

Abstract
The developing science called structural genomics has focused to date mainly on high-throughput expression of individual proteins, followed by their purification and structure determination. In contrast, the term structural biology is used to denote the determination of structures, often complexes of several macromolecules, that illuminate aspects of biological function. Here we bridge structural genomics to structural biology with a procedure for determining protein complexes of previously unknown function from any organism with a sequenced genome. From computational genomic analysis, we identify functionally linked proteins and verify their interaction in vitro by coexpression/copurification. We illustrate this procedure by the structural determination of a previously unknown complex between a PE and PPE protein from the Mycobacterium tuberculosis genome, members of protein families that constitute ≈10% of the coding capacity of this genome. The predicted complex was readily expressed, purified, and crystallized, although we had previously failed in expressing individual PE and PPE proteins on their own. The reason for the failure is clear from the structure, which shows that the PE and PPE proteins mate along an extended apolar interface to form a four-α-helical bundle, where two of the α-helices are contributed by the PE protein and two by the PPE protein. Our entire procedure for the identification, characterization, and structural determination of protein complexes can be scaled to a genome-wide level.
Keywords: computational biology, protein structure, functional linkages
Because cellular processes involve protein complexes, understanding function requires more efficient methods to identify and examine protein interactions at the molecular level. Useful experimental methods have been developed to identify protein interactions in vivo and in vitro, including the yeast two-hybrid (1, 2) and coaffinity purification methods (3, 4). Together these methods have enabled the identification of thousands of putative protein interactions in organisms ranging from yeast (1–4) to human (5). To complement these biochemical methods, computational procedures have been developed to infer linkages between proteins on a genome-wide scale. These techniques include the Rosetta stone (6), phylogenetic profile (7), conserved gene neighbor (8, 9), and operon/gene cluster methods (10–12). Protein linkages identified by these methods reveal proteins that participate in protein complexes, protein pathways, or serve related functions within the cell (13, 14). The question we address in this work is how to combine methods for inference of protein complexes with structure determination to give a more efficient procedure for learning biological function at the molecular level.
By using a combined procedure of inference of protein complexes followed by protein coexpression and cocrystallization, we targeted two large and poorly understood protein families in Mycobacterium tuberculosis (M.tb.), the PE and PPE families. These families, named for the conserved proline (P) and glutamate (E) residues near the N-terminal region of the encoded proteins, contain ≈100 PE members and >60 PPE members in the genome (15). Although no structure or precise function is known for any member of these families, it has been suggested that some PE proteins may play a role in immune evasion and antigenic variation (15–18), and some members have been found to associate with the cell wall (19, 20) and to influence interactions with other cells (20). Members of the PE and PPE families also have been linked to virulence (21, 22), and some PPE proteins have been found to be immunodominant antigens (23). Furthermore, because the PE and PPE genes are prevalent in M.tb., and absent in humans, they may serve as potential targets for the development of antituberculosis intervention strategies.
Results
Individual PE and PPE Proteins Fail to Express in Soluble Form.
Our efforts to determine structures for individual PE and PPE proteins were frustrated by our finding that they did not express well or expressed in insoluble or unfolded forms. Our attempts to individually express 17 PE and 11 PPE proteins are detailed in Table 1, which is published as supporting information on the PNAS web site. Of these 28 proteins, 27 either did not express in E. coli or were insoluble. Only 1 of the 28 individually expressed proteins, Rv3872, was soluble, but circular dichroism (CD) revealed that it was unfolded. These 28 proteins lack apparent transmembrane elements. Thus, a possible explanation for their failure to express on their own is that they need protein partners to fold. In fact, genomic analysis suggested to us that individual PE proteins are likely protein partners for PPE proteins, as explained below.
Combined Procedure to Identify Protein Complexes for Structural Determination.
Our procedure to identify protein complexes for structural determination is outlined in Fig. 1. First, four computational methods are used to infer functional linkages between proteins on a genome-wide basis (6–12). Previously, we reported application of these methods to discover protein functional modules in M.tb. (24). These methods are available for any sequenced genome (ProLinks: http//mysql5.mbi.ucla.edu/cgi-bin/functionator/pronav) (12) and in practice can be supplemented by information from two-hybrid (1, 2) coaffinity methods (3, 4) and other computational genomic servers (25).
Fig. 1.

Combined computational and biochemical procedure for the identification, characterization, and structural determination of protein complexes. Four computational methods are used to identify functionally linked proteins based on genomic analyses. Putative interacting proteins are cloned into a coexpression vector, where one protein is tagged with a His affinity tag and the others are not tagged. Genes are coexpressed, and interacting proteins are identified by affinity chromatography. If the proteins interact to form a long-lived protein complex, then the nontagged protein(s) copurifies with the tagged protein. Newly identified protein complexes are characterized and crystallized. We demonstrate this strategy by identifying, characterizing, and determining the crystal structure of a M.tb. PE/PPE protein complex.
Next, protein–protein interactions are verified by using a coexpression/copurification strategy. In this strategy, two genes are cloned into a coexpression vector, which has been modified to include two ribosome-binding sites and restriction sites for the insertion of two genes (26). The coexpression vector is transformed into competent E. coli cells, where induced genes are transcribed onto a polycistronic transcript. Translation results in the production of two proteins, only one of which is tagged with a histidine (His) affinity tag for purification. Long-lived protein complexes are identified by their copurification on a nickel affinity column. Identified protein complexes can be further purified by additional forms of chromatography for biophysical characterization and crystallization screens. In principle, the strategy can be extended to three or more interacting proteins.
Functional Linkage and Genomic Organization of the M.tb. PE and PPE Genes.
Our analysis of the PE and PPE genes by the operon/gene cluster method (10, 12) revealed that one PE gene is often functionally linked to one PPE gene. That is, the two genes tend to be in close chromosomal proximity on the M.tb. genome (10, 12).
Traditionally the PE gene family has been subdivided into two subfamilies, the PE–PGRS subfamily, which contains proteins with the conserved PE domain followed by long stretches of glycine and alanine-rich repeats, and the PE subfamily that encodes proteins that have either the conserved PE domain only or have the PE domain followed by a variable C-terminal domain (15). Based on our genomic analysis, we further subdivide the PE subfamily into three groups as shown in Fig. 5, which is published as supporting information on the PNAS web site: (i) PE genes that occur in putative operons with PPE genes (17 pairs of genes), (ii) PE genes that occur in putative operons with other PE genes (3 pairs of genes), and (iii) PE genes that are not adjacent to other PE or PPE genes (14 PE genes).
Further analysis suggested a PE–PPE pair for our study. We noticed that those PE genes in putative operons with PPE genes tend to encode proteins containing only the conserved, ≈100-aa, PE domain, whereas the PE genes of the other two subgroups tend to be longer and have extended C-terminal domains. In many cases, the PE genes that are located in putative operons with PPE members are separated by small intergenic distances. In addition to the linkage of PE and PPE proteins by the operon/gene cluster method, there is one case of a Rosetta Stone PE–PPE fusion protein in the Mycobacterium paratuberculosis genome, encoded by the MAP_1003c gene. Based on these linkages between the PE and PPE genes, as well as the distinctive domain size of ≈100 residues for PE proteins that occur in putative operons with PPE proteins, we hypothesized that each pair of PE and PPE proteins partner in a complex. To test our hypothesis, we chose the PPE protein Rv2430c, which is the smallest in this family but still contains the entire conserved PPE domain, and its putative partner PE protein Rv2431c.
Coexpression and Copurification of the PE and PPE Proteins.
We constructed a coexpression vector similar to that described by Chen et al. (26), by introducing a second ribosome-binding site into the multiple cloning site of a pET29b(+) vector. The PE gene, Rv2431c, and the PPE gene, Rv2430c, were PCR amplified from M.tb. genomic DNA and cloned into the coexpression vector as shown in Fig. 2a. The organization of genes in the coexpression vector mimics the genomic organization of the PE and PPE genes in M.tb.. The amplified PE gene encoded the full-length Rv2431c protein, and the PPE gene encoded the full-length Rv2430c protein fused to a C-terminal thrombin cleavable linker and His affinity tag. The PE/PPE coexpression plasmid was transformed into competent E. coli BL21(DE3) cells, and expression was induced. The strong expression of both proteins is shown in Fig. 6, which is published as supporting information on the PNAS web site: dominant bands corresponding to the molecular masses of both the PE and PPE proteins are observed.
Fig. 2.

Validation and characterization of the Rv2431c/Rv2430c PE/PPE protein complex. (a) The PE and PPE genes, Rv2431c and Rv2430c, were cloned and ligated into a coexpression vector. In this system the PPE protein is tagged with a His tag, whereas the PE protein is not tagged. Long-lived protein–protein interactions are identified by the coelution and copurification of the untagged PE protein with the tagged PPE protein. (b) Identification of interacting PE and PPE proteins. The soluble supernatant from the coexpressed PE and PPE experiments was bound to and eluted from a nickel affinity column. The untagged PE protein coelutes with the tagged PPE protein, suggesting a physical association of the two proteins. (c) Sedimentation equilibrium experiments suggested that the 10.7-kDa PE and 24.1-kDa His-tagged PPE proteins form a 1:1 heterodimer. (d) CD experiments show that the Rv2431c/Rv2430c PE/PPE protein complex is folded and mostly α-helical. This result is in contrast to the individually expressed PE protein, Rv3872, which is soluble but unfolded.
To determine whether the PE protein Rv2431c interacts with the PPE protein Rv2430c to form a long-lived protein complex, induced cells were lysed, the soluble supernatant was subjected to purification on a nickel affinity column, and fractions corresponding to the elution peak were assayed by SDS gel electrophoresis. Two dominant bands were observed in the elution peak fractions, as shown in Fig. 2b, corresponding to the molecular mass of the 10.7-kDa PE protein Rv2431c, and the 24.1-kDa His-tagged PPE protein Rv2430c. Because only the PPE protein Rv2430c was tagged, this result suggested that the smaller nontagged PE protein binds to the larger, tagged PPE protein. The identities of these bands were further verified by mass spectrometry and N-terminal protein sequencing.
To characterize the putative complex further, we performed sedimentation equilibrium and CD experiments. Sedimentation equilibrium revealed that the molecular mass of the PE/PPE protein complex is 35.2 kDa, as shown in Fig. 2c, suggesting that the two proteins form a 1:1 heterodimeric complex. CD revealed that the PE/PPE protein complex is folded and highly α-helical in nature, as shown in Fig. 2d. Because the individual PE and PPE proteins did not express well or fold, we conclude that protein partnering is necessary for these functions. Such a codependent folding has been seen with the M.tb. Esat-6/Cfp-10 proteins (27).
Crystal Structure of the PE/PPE Protein Complex.
Diffraction-quality protein crystals of the PE/PPE protein complex labeled with selenomethionine were grown, and the structure was determined at 2.2-Å resolution by multiwavelength anomalous dispersion. As expected from our solution experiments, the PE/PPE protein complex is highly α-helical and is heterodimeric, containing one PE and one PPE protein, as shown in Fig. 3. The PE protein is a two-helix bundle, which forms a four-helix bundle with two of the five helices of the PPE protein.
Fig. 3.

Crystal structure of the M.tb. PE/PPE protein complex. (a) Surface representation of the PE/PPE protein complex. The PE protein Rv2431c is shown in red, and the PPE protein Rv2430c is in blue. (b) The PE/PPE protein complex viewed down its longitudinal axis. (c) Ribbon diagram of the PE/PPE protein complex. The complex is composed of seven α-helices. Two α-helices of the PE protein interact with two helices of the PPE protein to form a four-helix bundle. Regions of high sequence conservation are indicated by arrows and discussed in the text. (d) Interface hydrophobicity of the PPE and PE proteins. The hydrophobicity of the interaction interface between the PPE and PE protein is color-coded: the most apolar regions are indicated in red, orange, and yellow, and the most polar regions are indicated in blue. Notice the extensive apolar regions that are shielded from solvent as the complex forms.
The PE protein is composed of two α-helices (residues 8–37 and 45–84) that run antiparallel to each other, connected by a loop (residues 38–44), with both the N and C termini at the top of the complex. This PE loop is stabilized by interactions with helices 2 and 5 of the PPE protein. The conserved proline–glutamate (PE) sequence motif, for which the PE proteins are named (15), is visible in the electron density map and is located at the N terminus of the PE protein (residues 8–9). The nearly 100 members of the M.tb. PE family are likely to share similar structural features.
The PPE protein, as shown in blue in Fig. 3c, is also almost entirely helical. The conserved proline–proline–glutamate (PPE) sequence motif, for which the PPE proteins are named (15), is visible in the electron density map and is located near the N-terminal “hook” of the PPE protein (residues 7–9). This hook cradles the interacting PE protein. Helices α2 (residues 21–53) and α3 (residues 58–103) of the PPE protein run antiparallel and form the interaction interface in contact with the PE protein.
Discussion
Formation of the Complex.
At the interface between the two long α-helices of the PE protein, and the long α-helices 2 and 3 of the PPE protein, there is both an exquisite steric (Fig. 4) and hydrophobic interaction (Fig. 3d). Extensive apolar regions thus are shielded from solvent as the complex forms, and it is easy to understand why neither the PE nor PPE protein might be stable on its own.
Fig. 4.

Interaction interface of the PE/PPE protein complex. (a) Two helices of the PE protein, α1 and α2, interact with two helices of the PPE protein, α2 and α3, to form a four-helix bundle. The four-helix-bundle is largely stabilized by hydrophobic interactions among apolar side chains, as seen in the core of the complex. In contrast, the outer surface is coated with polar residues. (b) The PE/PPE complex as viewed down the longitudinal axis.
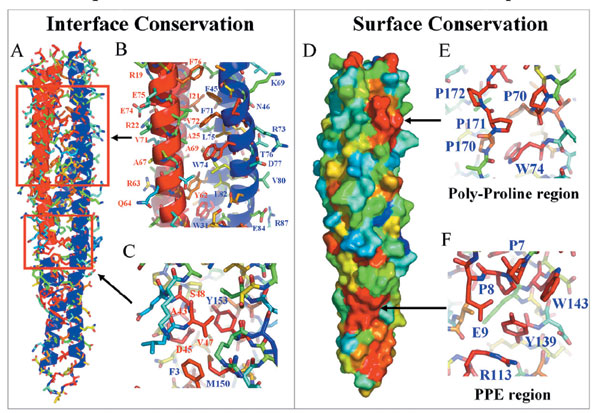
Regions of highly conserved residues are indicated by arrows in Fig. 3c and are shown in greater detail in the sequence alignment and the graphic display in, respectively, Figs 7 and 8, which are published as supporting information on the PNAS web site. The first region of high conservation is at the interface of the PE protein with the PPE protein, in the interior of the four-helix bundle (Fig. 4). Thus, the same sort of complex is likely to be conserved in the other PE–PPE pairs listed in Fig. 5. Also contributing to the conservation of the complex is the second region of conserved residues, residues in the PE loop that form part of the interaction surface with the PPE protein (Fig. 8). The third and fourth regions of highly conserved residues are on the surface of the complex and thus may be involved in interactions with other proteins. The third region includes the PPE sequence motif (residues P7, P8, and E9 of the PPE protein and the surrounding residues of the same protein R113, Y139, and W143). The tyrosine corresponding to Y139 of the PPE protein is one of the most conserved residues of the PPE protein family. The fourth region of high conservation is a polyproline-rich region, toward the C terminus of the PPE protein (Fig. 3c), including residues P170, P171, P172, and P70. The proline of the PE sequence motif is also highly conserved.
From Structure to Function.
The inference of function from structure is in its early days, but several approaches gave similar clues to function. An apolar stripe runs along one side of the complex, suggesting a docking site for another protein (see Fig. 9, which is published as supporting information on the PNAS web site). The metaserver ProKnow (28) (www.doe-mbi.ucla.edu/Services/ProKnow) used sequence and structure clues from the PE/PPE complex to infer possible functions for the complex. Possible functions are expressed as Gene Ontology (GO) terms, each given with a Bayesian weight. The highest scoring GO term for biological process is “signal transduction” with a probability of 75% (see Table 2, which is published as supporting information on the PNAS web site). A similar result came from the combinatorial extension program (29), which identifies protein structures with similar three-dimensional structures. The best match to the PPE protein of the PE/PPE protein complex was the cytoplasmic domain of a serine chemotaxis receptor (Tsr) (30). The cytoplasmic domain of Tsr forms an extended α-helix bundle (30) and functions as the cytoplasmic domain of a multidomain protein that senses extracellular signals and transmits them to the interior of the bacterium through a phosphorylation cascade. The domain organization of the Tsr protein is reminiscent of the domain organization proposed for the PE–PGRS proteins by Brennan et al. (20). These authors proposed that downstream from the conserved PE domain of PE–PGRS proteins is a putative transmembrane helix, followed by a glycine-and alanine-rich domain of variable length (20). In short, it is possible that some of the PE–PPE complexes may be involved in signal transduction, either as membrane-tethered proteins or as soluble proteins.
In summary, we present a procedure for inferring protein complexes encoded by any sequenced genome and a methodology for efficient determination of their structures. The procedure is capable of scale up and could narrow the present chasm between structural biology and structural genomics.
Materials and Methods
Coexpression Vector.
A pET29b(+) expression vector (Novagen) was modified to include a second ribosome binding site as described by Chen et al. (26). Two chemically synthesized oligonucleotides, corresponding to the ribosome-binding site sequence, were synthesized, annealed, and ligated between the KpnI and NcoI sites of the pET29b(+) vector.
Cloning.
The M.tb. Rv2431c and Rv2430c genes were amplified from M.tb. H37Rv genomic DNA by using the Advantage-GC Genomic PCR kit (Clontech). The following primers were used for PCR: Rv2430c fwd (containing a NcoI site, start codon underlined), 5’-GCCATGGCTTTCGAAGCGTACCCACCGGAGGTCAACTCC-3’; Rv2430c rev (containing a HindIII site, thrombin cleavage site underlined), 5’-AAGCTTAGAACCGCGTGGCACCAGAGTGTCTGTACGCGATGACG-3’; Rv2431c fwd (containing a NdeI site, start codon underlined), 5’-CCATATGTCTTTTGTGATCACAAATCCCGAGGCGTTGAC-3’; and Rv2431c rev (containing a KpnI site, stop codon underlined), 5’-CGGTACCTTAACTAAAGGTCTTGATGTTGTCGGCCTCGGC-3’. Boldface type in the primers indicates engineered restriction sites, cleavage sites, start codons, and stop codons.
PCR products were ligated into pCR-Blunt-TOPO vectors (Invitrogen) and then digested with the respective enzymes to generate 5’ and 3’ overhangs. Rv2430c was digested with NcoI and HindIII, and Rv2431c was digested with NdeI and KpnI. Rv2430c and Rv2431c were purified separately by agarose gel electrophoresis and ligated into the engineered coexpression vector in two steps.
First, the coexpression vector was digested with NcoI and HindIII and purified by agarose gel electrophoresis by using a gel extraction kit (Qiagen, Valencia, CA). Rv2430c was ligated into the digested coexpression vector at the NcoI and HindIII sites and transformed into NovaBlue competent cells (Novagen). The coexpression plasmid containing Rv2430c was purified by using a Qiagen spin miniprep kit.
Next, the coexpression vector was digested with NdeI and KpnI and purified by agarose gel electrophoresis. Rv2431c was ligated into the digested coexpression vector at the NdeI and KpnI sites and transformed into NovaBlue competent cells (Novagen). The coexpression plasmid containing both Rv2430c and Rv2431c was purified by using a Qiagen spin miniprep kit. Inserts were verified by gel electrophoresis and DNA sequencing.
Protein Coexpression and Copurification.
The coexpression plasmid containing Rv2430c and Rv2431c was transformed into BL21(DE3) competent cells (Novagen) and grown to an OD600 of ≈0.6 at 37°C. Protein expression was induced with 0.4 mM isopropyl β-d-thiogalactoside (IPTG) for 2–3 h. Cells were harvested by ultracentrifugation, and cell pellets were resuspended in 20 mM Hepes (pH 7.8), 150 mM NaCl, and 0.4 mM PMSF. Resuspended cells were lysed by lysozyme treatment and sonication. Cell lysates were centrifuged at 32,000 × g for 25 min, and the supernatant was filtered and loaded onto a Ni2+ charged HiTrap chelating column (Amersham Pharmacia). The column was washed with 20 mM Hepes (pH 7.8), 150 mM NaCl, and 10 mM imidazole and eluted with a linear gradient of imidazole from 10 to 250 mM in 20 mM Hepes (pH 7.8) and 150 mM NaCl. The fractions corresponding to the Rv2430c(PPE) and Rv2431c(PE) protein complex were pooled and concentrated and further purified on an Amersham Pharmacia Superdex 75 column equilibrated with 20 mM Hepes (pH 7.8) and 150 mM NaCl. Fractions corresponding to the Rv2430c(PPE) and Rv2431c(PE) complex were pooled and concentrated. Purified proteins of the PE/PPE complex were verified by SDS gel electrophoresis, mass spectrometry, and N-terminal protein sequencing.
Protein Complex Crystallization.
PE/PPE protein complexes of Rv2431c and Rv2430c were prepared for crystallization by coexpressing the proteins in E. coli grown in media containing selenomethionine (SeMet). SeMet proteins were copurified on a nickel affinity column, and fractions corresponding to the elution peak were pooled, concentrated, and subjected to a second purification on a Superdex 75 gel filtration column. Fractions corresponding to the dominant peak were verified to contain the protein complex and pooled. The His tag of the PPE protein was then cleaved with biotinylated thrombin, which was then removed by streptavidin beads. The purified complex was then passed through a second nickel column to remove all of the cut His tags. The purified PE/PPE protein complex was then dialyzed into a low-salt buffer containing 5 mM Hepes (pH 7.8) and 10 mM NaCl for crystallization experiments.
Diffraction-quality protein crystals of the PE/PPE protein complex were grown by using the hanging-drop vapor-diffusion method in 14% isopropanol, 0.07 M sodium acetate trihydrate (pH 4.6), 0.14 M calcium dehydrate, and 30% glycerol. Crystals were observed after 2 weeks. No additional cryoprotectant was needed for data collection because the crystals were grown in 30% glycerol. Crystals belong to space group P2221 with unit cell dimensions a = 41.0 Å, b = 47.2 Å, and c = 283.2 Å and two PE/PPE complexes in the asymmetric unit.
Structure Determination and Refinement.
A standard three-wavelength anomalous dispersion data set was collected on a selenomethionyl derivative at the Advance Light Source (ALS) beamline 8.2.2. An ADSC quantum 315 charge-coupled device detector (Area Detector Systems Corp., Poway, CA) was used to record the data. Data were processed by using denzo/scalepack (31) (see Table 3, which is published as supporting information on the PNAS web site). Six of 20 selenium sites were identified with the program shelxd (32). Initial phases were calculated with mlphare and later improved by density modification and twofold symmetry averaging with dm (33). Five additional selenium sites could be located later from an anomalous difference Fourier map and subsequently used to improve the phases (Table 3). The experimental electron density was lacking in detail (see Fig. 10A, which is published as supporting information on the PNAS web site) but was well connected, allowing an initial trace to be built by using the graphics program o (34). The model was refined by using conjugate gradient and simulated annealing algorithms as implemented by the program cns (35). Strong noncrystallographic symmetry (NCS) restraints were used throughout. Hydrogen-bond restraints were helpful in the early stages of refinement (36). This model was further refined with refmac (37), to introduce TLS parameters in the refinement. Later rounds of model building were performed with the graphics program coot (38). A higher-resolution (2.2 Å) data set was collected at ALS from a second selenomethionyl crystal and was used for the later stages of refinement.
This data set (as well as the earlier data sets used for phasing) was severely anisotropic, with diffraction limits of 2.2 Å along the a* and c* directions, but only 3.2 Å along the b* direction. For this reason, data were truncated that fell outside an ellipse centered at the reciprocal lattice origin and having vertices at 1/2.2, 1/3.2, and 1/2.2 Å along a*, b*, and c*, respectively. The anisotropic scale factor applied by refmac was used but was found to be inadequate because the positive B factor correction it applied along a* and c* components was so large and positive (to balance the negative B factor correction required along b*) that the electron density maps it produced looked relatively featureless. The lack of features made it difficult to improve the model by manual building and completely obscured the presence of any water molecules (Fig. 10B). To compensate, isotropy was approximated by applying a negative scale factor along b* (−14 Å2) and no correction along a* or c*. This anisotropically scaled data then were used for refinement with refmac. Many more details could be observed in the resulting maps, allowing the correction of side-chain rotamers and modeling of 72 water molecules (Fig. 10C). Data collection and refinement statistics are given in Table 3.
The geometric quality of the model was assessed with the structure validation tools errat (39), procheck (40), and whatif (41). procheck reported 95% of the residues fall in the most favored region of the Ramachandran plot, and 4% of the residues were in additionally allowed regions. errat reported an overall quality factor of 96%. Protein structures were illustrated by using the program pymol (42).
Sequence Conservation.
Multiple sequence alignments were constructed by using clustalx (43), and sequence conservation was mapped onto the protein structure by using the ProFunc server (44).
Supplementary Material
Acknowledgments
We thank Celia Goulding, Robert Riley, Arturo Medrano-Soto, Markus Kaufmann, Minmin Yu, and the ALS beamline 8.2.2 staff for discussion. This work was supported by the National Institutes of Health Protein Structure Inititative (Integrated Center for Structural and Functional Innovation Consortium).
Abbreviation
- M.tb.
Mycobacterium tuberculosis.
Footnotes
Conflict of interest statement: No conflicts declared.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2G38).
References
- 1.Uetz P., Giot L., Cagney G., Mansfield T. A., Judson R. S., Knight J. R., Lockshon D., Narayan V., Srinivasan M., Pochart P., et al. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 2.Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y. Proc. Natl. Acad. Sci. USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gavin A. C., Bosche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J. M., Michon A. M., Cruciat C. M., et al. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 4.Ho Y., Gruhler A., Heilbut A., Bader G. D., Moore L., Adams S. L., Millar A., Taylor P., Bennett K., Boutilier K., et al. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 5.Rual J. F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., et al. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 6.Marcotte E. M., Pellegrini M., Ng H. L., Rice D. W., Yeates T. O., Eisenberg D. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 7.Pellegrini M., Marcotte E. M., Thompson M. J., Eisenberg D., Yeates T. O. Proc. Natl. Acad. Sci. USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Overbeek R., Fonstein M., D’Souza M., Pusch G. D., Maltsev N. Proc. Natl. Acad. Sci. USA. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dandekar T., Snel B., Huynen M., Bork P. Trends Biochem. Sci. 1998;23:324–328. doi: 10.1016/s0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- 10.Strong M., Mallick P., Pellegrini M., Thompson M. J., Eisenberg D. Genome Biol. 2003;4:R59.1–R59.16. doi: 10.1186/gb-2003-4-9-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salgado H., Moreno-Haelsieb G., Smith T., Collado-Vides J. Proc. Natl. Acad. Sci. USA. 2000;97:6652–6657. doi: 10.1073/pnas.110147297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowers P. M., Pellegrini M., Thompson M. J., Fierro J., Yeates T. O., Eisenberg D. Genome Biol. 2004;5:R35. doi: 10.1186/gb-2004-5-5-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisenberg D., Marcotte E. M., Xenarios I., Yeates T. O. Nature. 2000;405:823–826. doi: 10.1038/35015694. [DOI] [PubMed] [Google Scholar]
- 14.Marcotte E. M., Pellegrini M., Thompson M. J., Yeates T. O., Eisenberg D. Nature. 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- 15.Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., III, et al. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 16.Banu S., Honore N., Saint-Joanis B., Philpott D., Prevost M. C., Cole S. T. Mol. Microbiol. 2002;44:9–19. doi: 10.1046/j.1365-2958.2002.02813.x. [DOI] [PubMed] [Google Scholar]
- 17.Brennan M. J., Delogu G. Trends Microbiol. 2002;10:246–249. doi: 10.1016/s0966-842x(02)02335-1. [DOI] [PubMed] [Google Scholar]
- 18.Delogu G., Brennan M. J. Infect. Immun. 2001;69:5606–5611. doi: 10.1128/IAI.69.9.5606-5611.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delogu G., Pusceddu C., Bua A., Fadda G., Brennan M. J., Zanetti S. Mol. Microbiol. 2004;52:725–733. doi: 10.1111/j.1365-2958.2004.04007.x. [DOI] [PubMed] [Google Scholar]
- 20.Brennan M. J., Delogu G., Chen Y., Bardarov S., Kriakov J., Alavi M., Jacobs W. R., Jr. Infect. Immun. 2001;69:7326–7333. doi: 10.1128/IAI.69.12.7326-7333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramakrishnan L., Federspiel N. A., Falkow S. Science. 2000;288:1436–1439. doi: 10.1126/science.288.5470.1436. [DOI] [PubMed] [Google Scholar]
- 22.Li Y., Miltner E., Wu M., Petrofsky M., Bermudez L. E. Cell Microbiol. 2005;7:539–548. doi: 10.1111/j.1462-5822.2004.00484.x. [DOI] [PubMed] [Google Scholar]
- 23.Choudhary R. K., Mukhopadhyay S., Chakhaiyar P., Sharma N., Murthy K. J., Katoch V. M., Hasnain S. E. Infect. Immun. 2003;71:6338–6343. doi: 10.1128/IAI.71.11.6338-6343.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strong M., Graeber T. G., Beeby M., Pellegrini M., Thompson M. J., Yeates T. O., Eisenberg D. Nucleic Acids Res. 2003;31:7099–7109. doi: 10.1093/nar/gkg924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Mering C., Jensen L. J., Snel B., Hooper S. D., Krupp M., Foglierini M., Jouffre N., Huynen M. A., Bork P. Nucleic Acids Res. 2005;33:D433–D437. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen F. E., Kempiak S., Huang D. B., Phelps C., Ghosh G. Protein Eng. 1999;12:423–428. doi: 10.1093/protein/12.5.423. [DOI] [PubMed] [Google Scholar]
- 27.Renshaw P. S., Panagiotidou P., Whelan A., Gordon S. V., Hewinson R. G., Williamson R. A., Carr M. D. J. Biol. Chem. 2002;277:21598–21603. doi: 10.1074/jbc.M201625200. [DOI] [PubMed] [Google Scholar]
- 28.Pal D., Eisenberg D. Structure (London) 2005;13:121–130. doi: 10.1016/j.str.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Shindyalov I. N., Bourne P. E. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 30.Kim K. K., Yokota H., Kim S.-H. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowsk Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Sheldrick G. M., Schneider T. R. In: Methods in Macromolecular Crystallography. Turk D., Johnson L., editors. Amsterdam: IOS; 2001. pp. 72–81. [Google Scholar]
- 33.Collaborative Computational Project, Number 4 Acta Crystallogr. D. 1994;50:760–763. [Google Scholar]
- 34.Jones T. A., Zou J.-Y., Cowan S. W., Kjeldgaard M. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 35.Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., et al. Acta Crystallogr. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Fabiola F., Bertram R., Korostelev A., Chapman M. S. Protein Sci. 2002;11:1415–1423. doi: 10.1110/ps.4890102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murshudov G. N., Vagin A. A., Dodson E. J. Acta Crystallogr. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P., Cowtan K. Acta Crystallogr. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Colovos C., Yeates T. O. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 41.Vriend G., Sander C. J. Appl. Crystallogr. 1993;26:47–60. [Google Scholar]
- 42.DeLano W. L. The pymol User's Manual. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 43.Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laskowski R. A., Watson J. D., Thornton J. M. Nucleic Acids Res. 2005;33:W89–W93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}