

Abstract
Epstein Barr virus (EBV) can establish distinct latency types with different growth-transforming properties. Type I latency and type III latency can be distinguished by the expression of EBNA2, which has been shown to be regulated, in part, by the EBNA1-dependent enhancer activity of the origin of replication (OriP). Here, we report that CTCF, a chromatin boundary factor with well-established enhancer-blocking activity, binds to EBV sequences between the OriP and the RBP-Jκ response elements of the C promoter (Cp) and regulates transcription levels of EBNA2 mRNA. Using DNA affinity, electrophoretic mobility shift assay, DNase I footprinting, and chromatin immunoprecipitation (ChIP), we found that CTCF binds both in vitro and in vivo to the EBV genome between OriP and Cp, with an ∼50-bp footprint at EBV coordinates 10515 to 10560. Deletion of this CTCF binding site in a recombinant EBV bacterial artificial chromosome (BAC) increased EBNA2 transcription by 3.5-fold compared to a wild-type EBV BAC. DNA affinity and ChIP showed more CTCF binding at this site in type I latency cell lines (MutuI and KemI) than in type III latency cell lines (LCL3456 and Raji). CTCF protein and mRNA expression levels were higher in type I than type III cell lines. Short interfering RNA depletion of CTCF in type I MutuI cells stimulated EBNA2 mRNA levels, while overexpression of CTCF in type III Raji cells inhibited EBNA2 mRNA levels. These results indicate that increased CTCF can repress EBNA2 transcription. We also show that c-MYC, as well as EBNA2, can stimulate CTCF mRNA levels, suggesting that CTCF levels may contribute to B-cell differentiation as well as EBV latency type determination.
Epstein-Barr virus (EBV) is a human gamma-1 herpesvirus that establishes a lifelong latency in over 90% of the world's population (27, 46). During latency, the virus resides as a chromatin-associated, multicopy episome, primarily in resting B lymphocytes with features of classical antigen-selected memory B cells (50, 55). Latent infection is also associated with several malignancies, including Burkitt's lymphoma, Hodgkin's disease, nasopharyngeal carcinoma, and lymphoproliferative disorders in the immunosuppressed. Different viral transcription patterns can be observed in each of these malignancies and have been referred to as latency types 0, I, II, and III (59). In endemic and sporadic Burkitt's lymphoma, EBV typically expresses a restricted, type I latency characterized by expression of the viral EBNA1, EBERS, and BART genes (48). In nasopharyngeal carcinoma, EBV is typically found with a characteristic type II latency associated with expression of additional viral genes, such as LMP1 and LMP2A (43). In lymphoproliferative diseases of the immunosuppressed, EBV expresses a type III latency program with a full panel of viral gene expression that includes the EBNA2 to -5 genes (58). Type 0 latency is reserved for quiescent, memory B cells where no viral genes are expressed (4). The unique functions of the EBV latency proteins are thought to affect B-cell development and the formation of distinct B-cell disorders.
Regulation of the C promoter, or Cp, is important to the biology of EBV because it is the key control point distinguishing type III latency, where Cp is active, from type I latency, where it is inactive (5). In established latent viruses, the polycistronic EBNA2-5 transcript initiates mainly from Cp (5). Known transcription factors that can bind elements around Cp to regulate its activity include CBF1 (or RBP-Jκ), CBF2 (or Auf1), NF-Y, C/EBP, Sp1, and Egr-1 (6, 17, 20, 22, 35). In addition, the viral protein products of Cp, EBNA-LP, EBNA2, and EBNA3A-C, are also known to act in conjunction with various transcription factors to autoregulate their expression (20, 30, 47, 62). Deletion mappings of the Cp region showed that sequences −433 to −245 upstream of the Cp initiation site (particularly around −370) are important for the EBNA2/CBF1/CBF2 response, with those at −119 to −112 important for C/EBP, those at −99 to −91 important for Sp1/Egr-1, and those at −71 to −63 important for NF-Y (16, 24, 35, 53).
The EBV latency transcripts are also subject to epigenetic regulatory events, like DNA methylation and histone modification (2). High levels of CpG DNA methylation of proximal promoter elements correlate with transcription repression of EBNA2 and LMP1 in type I latency (34, 39, 49). Posttranslational modifications of histone tails are also known to be important for EBNA2 regulation, since transcription activation correlates with promoter-specific histone hyperacetylation (1, 53).
The genomic organization of LMP1 and EBNA2 genes suggests that the common upstream region containing the origin of replication (OriP) and the EBERs (highly transcribed, noncoding RNAs) may be important in the coordinated regulation of these latency transcripts (33). Genetic evidence suggests that the family repeats (FR) element of OriP, upon binding by another viral protein, EBNA1, can function as an enhancer to regulate DNA methylation and transcription activity of the EBNA2 and LMP1 genes (18, 35, 41, 45, 52). It has been proposed that OriP and EBNA1 can influence the DNA methylation patterns at the EBNA2 promoter region by a DNA replication-independent mechanism (21).
It is known that chromatin is organized into discrete segments, or domains, characterized by distinct histone modifications (23). Open chromatin domains are typically characterized by high acetylation (of lysines 9 and 14 on histone H3 and lysines 5, 8, 12, and 16 on histone H4) and high methylation of lysine 4 on histone H3. Closed or compacted chromatin domains are typically characterized by low acetylation of histone H3 and H4 and methylation of histone H3 K9 (but not histone H3 K4). The boundaries of these domains are typically DNA elements, called insulators, which can insulate the genes of one domain from regulatory elements of adjacent domains (28, 56). Previously, we showed that the common upstream region of the EBNA2 and LMP1 promoters, which we referred to as the latency control region (LCR), is a higher-order, open chromatin domain that seems to initiate from OriP and ends at the boundaries of the EBNA2 and LMP1 promoters (8). Furthermore, we showed that the boundaries of this open chromatin domain are dynamic. In type I latency, the domain boundaries are limited to regions upstream of the promoter; in type III latency, the boundaries extended well beyond the C and LMP1 promoters. In order to understand the nature of the factors that regulate the boundaries of the EBV LCR, we investigated a number of protein factors typically involved in enhancer-blocking and insulator functions.
CTCF, or CCCTC-binding factor, is an 11-zinc-finger, DNA-binding nuclear phosphoprotein that was initially discovered as a factor involved in transcriptional repression of avian, mouse, and human MYC promoters by binding to multiple, different sequences (36). It was later characterized as being involved in enhancer blocking, chromatin insulation, gene activation, and imprinting on diverse genes, such as those coding for β-globin, c-MYC, and IGF2-H19 (13, 60). Combinatorial use of its multiple zinc fingers allows CTCF to bind dissimilar target sites with footprinting sequences spanning approximately 50 bp (25).
In this study, we looked for the presence of CTCF between OriP and Cp to ascertain a possible insulator site. We wanted to determine if CTCF can regulate Cp transcription and to determine if the presence of CTCF correlated with different latency types.
MATERIALS AND METHODS
Cell lines and antibodies.
MutuI and KemI are type I latency B-cell lines derived from Burkitt's lymphoma. Raji (ATCC) is a type III latency B-cell line derived from Burkitt's lymphoma. LCL3456-EBV is a type III latency B-cell line derived from primary lymphoblasts transformed with EBV strain B95-8. DG-75 (ATCC) is an EBV-negative, B-cell line derived from Burkitt's lymphoma. EREB 2.5 is a human B-cell line containing EBV (ΔEBNA2) in which the endogenous EBNA2 is replaced with an estrogen-inducible EBNA2-estrogen receptor (ER) fusion protein (26). These cell lines were maintained in RPMI supplemented with 10% fetal bovine serum, glutamine, penicillin, and streptomycin sulfate. 293 (ATCC) is a human kidney, epithelial cell line transformed with adenovirus 5 DNA, and IMR90-MYC-ER is a human fetal lung fibroblast cell line containing a tamoxifen-inducible human c-MYC-ER fusion protein (61). These cell lines were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, glutamine, penicillin, and streptomycin sulfate.
The following rabbit polyclonal antibodies were used: anti-CTCF (Upstate), anti-Orc2 (Pharmingen), anti-H3 (Upstate), and control rabbit immunoglobulin G (IgG) (Santa Cruz). Rabbit polyclonal anti-EBNA1 was raised against a recombinant full-length EBNA1. Mouse monoclonal anti-EBNA2 was obtained from DakoCytomation.
Plasmids.
N968 vector (pFASTBAC HTb-CTCF) was used to prepare baculoviral, recombinant His-tagged CTCF. Full-length human CTCF cDNA was prepared by PCR of pCI 7.1, a plasmid containing full-length human CTCF cDNA, using primers OPL1403 and OPL1404. The PCR product was then inserted into the XhoI and BamHI sites of pFASTBAC HTb (Invitrogen) to generate N968. Baculovirus His-tagged CTCF protein was expressed in Sf9 insect cells using standard BEV system protocols (Oxford Expression Technologies). The His-tagged CTCF protein was then purified using Ni-nitrilotriacetic acid (NTA) agarose beads (QIAGEN) according to the manufacturer's protocol.
N951 [pBKS-EBV(10393-10584)] was used for in vitro DNase I footprinting. EBV DNA fragment (coordinates 10393 to 10584) containing the CTCF binding site upstream of Cp was obtained by PCR of the EBV BamHI C BACmid using primers OPL1389 and OPL1390. The PCR product was inserted into pBluescript KS+ (Invitrogen) at the BamHI and HindIII sites to generate N951.
N1027 (p3xFlag-CTCF) was a kind gift from R. Shiekhattar, Wistar Institute, Philadelphia, PA. It was generated by inserting full-length, human CTCF cDNA into p3xFLAG-CMV-2a (Sigma).
All primer sequences used in this paper are available upon request.
Recombineering EBV BACs.
To generate a recombinant EBV bacterial artificial chromosome (BAC) with a deletion in the CTCF binding site upstream of the C promoter, bacterial recombineering was employed as previously described (29). In a PCR using pL452 plasmid as a template, two 75-bp homologous primers, OPL1721 and OPL1722 (5′ and 3′ primers), were used to generate a 2,700-bp PCR product. The PCR fragment contains a kanamycin resistance marker flanked by Floxed P sites and 50 bp of EBV DNA spanning the CTCF binding site upstream of Cp. The PCR fragment was gel purified and electroporated into EL350 Escherichia coli strain containing wild-type EBV BAC (also referred to as EBV-Hygro-green fluorescent protein [GFP] BAC or N1089) (9). Homologous insertion of the PCR product into EBV-Hygro-GFP BAC was obtained by first inducing expression of recombineering proteins in the EL350 strain by heating at 42°C prior to electroporation. The kanamycin-resistant EBV-Hygro-GFP BAC (N1195), containing the inserted PCR product, was verified by restriction enzyme digestion analysis. Next, the kanamycin resistance marker and Floxed P sites were removed via recombination by Cre protein, which was induced by adding arabinose to 0.1% in a growing culture of EL350 containing N1195. The resulting N1171 EBV-Hygro-GFP BAC contains a deletion of the CTCF binding site upstream of Cp. It was verified by restriction enzyme digestion analysis and DNA sequencing.
DNA affinity chromatography.
DNA affinity chromatography was performed as previously described (3). Soluble nuclear extract fractions were obtained from MutuI, KemI, Raji, and LCL3456 via the Dignam extraction method. Biotinylated DNA fragments of EBV were obtained by PCR using the BamHI C BACmid as a template. Control biotinylated DNA fragments were obtained by PCR using pBluescript KS+ as template. The DNA fragments were coupled to M-280 streptavidin-labeled magnetic Dynabeads (Dynal Biotech). The coupled beads were then washed and incubated with soluble nuclear extract for 1 h. The bound proteins and beads were then washed three times with D150 and three times with D300 buffers (20 mM HEPES, pH 7.9, 20% glycerol, 0.2 mM EDTA, 0.05% NP-40, 1 mM phenylmethylsulfonyl fluoride, 10 mM β-mercaptoethanol, protease inhibitor cocktail [Sigma P8340], and 150 mM KCl for D150 or 300 mM KCl for D300). A magnetic particle concentrator (Dynal Biotech) was used to collect the beads after each wash. The bound proteins were then eluted from the beads using 2× Laemmli protein loading buffer and heated at 95°C for 5 min. The proteins and beads were then cooled on ice for 5 min, centrifuged for 10 s at 14,300 rpm, and placed on a magnetic particle concentrator. The eluted proteins were then loaded and run on an 8 to 16% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel with care taken to avoid the bead pellet. Transfer of protein from gel to nitrocellulose membrane was done overnight for Western blotting.
EMSA.
For electrophoretic mobility shift assays (EMSAs), DNA probes covering different regions of the EBV genome were generated by PCR incorporation of [α-32P]dATP (3,000 Ci/mmol; Perkin-Elmer). The PCR mixture consisted of 30 μCi of [α-32P]dATP, 0.1 mM dCTP, 0.1 mM dGTP, 0.1 mM dTTP, 0.01 mM dATP, 1 μM primers, and 1 U of Taq polymerase (Roche) for 25 cycles. Unincorporated nucleotides were removed on a Microspin G50 column (Amersham Biosciences). In a 20-μl reaction mixture, purified baculoviral His6-tagged-CTCF (≈1 μg) was added to a phosphate-buffered saline (PBS) reaction mixture containing 0.5 μg poly(dI-dC), 5% glycerol, 0.1 mM ZnSO4, and 10,000 cpm of 32P-labeled DNA probe. Reaction mixtures were incubated for 30 min at 25°C, electrophoresed in a 5% nondenaturing, polyacrylamide gel at 110V, and visualized by PhosphorImager.
In vitro DNase I footprinting assay.
DNase I footprinting was performed as described previously (10). A DNA fragment covering the EBV genome (coordinates 10393 to 10584) was cut from N951 with BamHI and labeled in a 20-μl reaction mixture containing 30 μCi of [α-32P]dATP (6,000 Ci/mmol; Perkin-Elmer), 0.1 mM dGTP, 0.1 mM dTTP, 0.1 mM dCTP, and 2 U of Klenow fragment (Roche) for 30 min at 25°C. The second restriction digest was done with HindIII to generate an unlabeled 3′ end. Purified His-tagged CTCF was used, and the reaction mixture was PBS buffer containing 5 mM MgCl2, 0.1 mM ZnSO4, 1 mM dithiothreitol, 0.1% NP-40, 10% glycerol, 1 μg bovine serum albumin, 0.4 μg poly(dI-dC), and 10,000 cpm of radiolabeled probe. The protected probe was digested with different dilutions of DNase I (Sigma) and purified by phenol-chloroform extraction following proteinase K digestion. The DNA samples were then electrophoresed on a 7% denaturing, polyacrylamide sequencing gel at 33 mA and visualized by PhosphorImager.
ChIP assays.
The chromatin immunoprecipitation (ChIP) assay followed the protocol provided by Upstate Biotechnology, Inc., with minor modifications as previously described (10). Additional modifications are as follows. DNAs were sonicated to between 200- and 350-bp DNA fragments on a Diagenode Bioruptor according to manufacturer's protocol, and real-time PCR was performed with SYBER green probe in an ABI Prism 7000 using 1/100 to 1/2,500 of the ChIP DNA according to manufacturer's specified parameters.
Quantitative RT-PCR assays.
Reverse transcriptase PCR (RT-PCR) was done as previously described (8). Real-time PCR was performed with SYBER green probe in an ABI Prism 7000 using 1/100 to 1/2,500 of the cDNA according to the manufacturer's specified parameters.
For RT-PCR of EBV BAC-transfected 293 cells, 10 μg of N1089 or N1171 EBV BAC was used to transfect cells at 50 to 75% confluence in 10-cm2 plates. The cells were allowed to grow for 72 h and then harvested for RT-PCR.
For RT-PCR of EBV BAC-transfected DG-75, N1089 or N1171 EBV BACs were nucleofected according to the manufacturer's (Amaxa) protocol. Briefly, 5 × 106 cells (growing at 0.8 × 106 to 1 × 106 cells/ml) were centrifuged at 1,500 rpm for 5 min at 4°C, resuspended in 100 μl of T solution (Amaxa) containing 10 μg of vector, and electrophorated on an Amaxa Nucleofector using program A-23. After nucleofection, the cells were transferred to a flask containing 10 ml of fresh, complete RPMI 1640 medium and placed in a 37°C incubator. The cells were allowed to grow for 72 h and then harvested for RT-PCR.
To determine the effect of CTCF overexpression on EBNA2 transcription, Raji cells were transfected with control and pFlag-CTCF vectors. Briefly, 5 × 106 cells (growing at 0.8 × 106 to 1 × 106 cells/ml) were centrifuged at 1,500 rpm for 5 min at 4°C, resuspended in 100 μl of T solution (Amaxa) containing 10 μg of vector, and electrophorated on an Amaxa Nucleofector using program A-23. After nucleofection, the cells were transferred to a flask containing 10 ml of fresh, complete RPMI 1640 medium and placed in a 37°C incubator. After 24 h, the nucleofection was repeated. The cells were then allowed to grow for another 48 h and then harvested for quantitative RT-PCR.
To determine the effect of CTCF depletion on EBNA2 transcription, MutuI cells were transfected with short interfering RNA (siRNA) against CTCF using nucleofection. Per transfection, 1 × 107 cells (growing at 0.8 × 106 to 1 × 106 cells/ml) were resuspended in 100 μl of Nucleofector kit T solution. Fifty picomoles of siRNA against luciferase (Dharmacon) or siRNA against hCTCF (SMARTpool catalog no. M-020165-01; Dharmacon) combined with 2.5 μg of pmaxGFP vector (Amaxa) was electrophorated into the cells using program A-23. After nucleofection, the cells were then transferred to a flask containing 10 ml of fresh, complete RPMI 1640 medium and placed in a 37°C incubator. After 24 h, the cells were then sorted for GFP-positive cells using a DakoCytomation MoFlo Cell Sorter. The GFP-positive cells were then centrifuged, resuspended in appropriate volume of fresh, complete RPMI 1640 medium to obtain a density of 0.5 × 106 cells/ml, and placed in a 37°C incubator. The cells were allowed to grow for another 24 h and then harvested for quantitative RT-PCR.
For the time course study of IMR90-MYC-ER, cells at 50 to 75% confluence were induced with 200 nM hydroxytamoxifen (4-OHT) (Sigma). Cells were then collected at 0, 4, 12, 24, and 42 h following 4-OHT induction.
For the time-course study of EREB 2.5, cells growing in 1 μM β-estradiol (Sigma) at logarithmic growth phase (5 × 105 to 7 × 105 cells/ml) were centrifuged and resuspended in fresh complete RPMI 1640 medium without β-estradiol. Cells (5 × 106 cells/time point) were then collected at 0, 24, and 48 h. After 48 h, the cells were then resuspended in fresh complete RPMI 1640 medium with 1 μM β-estradiol. These cells were then collected at 8, 16, and 24 h.
RESULTS
CCCTC-binding factor, or CTCF, binds in vitro at a location between the OriP and the C promoter of EBV.
Previously, it had been shown that the FR of the OriP acts as an enhancer for the LMP and Cp of EBV (18, 41). Here, we showed that CTCF, a multi-zinc-finger nuclear protein with well-established enhancer-blocking activity, binds to an EBV DNA fragment between OriP and Cp (Fig. 1A). DNA affinity with various DNA fragments of EBV was used to determine the general location of CTCF binding (Fig. 1B). Using MutuI (type I) nuclear extracts, we found EBV fragment e, covering EBV coordinates 10041 to 12488, was bound by CTCF. Dividing this large fragment into three smaller fragments, we found that fragment b (covering 10041 to 10632, which is upstream of Cp) showed the strongest CTCF binding. In addition, there was also CTCF binding to fragment d (covering 12017 to 12488, which is within the W repeats). Using LCL3456 (type III) nuclear extract, we also found CTCF binding upstream of Cp (fragment b). However, there seems to be less CTCF binding when compared to DNA affinity using an equal amount of nuclear extract from MutuI. The second panel showed that our DNA affinity method is specific in that we were able to observe EBNA2 binding only in LCL3456 (type III) using fragment c (covering 10855 to 11399, where there is a well-established EBNA2 binding site) and fragment e (covering 10041 to 12488) (Fig. 1A).
FIG. 1.

DNA affinity pull-down of CTCF and EBNA2 with EBV DNA. (A) Western blots of CTCF and EBNA2 binding to different biotinylated DNA fragments of EBV using nuclear extracts from MutuI (type I EBV-positive cell line) and LCL3456 (type III EBV-positive cell line). Inputs represent 10% of total nuclear proteins used for DNA affinity. BKS is a random biotinylated DNA fragment of pBluescript KS+. Fragments a to e represent DNA affinity using different DNA segments of EBV encompassing a region between the DS and W repeats of EBV. αCTCF and αEBNA2, anti-CTCF and anti-EBNA2 antibodies, respectively. (B) Schematic diagram of the different DNA segments of EBV used in the DNA affinity assay. The segments are labeled a to e, and the regions of EBV covered by each segment are labeled with its EBV coordinates.
To confirm the binding of CTCF based on DNA affinity and to ascertain a more specific location of CTCF binding, we employed two additional assays: in vitro EMSA and in vitro DNase I footprinting. We purified His-tagged, full-length human CTCF from Sf9 insect cells, which gave a protein of approximately 130 to 140 kDa (Fig. 2A) that was recognized by specific anti-CTCF antibody (Fig. 2B). Using purified His-tagged CTCF and various EBV DNA probes (Fig. 2D) generated by PCR, we found CTCF bound fragment f (covering 10383 to 10594) and a smaller fragment, i (covering 10490 to 10594) (lane 2 and lanes 8 to 12 of Fig. 2C). Using the same His-tagged CTCF protein, we employed DNase I footprinting to determine the precise binding sequence of CTCF on this region of EBV (Fig. 3). Our DNase I footprinting revealed that CTCF weakly protected DNA at EBV sequence positions from 10515 to 10560 (Fig. 3A and B). We also observed that CTCF induced a strong DNase I-hypersensitive site at EBV coordinates 10498 and 10541 (Fig. 3A). CTCF binds to a variety of DNA sequences, and we find little similarity between our CTCF binding sequence on EBV and published CTCF binding sequences (Fig. 3B) (36). However, the pattern of CTCF (an 11-Zn-finger protein) protection covering approximately 50 bp with an intervening gap is reminiscent of other CTCF footprinting patterns (Fig. 3A) (14). These biochemical assays establish that a CTCF binding site on EBV DNA is located between OriP and Cp at EBV coordinates 10515 to 10560.
FIG. 2.

Electrophoretic mobility shift assay of CTCF with EBV DNA. (A) Coomassie gel of Ni-NTA purification of His-tagged CTCF expressed in Sf9 insect cells. Sf9 cell extracts expressing His-tagged CTCF were loaded onto Ni-NTA beads, washed with 20 mM imidazole buffer, eluted with 250 mM imidazole elution buffer, and dialyzed in PBS containing 20% glycerol. (B) Western blot of His-tagged CTCF. Different fractions of proteins during the purification process were electrophoresed on an 8 to 16% SDS-PAGE gel, transferred onto nitrocellulose membrane, and probed using a rabbit polyclonal anti-CTCF antibody (α-CTCF). (C) Autoradiogram of in vitro EMSA showing CTCF shifting of EBV DNA probes. Purified His-tagged CTCF was used for gel shift of various EBV 32P-labeled DNA probes. Lanes 1 and 2 represent EMSA with probe f, lanes 3 and 4 are EMSA with probe g, lanes 5 and 6 are EMSA with probe h, and lanes 7 to 12 are EMSA with probe i. Lanes 9 to 12 represent EMSA with specific (sp.) and nonspecific (nsp.) cold DNA competitors at 10- and 100-fold excess. (D) Schematic diagram of various EBV DNA probes used in EMSA. The probes cover regions upstream of the C promoter, between EBV coordinates 10383 to 10594 and 10904 to 11077.
FIG. 3.

DNase I footprinting of CTCF on EBV DNA. (A) Autoradiogram of in vitro DNase I footprinting gel showing CTCF protection on EBV DNA probe. Purified His-tagged CTCF was used for footprinting assay using EBV 32P-labeled DNA probe covering EBV coordinates 10393 to 10594. Nucleotide ladders A and A/G were generated by chemical cleavage using the Maxim-Gilbert method. For the CTCF lanes, + indicates prominent CTCF protection from DNase I digestion from EBV coordinates 10515 to 10560, whereas − lanes (no-CTCF controls) show no protection at the same region. The arrows indicate DNase I-hypersensitive sites. (B) Sequence of EBV protected by CTCF in DNase I footprinting assay. CTCF protection covers a DNA sequence from EBV coordinates 10515 to 10560. The top arrow indicates a DNase I-hypersensitivity site. The two bottom arrows show a possible inverted repeat.
CTCF binds in vivo at a location between the OriP and the C promoter of EBV.
ChIP coupled with quantitative PCR was used to determine if CTCF binds in vivo to EBV at the region predicted by our in vitro experiments (Fig. 4). We also compared CTCF binding patterns in two type I cell lines, MutuI and KemI, and two type III cell lines, Raji and LCL3456. The highest signals occurred when primers covering EBV coordinates 10401 to 10487 (labeled as the CTCF site) were used for ChIP of MutuI and KemI extracts (Fig. 4). Our ChIP had sonicated DNA that averaged 200 to 350 bp, and our CTCF binding site primers are within 100 bp of the exact site (10515 to 10560) predicted by DNase I footprinting (Fig. 3). ChIP of LCL3456 and Raji showed three- to fourfold less CTCF binding at this site compared to MutuI and KemI, respectively. This is interesting in that LCL3456 and Raji are type III cell lines that expressed EBNA2-5 using the C promoter while MutuI and KemI are type I cell lines that do not express EBNA2-5. Other regions tested include the dyad symmetry (DS), Rep*, Cp, and OriLyt regions. These regions showed less CTCF binding than the mapped CTCF site. The exception was Cp of MutuI, which showed a significant CTCF ChIP signal not predicted from our in vitro assays. IgG was used as an antibody control (Fig. 4).
FIG. 4.

ChIP of CTCF at various EBV sites in multiple EBV-positive cell lines. Results of real-time PCR analysis of ChIP assay with antibody specific to CTCF or control IgG are shown for type I EBV-positive cell lines (MutuI and KemI) and two type III EBV-positive cell lines. The EBV regions analyzed for CTCF binding are as follows: DS (primers covering EBV coordinates 8957 to 9043), Rep* (primers covering 9715 to 9793), CTCF site (primers covering 10401 to 10487), Cp (primers covering 10956 to 11030), and OriLyt (primers covering 52,654 to 52,797). Samples were analyzed in triplicate.
Role of CTCF binding sites in EBNA2 transcription.
To test the functional relevance of CTCF binding at our predicted site upstream of Cp, we used bacterial recombineering to delete the CTCF binding site (Fig. 5). We used a wild-type EBV BAC (N1089) as our template and generated a new recombinant EBV BAC (N1171) with a deletion of EBV sequence 10393 to 10590 that contains the CTCF binding site (Fig. 5A). The EBV BACs were verified by restriction enzyme digestion analysis (Fig. 5A). N1195 is the intermediate EBV BAC generated during the recombineering process. It showed a predicted 9.8-kb NheI fragment caused by insertion of the kanamycin marker PCR product. PCR using primers that span the CTCF binding site showed that there was a deletion when comparing N1089 and N1071 (lanes 2 and 3, Fig. 5B). PCR of other EBV regions showed similar size bands for DS, Cp, and the EBNA2 coding region, implying that there were no accidental deletions at these other regions (lanes 4 to 12, Fig. 5B). N1089 and N1171 were also sequenced with primers surrounding the deletion site to ensure correct site deletion (data not shown).
FIG. 5.

Differences in EBNA2 transcription level in wild-type EBV BAC and EBV BAC with CTCF binding site deletion. (A) Gel of NheI restriction enzyme digestion patterns of recombinant EBV BACs. N1089 represents the wild-type EBV BAC. N1195 is the wild-type EBV BAC after homologous recombination with a PCR fragment containing a kanamycin resistance marker flanked by Floxed P sites and 50 bp of EBV DNA spanning the CTCF binding site. A new NheI fragment is generated by insertion of the PCR product. N1171 is the EBV BAC generated after Cre-Lox recombination to obtain a CTCF site deletion spanning EBV coordinates 10393 to 10590. Each EBV BAC DNA was cut with NheI, electrophoresed on a 0.7% agarose gel, and stained with ethidium bromide. Below the gel, a schematic diagram of N1089 and N1171 illustrates the deletion generated in the EBV BAC. (B) PCR analysis of EBV BACs. DNAs from N1089 and N1171 and distilled water (Mock) were amplified by PCR using primers to various EBV regions. The following PCR regions were analyzed: CTCF binding site (covering EBV coordinates 10041 to 10632), DS (8587 to 9206), Cp (10855 to 11399), and the EBNA2 coding region (48504 to 49131). (C) RT-PCR of EBNA2 in wild-type EBV BAC (N1089) and EBV BAC with CTCF binding site deletion (N1171). N1089 and N1171 EBV BACs were transfected into 293 (left panel) or DG75 (right panel) cells. The cells were harvested 72 h later, and real-time RT-PCR was performed. EBNA2 mRNA levels were normalized to GFP. Samples were analyzed in triplicate.
The EBV BACs (N1089 and N1171) were then transfected into 293 and DG-75 cells and tested for EBNA2 transcription by RT-PCR, coupled with quantitative PCR (Fig. 5C). We found that EBNA2 levels increased by 3.5-fold in 293 cells when the CTCF binding site was deleted in EBV BAC (Fig. 5C, left panel). EBNA2 levels increased by 6.3-fold in DG-75 cells when the CTCF binding site was deleted in EBV BAC (Fig. 5C, right panel). We used GFP as a normalization factor because it is contained in the EBV BAC as a selection marker, and normalization with GFP is a convenient way to normalize EBNA2 expression levels to equal numbers of transfected EBV BACs that are also transcriptionally competent.
CTCF protein levels are more abundant in type I EBV-positive cell lines than type III EBV-positive cell lines.
One possible explanation for the differences in CTCF binding among different EBV-positive cell lines, based on DNA affinity and ChIP assays, is that CTCF protein levels are different among the cell lines. Western blotting using total cell lysates from MutuI, KemI, Raji, and LCL3456 showed that CTCF expression levels were substantially higher in type I cell lines, MutuI and KemI, than type III cell lines, Raji and LCL3456 (Fig. 6A). Western blots with specific antibodies to EBNA2 showed expression only in Raji and LCL3456, as expected. EBNA1 was present in all cell lines, with higher levels in Raji and LCL3456 than MutuI and KemI. Orc2 and histone H3 were controls that showed equal loading of proteins (Fig. 6A). This result suggests that the differential regulation of CTCF and EBNA2 in type III latency might result from distinct patterns of transcriptional regulation in the two latency types. In order to determine whether the difference in CTCF and EBNA2 was at the level of protein stability or transcription, mRNA levels were assayed. RT-PCR showed higher expression of CTCF levels (normalized to β-actin) in MutuI and KemI, with KemI being an average of 5.3- and 5.7-fold higher than Raji or LCL3456, respectively, and MutuI being an average of 2.5- and 2.7-fold higher than Raji and LCL3456, respectively (Fig. 6B). EBNA2 mRNA levels (normalized to β-actin) were almost undetectable in MutuI and KemI, high in Raji, and highest in LCL3456 cells (Fig. 6B).
FIG. 6.

Differences in CTCF protein levels in MutuI, KemI, Raji, and LCL3456. (A) Western blots of CTCF, EBNA2, EBNA1, Orc2, and histone H3 in different EBV-positive cell lines. Total cell lysates from MutuI (type I), KemI (type I), Raji (type III), and LCL3456 (type III) cells were loaded on an 8 to 15% SDS-PAGE gel, transferred to nitrocellulose membrane, and probed with antibodies specific to CTCF, EBNA2 and EBNA1, Orc2, and histone H3. (α-CTCF, α-EBNA2 and α-EBNA1, α-ORC2, and α-H3, respectively) (B) RT-PCR of CTCF and EBNA2 mRNA levels in different EBV-positive cell lines. Cells (MutuI, KemI, Raji, and LCL3456) were collected at logarithmic growth phase (5 × 105 to 7.5 × 105 cells/ml) for real-time RT-PCR. CTCF and EBNA2 mRNA levels were normalized to β-actin. Samples were analyzed in triplicate.
Depletion and overexpression of CTCF affect EBNA2 mRNA levels.
To determine if CTCF levels in cells can influence EBNA2 mRNA levels (presumably by influencing transcription from the nearby C promoter), nucleofection of siRNA against CTCF (siCTCF) or control luciferase (siLUC) in type I, MutuI was carried out (Fig. 7). By real-time RT-PCR, we determined that siCTCF nucleofection was able to decrease CTCF mRNA (normalized to β-actin) by an average of 2.2-fold and that correlated with an average 1.9-fold increase in EBNA2 mRNA levels (normalized to β-actin) (Fig. 7A). Nucleofection of Flag-CTCF and control Flag vector in Raji cells was carried out to determine the effect on EBNA2 mRNA levels with CTCF overexpression (Fig. 7B). By real-time RT-PCR, we determined that we could increase CTCF mRNA levels (normalized to β-actin) by an average of 6.0-fold and that correlated with an average 4.1-fold decrease in EBNA2 mRNA levels (normalized to β-actin) (Fig. 7B). We used primers that can recognize Flag-CTCF, as well as endogenous CTCF.
FIG. 7.
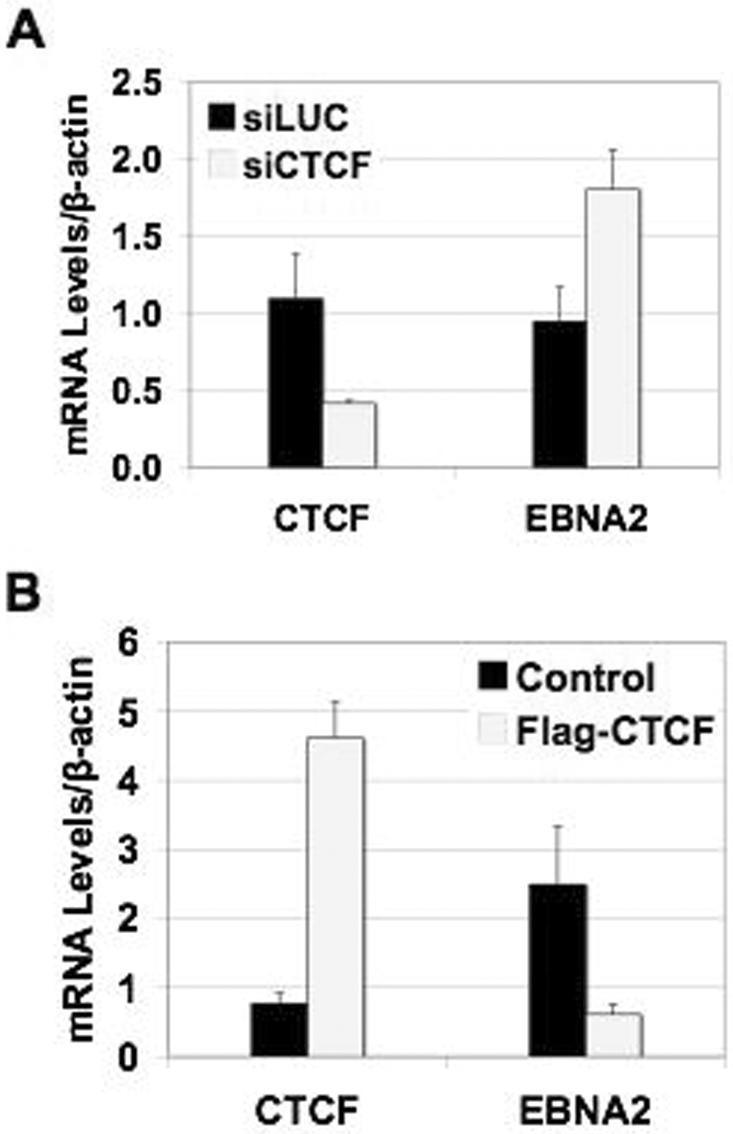
Regulation of EBNA2 transcription by CTCF. (A) RT-PCR analysis of EBNA2 mRNA levels after depletion of CTCF in type I MutuI cells. MutuI cells were cotransfected with GFP and siRNA against CTCF or luciferase (control) using nucleofection. After 48 h, cells were sorted for GFP expression. GFP-positive cells were collected and allowed to grow for another 24 h. GFP-positive cells were then harvested for RT-PCR. EBNA2 and CTCF mRNA levels were analyzed by real-time PCR and normalized to β-actin. Samples were analyzed in triplicate. (B) RT-PCR analysis of EBNA2 mRNA levels after overexpression of CTCF in type III Raji cells. Raji cells were transfected with either Flag-CTCF vector or the control (Flag vector) using double nucleofections. After 48 h, cells were harvested for RT-PCR. EBNA2 and CTCF mRNA levels were analyzed by real-time PCR and normalized to β-actin. Samples were analyzed in triplicate.
c-MYC induces CTCF mRNA levels.
MutuI and KemI are type I, EBV-positive cell lines derived from Burkitt's lymphoma, which are characterized as having a translocated c-myc gene that allows for high levels of MYC expression. To determine if high levels of MYC can be a factor in determining the levels of CTCF in different cells, we used a tamoxifen-inducible MYC-ER fibroblast cell line, IMR-90-MYC-ER. Samples were collected at 0, 2, 4, 8, 24, and 42 h after MYC-ER induction with 200 nM 4-OHT. RT-PCR was performed for CTCF, ornithine decarboxylase (ODC), cyclin D2 (cycD2), and elongation factor 1 alpha (ELF1α) mRNA levels (Fig. 8A). CTCF showed a slight increase in expression at 4 h and reached a threefold induction by 24 h before leveling off (Fig. 8A). The well-characterized c-myc gene targets ODC, and cycD2 showed similar increases in expression, while ELF1α, which is not regulated by c-MYC, showed no increases in expression following MYC-ER induction (Fig. 8A).
FIG. 8.

Correlations between CTCF, c-MYC, and EBNA2 levels. (A) RT-PCR of MYC-ER induction time course using IMR-90-MYC-ER cells. Cells were collected at 0, 2, 4, 8, 24, and 42 h after induction with 200 nM 4-OHT. CTCF, ODC, cycD2, and ELF1α mRNA levels were analyzed by real-time PCR and normalized to β-actin. Samples were analyzed in triplicate. (B) RT-PCR of EBNA2-ER induction time course using EREB 2.5 cells. EREB 2.5 cells were grown to a density of 5 × 105 to 7.5 × 105 cells/ml in 1 μM β-estradiol. Then, β-estradiol was removed from the cells by placing the cells in fresh, complete RPMI 1640 medium without β-estradiol. Samples were then collected at 0, 24, and 48 h after β-estradiol removal (time points labeled as −E). After 48 h without β-estradiol, the cells were then resuspended in fresh, complete RPMI 1640 medium with 1 μM β-estradiol at a density of 5 × 105 to 7.5 × 105 cells/ml. Cells were collected at 8, 12, and 24 h after addition of 1 μM β-estradiol (time points labeled as +E). CTCF, c-MYC, and LMP1 mRNA levels were analyzed by real-time PCR and normalized to β-actin. All samples were done in triplicate.
EBNA2 induces c-MYC and CTCF expression levels.
EBNA2 has been shown to induce c-MYC and LMP1 expression levels in B cells by using an estrogen-inducible, EBNA2-ER cell line, EREB 2.5. We tested whether EBNA2, by inducing LMP1 and c-MYC expression levels, also induced CTCF levels (Fig. 8B). EREB 2.5 cells were grown to a density of 5 × 105 to 7.5 × 105 cells/ml in 1 μM β-estradiol. Then, β-estradiol was removed from the cells by placing the cells in fresh, complete RPMI-1640 medium without β-estradiol. Quantitative PCR of samples collected at 0, 24, and 48 h after β-estradiol removal (time points labeled as −E) showed a decrease in LMP1, c-MYC, and CTCF mRNA levels (Fig. 8B). After 48 h without β-estradiol, the cells were then resuspended in fresh, complete RPMI 1640 medium containing 1 μM β-estradiol at a density of 5 × 105 to 7.5 × 105 cells/ml, and RT-PCR samples were collected at 8, 12, and 24 h after addition of 1 μM β-estradiol (time points labeled as +E). There was a 4.9-fold increase in c-MYC and a 63.0 fold increase in LMP1 mRNA levels at 8 h after readdition of β-estradiol when compared to the c-MYC mRNA levels at 48 h after β-estradiol removal (8 h/+E versus 48 h/−E, Fig. 8B). CTCF, LMP1, and c-MYC mRNA levels peaked at 16 h following β-estradiol addition (16 h/+E, Fig. 8B) and returned to levels that were similar to those in cells grown continuously in β-estradiol (0 h/−E, Fig. 8B) by the 24 h following β-estradiol addition (24 h/+E, Fig. 8B).
DISCUSSIONS
CTCF binds in vitro and in vivo to EBV between OriP and the C promoter.
In this study, we investigated and found CTCF binding to an ≈45-bp region (EBV coordinates 10515 to 10560) between OriP and Cp. Initially, DNA affinity was used to determine the general location of CTCF binding (Fig. 1B). Confirmation of CTCF binding was provided by EMSA, and DNase I footprinting narrowed the binding to EBV coordinates 10515 to 10560. CTCF binds to a variety of DNA sequences, and we find little similarity between the CTCF binding sequence on EBV and published CTCF binding sequences (Fig. 3B) (14, 36). The ChIP assay indicated that CTCF binds in vivo to EBV between OriP and Cp (Fig. 4). We designated the binding region as the “CTCF site” (Fig. 4). These biochemical assays establish a CTCF binding site on EBV DNA between OriP and Cp.
In addition to the identification of a CTCF binding site upstream of Cp, these studies also provided a comparison of CTCF binding from different EBV-positive cell lines with different latencies (type I versus type III). From the DNA affinity assay, more CTCF protein was bound to EBV DNA using equal amounts of proteins from nuclear extracts of MutuI (type I) than LCL3456 (type III) (Fig. 1). ChIP of MutuI and KemI showed three- to fourfold higher CTCF binding at this site when compared to LCL3456 and Raji, respectively. This is interesting in that LCL3456 and Raji are type III cell lines that expressed EBNA2-5 using the C promoter while MutuI and KemI are type I cell lines that do not express EBNA2-5.
CTCF binding on EBV represses EBNA2 transcription.
Others have found that deletion of a 262-bp region (EBV coordinates 10221 to 10482) containing the glucocorticoid response elements produced an unexpected increase in Cp transcription (12). We used a bacterial recombineering process to delete the CTCF binding site (Fig. 5) in a wild-type EBV BAC (N1089) and generated a new recombinant EBV BAC (N1171) with a deletion of EBV coordinates 10393 to 10590 that contains the CTCF binding site (Fig. 5A). EBNA2 mRNA levels increased by an average of 3.5-fold in 293 cells and 6.3-fold in DG-75 cells when the CTCF binding site was deleted in EBV BAC (Fig. 5C).
CTCF sites have been implicated in chromatin boundary elements and enhancer blocking activity (60). Cp transcription is known to be regulated by the OriP enhancer, and it is tempting to speculate that the CTCF site may function to insulate OriP enhancer activation of Cp in type I latency where CTCF binding is elevated. We have also reported previously that histone H3 mK4 modification is elevated at the OriP and EBER region and that this domain expands in type III latency to the Cp and W repeats (8). We further speculate that CTCF binding may prevent the spreading of the H3mK4 modification to the Cp region and therefore restrict chromatin access at Cp in type I latency. The glucocorticoid elements are adjacent to the CTCF binding sites, and deletion of either element would increase Cp transcription. This is similar to reported CTCF sites being adjacent to thyroid hormone receptor sites (7). It would be interesting to see if these elements can combine to mediate synergistic repression of Cp, as well as combining to form a complete, boundary insulator since it has been noted that a CTCF site alone may not form a complete, boundary insulator (57).
CTCF levels and the regulation of latency type.
Our data suggest that CTCF may be a critical repressor of type III latency transcription by inhibiting OriP enhancer activation of Cp. In this respect, CTCF may be a critical factor in determining latency types. We found that CTCF protein and mRNA levels were themselves subject to cell type differences that correlated with latency type. In particular, we found that CTCF protein and mRNA levels were elevated in type I cells where Cp transcription is repressed, relative to type III cells where Cp transcription is active (Fig. 6B). Manipulation of CTCF levels by overexpression or by siRNA depletion altered EBNA2 mRNA levels. Depletion of CTCF mRNA by siRNA led to an increase in EBNA2 mRNA levels in MutuI (type I) cells (Fig. 7A). Conversely, overexpression of Flag-CTCF in Raji (type III) cells decreased EBNA2 mRNA levels (Fig. 7B). These observations suggest that CTCF can repress transcription of EBNA2 and that levels of CTCF in different EBV-positive cell lines can determine the type of EBV latency, with high levels of CTCF correlating with type I and low levels of CTCF correlating with type III.
Since changes in CTCF levels may have effects on other cellular genes, we cannot rule out an indirect effect of CTCF on EBNA2 transcription. However, given the observation that CTCF binds directly to the upstream region of Cp and that deletion of this region leads to an increase in EBNA2 transcription, it seems more likely that this effect is mediated directly by CTCF's enhancer-blocking function of FR to Cp or W promoter.
Regulation of CTCF by c-MYC and EBNA2.
To better understand the cellular factors that determine latency type, we examined the effects of two known regulators of B-cell proliferation and differentiation, namely c-MYC and virally encoded EBNA2 (Fig. 8). We found that both c-MYC activation and EBNA2 activation led to a corresponding increase in CTCF mRNA levels (Fig. 8A and B). It was previously established that EBNA2 induces c-MYC expression levels either directly or indirectly through LMP1 (11). Our data suggest that EBNA2 induces CTCF directly and also indirectly through its activation of c-MYC and LMP1. These findings suggest a pathway whereby an ectopic increase in c-MYC levels due to c-MYC/Ig translocation in Burkitt's lymphoma (54) or a transient increase in c-MYC levels as a naïve B cell enters the germinal center stage of B-cell development (15, 31, 32) will correspond with an increase in CTCF levels. In turn, the increase in CTCF levels can function, in conjunction with other factors, to cause global changes in chromatin structures of the B cells (19). The results of these changes will likely include repression of EBNA2 expression, as observed in this study, and repression of c-MYC (42, 44). This model is consistent with the proposed complicity of EBV infection in promoting B-cell germinal center development (55). However, although it has been demonstrated that elevated c-MYC levels can induce a Burkitt's lymphoma phenotype in EBV-transformed lymphoblastoid cells, elevated c-MYC levels alone cannot induce a type III-to-type I switch in lymphoblastoid cells (37, 38, 40, 51). It is also clear that sustained expression of EBNA2 in type III cells does not necessarily lead to an increase in CTCF levels sufficient for the down regulation of Cp and the transition to type I latency. More likely, other immune factors, in combination with c-MYC elevation, will be needed to cause this change. Nonetheless, the initial studies here provide an important component to the overall mechanism by which EBV latency types can be determined during the course of EBV infection and B-cell development.
Acknowledgments
We are grateful to R. Gregory and R. Shiekhattar (Wistar Institute, Philadelphia, PA) for the p3xFlag-CTCF vector; D. Herlyn (Wistar Institute) for the LCL3456-EBV cell line; B. Kempkes (GSF-NRC, Munich, Germany) for the EREB 2.5 cell line; W. Hammerschmidt (GSF-NRC) for the EBV-Hygro-GFP BAC; B. Sugden (University of Wisconsin, Madison) for the EBV BamHI C BACmid; and The Wistar Institute Cancer Core Facilities for baculoviral protein generation, FACS analysis, and cell sorting. We also thank Pegah Johansson and L. Rymo's lab (Gothenburg University, Gothenburg, Sweden) for guidance in using the EREB 2.5 cell line. Finally, we thank J. Kissel (Wistar Institute) for the EL350 E. coli strain, pL452 Cre-Lox plasmid, and guidance in generating the recombinant EBV BACs.
This work was funded in part by grants from the NIH to P.M.L. (CA93606 and CA05678) and to S.B.M. (CA090465), the Pennsylvania Department of Health, and a Wistar Institute NIH Post-Doctoral Training Fellowship to C.M.C.
REFERENCES
- 1.Alazard, N., H. Gruffat, E. Hiriart, A. Sergeant, and E. Manet. 2003. Differential hyperacetylation of histones H3 and H4 upon promoter-specific recruitment of EBNA2 in Epstein-Barr virus chromatin. J. Virol. 77:8166-8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambinder, R. F., K. D. Robertson, and Q. Tao. 1999. DNA methylation and the Epstein-Barr virus. Semin. Cancer Biol. 9:369-375. [DOI] [PubMed] [Google Scholar]
- 3.Atanasiu, C., L. Lezina, and P. M. Lieberman. 2005. DNA affinity purification of Epstein-Barr virus OriP-binding proteins. Methods Mol. Biol. 292:267-276. [DOI] [PubMed] [Google Scholar]
- 4.Babcock, G. J., D. Hochberg, and A. D. Thorley-Lawson. 2000. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 13:497-506. [DOI] [PubMed] [Google Scholar]
- 5.Bodescot, M., M. Perricaudet, and P. J. Farrell. 1987. A promoter for the highly spliced EBNA family of RNAs of Epstein-Barr virus. J. Virol. 61:3424-3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boreström, C., H. Zetterberg, K. Liff, and L. Rymo. 2003. Functional interaction of nuclear factor Y and Sp1 is required for activation of the Epstein-Barr virus C promoter. J. Virol. 77:821-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke, L. J., R. Zhang, M. Lutz, and R. Renkawitz. 2002. The thyroid hormone receptor and the insulator protein CTCF: two different factors with overlapping functions. J. Steroid Biochem. Mol. Biol. 83:49-57. [DOI] [PubMed] [Google Scholar]
- 8.Chau, C. M., and P. M. Lieberman. 2004. Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J. Virol. 78:12308-12319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delecluse, H. J., T. Hilsendegen, D. Pich, R. Zeidler, and W. Hammerschmidt. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 95:8245-8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng, Z., L. Lezina, C. J. Chen, S. Shtivelband, W. So, and P. M. Lieberman. 2002. Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of plasmid replication. Mol. Cell 9:493-503. [DOI] [PubMed] [Google Scholar]
- 11.Dirmeier, U., R. Hoffmann, E. Kilger, U. Schultheiss, C. Briseno, O. Gires, A. Kieser, D. Eick, B. Sugden, and W. Hammerschmidt. 2005. Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 24:1711-1717. [DOI] [PubMed] [Google Scholar]
- 12.Evans, T. J., P. J. Farrell, and S. Swaminathan. 1996. Molecular genetic analysis of Epstein-Barr virus Cp promoter function. J. Virol. 70:1695-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fedoriw, A. M., P. Stein, P. Svoboda, R. M. Schultz, and M. S. Bartolomei. 2004. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science 303:238-240. [DOI] [PubMed] [Google Scholar]
- 14.Filippova, G. N., A. Lindblom, L. J. Meincke, E. M. Klenova, P. E. Neiman, S. J. Collins, N. A. Doggett, and V. V. Lobanenkov. 1998. A widely expressed transcription factor with multiple DNA sequence specificity, CTCF, is localized at chromosome segment 16q22.1 within one of the smallest regions of overlap for common deletions in breast and prostate cancers. Genes Chromosomes Cancer 22:26-36. [PubMed] [Google Scholar]
- 15.Fischer, G., S. C. Kent, L. Joseph, D. R. Green, and D. W. Scott. 1994. Lymphoma models for B cell activation and tolerance. X. Anti-mu-mediated growth arrest and apoptosis of murine B cell lymphomas is prevented by the stabilization of myc. J. Exp. Med. 179:221-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuentes-Panana, E. M., and P. D. Ling. 1998. Characterization of the CBF2 binding site within the Epstein-Barr virus latency C promoter and its role in modulating EBNA2-mediated transactivation. J. Virol. 72:693-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuentes-Panana, E. M., R. Peng, G. Brewer, J. Tan, and P. D. Ling. 2000. Regulation of the Epstein-Barr virus C promoter by AUF1 and the cyclic AMP/protein kinase A signaling pathway. J. Virol. 74:8166-8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gahn, T. A., and B. Sugden. 1995. An EBNA-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Esptein-Barr virus LMP gene. J. Virol. 69:2633-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrett, F. E., A. V. Emelyanov, M. A. Sepulveda, P. Flanagan, S. Volpi, F. Li, D. Loukinov, L. A. Eckhardt, V. V. Lobanenkov, and B. K. Birshtein. 2005. Chromatin architecture near a potential 3′ end of the Igh locus involves modular regulation of histone modifications during B-cell development and in vivo occupancy at CTCF sites. Mol. Cell. Biol. 25:1511-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grossman, S. R., E. Johannsen, X. Tong, R. Yalamanchili, and E. Kieff. 1994. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 91:7568-7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsieh, C.-L. 1999. Evidence that protein binding specifies sites of DNA demethylation. Mol. Cell. Biol. 19:46-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh, J. J., and S. D. Hayward. 1995. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein-Barr virus EBNA2. Science 268:560-563. [DOI] [PubMed] [Google Scholar]
- 23.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science 293:1074-1080. [DOI] [PubMed] [Google Scholar]
- 24.Jin, X. W., and S. H. Speck. 1992. Identification of critical cis elements involved in mediating Epstein-Barr virus nuclear antigen 2-dependent activity of an enhancer located upstream of the viral BamHI C promoter. J. Virol. 66:2846-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanduri, C., V. Pant, D. Loukinov, E. Pugacheva, C. F. Qi, A. Wolffe, R. Ohlsson, and V. V. Lobanenkov. 2000. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol. 10:853-856. [DOI] [PubMed] [Google Scholar]
- 26.Kempkes, B., D. Spitkovsky, P. Jansen-Durr, J. W. Ellwart, E. Kremmer, H. J. Delecluse, C. Rottenberger, G. W. Bornkamm, and W. Hammerschmidt. 1995. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J. 14:88-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kieff, E. 1996. Epstein-Barr virus and its replication, p. 2343-2396. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Field's virology, 3rd ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, Pa. [Google Scholar]
- 28.Kuhn, E. J., and P. K. Geyer. 2003. Genomic insulators: connecting properties to mechanism. Curr. Opin. Cell Biol. 15:259-265. [DOI] [PubMed] [Google Scholar]
- 29.Lee, E. C., D. Yu, J. Martinez de Velasco, L. Tessarollo, D. A. Swing, D. L. Court, N. A. Jenkins, and N. G. Copeland. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56-65. [DOI] [PubMed] [Google Scholar]
- 30.Ling, P. D., R. S. Peng, A. Nakajima, J. H. Yu, J. Tan, S. M. Moses, W. H. Yang, B. Zhao, E. Kieff, K. D. Bloch, and D. B. Bloch. 2005. Mediation of Epstein-Barr virus EBNA-LP transcriptional coactivation by Sp100. EMBO J. 24:3565-3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez-Valdez, H., C. Guret, O. de Bouteiller, I. Fugier, J. Banchereau, and Y. J. Liu. 1996. Human germinal center B cells express the apoptosis-inducing genes Fas, c-myc, P53, and Bax but not the survival gene bcl-2. J. Exp. Med. 183:971-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCormack, J. E., V. H. Pepe, R. B. Kent, M. Dean, A. Marshak-Rothstein, and G. E. Sonenshein. 1984. Specific regulation of c-myc oncogene expression in a murine B-cell lymphoma. Proc. Natl. Acad. Sci. USA 81:5546-5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niller, H. H., D. Salamon, F. Banati, F. Schwarzmann, H. Wolf, and J. Minarovits. 2004. The LCR of EBV makes Burkitt's lymphoma endemic. Trends Microbiol. 12:495-499. [DOI] [PubMed] [Google Scholar]
- 34.Niller, H. H., D. Salamon, M. Takacs, J. Uhlig, H. Wolf, and J. Minarovits. 2001. Protein-DNA interaction and CpG methylation at rep*/vIL-10p of latent Epstein-Barr virus genomes in lymphoid cell lines. Biol. Chem. 382:1411-1419. [DOI] [PubMed] [Google Scholar]
- 35.Nilsson, T., H. Zetterberg, Y. C. Wang, and L. Rymo. 2001. Promoter-proximal regulatory elements involved in oriP-EBNA1-independent and -dependent activation of the Epstein-Barr virus C promoter in B-lymphoid cell lines. J. Virol. 75:5796-5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohlsson, R., R. Renkawitz, and V. Lobanenkov. 2001. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 17:520-527. [DOI] [PubMed] [Google Scholar]
- 37.Pajic, A., A. Polack, M. S. Staege, D. Spitkovsky, B. Baier, G. W. Bornkamm, and G. Laux. 2001. Elevated expression of c-myc in lymphoblastoid cells does not support an Epstein-Barr virus latency III-to-I switch. J. Gen. Virol. 82:3051-3055. [DOI] [PubMed] [Google Scholar]
- 38.Pajic, A., M. S. Staege, D. Dudziak, M. Schuhmacher, D. Spitkovsky, G. Eissner, M. Brielmeier, A. Polack, and G. W. Bornkamm. 2001. Antagonistic effects of c-myc and Epstein-Barr virus latent genes on the phenotype of human B cells. Int. J. Cancer 93:810-816. [DOI] [PubMed] [Google Scholar]
- 39.Paulson, E. J., J. D. Fingeroth, J. L. Yates, and S. H. Speck. 2002. Methylation of the EBV genome and establishment of restricted latency in low-passage EBV-infected 293 epithelial cells. Virology 299:109-121. [DOI] [PubMed] [Google Scholar]
- 40.Polack, A., K. Hortnagel, A. Pajic, B. Christoph, B. Baier, M. Falk, J. Mautner, C. Geltinger, G. W. Bornkamm, and B. Kempkes. 1996. c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proc. Natl. Acad. Sci. USA 93:10411-10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puglielli, M. T., M. Woisetschlaeger, and S. H. Speck. 1996. oriP is essential for EBNA gene promoter activity in Epstein-Barr virus-immortalized lymphoblastoid cell lines. J. Virol. 70:5758-5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qi, C. F., A. Martensson, M. Mattioli, R. Dalla-Favera, V. V. Lobanenkov, and H. C. Morse III. 2003. CTCF functions as a critical regulator of cell-cycle arrest and death after ligation of the B cell receptor on immature B cells. Proc. Natl. Acad. Sci. USA 100:633-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raab-Traub, N. 2002. Epstein-Barr virus in the pathogenesis of NPC. Semin. Cancer Biol. 12:431-441. [DOI] [PubMed] [Google Scholar]
- 44.Rasko, J. E., E. M. Klenova, J. Leon, G. N. Filippova, D. I. Loukinov, S. Vatolin, A. F. Robinson, Y. J. Hu, J. Ulmer, M. D. Ward, E. M. Pugacheva, P. E. Neiman, H. C. Morse III, S. J. Collins, and V. V. Lobanenkov. 2001. Cell growth inhibition by the multifunctional multivalent zinc-finger factor CTCF. Cancer Res. 61:6002-6007. [PubMed] [Google Scholar]
- 45.Reisman, D., and B. Sugden. 1986. trans activation of an Epstein-Barr viral transcriptional enhancer by the Epstein-Barr viral nuclear antigen 1. Mol. Cell. Biol. 6:3838-3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rickinson, A. B., and E. Kieff. 1996. Epstein-Barr virus, p. 2397-2446. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, Pa. [Google Scholar]
- 47.Robertson, E. S., J. Lin, and E. Kieff. 1996. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJκ. J. Virol. 70:3068-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rowe, M., D. T. Rowe, C. D. Gregory, L. S. Young, P. J. Farrell, H. Rupani, and A. B. Rickinson. 1987. Differences in B-cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt's lymphoma cells. EMBO J. 6:2743-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salamon, D., M. Takacs, D. Ujvari, J. Uhlig, H. Wolf, J. Minarovits, and H. H. Niller. 2001. Protein-DNA binding and CpG methylation at nucleotide resolution of latency-associated promoters Qp, Cp, and LMP1p of Epstein-Barr virus. J. Virol. 75:2584-2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Souza, T. A., B. D. Stollar, J. L. Sullivan, K. Luzuriaga, and D. A. Thorley-Lawson. 2005. Peripheral B cells latently infected with Epstein-Barr virus display molecular hallmarks of classical antigen-selected memory B cells. Proc. Natl. Acad. Sci. USA 102:18093-18098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Staege, M. S., S. P. Lee, T. Frisan, J. Mautner, S. Scholz, A. Pajic, A. B. Rickinson, M. G. Masucci, A. Polack, and G. W. Bornkamm. 2002. MYC overexpression imposes a nonimmunogenic phenotype on Epstein-Barr virus-infected B cells. Proc. Natl. Acad. Sci. USA 99:4550-4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugden, B., and N. Warren. 1989. A promoter of Epstein-Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J. Virol. 63:2644-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sung, N. S., S. Kenney, D. Gutsch, and J. S. Pagano. 1991. EBNA-2 transactivates a lymphoid-specific enhancer in the BamHI C promoter of Epstein-Barr virus. J. Virol. 65:2164-2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taub, R., I. Kirsch, C. Morton, G. Lenoir, D. Swan, S. Tronick, S. Aaronson, and P. Leder. 1982. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 79:7837-7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorley-Lawson, D. A., and A. Gross. 2004. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 350:1328-1337. [DOI] [PubMed] [Google Scholar]
- 56.West, A. G., M. Gaszner, and G. Felsenfeld. 2002. Insulators: many functions, many mechanisms. Genes Dev. 16:271-288. [DOI] [PubMed] [Google Scholar]
- 57.West, A. G., S. Huang, M. Gaszner, M. D. Litt, and G. Felsenfeld. 2004. Recruitment of histone modifications by USF proteins at a vertebrate barrier element. Mol. Cell 16:453-463. [DOI] [PubMed] [Google Scholar]
- 58.Young, L., C. Alfieri, K. Hennessy, H. Evans, C. O'Hara, K. C. Anderson, J. Ritz, R. S. Shapiro, A. Rickinson, E. Kieff, et al. 1989. Expression of Epstein-Barr virus transformation-associated genes in tissues of patients with EBV lymphoproliferative disease. N. Engl. J. Med. 321:1080-1085. [DOI] [PubMed] [Google Scholar]
- 59.Young, L. S., and A. B. Rickinson. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4:757-768. [DOI] [PubMed] [Google Scholar]
- 60.Yusufzai, T. M., H. Tagami, Y. Nakatani, and G. Felsenfeld. 2004. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol. Cell 13:291-298. [DOI] [PubMed] [Google Scholar]
- 61.Zhang, X. Y., L. M. DeSalle, J. H. Patel, A. J. Capobianco, D. Yu, A. Thomas-Tikhonenko, and S. B. McMahon. 2005. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc. Natl. Acad. Sci. USA 102:13968-13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao, B., D. R. Marshall, and C. E. Sample. 1996. A conserved domain of the Epstein-Barr virus nuclear antigens 3A and 3C binds to a discrete domain of Jκ. J. Virol. 70:4228-4236. [DOI] [PMC free article] [PubMed] [Google Scholar]