

Abstract
Most strains of murine coronavirus mouse hepatitis virus (MHV) express a cleavable spike glycoprotein that mediates viral entry and pH-independent cell-cell fusion. The MHV type 2 (MHV-2) strain of murine coronavirus differs from other strains in that it expresses an uncleaved spike and cannot induce cell-cell fusion at neutral pH values. We show here that while infection of the prototype MHV-A59 strain is not sensitive to pretreatment with lysosomotropic agents, MHV-2 replication is significantly inhibited by these agents. By use of an A59/MHV-2 chimeric virus, the susceptibility to lysosomotropic agents is mapped to the MHV-2 spike, suggesting a requirement of acidification of endosomes for MHV-2 spike-mediated entry. However, acidification is likely not a direct trigger for MHV-2 spike-mediated membrane fusion, as low-pH treatment is unable to overcome ammonium chloride inhibition, and it also cannot induce cell-cell fusion between MHV-2-infected cells. In contrast, trypsin treatment can both overcome ammonium chloride inhibition and promote cell-cell fusion. Inhibitors of the endosomal cysteine proteases cathepsin B and cathepsin L greatly reduce MHV-2 spike-mediated entry, while they have little effect on A59 entry, suggesting that there is a proteolytic step in MHV-2 entry. Finally, a recombinant virus expressing a cleaved MHV-2 spike has the ability to induce cell-cell fusion at neutral pH values and does not require low pH and endosomal cathepsins during infection. These studies demonstrate that endosomal proteolysis by cathepsins is necessary for MHV-2 spike-mediated entry; this is similar to the entry pathway recently described for severe acute respiratory syndrome coronavirus and indicates that coronaviruses may use multiple pathways for entry.
Enveloped viruses attach to receptors on the host cell through surface glycoproteins and enter cells by fusing with cellular membranes. The fusion event can occur either directly at the plasma membrane, as in the case of human immunodeficiency virus (HIV), or at the endosomal membrane following receptor-mediated endocytosis, as exemplified by the influenza A virus (recently reviewed in references 33 and 37). Viruses that enter by the latter route are believed to require the low-pH environment in the endosome to trigger conformational changes in the viral glycoprotein that lead to the exposure and insertion of the fusion peptide into the cellular membrane. Viral glycoproteins are thus classified as pH independent (such as HIV gp120/gp41) or pH dependent (such as influenza A virus hemagglutinin [HA]) based on the trigger required to activate their membrane fusion potential (36). However, it has been recently recognized that proteolysis by endosomal proteases is critical to activate some pH-dependent viral glycoproteins (5, 6, 10, 17, 34). For example, digestion of viral glycoprotein (GP) GP1 by endosomal cysteine proteases cathepsin B and L is proposed to be necessary to initiate membrane fusion and viral entry after the internalization of Ebola virus into the endosome (6). It is thus believed to be a new mechanism by which a viral glycoprotein is triggered to become fusogenic.
Coronaviruses comprise a large and diverse family of avian and mammalian enveloped, positive-stranded RNA viruses, which have the largest viral RNA genomes known (approximately 30 kb) (16). Murine coronavirus mouse hepatitis virus (MHV) is the prototype of group II coronaviruses; the MHV strains used in this study infect primarily the liver and the brain and thus provide animal models for encephalitis and hepatitis as well as demyelinating disease (recently reviewed in reference 43). The MHV spike protein (S protein), which forms the peplomers on viral particles, is responsible for receptor binding, for virus-cell fusion during entry, and also for cell-cell fusion during infection (11). S protein is a class I glycoprotein of 180 kDa; for most MHV strains, it is cleaved into two noncovalently associated subunits (S1 and S2) of about 90 kDa each by furin-like enzymes during processing in the Golgi (3, 8). The proteolytic cleavage motif is BBXBB (B stands for basic residue). For instance, the cleavage sites of MHV-A59 and MHV-JHM S protein are RRAHR and RRARR, respectively. The N-terminal S1 subunit contains a receptor binding domain within the first 330 amino acids (39); while the C-terminal S2 subunit mediates virus-cell membrane fusion, and contains two heptad repeat domains as well as the transmembrane domain. Unlike the fusion peptides of HIV (gp41) or influenza A virus (HA2), which are immediately adjacent to the cleavage site at the N-terminal of their S2-like subunits, the putative fusion peptide of MHV is internal, located near or within the first heptad repeat domain of the S2 subunit (3, 21).
The relationship between cell fusion activity and MHV spike has been studied intensively but with inconsistent results. Generally, it is believed that MHV strains expressing cleaved spike can induce cell-cell fusion, and the extent of spike cleavage correlates with its ability to mediate fusion. MHV-2, which expresses an uncleaved spike, is the only identified MHV strain that cannot induce syncytia. However, an MHV-2 variant with a functional proteolytic cleavage site in the spike causes cell-cell fusion, suggesting that the inability of MHV-2 to induce fusion is due to the lack of cleavage of its spike (44). Consistent with this observation, when cleavage of the MHV-A59 S protein is minimized by mutation of the protease recognition site, syncytium formation induced by these mutant viruses is delayed (13, 14). The observations described above suggest that cleavage, at least to some extent, is a prerequisite for fusion. In contrast, it was demonstrated using a vaccinia virus expression system that mutant spikes of MHV-A59 and MHV-JHM, which are not cleaved to a detectable level, can induce cell-cell fusion, although with a delay compared to cells expressing wild-type spike (1, 40).
Several lines of evidence indicate that MHV viral entry and cell-cell fusion have different requirements. For example, mutant spikes of MHV-A59 that are poorly cleaved mediate entry into susceptible cells with efficiency similar to that of the wild type (8, 13), despite the fact that they carry out cell-cell fusion very inefficiently. A study of MHV-A59 virus-like particles also demonstrated that spike cleavage is not required for viral infectivity (2). Similarly, inhibition of the cleavage of spike by a specific furin protease inhibitor also appears to have no effect on the entry kinetics of MHV-A59 (8).
The mechanism of MHV entry into cells is not well understood. However, for most strains, it appears to occur through a pH-independent pathway. This is supported by the observations that MHV has the ability to induce cell-cell fusion at neutral pH values and entry is not significantly affected by lysosomotropic agents (19, 26). However, there are some data suggesting that an endosomal route may be utilized by some MHV isolates. For instance, while wild-type MHV-JHM enters cells in culture by a pH-independent pathway, persistent JHM infection of a neuronal cell line (OBL21a) gave rise to a low-pH-dependent fusion variant OBLV60 (12). OBLV60 is dependent on an acidic pH to fuse host cells and is believed to enter cells through the endosomal pathway (26). MHV-3 has also been reported to be sensitive to lysosomotropic agents, which suggests that it may enter cells though the endosome (20). In this report, we have further investigated the mechanism of MHV entry, focusing on the MHV-2 strain. MHV-2 is the only wild-type MHV strain known to express an uncleaved spike. Although MHV-2 lacks the ability to induce cell-cell fusion, it can infect susceptible cells efficiently in culture and causes severe hepatitis in mice (31). By comparing MHV-2 with A59, which expresses a cleaved spike, our goal was to understand how entry mediated by uncleaved MHV spike may differ from that mediated by cleaved spike. Furthermore, severe acute respiratory syndrome coronavirus (SARS-CoV), a coronavirus related to MHV, expresses an uncleaved spike and enters cells through a pH-dependent, endosomal route (22, 35). Interestingly, the low-pH requirement for entry of SARS-CoV is not due to a direct effect on the conformational change of the spike but rather due to proteolysis by the acidic endosomal cathepsin L (34). We demonstrate here that uncleaved MHV-2 spike may mediate entry via a similar pathway that requires proteolysis by endosomal cathepsins.
MATERIALS AND METHODS
Cell lines and viruses.
Murine fibroblast L2 and 17CL-1 cells were maintained in Dulbecco's modified eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; HyClone), 1% antibiotic-antimycotic (penicillin-streptomycin-amphotericin B; Invitrogen), and 10 mM HEPES buffer solution (Invitrogen).
The following viruses were used in this study: wild-type MHV-2 (31), recombinant wild-type MHV-A59 (RA59) (30), recombinant MHV-A59 with the spike gene of MHV-2 (RA59/MHV-2S, previously referred to as Penn98-1 [7]), RA59/MHV-2S containing a substitution in the cleavage site (RA59/MHV-2SS757R), and enhanced green fluorescent protein (EGFP)-expressing viruses RA59EGFP (32) and RA59EGFP/MHV-2S. EGFP is expressed in the place of the nonessential gene 4. The viruses were propagated in 17 CL-1 cells and titrated in L2 cell monolayers by plaque assay as previously described (30). The selection of the recombinant viruses by targeted RNA recombination has previously been described (30). Selection of RA59/MHV-2SS757R and RA59EGFP/MHV-2S is described below.
The S757R substitution was generated by two-step PCR mutagenesis. Primer pair 2011F (CTTATATGATGTTAATGGC) and CSR (2) (GACAGAGCGGCGAGC) and primer pair CSF (2) (GCTCGCCGCTCTGTC) and 3418R (TAATTGAAAGAGATATCGG) were used to generate overlapping DNA fragments containing an A-to-C nucleotide substitution in the cleavage site of the MHV-2 spike gene (the relevant nucleotides in the cleavage signal primers are underlined). Using the fragments as the template and the primers 2011F and 3418R, a 1.3-kb fragment was amplified and subsequently cloned into pMH54-MHV-2S (7). This plasmid was used in targeted recombination to generate the RA59/MHV-2SS757R virus, which contains a single substitution (S757R) that replaced the wild-type MHV-2 spike cleavage site (HRARS) with the functional motif HRARR.
For the EGFP-expressing virus, RA59EGFP/MHV-2S, the MHV-2 spike gene was cleaved with AvrII and SbfI from pMH54-MHV-2S (7) and inserted into pMH54EGFP, which was derived previously during the generation of RA59EGFP (32), in place of the A59 spike gene. This plasmid was used in targeted RNA recombination to select the RA59EGFP/MHV-2S virus. Sequence and restriction analysis was performed using MacVector (Accelrys, San Diego, Calif.).
Treatment with lysosomotropic agents.
Lysosomotropic agents were obtained from Sigma. Ammonium chloride (NH4Cl) and chloroquine stock solutions were adjusted to pH 7.4 before use. L2 cell monolayers in six-well tissue culture plates were pretreated with 40 mM NH4Cl, 50 μM chloroquine, or 100 nM bafilomycin A1 at 37°C 1 h prior to infection. Viruses, in the presence of inhibitors, were adsorbed for 60 min at 4°C at a multiplicity of infection (MOI) of 1 PFU/cell. Virus inocula were removed and the cells were incubated in growth medium containing the same agents for another 1 h, followed by overnight incubation at 37°C with fresh growth medium without inhibitors. As a control, the infected cells were treated with the agents from 1 h to 3 h postinfection. Viral titers in the supernatants at 16 h postinfection were determined by plaque assay on L2 cells (30).
Trypsin/low-pH treatment.
L2 cell monolayers were pretreated with 40 mM NH4Cl, followed by adsorption of RA59EGFP/MHV-2S at 4°C. After adsorption, cells were incubated with phosphate-buffered saline (PBS) (pH 7.4) at 37°C for 15 min (24) and then treated either with pH 5.0 buffer (PBS adjusted with HCl) for 15 min or with 10 μg/ml TPCK (tosylsulfonyl phenylalanyl chloromethyl ketone, l-1-tosylamide-2-phenylmethyl chloromethyl ketone)-treated trypsin (Sigma) in PBS for 5 min and then washed thoroughly with PBS. NH4Cl was present during the entire process and for another hour after low-pH or trypsin treatment. Images were recorded with a Nikon fluorescence microscope after a subsequent 7-h incubation at 37°C in fresh medium without NH4Cl.
Cell-cell fusion.
L2 cell monolayers were inoculated with RA59, RA59/MHV-2S, or RA59/MHV-2SS757R at an MOI of 1 PFU/cell. Viruses were allowed to adsorb onto the monolayers at 4°C for 1 h, then the monolayers were rinsed, fresh medium was added, and the monolayers were incubated at 37°C. Syncytium formation was observed at 9 and 24 h postinfection using an inverted Nikon microscope.
To determine the effects of low pH and trypsin on cells infected with RA59/MHV-2S, at 24 h postinfection cells were washed with PBS and incubated for 5 min at 37°C with PBS at pH 5.0 (adjusted with HCl) or PBS at pH 7.4 containing 0, 5, or 20 μg/ml TPCK-treated trypsin. PBS was then replaced with fresh medium, and the monolayers were examined for syncytium formation after an additional hour of incubation at 37°C.
Western blot analysis.
Cell lysates were prepared from infected and uninfected monolayers by addition of 200 μl of lysis buffer (50 mM Tris buffer, pH 7 containing 0.15 M NaCl and 1% Nonidet P-40) to approximately 1 to 3 × 106 cells. MHV spike proteins were detected by immunoblotting using polyclonal goat anti-MHV-S serum AO4 (kindly provided by K. Holmes) and a chemiluminescence detection system (Santa Cruz Biotechnology, CA). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transfer to nitrocellulose membranes were performed using the NuPAGE 7% Tris-acetate gel system, following the manufacturer's recommended protocol (Invitrogen, CA).
Protease inhibitor assays.
Protease inhibitors E-64, CA-074 (Sigma), and Z-FY-DMK (Calbiochem) were dissolved in dimethyl sulfoxide (DMSO), and diluted into 2% FBS-DMEM containing 1% DMSO immediately before use. L2 cell monolayers in 48-well plates were pretreated with 500 μl medium with different concentration of protease inhibitors at 37°C for 3 h. Forty microliters of serially diluted EGFP-expressing viruses was then added directly to the culture medium containing DMSO/inhibitor. After one-hour adsorption at 4°C, the viral inocula were removed and the cells were incubated with medium containing the same drugs for another 1 h. Then the cells were incubated at 37°C with fresh growth medium without drugs, and levels of infectivity of RA59EGFP and RA59EGFP/MHV-2S were determined by counting EGFP-positive cells under a Nikon fluorescence microscope at 8 h after infection. Infectivity values were reported as infectivity units (iu)/ml of virus, where one infectivity unit corresponds to one EGFP-positive cell. Each measurement was obtained from wells containing 20 to 200 EGFP-positive cells by multiplying the number of EGFP-positive cells by the reciprocal of the dilution for that well. Infectivity of EGFP-expressing viruses in the presence of inhibitors was expressed as a percentage of the infectivity observed in mock-treated cells using the following formula:
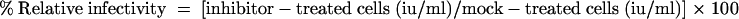 |
Alternatively, cells were pretreated with E-64 (100 μM) or Z-FY-DMK (10 μM) in 2% FBS-DMEM containing 1% DMSO for 3 h and then infected with RA59/MHV-2S or RA59/MHV-2SS757R at an MOI of 1 PFU/cell for 1 h in the presence of inhibitors. Cultures were then washed thoroughly, and incubated with fresh growth medium at 37°C. At indicated times (10, 14, and 18 h), supernatants were harvested. Infectious viruses in the supernatants were titrated by plaque assay on L2 cells.
RESULTS
Lysosomotropic agents block an early step in MHV-2 replication in L2 cells and their effect maps to the spike protein.
Low-pH-dependent viruses enter cells through endocytosis and their entry is blocked by lysosomotropic agents which prevent endosomal acidification (24). The sensitivity of murine coronavirus strains A59 and MHV-2 to lysosomotropic agents was assessed on murine L2 cells. Three agents were used to pretreat cells, including two weak bases, ammonium chloride (NH4Cl) and chloroquine, and an inhibitor of vacuolar H+-ATPase, bafilomycin A1. To avoid possible inhibition of MHV at a later stage of replication, the drugs were washed away 1 h postinfection (26). Replication of different viruses at 16 h postinfection was measured by titrating infectious virus in the supernatant by plaque assay. MHV-2 infection was strongly inhibited by pretreatment with NH4Cl (Fig. 1A) and bafilomycin A1 (Fig. 1B), reducing viral titers by 1.7 to 2.3 log10, while replication of RA59 was not affected. Pretreatment with chloroquine had similar results (data not shown). Also shown in Fig. 1, when the lysosomotropic agents were added 1 h postinfection, the inhibition of MHV-2 infection was not observed. These data demonstrate that while MHV-A59 is not sensitive to treatment with several lysosomotropic agents, MHV-2 replication is significantly inhibited by these agents. Furthermore, these data imply that the low-pH-dependent step required for MHV-2 infection is an early event in viral replication. In contrast, it appears that MHV-A59 enters cells through a pH-independent pathway as would be expected, since this virus can induce syncytia at neutral pHs on L2 cells (see Fig. 6A).
FIG. 1.

Effect of ammonium chloride (NH4Cl, a weak base) (A) and bafilomycin A1 (BAF, a vacuolar H+/ATPase inhibitor) (B) on viral replication in L2 cells. Cells were either pretreated from 1 h before virus infection to 1 h postinfection (p.i.) or treated from 1 to 3 h p.i. with 40 mM NH4Cl or 100 nM BAF. After treatment, medium containing the agents was removed from cells and replaced with fresh growth medium. Culture supernatants were collected at 16 h p.i., and viral titers were determined by plaque assay. Error bars represent the standard deviations from three replicates.
FIG. 6.

(A) Effect of S757R substitution in MHV-2 spike on cell-cell fusion. L2 cells were infected with isogenic A59 recombinant viruses expressing spike of MHV-2, MHV-2S757R, and A59 as labeled in the figure. Images were recorded at 9 h and 24 h postinfection. The bar represents 50 μm. (B) Immunoblot of MHV spike proteins in lysates of cells infected with the viruses described for panel A. In addition to the uncleaved and cleaved spike bands, an extra spike-related band of about 120 kDa, which has been previously observed (13, 14), was detected in lysates of cells infected with A59.
To further investigate whether the low-pH requirement for the replication of MHV-2 is indeed due to the entry step, we used a chimeric A59 recombinant virus, RA59/MHV-2S, in which the MHV-2 spike protein is expressed within the background of the pH-independent parent A59 (Fig. 2) (7). Viral replication in the presence of lysosomotropic agents was carried out to compare RA59 and RA59/MHV-2, A59 recombinant viruses isogenic except for the viral spike protein. Analysis of supernatant viral titers at 16 h postinfection revealed that replication of RA59 was not affected; however, pretreatment with NH4Cl (Fig. 1A) and bafilomycin A1 (Fig. 1B) reduced the titers of RA59/MHV-2 by 1.6 to 2.3 log10, the same order of magnitude as the inhibition of wild-type MHV-2 replication. Similar results were obtained using a pair of EGFP-expressing viruses (RA59EGFP and RA59EGFP/MHV-2S) that again were isogenic except for their spike proteins (32). In this assay, virus-infected cells express EGFP and appear green under the fluorescence microscope. Inhibition of viral infection can be directly observed at 8 h postinfection, which is earlier than titrating infectious virus in the supernatants at 16 h postinfection. Pretreatment of L2 cells with NH4Cl significantly decreased the numbers of EGFP-positive cells observed following infection with RA59EGFP/MHV-2S (Fig. 3A and B), while it had no effect on numbers of infected cells observed upon infection with RA59EGFP (data not shown). The experiments with the two pairs of isogenic A59 recombinant viruses expressing either MHV-A59 spike or MHV-2 spike consistently demonstrate that the low-pH requirement segregates with the MHV-2 spike, and also provide further evidence that the low pH is acting at the entry stage rather than at a later stage of viral uncoating or replication.
FIG. 2.

Schematic diagram of chimeric isogenic A59 recombinant viruses expressing A59 (white) or MHV-2 (gray) spike genes. A59 spike is cleaved by furin-like enzymes at a site with dibasic amino acids (RRAHR) into S1 and S2 subunits. However, with one less basic residue, the proteolytic cleavage site of MHV-2 is not functional (HRARS) and the spike is not cleaved. Using targeted RNA recombination, chimeric A59 recombinant viruses expressing MHV-2 spike and MHV-2 spike with a S757R substitution (HRARR) were selected, termed RA59/MHV-2S and RA59/MHV-2SS757R, respectively. The relevant substitution is underlined. The MHV-2 genome shown in gray is not a recombinant. Genomes are not shown to scale.
FIG. 3.

(A and B) Pretreatment with NH4Cl diminishes RA59/MHV-2SEGFP infection. L2 cell monolayers were pretreated from 1 h before virus infection to 1 h postinfection with 40 mM NH4Cl. After treatment, medium containing NH4Cl was removed from cells and replaced with fresh growth medium. Images were recorded at 8 h postinfection. (C and D) Trypsin, but not low-pH treatment, overcomes the NH4Cl block. After viral adsorption in the presence of NH4Cl, cells were incubated with PBS (pH 7.4) at 37°C for 15 min and then treated either with pH 5.0 buffer for 15 min (C) or with 10 μg/ml trypsin for 5 min (D) and then washed thoroughly with PBS. NH4Cl was present during the entire process and for another 1 h after low-pH or trypsin treatment. Images were recorded after subsequent 7-h incubation in fresh medium without NH4Cl.
Trypsin, but not low-pH treatment, activates MHV-2 spike-mediated membrane fusion and overcomes the NH4Cl block.
To verify that the inhibition of MHV-2 spike-mediated entry in the presence of these agents was directly due to a failure of acidification, we attempted to determine whether the NH4Cl block could be overcome by incubating cells with bound virus at a low pH. The experiment was performed with RA59EGFP/MHV-2S, the lysosomotropic-agent-sensitive EGFP-expressing A59 recombinant virus bearing MHV-2 spike. Virus was adsorbed onto L2 cells at 4°C for 1 h; the medium was replaced with pH 7.4 PBS buffer at 37°C for 15 min and then with pH 5.0 buffer for 15 min (24). NH4Cl was present the whole time. Surprisingly, the low-pH treatment was unable to promote infection of RA59EGFP/MHV-2S (Fig. 3C) which indicates that low pH at the plasma membrane cannot overcome the lysosomotropic agent inhibition of MHV-2 spike-mediated entry. This suggests the possibility that a critical host factor for MHV-2 entry, likely within the endosome, is dependent on acidic pH.
MHV-2, which expresses an uncleaved spike, is the only identified MHV strain that cannot induce cell-cell fusion. It is presumed that the inability of MHV-2 to induce fusion is due to the lack of cleavage of its S protein (44). It has also been reported that trypsin treatment of MHV-2-infected cells can induce fusion-type plaques (45). Together with our observations with the lysosomotropic agents, we hypothesized that proteolysis of MHV-2 spike may be necessary to activate its membrane fusion potential. To test this hypothesis, we investigated whether trypsin could trigger syncytium formation in RA59/MHV-2S-infected cells. L2 cells were infected with RA59/MHV-2S, and at 24 h postinfection, cell-cell fusion was not observed (Fig. 4A). The infected cells were then treated with various concentrations of TPCK-treated trypsin or pH 5.0 buffer for 5 min. After cell treatment, PBS was replaced with fresh medium. Consistent with the result that low-pH treatment cannot overcome the NH4Cl block to MHV-2 spike-mediated entry, the low-pH treatment failed to induce syncytium formation in RA59/MHV-2S-infected cells. In contrast, after trypsin treatment, cell-cell fusion was observed; the amount of fusion was dependent on the dose of trypsin (Fig. 4A). Western blot analysis of lysates of trypsin-treated cells demonstrated that cleavage of the MHV-2 spike had occurred at a site near the middle of the protein, generating a band with electrophoretic mobility similar to those of the S1 and S2 subunits of A59 spike protein (Fig. 4B). Thus, MHV-2 spike-induced cell-cell fusion was accompanied by a cleavage event at a site equivalent to or close to the furin cleavage site utilized in other MHV spikes.
FIG. 4.

(A) Trypsin, but not low-pH treatment, induces MHV-2 spike-mediated cell-cell fusion. L2 cells were infected with RA59/MHV-2S. At 24 h postinfection, cells were washed with PBS and incubated for 5 min with PBS at pH 5.0 or PBS at pH 7.4 containing 0, 5, or 20 μg/ml trypsin. PBS was then replaced with fresh medium, and images were recorded after 1 h of incubation. The bar represents 50 μm. (B) Immunoblot of MHV spike proteins in infected cell lysates following trypsin or low-pH treatment as described in the legend to panel A.
To further evaluate the role of proteolysis in MHV-2 spike-mediated membrane fusion, we adsorbed RA59EGFP/MHV-2S onto L2 cells in the presence of NH4Cl and treated these cells with 10 μg/ml TPCK-treated trypsin instead of a low-pH buffer as described above. As shown in Fig. 3D, trypsin treatment allowed the virus to replicate and to overcome the inhibition mediated by NH4Cl. Thus, proteolysis of MHV-2 spike by trypsin bypasses the requirement for acidic pH during the viral entry process. Taken together, the lysosomotropic agent inhibition studies and trypsin activation experiments suggest that MHV-2 enters cell through an endosomal pathway and that proteolysis of MHV-2 spike is necessary to activate its membrane fusion potential.
Cathepsin B and cathepsin L/B inhibitors prevent entry mediated by MHV-2 spike.
The ability of trypsin cleavage to overcome the NH4Cl inhibition of entry suggests a requirement for endosomal proteases in MHV-2 spike-mediated infection. To test this hypothesis and identify candidate proteases, we assessed the ability of a series of protease inhibitors to block infection by RA59EGFP/MHV-2S using RA59EGFP as a control. L2 cells were pretreated with various concentrations of protease inhibitors for 3 h before adding serially diluted EGFP-expressing viruses. After 1 h of adsorption, the viral inocula were removed and the cells were incubated with medium containing the same drug for another hour. The cells were then incubated with fresh growth medium without drugs, and infectivity was measured at 8 h postinfection by counting EGFP-positive cells. The cysteine protease inhibitor E-64 at 100 μM nearly eliminated RA59EGFP/MHV-2S infection in L2 cells, reducing the infectivity to 0.01% of the control (Fig. 5A). The infectivity of RA59EGFP decreased only slightly to 80% at 100 μM E-64, which is possibly due to inhibition of the virally encoded cysteine proteases, consistent with the observation that E-64d, a membrane-permeable derivative of E-64, can inhibit MHV-A59 protein processing and RNA synthesis (18). However, since RA59EGFP/MHV-2S is the isogenic with RA59EGFP except for its spike, the extreme sensitivity to E-64 of RA59EGFP/MHV-2S infection must be attributed to the MHV-2 spike.
FIG. 5.

Effect of endosomal cysteine protease inhibitors on infectivity of RA59EGFP and RA59/MHV-2SEGFP in L2 cells. Cells were pretreated with various concentrations of protease inhibitors E-64 (cysteine protease inhibitor) (A), CA-074 (specific cathepsin B inhibitor) (B), and Z-FY-DMK (cathepsin L/B inhibitor) (C) for 3 h before adding serially diluted EGFP-expressing viruses. After 1 h of adsorption, the viral inocula were removed and the cells were incubated with medium containing the same drug for another hour. The cells were then incubated with fresh growth medium without drugs, and infectivity was measured at 8 h postinfection by counting EGFP-positive cells. Infectivity values are reported as infectivity units (iu)/ml virus, where one infectivity unit corresponds to one EGFP-positive cell. Infectivity of EGFP-expressing viruses in the presence of inhibitors was expressed as a percentage of infectivity observed in mock-treated cells. Error bars represent the standard deviations from at least three replicates.
Cathepsin B and cathepsin L are E-64-sensitive cysteine proteases that are the most abundant proteases present in endosomes and lysosomes and are active at acidic pHs in a broad range of mammalian cells (28, 42). To further examine the roles of these enzymes in MHV-2 spike-mediated infection, the effects of a specific cathepsin B inhibitor (CA-074) and a cathepsin L/B inhibitor (Z-FY-DMK) on viral replication were evaluated. CA-074, which inhibits cathepsin B enzyme activity selectively, efficiently reduced infectivity of RA59EGFP/MHV-2S to 10% of the control (Fig. 5B), suggesting a role of cathepsin B in MHV-2 spike-mediated infection. However, Z-FY-DMK, which inhibits both cathepsin L and cathepsin B in the concentration range of 1 to 10 μM, reduced the infectivity to 1% of the control (Fig. 5C). The greater efficiency of Z-FY-DMK compared to CA-074 implies that cathepsin L may also play a role in MHV-2 spike-mediated infection. Infectivity of RA59EGFP was not affected significantly by any of the inhibitors, indicating that the cathepsin inhibitors did not alter the infectivity of viruses entering through a pH-independent pathway. These results with protease inhibitors suggest that acid-dependent endosomal cysteine proteases in L2 cells, including cathepsin B and cathepsin L, are important for MHV-2 spike-mediated entry.
A recombinant virus with an S757R substitution in the MHV-2 S expresses a cleaved spike, induces cell-cell fusion at neutral pH and is insensitive to both lysosomotropic agents and cathepsin inhibitors.
Taguchi et al. (44) reported that an MHV-2 variant with mutations in the proteolytic cleavage site (S757R) and the signal sequence of the spike acquired the ability to induce cell-cell fusion at neutral pHs. This observation suggests that the inability of MHV-2 to induce syncytia is due to the lack of cleavage of its spike. Using targeted RNA recombination, we selected a recombinant virus, RA59/MHV-2SS757R, with the S757R substitution in the cleavage site of the MHV-2 spike gene. This mutation (HRARS to HRARR) created a sequence consistent with a known furin cleavage site. In contrast to RA59/MHV-2S, the new recombinant virus expresses a cleaved spike (Fig. 6B) and is able to cause cell-cell fusion at neutral pHs (Fig. 6A). These data clearly prove the previous hypothesis that the lack of spike cleavage is responsible for the inability of MHV-2 to produce syncytia. However, the kinetics of syncytia formation induced by the mutant virus is delayed compared to A59; this is probably due to the slower replication kinetics of the mutant in tissue culture compared with RA59/MHV-2S (data not shown). Furthermore, the S757R mutant virus is also highly attenuated and replicates poorly in the brain and liver following intracerebral inoculation of C57BL/6 mice (data not shown).
As RA59/MHV-2SS757R-infected cells form syncytia at neutral pH values, we hypothesized that this mutant virus enters L2 cells directly through the cell surface instead of by an endosomal pathway. To test this hypothesis, we measured the sensitivity of RA59/MHV-2SS757R infection in L2 cells to lysosomotropic agents. Cells were pretreated with NH4Cl or bafilomycin A1 and then infected with RA59/MHV-2SS757R in the presence of the agents. As shown in Fig. 7, replication of this virus was not dramatically affected by NH4Cl pretreatment even though RA59/MHV-2S infection was strongly inhibited. Pretreatment with bafilomycin A1 had similar results (data not shown). These data indicate that the S757R amino acid substitution and consequent cleavage of MHV-2 spike eliminate the entry requirement of endosomal acidification in L2 cells.
FIG. 7.

Effect of NH4Cl on RA59/MHV-2SS757R replication in L2 cells. Cells were either pretreated from 1 h before virus infection to 1 h p.i. or treated from 1 to 3 h p.i. with 40 mM NH4Cl. After treatment, medium containing NH4Cl was removed from cells and replaced with fresh growth medium. Culture supernatants were collected at 16 h p.i., and viral titers were determined by plaque assay. Error bars represent the standard deviations from three replicates.
To further demonstrate that endosomal cysteine proteases are not necessary for infection by the S757R mutant virus, we assessed the effects of E-64 and Z-FY-DMK on growth of RA59/MHV-2S and RA59/MHV-2SS757R. L2 cells were pretreated with E-64 (100 μM) or Z-FY-DMK (10 μM) for 3 h and then infected with RA59/MHV-2S or RA59/MHV-2SS757R in the presence of inhibitors. Cultures were then washed thoroughly and incubated with fresh growth medium without drugs. Supernatants were harvested at 10, 14, and 18 h postinfection and infectious viruses were titrated by plaque assay. By 18 h postinfection, the yield of infectious RA59/MHV-2S was significantly decreased by 2 log10 in inhibitor-treated cells, while RA59/MHV-2SS757R was not affected (Fig. 8). This experiment demonstrates that whereas RA59/MHV-2S infection is sensitive to the treatment of cysteine protease inhibitors, in particular, cathepsin L/B inhibitors, the S757R substitution allows RA59/MHV-2SS757R to replicate without requiring active endosomal cysteine proteases.
FIG. 8.

Effect of endosomal cysteine protease inhibitors on RA59/MHV-2S (A) and RA59/MHV-2SS757R (B) replication in L2 cells. Cells were pretreated with 100 μM E-64 (to inactivate endosomal cysteine proteases) or 10 μM Z-FY-DMK (to specifically inactivate cathepsin B and L) for 3 h, and then infected with viruses in the presence of inhibitor for 1 h. Growth medium containing inhibitors was then removed from cells and replaced with fresh medium without inhibitors. At indicated times (10, 14, and 18 h), supernatants were harvested and viral yields were determined by plaque assay.
DISCUSSION
We have demonstrated that a nonfusogenic murine coronavirus strain, MHV-2, uses a pH-dependent entry pathway. However, our data indicate that acidification is not the direct trigger for MHV-2 spike-mediated fusion, but rather proteolysis by endosomal cathepsins following endocytosis is necessary for MHV-2 spike-mediated entry. Lysosomal cathepsins are a group of cysteine proteases which are responsible for protein digestion in the late endosome and lysosome (28, 42). In addition to intracellular nonspecific protein degradation, cathepsins are also involved in a wide range of physiological and pathological processes, including antigen presentation, prohormone processing, tumor invasion, tumor angiogenesis and apoptosis (28, 42). For example, cathepsin L is necessary for the degradation of invariant chain (Ii) in cortical thymic epithelial cells, which is a critical step in Major histocompatibility complex class II-restricted antigen presentation. As a result, cathepsin L-deficient mice are immunodeficient due to a defect in positive selection of T cells (25).
Cathepsins have also been reported to play an important role in the life cycle of several viruses, especially for viral entry. The role of cathepsins in nonenveloped reovirus entry and disassembly has been well defined (5, 10). After receptor-mediated endocytosis, the reovirus virion is converted to an infectious subvirion through proteolysis of outer capsid σ3 by endosomal cathepsin B and cathepsin L. Digestion of the protector protein σ3 exposes surface μ1 protein, which then mediates viral penetration through the endosomal membrane into the cytoplasm. In addition, proteolytic digestion by endosomal cathepsins has been recently described during entry of two enveloped viruses, Ebola virus and SARS coronavirus (6, 34). It has been demonstrated that infection mediated by Ebola virus GP depends primarily on cathepsin B and, to a lesser extent, on cathepsin L (6); SARS-CoV spike-mediated entry depends on cathepsin L (34). Interestingly, cathepsin L is also proposed to play a role in proteolytic maturation of Hendra virus F protein during recycling of F protein between the endosome and the cell surface prior to budding of the virion from the cell surface (29).
The most likely possibility for the roles of cathepsin B and L in MHV-2 spike-mediated entry is cleavage of the spike. MHV-2 spike, unlike the spikes encoded by other MHV strains, is not cleaved into S1 and S2 subunits. Cleavage by trypsin into similarly sized subunits suggests that this proteolysis activates MHV-2 spike membrane fusion potential. Similar observations have also been made for SARS-CoV, in that proteases such as trypsin facilitate viral entry from the cell surface (22, 35). Thus, it is likely that cathepsin B and L digest the MHV-2 spike in the endosome, triggering membrane fusion and entry. Furthermore, the S757R mutant virus contains a functional dibasic furin-like cleavage site in the spike and is able to infect cells in the presence of cathepsin inhibitors, consistent with the conclusion that cleavage of wild-type MHV-2 spike by cathepsins is necessary for infection. However, it is still not clear how cathepsin B and L cleavage of spike facilitates viral entry, or what the specific roles each of the cathepsins plays in facilitating entry. More specifically, the two activities may be functionally redundant, or there may be a multistep process as proposed for Ebola virus GP-mediated entry (6).
Given that the MHV-2 spike with the S757R substitution and other cleaved MHV spikes are fusogenic, one hypothesis is that cathepsins cleave the MHV-2 spike near the S1-S2 boundary, which is enough to trigger the necessary conformational changes to expose the hydrophobic fusion peptide and insert it into the cellular membrane. Another possibility is that after binding to the receptor, the spike is physically constrained from undergoing the necessary conformational changes required for fusion peptide insertion. Because of the rather nonspecific character of cathepsins, cleavage by cathepsins at any sites exposed by receptor-binding could either relieve such constrains or induce conformational rearrangements leading to fusion peptide insertion. In this model, MHV-2 spike cleavage by cellular cathepsin B and L is functionally equivalent to fusion-triggering signals for other viruses, such as binding of retroviruses to receptors (24) and the exposure of influenza virus HA to acidic pH (4).
Most MHV strains enter cells via direct fusion with the plasma membrane. In addition to MHV-2, the OBLV60 variant of JHM has been demonstrated to be dependent on acidic pH to induce cell-cell fusion and for entry, presumably through an endosomal pathway (12, 26). However, the low-pH requirement of OBLV60 appears to be different from that of MHV-2. A low-pH treatment can activate spike-mediated cell-cell fusion of OBLV60 (12), but not MHV-2. The change from neutral pH-induced fusion to low-pH-induced fusion of OBLV60 is dependent on three amino acid substitutions, Q1067H, Q1094H, and L1114R, all within the first heptad repeat (HR1) of the S2 subunit, while the S1/S2 cleavage site remains intact. Thus, it was suggested that the acquisition of a titratable proton and/or positive charge, as a result of the Q-to-H substitution, could result in the requirement for a low-pH (pH 6) environment for the spike structural rearrangements that activate membrane fusion (12, 26, 41). Despite the observation that both MHV-2 and OBLV60 are pH-dependent murine coronaviruses, they apparently utilize two different pH-dependent entry mechanisms. For OBLV60, like other pH-dependent viruses, such as influenza virus, low pH may directly trigger conformational changes of the spike and facilitate fusion peptide insertion. In the case of MHV-2, the dependence on acidification is due to a requirement for acidic endosomal protease activation leading to cleavage of the spike.
It has been established that a large number of viruses express glycoproteins that are posttranslationally cleaved; these include influenza virus, HIV type 1, herpesviruses, filoviruses, paramyxoviruses, and some coronaviruses (38). The presence of an R-X-R/K-R consensus sequence motif among the glycoproteins of these viruses indicates that cleavage activation by furin-like enzymes is a general phenomenon. In many cases cleavage has been demonstrated as a requirement for activation of membrane fusion potential and infectivity (9, 23). The availability of cellular enzymes capable of processing the glycoprotein precursors can be a major determinant of viral tropism and pathogenicity. For instance, the HA of certain avirulent strains of influenza virus can be efficiently processed only by the proteases present in a limited number of cell or tissue types, so these viruses normally cause localized infections in, for example, the respiratory tract of mammals or intestinal tract of wild birds. In pathogenic viral strains, introduction of a polybasic cleavage site into HA renders the glycoprotein susceptible to proteolytic processing by a family of widely expressed host proteases, thereby extending tissue tropism. It is believed that this expanded tropism is a major determinant of the increased virulence of these viruses (38). In contrast to these viral systems, the virulence of murine coronaviruses appears to be independent of efficient spike cleavage and the ability to induce cell-cell fusion (14). MHV-2, a naturally nonfusogenic strain, can cause severe hepatitis in mice, demonstrating that entry and efficient replication of this strain is independent of spike cleavage by furin-like enzymes. The data reported here suggest that endosomal proteolysis by cathepsins activates MHV-2 spike-mediated entry and allows MHV-2 to be virulent in the mouse. The main target of MHV-2 infection is the liver, where it causes severe hepatitis (31); the liver is a site of high activity of cathepsin B and L (28, 42), perhaps facilitating viral infection. Interestingly, when a dibasic proteolytic cleavage site is created by the S757R amino acid substitution in the MHV-2 spike, the recombinant virus expressing a partially cleaved spike, does not replicate efficiently in either tissue culture or in vivo (data not shown). Thus, adding a cleavage site to MHV-2 likely changes the conformation of the spike such that the virus expressing this spike is not quite as fit as the wild-type virus, and this is enough to compromise replication and pathogenesis.
In the case of the fusogenic A59 strain of MHV, we have demonstrated that the H716D substitution in MHV-A59 spike cleavage site (RRAHR to RRADR) can significantly reduce spike cleavage in cell culture and cause a fusion-delayed phenotype (13). Interestingly, we have recently shown that a recombinant MHV-A59 expressing a spike with the H716D substitution causes lethal hepatitis, higher viral load, and enhanced virus spread following intrahepatic inoculation of mice (27). This demonstrates again that the virulence of murine coronaviruses is independent of efficient cleavage of the virion-associated spike proteins and cell-cell fusion (14). Intriguingly, we previously found that spike protein analyzed from brain and liver lysates of mice infected with wild-type A59 is not cleaved into S1 and S2 (15), suggesting that replication in these organs does not involve cleavage during intracellular processing and that A59 infection in vivo may also involve an endosomal route of entry. In addition, Rottier et al. (8) recently reported that inhibitors of endocytosis have an inhibitory effect on MHV-A59 entry when cells were treated with furin inhibitors. These observations, along with the data reported here on the role of cathepsins in MHV-2 spike-mediated entry, suggest a role for an endocytic compartment in the MHV infection process when the spikes are not cleaved. We speculate that when the spike of an MHV is not cleaved by furin-like enzymes during intracellular maturation, as in the case of MHV-2, the virus still can be pathogenic through an alternative entry pathway, involving receptor-mediated endocytosis and fusion triggered through proteolysis by endosomal cathepsins.
Acknowledgments
This work was supported by NIH grants AI60021 (S.R.W.) and AI059172 (P.B.).
REFERENCES
- 1.Bos, E. C., L. Heijnen, W. Luytjes, and W. J. Spaan. 1995. Mutational analysis of the murine coronavirus spike protein: effect on cell-to-cell fusion. Virology 214:453-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bos, E. C., W. Luytjes, and W. J. Spaan. 1997. The function of the spike protein of mouse hepatitis virus strain A59 can be studied on virus-like particles: cleavage is not required for infectivity. J. Virol. 71:9427-9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosch, B. J., R. van der Zee, C. A. de Haan, and P. J. Rottier. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77:8801-8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boulay, F., R. W. Doms, I. Wilson, and A. Helenius. 1987. The influenza hemagglutinin precursor as an acid-sensitive probe of the biosynthetic pathway. EMBO J. 6:2643-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandran, K., and M. L. Nibert. 2003. Animal cell invasion by a large nonenveloped virus: reovirus delivers the goods. Trends Microbiol. 11:374-382. [DOI] [PubMed] [Google Scholar]
- 6.Chandran, K., N. J. Sullivan, U. Felbor, S. P. Whelan, and J. M. Cunningham. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das Sarma, J., L. Fu, J. C. Tsai, S. R. Weiss, and E. Lavi. 2000. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J. Virol. 74:9206-9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Haan, C. A., K. Stadler, G. J. Godeke, B. J. Bosch, and P. J. Rottier. 2004. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J. Virol. 78:6048-6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong, J. Y., J. W. Dubay, L. G. Perez, and E. Hunter. 1992. Mutations within the proteolytic cleavage site of the Rous sarcoma virus glycoprotein define a requirement for dibasic residues for intracellular cleavage. J. Virol. 66:865-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebert, D. H., J. Deussing, C. Peters, and T. S. Dermody. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 277:24609-24617. [DOI] [PubMed] [Google Scholar]
- 11.Gallagher, T. M., and M. J. Buchmeier. 2001. Coronavirus spike proteins in viral entry and pathogenesis. Virology 279:371-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallagher, T. M., C. Escarmis, and M. J. Buchmeier. 1991. Alteration of the pH dependence of coronavirus-induced cell fusion: effect of mutations in the spike glycoprotein. J. Virol. 65:1916-1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gombold, J. L., S. T. Hingley, and S. R. Weiss. 1993. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J. Virol. 67:4504-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hingley, S. T., I. Leparc-Goffart, S. H. Seo, J. C. Tsai, and S. R. Weiss. 2002. The virulence of mouse hepatitis virus strain A59 is not dependent on efficient spike protein cleavage and cell-to-cell fusion. J. Neurovirol. 8:400-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hingley, S. T., I. Leparc-Goffart, and S. R. Weiss. 1998. The spike protein of murine coronavirus mouse hepatitis virus strain A59 is not cleaved in primary glial cells and primary hepatocytes. J. Virol. 72:1606-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmes, K. V. 2001. Coronaviruses, p. 1187-1203. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lipincott Williams & Wilkins, New York, N.Y.
- 17.Huang, I. C., B. J. Bosch, F. Li, W. Li, K. H. Lee, S. Ghiran, N. Vasilieva, T. S. Dermody, S. C. Harrison, P. R. Dormitzer, M. Farzan, P. J. Rottier, and H. Choe. 2006. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 281:3198-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim, J. C., R. A. Spence, P. F. Currier, X. Lu, and M. R. Denison. 1995. Coronavirus protein processing and RNA synthesis is inhibited by the cysteine proteinase inhibitor E64d. Virology 208:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kooi, C., M. Cervin, and R. Anderson. 1991. Differentiation of acid-pH-dependent and -nondependent entry pathways for mouse hepatitis virus. Virology 180:108-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krzystyniak, K., and J. M. Dupuy. 1984. Entry of mouse hepatitis virus 3 into cells. J. Gen. Virol. 65:227-231. [DOI] [PubMed] [Google Scholar]
- 21.Luo, Z., and S. R. Weiss. 1998. Roles in cell-to-cell fusion of two conserved hydrophobic regions in the murine coronavirus spike protein. Virology 244:483-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuyama, S., M. Ujike, S. Morikawa, M. Tashiro, and F. Taguchi. 2005. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 102:12543-12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCune, J. M., L. B. Rabin, M. B. Feinberg, M. Lieberman, J. C. Kosek, G. R. Reyes, and I. L. Weissman. 1988. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 53:55-67. [DOI] [PubMed] [Google Scholar]
- 24.Mothes, W., A. L. Boerger, S. Narayan, J. M. Cunningham, and J. A. Young. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103:679-689. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa, T., W. Roth, P. Wong, A. Nelson, A. Farr, J. Deussing, J. A. Villadangos, H. Ploegh, C. Peters, and A. Y. Rudensky. 1998. Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science 280:450-453. [DOI] [PubMed] [Google Scholar]
- 26.Nash, T. C., and M. J. Buchmeier. 1997. Entry of mouse hepatitis virus into cells by endosomal and nonendosomal pathways. Virology 233:1-8. [DOI] [PubMed] [Google Scholar]
- 27.Navas-Martin, S., S. T. Hingley, and S. R. Weiss. 2005. Murine coronavirus evolution in vivo: functional compensation of a detrimental amino acid substitution in the receptor binding domain of the spike glycoprotein. J. Virol. 79:7629-7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otto, H. H., and T. Schirmeister. 1997. Cysteine proteases and their inhibitors. Chem. Rev. 97:133-172. [DOI] [PubMed] [Google Scholar]
- 29.Pager, C. T., and R. E. Dutch. 2005. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12714-12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phillips, J. J., M. M. Chua, E. Lavi, and S. R. Weiss. 1999. Pathogenesis of chimeric MHV4/MHV-A59 recombinant viruses: the murine coronavirus spike protein is a major determinant of neurovirulence. J. Virol. 73:7752-7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarma, J. D., L. Fu, S. T. Hingley, and E. Lavi. 2001. Mouse hepatitis virus type-2 infection in mice: an experimental model system of acute meningitis and hepatitis. Exp. Mol. Pathol. 71:1-12. [DOI] [PubMed] [Google Scholar]
- 32.Sarma, J. D., E. Scheen, S. H. Seo, M. Koval, and S. R. Weiss. 2002. Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. J. Neurovirol. 8:381-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sieczkarski, S. B., and G. R. Whittaker. 2005. Viral entry. Curr. Top. Microbiol. Immunol. 285:1-23. [DOI] [PubMed] [Google Scholar]
- 34.Simmons, G., D. N. Gosalia, A. J. Rennekamp, J. D. Reeves, S. L. Diamond, and P. Bates. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 102:11876-11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simmons, G., J. D. Reeves, A. J. Rennekamp, S. M. Amberg, A. J. Piefer, and P. Bates. 2004. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA 101:4240-4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531-569. [DOI] [PubMed] [Google Scholar]
- 37.Smith, A. E., and A. Helenius. 2004. How viruses enter animal cells. Science 304:237-242. [DOI] [PubMed] [Google Scholar]
- 38.Steinhauer, D. A. 1999. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258:1-20. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki, H., and F. Taguchi. 1996. Analysis of the receptor-binding site of murine coronavirus spike protein. J. Virol. 70:2632-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taguchi, F. 1993. Fusion formation by the uncleaved spike protein of murine coronavirus JHMV variant cl-2. J. Virol. 67:1195-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai, J. C., L. de Groot, J. D. Pinon, K. T. Iacono, J. J. Phillips, S. H. Seo, E. Lavi, and S. R. Weiss. 2003. Amino acid substitutions within the heptad repeat domain 1 of murine coronavirus spike protein restrict viral antigen spread in the central nervous system. Virology 312:369-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turk, B., D. Turk, and V. Turk. 2000. Lysosomal cysteine proteases: more than scavengers. Biochim. Biophys. Acta 1477:98-111. [DOI] [PubMed] [Google Scholar]
- 43.Weiss, S. R., and S. Navas-Martin. 2005. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 69:635-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamada, Y. K., K. Takimoto, M. Yabe, and F. Taguchi. 1997. Acquired fusion activity of a murine coronavirus MHV-2 variant with mutations in the proteolytic cleavage site and the signal sequence of the S protein. Virology 227:215-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshikura, H., and S. Tejima. 1981. Role of protease in mouse hepatitis virus-induced cell fusion. Studies with a cold-sensitive mutant isolated from a persistent infection. Virology 113:503-511. [DOI] [PMC free article] [PubMed] [Google Scholar]