

Abstract
Early onset torsion dystonia is characterized by involuntary movements and distorted postures and is usually caused by a 3-bp (GAG) deletion in the DYT1 (TOR1A) gene. DYT1 codes for torsinA, a member of the AAA+ family of proteins, implicated in membrane recycling and chaperone functions. A close relative, torsinB may be involved in similar cellular functions. We investigated torsinA and torsinB message and protein levels in the developing mouse brain. TorsinA expression was highest during prenatal and early postnatal development (until postnatal day 14; P14), whereas torsinB expression was highest during late postnatal periods (from P14 onwards) and in the adult. In addition, significant regional variation in the expression of the two torsins was seen within the developing brain. Thus, torsinA expression was highest in the cerebral cortex from embryonic day 15 (E15)–E17 and in the striatum from E17–P7, while torsinB was highest in the cerebral cortex between P7–P14 and in the striatum from P7–P30. TorsinA was also highly expressed in the thalamus from P0–P7 and in the cerebellum from P7–P14. Although functional significance of the patterns of torsinA and B expression in the developing brain remains to be established, our findings provide a basis for investigating the role of torsins in specific processes such as neurogenesis, neuronal migration, axon/dendrite development, and synaptogenesis.
Keywords: Torsion dystonia, Brain development
1. Introduction
Early onset torsion dystonia is a movement disorder caused by the loss of 3-bp (GAG) encoding a glutamic acid residue near the carboxyl terminal of the DYT1 gene (Ozelius et al., 1997). The disease results in twisted or contorted postures that begin usually in the arms or legs and spread gradually to other parts of the body (Bressman et al., 2000). The disorder is believed to result from an imbalance in basal ganglia neurotransmission without neurodegeneration (Hedreen et al., 1988; Sanghera et al., 2004).
DYT1 codes for torsinA, a member of the AAA+ superfamily of proteins, which typically forms 6-member oligomeric complexes and associates with additional protein species. The torsin family of proteins consists of torsinA, torsinB, torp2a, and torp3a in mammals (Ozelius et al., 1999; Neuwald et al., 1999). Three related genes exist in nematodes and one in Drosophila. TorsinA is highly homologous (70%) to torsinB, a product of the TOR1B gene, located adjacent to DYT1 on chromosome 9 (Ozelius et al., 1997). The AAA+ superfamily members share Mg++ ATP domains (Breakefield et al., 2001) and therefore may function as ATP-dependent chaperones in membrane trafficking and vesicle fusion (Vale, 2000; Neuwald et al., 1999; Toninelli et al., 2003).
As the name suggests, early onset torsion dystonia manifests typically between 5 and 20 years of age (Bressman et al., 2000). If carriers of the DYT1 mutation reach the age of 28 years without showing symptoms, they escape the disorder permanently, implying a role for other genetic or environmental factors in triggering the disease. Several lines of evidence corroborate a developmental component in the pathophysiology of this disorder. TorsinA mRNA and protein expression begins early in the prenatal period in the rodent and human brain (Xiao et al., 2004; Ferrari-Toninelli et al., 2004; Siegert et al., 2005). Transgenic mice overexpressing mutant human torsinA show motor abnormalities ranging from hyperkinesia to delayed motor learning, some of which begin in the early postnatal period (Goodchild and Dauer, 2004; Shashidharan et al., 2005; Sharma et al., 2005). Other reports indicate that DYT1 gene knockout mice die at birth (Dauer and Goodchild, 2004). The function of torsin is unknown, but it is implicated in neurite extension and processing of proteins critical to synaptic function. Therefore, a role for torsinA and torsinB in brain development appears likely. Here, we investigated torsinA and torsinB mRNA and protein expression in the developing mouse brain to explore possible associations between torsin function during brain development and disease onset or progression.
2. Results
To establish the patterns of expression of torsinA and torsinB in the embryonic and postnatal brain, we quantified message and protein levels in the whole brain first and then in selected brain regions.
2.1. Examination of torsinA and torsinB in the whole brain
Northern blots showed a single prominent torsinA message of 1.5 kb (Fig. 1A). This expression pattern is different from that in the human tissue, in which additional less abundant messages suggestive of splice variants or alternative transcription start sites were reported (Ozelius et al., 1997). A prominent 3.4-kb message was found for torsinB, along with other less abundant messages (Fig. 1B), a pattern similar to that reported for human torsinB (Hewett et al., 2004). TorsinA and B messages were detected in all embryonic and postnatal brain samples examined, even as early as E11. TorsinA transcript level increased during the embryonic and early postnatal period and reached its highest level at P14, rising by about 2.9-fold between E11 and P14 (Fig. 1C). It declined after P14, by about 50%, until P30, the oldest age examined (Fig. 1C). TorsinB mRNA levels also increased during the prenatal period in step with torsinA mRNA levels (Fig. 1C). During the postnatal period, torsinB mRNA levels increased till P14, again in step with torsinA mRNA. However, between P14 and P30, torsinA and torsinB mRNA showed opposite expression patterns: While torsinA mRNA levels declined, torsinB mRNA levels continued to be maintained until P30 (Fig. 1C).
Fig. 1.

Northern blots of torsinA and torsinB mRNA in E11–P30 mice. RNA blots were hybridized to probes corresponding to the 3'UTR of (A) mouse torsinA and (B) mouse torsinB. A single prominent message of 1.5 kb was found for torsinA, while a prominent 3.4-kb message was found for torsinB, along with other less abundant messages. A DNA probe for GAPDH, which detects a 1.4-kb message was used as loading control. Marker sizes are indicated. (C) TorsinA and torsinB signal intensities are expressed as a proportion of GAPDH (loading control) signal. For quantification of the torsinB blot, only the signal intensity of the primary 3.4-kb band was expressed as a ratio to GAPDH. The intensities of the fainter bands were not quantified.
We examined the specificity of torsinA and torsinB antibodies by Western blot before proceeding with developmental analysis of protein expression. Five antibodies for torsinA and torsinB were examined (DM2A8, TA1, TA2, TAB1, and TB2). The monoclonal antibody, DM2A8 and the polyclonal antibody TB2 that recognize human torsinA and B, respectively produced the highest level of specificity. These two antibodies have been well characterized using human tissue and mouse cell lines (Hewett et al., 2004). In mouse brain, DM2A8 showed a 37-kDa torsinA band, another prominent band around 50 kDa and other less prominent bands (Fig. 2A). CAD cells overexpressing wild-type human torsinA (Hewett et al., 2000) was used as a positive control in all torsinA blots (refer to Figs. 3A and 4A). TB2 showed a 38-kDa torsinB band as well as an unknown band of about 45 kDa (Fig. 2B). Both the 50-kDa band in torsinA blots and the 45-kDa band in torsinB blots are not sensitive to de-glycosylation treatments with EndoH and PNGaseF (data not shown). We quantitatively analyzed the 37-kDa and the 38-kDa bands of torsinA and B respectively, the known sizes of these proteins in all the samples. Due to the presence of the non-specific bands, we were unable to perform immunohistochemical analyses.
Fig. 2.
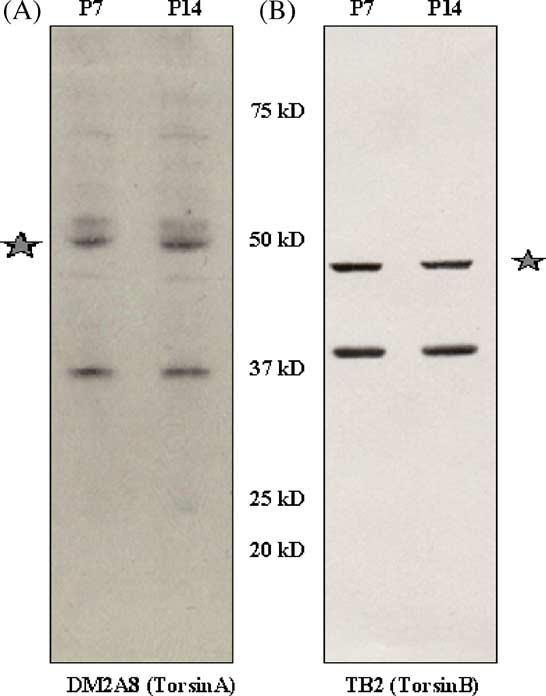
Specificity of torsinA and B antibodies was determined by western blot of mouse brain. Blots were probed with (A) the monoclonal antibody DM2A8, which recognizes the torsinA band at 37 kDa and (B) polyclonal antibody TB2, which recognizes the 38-kDa band of torsinB. Prominent bands of unknown origin (asterisk) at 50 kDa and 45 kDa were also seen with DM2A8 and TB2 respectively. Molecular weight markers are indicated.
Fig. 3.
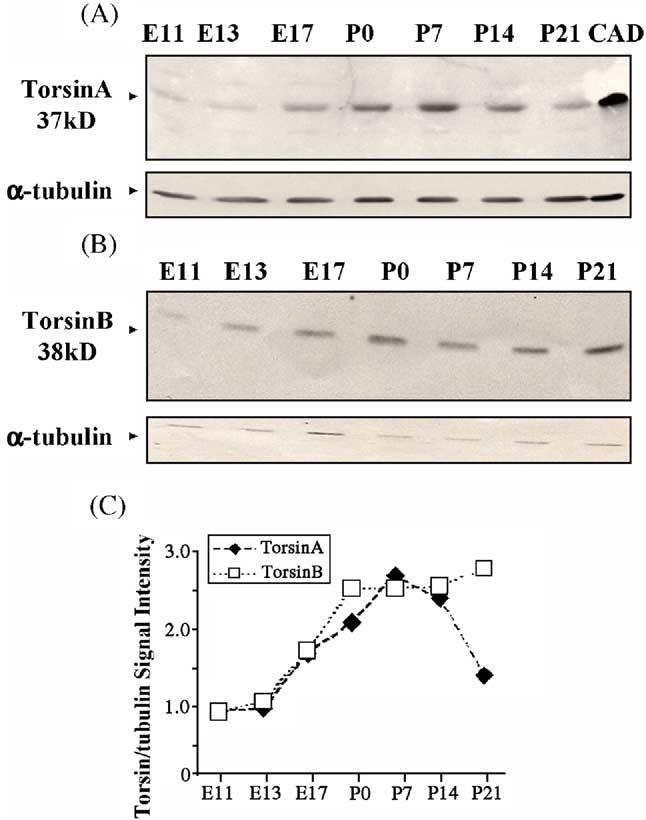
Western blots of torsinA (A) and torsinB (B) protein in whole brains of E11–P21 mice. CAD cells expressing wild-type human torsinA protein were used as positive controls in panel (A). Arrowheads indicate position and size of torsinA and torsinB. α-Tubulin was used as a loading control. (C) TorsinA and torsinB signal intensities are expressed as a proportion of α-tubulin (loading control) signal.
Fig. 4.

Western blot of torsinA protein in selected regions of the mouse brain at various prenatal and postnatal stages of development. The hindbrain and cerebellum samples contain only the hindbrain on E15 and E17 and contain also the cerebellum from P0 onwards. (A) TorsinA protein from the different brain regions was examined from E15–P30. CAD cells expressing wild-type human torsinA protein were used as positive controls. (B) and (C) Quantitative representation of torsinA expression levels normalized to α-tubulin levels.
TorsinA and torsinB proteins could be detected in the brain as early as by E11 (Figs. 3A, B). The protein expression profiles were similar to the mRNA profiles during development in whole brain homogenates. Both the proteins increased from E11 to P7 (Fig. 3C). TorsinA levels declined after P7, decreasing by about 50% by P21. TorsinB protein levels continued to be maintained beyond P7 till P21 (Fig. 3C).
2.2. Regional expression profiles for torsinA and torsinB
TorsinA expression was high in the cerebral cortex between E15 and E17 and then decreased. The decrease from P0 to P14 was gradual compared to that from P14 to P21 (Fig. 4B). In the striatum, a similar trend was seen, but the highest expression was between E17 and P7 with a decline from P7 to P21 (Fig. 4B). TorsinA levels increased in the hippocampal formation from P0 to P7 and decreased from P7 to P30 (Fig. 4B). TorsinA expression in the thalamus and midbrain rose from E15 to P7 and declined from P7 to P30 (Fig. 4C). In the cerebellum and hindbrain, torsinA was more or less uniform from E15 to P7. It rose between P7 and P14 and declined from P14 to P30 (Fig. 4C).
In the cerebral cortex, torsinB increased from E17 to P14 and then declined (Fig. 5B). In the striatum, expression increased until P14 and declined only slightly between P14 and P30. Thus, striatal torsinB levels were higher than cerebral cortical levels during the postnatal period. In the hippocampus, torsinB level was uniform from P0 to P14, rose from P14 to P21, and remained high at P30 (Fig. 5B). TorsinB expression levels in the thalamus and midbrain increased from E15 to P0 and remained unchanged thereafter (Fig. 5C). In the cerebellum and hindbrain, a marked increase was seen from P0 to P7, a steady state from P7 to P14 and another increase from P14 to P30 (Fig. 5C).
Fig. 5.

Western blot of torsinA protein in selected regions of the mouse brain at various prenatal and postnatal stages of development. The hindbrain and cerebellum samples contain only the hindbrain on E15 and E17 and contain also the cerebellum from P0 onwards. (A) TorsinB protein from the different brain regions was examined from E15–P30. (B) and (C)
3. Discussion
Our data show that both torsinA and torsinB are expressed in the brain beginning early in the embryonic period, and that torsinA expression declines in the early postnatal period, whereas torsinB expression remains high. We also observed significant regional variation in the patterns of expression of torsinA and torsinB in the developing brain. These data suggest that torsinA and torsinB may play a role in the developing brain, and that mutations in these proteins may affect some regions of the developing and adult brain more significantly than others.
TorsinA and torsinB mRNA and protein levels in whole brain increased in comparable fashion in the prenatal period, and it was only in the second postnatal week that their expression profiles diverged. TorsinA mRNA levels declined sharply after P14 and protein levels after P7, while torsinB mRNA and protein levels continued to rise until P30. These data are consistent with previous reports concerning torsinA expression profiles during development in rat (Xiao et al., 2004) and human (Siegert et al., 2005) and suggest a role for both the torsins during prenatal development and a predominant role for torsinB during postnatal development. However, more complex expression profiles of torsinA and torsinB that could not have been predicted from the analysis of the whole brain samples emerged when specific regions of the developing brain were analyzed separately. We emphasize that due to the difficulty in sampling the different brain regions from E11 and E13 brains, the regional analyses were performed on E15 and older samples unlike the whole brain analyses, which included E11 and E13 samples also.
The telencephalic derivative cerebral cortex, striatum and hippocampus showed high levels of torsinA until P7 and a decline thereafter. In the cerebral cortex, torsinA expression was the highest during E15–E17 and in the striatum from E17 to P7. Since cortical-basal ganglia dysfunction is implicated in early onset torsion dystonia, we compared the expression profiles of torsinA and torsinB in the cerebral cortex and striatum to certain developmental milestones (Fig. 6). The relatively high prenatal expression coincides with neurogenesis, neuronal migration and formation of initial synaptic connections. The extended period of high expression (until P7) in the striatum may be associated with the relatively delayed maturation of synaptic connections and elaboration of electrophysiological properties of medium spiny neurons in this region (Tepper et al., 1998). The first postnatal week also coincides with the maturation of dendritic arbors of striatal medium spiny neurons (Tepper et al., 1998) and the establishment of cortico-striate connectivity (Sheth et al., 1998). Another developmental parameter is the onset and maturation of nigro-striate dopaminergic pathway. Midbrain dopaminergic axons arrive in the embryonic striatum by E13 when striatal dopamine levels begin to increase (Ohtani et al., 2003; Popolo et al., 2004). By E17, dopaminergic innervation is dense in the dorsal striatum, and during the first postnatal week, it continues into medial areas with the formation of extensive dopaminergic synapses (Riddle and Pollock, 2003; Specht et al., 1981; Moon Edley and Herkenham, 1984). Imbalance in the striatal dopaminergic system is associated with generation of dystonic syndromes (Kuner et al., 2004). A transgenic mouse model of dystonia with severe behavioral abnormalities showed marked decrease in striatal dopamine levels (Shashidharan et al., 2005). Therefore, it is plausible that torsinA and torsinB play a role in the establishment of nigro-striate dopaminergic pathways.
Fig. 6.
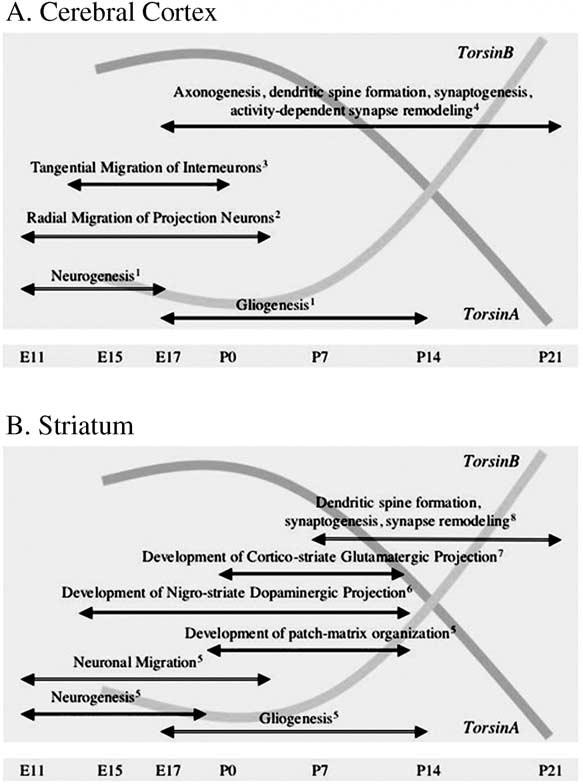
Graphical representation of overlap between developmental changes in torsinA and torsinB expression and salient developmental events in the rodent cerebral cortex (A) and striatum (B). 1—(Caviness et al., 1995; Cameron and Rakic, 1991; Parnavelas et al., 1983); 2—(Parnavelas, 2000; Gleeson and Walsh, 2000; Kriegstein and Noctor, 2004); 3—(Parnavelas, 2000; Anderson et al., 1999); 4—(Blue and Parnavelas, 1983; Parnavelas et al., 1977; Katz and Shatz, 1996); 5—(Graybiel and Hickey, 1982; Van Der Kooy and Fishell, 1986; Bayer, 1984; Sadikot and Sasseville, 1997); 6—(Ohtani et al., 2003); 7—(Sheth et al., 1998); 8—(Tepper et al., 1998).
In the hippocampus, torsinA expression increased from P0 to P7. Hippocampal neurogenesis begins around E10 and is completed before birth, while neurogenesis in the dentate gyrus continues postnatally. The high level of torsinA expression in the hippocampal formation in the postnatal period overlaps with the period of synaptogenesis and dentate gyrus neurogenesis (Bayer, 1982; Cameron et al., 1993; Gage et al., 1998; Altman and Bayer, 1990).
TorsinA expression was two-fold higher in the thalamus by E15 and nearly three-fold by P0-P7, when compared to the adult. The thalamus and cortex develop synchronously and link with each other through reciprocal connections between E13 and E18. By P8 thalamo-cortical arborization is nearly complete (Lopez-Bendito et al., 2002; Ghosh and Shatz, 1992, 1993). TorsinA expression at these specific developmental stages might suggest a role in the establishment of thalamo-cortical connectivity.
Cerebellar Purkinje cells are generated prenatally from the ventricular zone and the granule cells postnatally from the external granule cell layer (Altman, 1972). TorsinA expression was highest between P7 and P14, a period when cerebellar neurons are undergoing extensive axonal outgrowth, dendritic branching, and synaptic remodeling.
TorsinB expression was high in all regions of the brain between P7 and P30. Although torsinB has not been linked to any aspect of early onset dystonia, similarities of its sequence (even in the critical GAG region), subcellular localization (Hewett et al., 2004) and expression patterns (present data) to torsinA suggest complementary functions.
Many mouse models of torsinA dysfunction have been generated, but each shows somewhat different phenotype (Goodchild and Dauer, 2004; Shashidharan et al., 2005; Sharma et al., 2005) emphasizing the difficulties associated with recapitulation of the disease in a mouse model. A prominent feature of the disorder is impaired motor coordination and motor learning, both of which require functional integration of multiple circuits such as frontoparietal and motor cortices, cerebellum and basal ganglia. Thus, dystonia is likely to be a disorder involving multiple brain regions. The lack of neuronal degeneration in torsion dystonia patients suggests the possibility of functional defects, possibly caused by subtle changes in connectivity such as those reported in sensorimotor cortex by diffusion tensor magnetic resonance imaging (Carbon et al., 2004). Though there is no evidence of neuronal degeneration, torsinA, laminA, and ubiquitin inclusions have been reported in midbrain of DYT1 patients (McNaught et al., 2004). Manifesting and non-manifesting carriers of DYT1 mutation show abnormal electrical excitability in the cerebral cortex and asymptomatic carriers of DYT1 mutation show altered resting glucose utilization and decreased D2 receptors in several brain areas as well as delayed motor learning (Eidelberg et al., 1998). At present, it is difficult to correlate these functional changes in the human brain with specific patterns of torsinA or torsinB expression in the different brain regions. Once the important developmental stages and regions where torsin expression is high are well defined, region-specific DYT1 knockouts, knock-in, and knockdown models can be generated, which may be more appropriate for understanding the disorder. The present study is a first step toward defining the role of torsinA and torsinB in the development of different regions of the brain.
4. Experimental procedure
4.1. Animals
Timed-pregnant CD1 mice (Charles River Laboratories; Wilmington, MA) were housed in the Institutional animal facility. All of the experimental procedures were approved by Institutional Animal Care and Use Committee and conform to the NIH guidelines.
4.2. Western blot analysis
Embryos were removed by hysterotomy of anesthetized mice (ketamine, i.p, 50 mg/kg body weight and xylazine i.p. 10 mg/kg body weight) and decapitated. Whole brains were collected from embryonic day 11 (E11; day of conception is E0), E13 and E17 mice, and postnatal day P0 (day of birth), P7, P14, P21 and P30 mice. Brains from E15, E17, P0, P7, P14, P21, and P30 mice were dissected in ice-cold phosphate buffer saline (PBS), pH 7.2, under a dissecting microscope to obtain samples of the cerebral cortex, striatum, hippocampus, midbrain, thalamus, hindbrain, and cerebellum. We could not distinguish between the thalamus and midbrain consistently in the E15 or E17 samples. Therefore, to facilitate uniform sampling across ages, we combined these two regions at all the ages examined. For the same reason, we combined samples of hindbrain and cerebellum. The relatively small size of E15–P7 micro-dissected tissues required pooling of each type of sample from brains of a whole litter (8–10 embryos). In the case of P14–P30 age groups, we pooled samples from 4 mice. We extracted 0.2 g of tissue in 800 μl of 0.05 M Tris HCl, pH 7.4, 0.1 M NaCl and 2% SDS containing protease inhibitor cocktail (Roche) for 30 min at 4°C. The supernatant was clarified by centrifugation (17,700×g, 30 min). The protein content of the extracts was determined by using the BCA protein assay (Biorad Laboratories). Equal amounts of protein (200 μg) were resolved on 12% polyacrylamide gels and transferred to nitrocellulose membrane (Osmonics). Blots were probed with the torsinA-specific monoclonal antibody D-M2A8 (1:100) and the torsinB-specific polyclonal antibody TB2 (1:50) (Hewett et al., 2004). CAD cells overexpressing wild-type human torsinA (Hewett et al., 2000) was used as a positive control in torsinA blots. Blots were stripped and re-probed with anti-α-tubulin antibody (1:5000, Abcam) to estimate total protein levels. Quantitative analysis of band intensities was performed using a Kodak 1-D System (Eastman Kodak, Rochester, NY). In each experiment, the signal intensities of torsinA and torsinB were normalized by comparison to signal intensity of α-tubulin.
4.3. Northern analysis
Northern blot filters containing 2 μg of Poly A+ RNA from brains of E11, E13, E17, P0, P7, P14, P21, and P30 mice were used (Zyagen). Blots were hybridized to PCR probes corresponding to the 3′UTR of mouse torsinA and B messages. A genomic clone containing the 3′UTR sequence of both torsinA and B genes (kindly provided by Dr. Laurie Ozelius, Albert Einstein College of Medicine, New York) was used for the PCR. Primers used to amplify the torsinA and B fragments were as follows. TorsinA primers were 5′-AGAAGGTCTTCTCTGACAAGG-3′and 5′-AAACGTTATCACATTTGC-3′. TorsinB primers were 5′-AGATCTACTCAGACAAGGG-3′ and 5′-TCATCCTGATGCAAGCAGG-3′. A mouse GAPDH probe (Ambion) was used as a loading control. The signal intensities of torsinA and B messages were normalized by comparison to signal intensity of GAPDH messages. Probes were labeled with [α32P]dCTP (Perkin Elmer) by random priming using the Ladderman™ Labelling Kit (TaKaRa Shuzo Co., Ltd.). Hybridization was performed overnight with ULTRAhyb™ hybridization buffer Amersham) at 65°C. After high stringency washing, the blots were exposed to film. Signal intensities were quantified using the Kodak 1-D System.
Acknowledgments
We are grateful to Dr. Vijaya Ramesh for the torsin antibodies and Dr. Nutan Sharma for the advice on the use of densitometry. This work was supported by grants from the NIH (P01 NS37409) and the Bachmann-Strauss Dystonia and Parkinson Foundation, Inc. New York.
REFERENCES
- Altman J. Postnatal development of the cerebellar cortex in the rat: I. The external germinal layer and the transitional molecular layer. J. Comp. Neurol. 1972;145:353–397. doi: 10.1002/cne.901450305. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J. Comp. Neurol. 1990;301:365–381. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Mione MC, Yun K, Rubenstein JLR. Differential origins of neocortical projection and local circuit neurons: role of Dlx genes in neocortical interneuronogenesis. Cereb. Cortex. 1999;9:646–654. doi: 10.1093/cercor/9.6.646. [DOI] [PubMed] [Google Scholar]
- Bayer SA. Changes in the total number of dentate granule cells in juvenile and adult rats: a correlated volumetric and 3H-thymidine autoradiographic study. Exp. Brain Res. 1982;46:315–323. doi: 10.1007/BF00238626. [DOI] [PubMed] [Google Scholar]
- Bayer SA. Neurogenesis in the rat neostriatum. Int. J. Dev. Neurosci. 1984;2:163–175. doi: 10.1016/0736-5748(84)90008-X. [DOI] [PubMed] [Google Scholar]
- Blue ME, Parnavelas JG. The formation and maturation of synapses in the visual cortex of the rat: II. Quantitative analysis. J. Neurocytol. 1983;12:697–712. doi: 10.1007/BF01181531. [DOI] [PubMed] [Google Scholar]
- Breakefield XO, Kamm C, Hanson PI. TorsinA: movement at many levels. Neuron. 2001;31:9–12. doi: 10.1016/s0896-6273(01)00350-6. [DOI] [PubMed] [Google Scholar]
- Bressman SB, Sabatti C, Raymond D, De Leon D, Klein C, Kramer PL, Brin MF, Fahn S, Breakefield X, Ozelius LJ, Risch NJ. The DYT1 phenotype and guidelines for diagnostic testing. Neurology. 2000;54:1746–1752. doi: 10.1212/wnl.54.9.1746. [DOI] [PubMed] [Google Scholar]
- Cameron RS, Rakic P. Glial cell lineage in the cerebral cortex: a review and synthesis. Glia. 1991;4:124–137. doi: 10.1002/glia.440040204. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Woolley CS, Mcewen BS, Gould E. Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience. 1993;56:337–344. doi: 10.1016/0306-4522(93)90335-d. [DOI] [PubMed] [Google Scholar]
- Carbon M, Kingsley PB, Su S, Smith GS, Spetsieris P, Bressman S, Eidelberg D. Microstructural white matter changes in carriers of the DYT1 gene mutation. Ann. Neurol. 2004;56:283–286. doi: 10.1002/ana.20177. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr., Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. TINS. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- Dauer W, Goodchild R. Mouse models of torsinA dysfunction. Adv. Neurol. 2004;94:67–72. [PubMed] [Google Scholar]
- Eidelberg D, Moeller JR, Antonini A, Kazumata K, Nakamura T, Dhawan V, Spetsieris P, Deleon D, Bressman SB, Fahn S. Functional brain networks in DYT1 dystonia. Ann. Neurol. 1998;44:303–312. doi: 10.1002/ana.410440304. [DOI] [PubMed] [Google Scholar]
- Ferrari-Toninelli G, Paccioretti S, Francisconi S, Uberti D, Memo M. TorsinA negatively controls neurite outgrowth of SH-SY5Y human neuronal cell line. Brain Res. 2004;1012:75–81. doi: 10.1016/j.brainres.2004.02.080. [DOI] [PubMed] [Google Scholar]
- Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. J. Neurobiol. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Shatz CJ. Pathfinding and target selection by developing geniculocortical axons. J. Neurosci. 1992;12:39–55. doi: 10.1523/JNEUROSCI.12-01-00039.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Shatz CJ. A role for subplate neurons in the patterning of connections from thalamus to neocortex. Development. 1993;117:1031–1047. doi: 10.1242/dev.117.3.1031. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23:352–359. doi: 10.1016/s0166-2236(00)01607-6. In Process Citation. [DOI] [PubMed] [Google Scholar]
- Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc. Natl. Acad. Sci. U. S. A. 2004;101:847–852. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Hickey TL. Chemospecificity of ontogenetic units in the striatum: demonstration by combining [3H] thymidine neuronography and histochemical staining. Proc. Natl. Acad. Sci. U. S. A. 1982;79:196–202. doi: 10.1073/pnas.79.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedreen JC, Zweig RM, Delong MR, Whitehouse PJ, Price DL. Primary dystonias: a review of the pathology and suggestions for new directions of study. Adv. Neurol. 1988;50:123–132. [PubMed] [Google Scholar]
- Hewett JW, Gonzales-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum. Mol. Genet. 2000;9:1403–1413. doi: 10.1093/hmg/9.9.1403. [DOI] [PubMed] [Google Scholar]
- Hewett JW, Kamm C, Boston H, Beauchamp R, Naismith T, Ozelius L, Hanson PI, Breakefield XO, Ramesh V. TorsinB-perinuclear location and association with torsinA. J. Neurochem. 2004;89:1186–1194. doi: 10.1111/j.1471-4159.2004.02404.x. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Kuner R, Teismann P, Trutzel A, Naim J, Richter A, Schmidt N, Bach A, Ferger B, Schneider A. TorsinA, the gene linked to early-onset dystonia, is upregulated by the dopaminergic toxin MPTP in mice. Neurosci. Lett. 2004;355:126–130. doi: 10.1016/j.neulet.2003.10.069. [DOI] [PubMed] [Google Scholar]
- Lopez-Bendito G, Chan CH, Mallamaci A, Parnavelas J, Molnar Z. Role of Emx2 in the development of the reciprocal connectivity between cortex and thalamus. J. Comp. Neurol. 2002;451:153–169. doi: 10.1002/cne.10345. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Kapustin A, Jackson T, Jengelley TA, Jnobaptiste R, Shashidharan P, Perl DP, Pasik P, Olanow CW. Brainstem pathology in DYT1 primary torsion dystonia. Ann. Neurol. 2004;56:540–547. doi: 10.1002/ana.20225. [DOI] [PubMed] [Google Scholar]
- Moon Edley S, Herkenham M. Comparative development of striatal opiate receptors and dopamine revealed by autoradiography and histofluorescence. Brain Res. 1984;305:27–42. doi: 10.1016/0006-8993(84)91116-8. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- Ohtani N, Goto T, Waeber C, Bhide PG. Dopamine modulates cell cycle in the lateral ganglionic eminence. J. Neurosci. 2003;23:2840–2850. doi: 10.1523/JNEUROSCI.23-07-02840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, De Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Page CE, Klein C, Hewett JW, Mineta M, Leung J, Shalish C, Bressman SB, De Leon D, Brin MF, Fahn S, Corey DP, Breakefield XO. The TOR1A (DYT1) gene family and its role in early onset torsion dystonia. Genomics. 1999;62:377–384. doi: 10.1006/geno.1999.6039. [DOI] [PubMed] [Google Scholar]
- Parnavelas JG. The origin and migration of cortical neurones: new vistas. Trends Neurosci. 2000;23:126–131. doi: 10.1016/s0166-2236(00)01553-8. [DOI] [PubMed] [Google Scholar]
- Parnavelas JG, Sullivan K, Lieberman AR, Webster KE. Neurons and their synaptic organization in the visual cortex of the rat. Electron microscopy of Golgi preparations. Cell Tissue Res. 1977;183:499–517. doi: 10.1007/BF00225663. [DOI] [PubMed] [Google Scholar]
- Parnavelas JG, Luder R, Pollard SG, Sullivan K, Lieberman AR. A qualitative and quantitative ultrastructural study of glial cells in the developing visual cortex of the rat. Philos. Trans. R Soc. Lond., B Biol. Sci. 1983;301:55–84. doi: 10.1098/rstb.1983.0022. [DOI] [PubMed] [Google Scholar]
- Popolo M, Mccarthy DM, Bhide PG. Influence of dopamine on precursor cell proliferation and differentiation in the embryonic mouse telencephalon. Dev. Neurosci. 2004;26:229–244. doi: 10.1159/000082140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle R, Pollock JD. Making connections: the development of mesencephalic dopaminergic neurons. Brain Res. Dev. Brain Res. 2003;147:3–21. doi: 10.1016/j.devbrainres.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Sadikot AF, Sasseville R. Neurogenesis in the mammalian neostriatum and nucleus accumbens: parvalbumin-immunoreactive GABAergic interneurons. J. Comp. Neurol. 1997;389:193–211. [PubMed] [Google Scholar]
- Sanghera MK, Grossman RG, Kalhorn CG, Hamilton WJ, Ondo WG, Jankovic J. Basal ganglia neuronal discharge in primary and secondary dystonia. Adv. Neurol. 2004;94:29–36. [PubMed] [Google Scholar]
- Sharma N, Baxter MG, Petravicz J, Bragg DC, Schienda A, Standaert DG, Breakefield XO. Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J. Neurosci. 2005;25:5351–5355. doi: 10.1523/JNEUROSCI.0855-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashidharan P, Sandu D, Potla U, Armata IA, Walker RH, Mcnaught KS, Weisz D, Sreenath T, Brin MF, Olanow CW. Transgenic mouse model of early-onset DYT1 dystonia. Hum. Mol. Genet. 2005;14:125–133. doi: 10.1093/hmg/ddi012. [DOI] [PubMed] [Google Scholar]
- Sheth AN, Mckee ML, Bhide PG. The sequence of formation and development of corticostriate connections in mice. Dev. Neurosci. 1998;20:98–112. doi: 10.1159/000017306. [DOI] [PubMed] [Google Scholar]
- Siegert S, Bahn E, Kramer ML, Schulz-Schaeffer WJ, Hewett JW, Breakefield XO, Hedreen JC, Rostasy KM. TorsinA expression is detectable in human infants as young as 4 weeks old. Brain Res. Dev. Brain Res. 2005;157:19–26. doi: 10.1016/j.devbrainres.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Specht LA, Pickel VM, Joh TH, Reis DJ. Light microscopic immunocytochemical localization of tyrosine hydroxylase in prenatal rat brain: I. Early ontogeny. J. Comp. Neurol. 1981;199:233–254. doi: 10.1002/cne.901990207. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Sharpe NA, Koos TZ, Trent F. Postnatal development of the rat neostriatum: electrophysiological, light- and electron-microscopic studies. Dev. Neurosci. 1998;20:125–145. doi: 10.1159/000017308. [DOI] [PubMed] [Google Scholar]
- Toninelli GF, Spano P, Memo M. TorsinA, microtubules and cell polarity. Funct. Neurology. 2003;18:7–10. [PubMed] [Google Scholar]
- Vale RD. AAA proteins. Lords of the ring. J. Cell Biol. 2000;150:F13–F19. doi: 10.1083/jcb.150.1.f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Kooy D, Fishell G. Neuronal birthdate underlies the development of striatal compartments. Brain Res. 1986;401:155–161. doi: 10.1016/0006-8993(87)91176-0. [DOI] [PubMed] [Google Scholar]
- Xiao J, Gong S, Zhao Y, Ledoux MS. Developmental expression of rat torsinA transcript and protein. Brain Res. Dev. Brain Res. 2004;152:47–60. doi: 10.1016/j.devbrainres.2004.05.012. [DOI] [PubMed] [Google Scholar]