradient PhastGel (Amersham Pharmacia) under native conditions. The gel was rinsed with buffer B at room temperature for 15 min, then stained with 5 μM BODIPY 493/503 methyl bromide (Invitrogen) in the same buffer at room temperature for 30 min. After staining, the gel was washed with buffer B at room temperature for 15 min. The gel was then scanned for fluorescence by using Molecular Imager FX (Bio-Rad) equipped with an external laser.
Supplementary Material
Acknowledgments
We thank David M. J. Lilley (University of Dundee, Dundee, U.K.) for providing the clone of T7EI E65K mutant, Melanie Bennett for discussion, Martin Phillips at University of California, Los Angeles–Department of Energy Biochemistry Instrumentation Facility for assistance, and Mari Gingery for help with electron microscopy. This work was supported by the National Science Foundation, the National Institutes of Health, and the Howard Hughes Medical Institute.
Abbreviations
- T7EI
T7 endonuclease I
- TCEP
tris(2-carboxyethyl)phosphine.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Dobson C. M. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Tycko R. Curr. Opin. Struct. Biol. 2004;14:96–103. doi: 10.1016/j.sbi.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe D. J. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 4.Cohen A. S., Calkins E. Nature. 1959;183:1202–1203. doi: 10.1038/1831202a0. [DOI] [PubMed] [Google Scholar]
- 5.Puchtler H., Sweat F., Levine M. J. Histochem. Cytochem. 1962;10:355–364. [Google Scholar]
- 6.Vassar P. S., Culling C. F. A. Arch. Pathol. 1959;68:487–498. [PubMed] [Google Scholar]
- 7.Eanes E. D., Glenner G. G. J. Histochem. Cytochem. 1968;16:673–677. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- 8.Nelson R., Sawaya M. R., Balbirnie M., Madsen A. O., Riekel C., Grothe R., Eisenberg D. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sunde M., Serpell L. C., Bartlam M., Fraser P. E., Pepys M. B., Blake C. C. F. J. Mol. Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 10.Bennett M. J., Choe S., Eisenberg D. Proc. Natl. Acad. Sci. USA. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y., Eisenberg D. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett M. J., Schlunegger M. P., Eisenberg D. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korth C., Kaneko K., Groth D., Heye N., Telling G., Mastrianni J., Parchi P., Gambetti P., Will R., Ironside J., et al. Proc. Natl. Acad. Sci. USA. 2003;100:4784–4789. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krebs M. R. H., Morozova-Roche L. A., Daniel K., Robinson C. V., Dobson C. M. Protein Sci. 2004;13:1933–1938. doi: 10.1110/ps.04707004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammarström P., Wiseman R. L., Powers E. T., Kelly J. W. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 16.Kammerer R. A., Kostrewa D., Zurdo J., Detken A., Garcia-Echeverria C., Green J. D., Muller S. A., Meier B. H., Winkler F. K., Dobson C. M., et al. Proc. Natl. Acad. Sci. USA. 2004;101:4435–4440. doi: 10.1073/pnas.0306786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferguson N., Berriman J., Petrovich M., Sharpe T. D., Finch J. T., Fersht A. R. Proc. Natl. Acad. Sci. USA. 2003;100:9814–9819. doi: 10.1073/pnas.1333907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abrahamson M., Grubb A. Proc. Natl. Acad. Sci. USA. 1994;91:1416–1420. doi: 10.1073/pnas.91.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janowski R., Kozak M., Jankowska E., Grzonka Z., Grubb A., Abrahamson M., Jaskolski M. Nat. Struct. Biol. 2001;8:316–320. doi: 10.1038/86188. [DOI] [PubMed] [Google Scholar]
- 20.Janowski R., Abrahamson M., Grubb A., Jaskolski M. J. Mol. Biol. 2004;341:151–160. doi: 10.1016/j.jmb.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Nilsson M., Wang X., Rodziewicz-Motowidlo S., Janowski R., Lindstrom V., Onnerfjord P., Westermark G., Grzonka Z., Jaskolski M., Grubb A. J. Biol. Chem. 2004;279:24236–24245. doi: 10.1074/jbc.M402621200. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y., Gotte G., Libonati M., Eisenberg D. Nat. Struct. Biol. 2001;8:211–214. doi: 10.1038/84941. [DOI] [PubMed] [Google Scholar]
- 23.Staniforth R. A., Giannini S., Higgins L. D., Conroy M. J., Hounslow A. M., Jerala R., Craven C. J., Waltho J. P. EMBO J. 2001;20:4774–4781. doi: 10.1093/emboj/20.17.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanders A., Jeremy Craven C., Higgins L. D., Giannini S., Conroy M. J., Hounslow A. M., Waltho J. P., Staniforth R. A. J. Mol. Biol. 2004;336:165–178. doi: 10.1016/j.jmb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 25.Knaus K. J., Morillas M., Swietnicki W., Malone M., Surewicz W. K., Yee V. C. Nat. Struct. Biol. 2001;8:770–774. doi: 10.1038/nsb0901-770. [DOI] [PubMed] [Google Scholar]
- 26.Lee S., Eisenberg D. Nat. Struct. Biol. 2003;10:725–730. doi: 10.1038/nsb961. [DOI] [PubMed] [Google Scholar]
- 27.Klafki H. W., Pick A. I., Pardowitz I., Cole T., Awni L. A., Barnikol H. U., Mayer F., Kratzin H. D., Hilschmann N. Biol. Chem. Hoppe-Seyler. 1993;374:1117–1122. doi: 10.1515/bchm3.1993.374.7-12.1117. [DOI] [PubMed] [Google Scholar]
- 28.Sambashivan S., Liu Y., Sawaya M. R., Gingery M., Eisenberg D. Nature. 2005;437:266–269. doi: 10.1038/nature03916. [DOI] [PubMed] [Google Scholar]
- 29.Center M. S., Studier F. W., Richards C. C. Proc. Natl. Acad. Sci. USA. 1970;65:242–248. doi: 10.1073/pnas.65.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hadden J. M., Convery M. A., Declais A. C., Lilley D. M. J., Phillips S. E. V. Nat. Struct. Biol. 2001;8:62–67. doi: 10.1038/83067. [DOI] [PubMed] [Google Scholar]
- 31.Thornton J. M. J. Mol. Biol. 1981;151:261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- 32.Duckett D. R., Murchie A. I., Diekmann S., von Kitzing E., Kemper B., Lilley D. M. Cell. 1988;55:79–89. doi: 10.1016/0092-8674(88)90011-6. [DOI] [PubMed] [Google Scholar]
- 33.Duckett D. R., Panis M.-J. E. G., Lilley D. M. J. J. Mol. Biol. 1995;246:95–107. doi: 10.1006/jmbi.1994.0069. [DOI] [PubMed] [Google Scholar]
- 34.Déclais A. C., Fogg J. M., Freeman A. D. J., Coste F., Hadden J. M., Phillips S. E. V., Lilley D. M. J. EMBO J. 2003;22:1398–1409. doi: 10.1093/emboj/cdg132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rousseau F., Schymkowitz J. W. H., Wilkinson H. R., Itzhaki L. S. Proc. Natl. Acad. Sci. USA. 2001;98:5596–5601. doi: 10.1073/pnas.101542098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prusiner S. B. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly J. W. Curr. Opin. Struct. Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 38.Uversky V. N., Fink A. L. Biochim. Biophys. Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Kelly J. W. Curr. Opin. Struct. Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 40.Eakin C. M., Attenello F. J., Morgan C. J., Miranker A. D. Biochemistry. 2004;43:7808–7815. doi: 10.1021/bi049792q. [DOI] [PubMed] [Google Scholar]
- 41.Déclais A. C., Hadden J., Phillips S. E. V., Lilley D. M. J. J. Mol. Biol. 2001;307:1145–1158. doi: 10.1006/jmbi.2001.4541. [DOI] [PubMed] [Google Scholar]
- 42.Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riddles P. W., Blakeley R. L., Zerner B. Anal. Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.