

Abstract
Antiretroviral nucleoside analogs used in highly active antiretroviral therapy (HAART) are associated with cardiovascular and other tissue toxicity associated with mitochondrial DNA depletion, suggesting a block in mitochondrial (mt)-DNA replication. Because the triphosphate forms of these analogs variably inhibit mt-DNA polymerase, this enzyme has been promoted as the major target of toxicity associated with HAART. We have used isolated mitochondria from rat heart to study the mitochondrial transport and phosphorylation of thymidine and AZT (azidothymidine, or zidovudine), a component used in HAART. We demonstrate that isolated mitochondria readily transport thymidine and phosphorylate it to thymidine 5′-triphosphate (TTP) within the matrix. Under identical conditions, AZT is phosphorylated only to AZT-5′-monophosphate (AZT-MP). The kinetics of thymidine and AZT suggest negative cooperativity of substrate interaction with the enzyme, consistent with work by others on mitochondrial thymidine kinase 2. Results show that TMP and AZT-MP are not transported across the inner membrane, suggesting that AZT-MP may accumulate with time in the matrix. Given the lack of AZT-5′-triphosphate (AZT-TP), it seems unlikely that the toxicity of AZT in the heart is mediated by AZT-TP inhibition of DNA polymerase γ. Rather, our work shows that AZT is a potent inhibitor of thymidine phosphorylation in heart mitochondria, having an inhibitory concentration (IC)50 of 7.0 ± 0.9 μM. Thus, the toxicity of AZT in some tissues may be mediated by disrupting the substrate supply of TTP for mt-DNA replication.
Keywords: Heart mitochondria, AZT cardiotoxicity, thymidine kinase, TMP kinase, mitochondrial DNA depletion
Introduction
Zidovudine (3′-azido-3′-deoxythymidine, AZT) is a thymidine analog pro-drug that is phosphorylated in the host to AZT-triphosphate, which functions by inhibiting viral reverse transcriptase. The drug was the first to be approved for the treatment of acquired immune deficiency syndrome (AIDS) and is still part of the highly active antiretroviral therapy (HAART) regimen used in the present treatment of AIDS. However, the utility of the drug has been limited because of its mitochondrial toxicity. In early reviews of toxicity, when AZT was given alone in high doses (1000 mg or more), 21% of AIDS patients developed hematological disorders related to depressed synthesis of blood components, and 15–20% developed a mitochondrial myopathy (myalgia, fatigue, weakness, and ragged red fibers) or dilated cardiomyopathy leading to congestive heart failure (1,2). These side effects were caused by long-term AZT treatment and were not caused by the AIDS disorder (1,2). Discontinuing the drug usually resulted in amelioration of the side effect. With the advent of HAART, AZT is now used in much lower doses as one component of AIDS therapy, and heart and muscle toxicity associated with AZT is now relatively rare; however, gastrointestinal and hematological toxicity is still observed (3). The prevailing hypothesis for AZT toxicity is the inhibition of mitochondrial DNA polymerase γ by AZT-5′-triphosphate (TP) (4–9). Because only mitochondrial biogenesis is affected, long-term treatment with AZT is required before mitochondria are depleted enough to significantly reduce ATP synthesis. This might have a more rapid time-course in mitotic versus nonmitotic tissue. Long-term inhibition of mitochondrial DNA replication and thus biogenesis is suggested by a marked decrease in mt-DNA in muscles from AZT-treated AIDS patients with myopathy (4); decreased DNA replication in isolated mitochondria in response to long-term AZT treatment (5); decreased mt-DNA in tissues from animals treated with AZT (6); and finally, direct inhibition of purified DNA polymerase γ by AZT-TP (IC50 = 100–200 μM) and AZT-MP (IC50 = 2–3 mM) (7–9). In summary, there is a strong correlation between mitochondrial DNA depletion and AZT toxicity. However, the data that suggest that DNA depletion is mediated by AZT-TP inhibition of DNA polymerase γ is not as compelling. At least one group studying purified DNA polymerase γ reported that although the kinetics of AZT-TP on the enzyme were complex, the simplest interpretation of their results was that AZT-TP was not an inhibitor of the purified polymerase (10). The case is further weakened by the lack of evidence that AZT-TP ever reaches levels (>100 μM) in the mitochondrial matrix of cells consistent with inhibition (11,12). This “bottleneck” of AZT activation has been well described and is caused by the fact that AZT-MP is not a very good substrate for cytosolic thymidylate kinase (13). It is perhaps more likely that AZT-MP is the toxic product that inhibits DNA polymerase γ, even though it has a >20-fold higher IC50. AZT-MP levels are usually much higher than AZT-TP levels in cells and AZT-MP levels in the mM range have been reported in cultured cells (14), although not necessarily in the mitochondrial matrix. Levels of phosphorylated AZT intermediates have not been reported in postmitotic tissues such as the heart.
Other in vitro work has demonstrated that AZT toxicity is more complex, with events occurring in some cells before any significant depletion of mt-DNA. These events include an inhibition of oxidative phosphorylation in Friend murine erythroleukemic cells (15), inhibition of adenylate kinase, and inhibition of the ADP/ATP translocator (3).
Postmitotic tissues obtain all of their deoxynucleotides by transport of deoxynucleosides across the plasma membrane with phosphorylation and salvage occurring within cells. The most important reactions in ribonucleoside salvage appear to involve the base rather than the nucleoside (base sugar) and use cytosolic phosphoribosyl transferases such as hypoxanthine-guanine phosphoribosyl transferase to generate the monophosphates. This is not a viable route for deoxynucleosides, as the substrate for the transferases (PRPP) is not a deoxy sugar. For deoxynucleosides, the first phosphorylation reaction is irreversible and usually the rate-limiting reaction. It is catalyzed by deoxyribonucleoside kinases (for review, see ref. 16). Four enzymes specific for salvage of deoxynucleosides have been characterized, cloned, and sequenced from a variety of mammalian species, including humans. Two enzymes are cytoplasmic: deoxycytidine kinase (dCK), which phosphorylates deoxycytidine, deoxyadenosine, and deoxyguanosine; and thymidine kinase 1 (TK1), which phosphorylates thymidine and deoxyuridine. The other two enzymes are mitochondrial: deoxyguanosine kinase (dGK), which phosphorylates deoxyadenosine, deoxyguanosine, and deoxyinosine (17); and thymidinekinase 2 (TK2), which phosphorylates thymidine, deoxyuridine, and deoxycytidine (18). With these four enzymes, it is possible to generate all four of the naturally occurring deoxynucleoside monophosphates in either the cytoplasm or the mitochondria. Of these four enzymes, the level of TK1 is regulated by the cell cycle and is expressed mostly at the G1/S boundary (16). The other three enzymes appear to be constitutively expressed in most tissues and reflect mitochondrial levels. Once the deoxynucleoside monophosphates are formed, they can take part in two different reactions. They can be cleaved back to the deoxynucleoside by 5′-nucleotidases that are located in both the cytoplasm and the mitochondria (19), or they can be phosphorylated again to the diphosphate via nucleoside monophosphate kinases (NMPKs) that appear to function with both ribo-monophosphates and deoxyribo-monophosphates. The NMPK enzymes have been recently reviewed (20) and include thymidylate kinase (TMPK), which phosphorylates TMP and dUMP; uridylate-cytidylate kinase, which phosphorylates UMP, CMP, and dCMP with high efficiency, and dUMP, AMP, and dAMP with lower efficiency; several guanylate kinases that phosphorylate GMP and dGMP; and five adenylate kinases, all of which phosphorylate AMP and several kinases (AK3 and AK5) that phosphorylate dAMP. Of this group of monophosphate kinases, only AK3 and AK4 are known to be mitochondrial; the others are either clearly cytosolic or assumed to be cytosolic from sequence analysis. Cytosolic TMPK appears to be regulated by the cell cycle like TK1 (20) and is the only enzyme of this group that has been reported to phosphorylate AZT-MP, although not very efficiently (13). Finally, the addition of the third phosphate is a function of nucleotide diphosphate kinases (NDPKs) that are present in the cytosol and in the inner membrane space and matrix of the mitochondria (21). The NDPKs use ATP as a phosphate donor to phosphorylate a wide spectrum of nucleotide diphosphates.
The phosphorylation of thymidine and AZT in mitotic cells is initiated by cytoplasmic TK1 followed by a cytosolic TMPK and an NDPK to TTP and AZT-TP. Once formed in the cytosol, the triphosphates can be transported into the matrix on an inner membrane transporter specific for deoxynucleotide triphosphates (22). However, in postmitotic tissues such as the heart, TK1 is not expressed (16) and AZT and thymidine must be transported across the mitochondrial inner membrane and phosphorylated by the mitochondrial matrix TK2 (23). Thus, TMP and AZT-MP are produced in the matrix in tissues such as the heart. At this point, little is known concerning subsequent steps. The monophosphates could be transported out of the matrix and phosphorylated by a continuation of the cytosolic pathway, or could be further phosphorylated to the triphosphate in the matrix, or both. Neither of these activities has been previously described.
While cardiovascular toxicity of AZT in its present use in HAART is relatively rare, it is nevertheless crucial to understand the primary target of toxicity of AZT in developing new drugs. The goal of the present work is to characterize the phosphorylation pathway for thymidine and the thymidine analog AZT in isolated rat heart mitochondria. We will demonstrate that [3H]-thymidine is phosphorylated to labeled TMP, thymidine 5′-diphosphate (TDP), and TTP in the matrix of isolated rat heart mitochondria, implying the expected activity of TK2 and NDPK as well as a previously undescribed matrix TMPK activity. Under the same conditions, [3H]-AZT was converted by isolated heart mitochondria to the monophosphate form only, demonstrating that AZT-MP is not a substrate for the matrix TMPK. Evidence will be presented demonstrating that TMP and AZT-MP are not readily transported across the inner mitochondrial membrane and thus cannot be substrates for the cytosolic form of TMPK, which may not be expressed in heart at all. This result provides strong evidence that the toxicity of AZT noted in the adult rat heart is not likely to be mediated by AZT-TP inhibition of polymerase γ and rather must be mediated either directly by AZT, or perhaps by AZT-MP, which appears to slowly accumulate in the matrix.
AZT was originally developed as a chemotherapeutic agent and was later found to be useful in AIDS treatment. As such, AZT is not a natural substrate for the salvage pathway enzymes, and because the mitochondrial pathway appears to be required for TTP synthesis in the heart, we have developed an alternative hypothesis of AZT toxicity. We will demonstrate that AZT is a potent inhibitor of the thymidine salvage pathway, disrupting the synthesis of TTP that supplies mitochondrial DNA replication. Recent work has shown that an inherited deficiency in TK2 leads to a severe myopathic mitochondrial deletion disease, clearly illustrating the importance of this enzyme in TTP synthesis and DNA replication (24). The present study offers direct evidence that impaired mitochondrial pyrimidine metabolism is also a player in the complex process of mitochondrial toxicity associated with AZT.
Materials and Methods
Isolation and Incubation of Rat Heart Mitochondria
Tightly coupled mitochondria were isolated from hearts of control rats (Harlan Sprague Dawley) exactly as described previously (25). Intactness was monitored with an oxygen electrode by determining the respiratory control ratio using glutamate as the substrate (25). Only mitochondria with respiratory control values >6 were used in these experiments. Isolated heart mitochondria were incubated at 30°C in a medium originally defined for mitochondrial translation experiments (25) containing mitochondria at 4 mg protein/mL in 25 mM MOPS buffer (pH 7.2), 90 mM KCl, 4 mM magnesium sulfate, 5 mM potassium phosphate, 0.4 mM EGTA, 44 mM mannitol, 14 mM sucrose, BSA (1 mg/mL); 2–4 mM ADP or ATP, 20 mM glutamate, and 0.1 mM each of the 20 amino acids. Additions and changes in this standard medium are outlined where appropriate.
Mitochondrial Protein Synthesis
The rate of mitochondrial protein synthesis in isolated rat heart mitochondria was determined as described previously (25) in the presence of increasing concentrations of AZT, AZT-MP, or AZT-TP.
Mitochondrial Compartmentalization of Thymidine and AZT Phosphorylation Reactions and Intermediates
Centrifugation Through Silicone Oil
To quantitate labeled thymidine, AZT, and phosphorylated intermediates in the mitochondrial matrix when they are also present in the medium requires the careful and complete separation of mitochondria from the medium. This was accomplished as described previously (26,27). Briefly, after an appropriate incubation period in the medium described in “Isolation and Incubation of Rat Heart Mitochondria,” a 500-μL sample is layered over 600 μL of cold silicone oil, which is layered over 500 μL of cold 12% trichloro-acetic acid (TCA). The mitochondria were rapidly (seconds) centrifuged (Fisher microfuge, 13,000g) through the oil into the TCA. After centrifugation, a measured volume of the solution above the oil (the medium) was removed, adjusted to 5% TCA, and both the medium and the solution below the oil (mitochondrial extract) were neutralized by addition of the resin AG-11A8. The neutralized mitochondrial extracts and the medium were filtered and analyzed by high-pressure liquid chromatography (HPLC) as described in “HPLC Analysis of Thymidine, AZT, and Phosphorylated Nucleotides.”
When intact mitochondria were used in the above method described in this section, both the intermembrane space and the matrix space were precipitated into the lower acid fraction. In order to determine the concentrations of deoxynucleotides in the matrix space it was necessary to subtract the deoxynucleotide contribution from the intermembrane space. This was done by assuming that the concentration of deoxynucleotide in the intermembrane space was the same as in the medium, because the outer membrane is freely permeable to nucleotides. In earlier work, the size of the intermembrane space and the matrix space were determined by measuring the uptake of [3H]-water and [14C]-sucrose (not transported to the matrix) for a typical isolated heart mitochondrial preparation (26,27). The water space consistently averaged about 3.0 ± 0.2 μL/mg mitochondrial protein, and the sucrose space consistently averaged 2.0 + 0.2 μL, leaving a matrix space of about 1 μL.
Mitochondrial Phosphorylation of Thymidine and AZT: Direct Precipitation
The goal of the work in this section was to quantitate the rate and kinetics at which isolated mitochondria were capable of phosphorylating labeled thymidine or AZT without regard to mitochondrial compartment. This was accomplished by removing a 0.1-mL aliquot of the incubation suspension (described previously) at various time-points and mixing the aliquot with an equal volume of cold 10% TCA. The extract was kept on ice and centrifuged, and a measured volume was removed and neutralized by adding a resin (AG-11A8). The neutralized extract was filtered and analyzed by HPLC as described in the next section.
This method yielded the total of the medium and the acid-soluble matrix nucleoside and nucleotide components. The rate of appearance of the mono-, di-, and triphosphoforms of a labeled precursor can be measured as a function of time or substrate concentration. Although this method does not identify the mitochondrial compartment carrying out the reaction, it requires less sample, is easier to process, and provides more reliable results than the silicone oil method of fractionation described in the previous section.
HPLC Analysis of Thymidine, AZT, and Phosphorylated Nucleotides
[3H]-thymidine, [3H]-AZT, and their phosphorylated forms from heart mitochondria were extracted, neutralized, and filtered as described in the previous two sections.
The peaks were identified and quantitated from samples by reverse-phase HPLC over an Alltech nucleotide/nucleoside column as described previously (26, 27) connected in-line to a UV monitor (254 nm) and a Radiomatic flow-through scintillation counter. All thymidine, AZT, and adenine nucleotides were separated and identified by this method (see “Results” and Fig. 2).
Fig. 2.
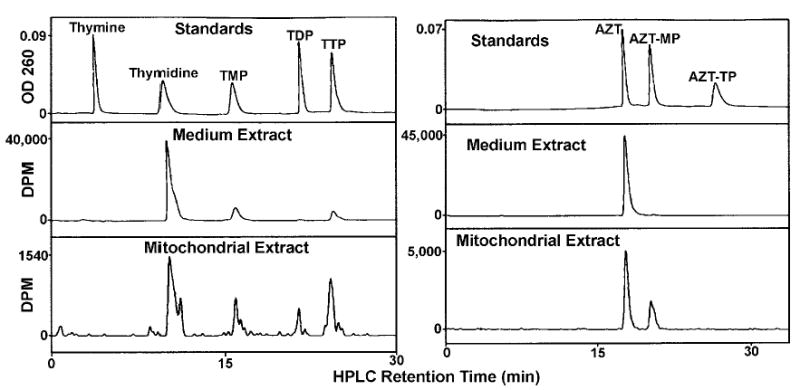
HPLC analysis of thymidine and AZT nucleosides and nucleotides in the medium and in the mitochondrial extract. Mitochondria were incubated for 2 h in the presence of [3H]-thymidine, left (10 μCi/mL, 1 μM), or [3H]-AZT, right (10 μCi/mL, 1 μ,M), and the mitochondria layered over silicone oil for rapid separation by centrifugation. Medium and mitochondrial extracts were prepared as described and analyzed by HPLC. (Top) Thymidine and AZT standards detected by absorption at 254 nm. (Middle) Medium extract detected by an in-line scintillation counter. (Bottom) Mitochondrial extract detected by an in-line scintillation counter.
Results
Effect of AZT, AZT-MP, and AZT-TP on Mitochondrial Protein Synthesis
As noted in the Introduction, AZT toxicity is complex and a variety of affects of AZT that occur more rapidly than can be accounted for by mt-DNA depletion have been reported in in vitro studies (3,15). Lewis et. al. (6) have reported a 40% decrease in mitochondrial protein synthesis secondary to mitochondrial DNA depletion associated with long-term AZT treatment (6). This led to our initial experiments aimed at determining whether AZT or its phosphorylated intermediates had direct effects on mitochondrial protein synthesis in isolated intact rat heart mitochondria. The results shown in Fig. 1 demonstrate that AZT and AZT-MP had little effect on translation in isolated heart mitochondria. In contrast, AZT-TP inhibited translation with an IC50 of 0.72 mM. Levels of AZT-TP have not been reported in the heart, but typical treatment levels in other cells are 5–20 μM (11,12,14). Thus the inhibition of translation would not appear to be relevant to the mitochondrial toxicity of AZT. However, the differential effect of AZT vs AZT-TP in these experiments suggests that isolated mitochondria were not particularly adept at converting AZT to AZT-TP. Consequently, the mechanism of cardiotoxicity of AZT by AZT-TP inhibition of mitochondrial DNA polymerase γ needed to be further examined. We therefore began studies to directly investigate the phosphorylation of AZT in isolated heart mitochondria and to compare AZT phosphorylation to that of the naturally occurring substrate thymidine.
Fig. 1.
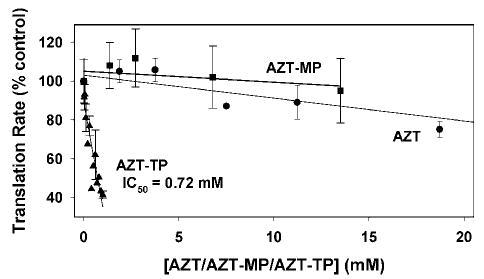
Effect of AZT, AZT-MP, and AZT-TP on mitochondrial protein synthesis in isolated heart mitochondria. Mitochondria were incubated as described in Materials and Methods, except that 0.2 mCi/mL of [35S]-methionine at 20 μM was added with varying amounts of AZT (circles), AZT-MP (squares), and AZT-TP (triangles). Aliquots were taken at 20-, 40-, and 60-min incubation and incorporation of label into protein was measured by a filter paper assay as described previously. Each time-course was converted to a best-fit slope with the results presented as a percent of control ± SEM of five experiments.
Mitochondrial Compartmentalization of Thymidine and AZT Phosphorylation Reactions and Intermediates
Mitochondria were incubated with [3H]-thymidine or [3H]-AZT for 2 h and the mitochondria rapidly separated from the medium over silicone oil as described in “Materials and Methods”. Neutralized extracts of the mitochondria and medium were separated by HPLC and the peaks quantitated as described in “Materials and Methods”. The results are shown in Fig. 2. The top panel represents the separation of standard thymidine (left) and AZT (right) nucleosides and nucleotides used to identify the peaks in the medium (middle panel) and mitochondrial extracts (bottom panel). Clearly, [3H]-thymidine is converted by isolated heart mitochondria to labeled TMP, TDP, and TTP (left), whereas labeled AZT is converted only to AZT-MP (right). After correction for volumes, the concentrations of [3H]-thymidine and [3H]-AZT in the mitochondrial extract corresponded to their concentrations in the medium, indicating equilibration of both labeled compounds across the inner membrane. However, the concentrations of the phosphorylated thymidine compounds are much higher in the matrix than in the medium, implying that the phosphorylations are matrix reactions. The labeled thymidine data (Fig. 2, left) demonstrate that the heart mitochondrial matrix contains a thymidine kinase (TK2), a TMPK, not previously described in mitochondria, and a diphosphate kinase. The peaks of TMP and TTP in the medium presumably represent the transport of TTP out of the matrix and dephosphorylation to TMP, because experiments with [3H]-TMP (see “Compartmentalization of TMP”) suggest that TMP is not readily transported across the inner membrane. In the experiment with labeled AZT (Fig. 2, right), AZT is a substrate for the mitochondrial matrix TK2, but AZT-MP is not a substrate for TMPK. AZT-MP is absent from the medium, suggesting that AZT-MP is also not transported out of the matrix. AZT-MP may be an accumulating dead-end compound that cannot be further phosphorylated nor transported out of the matrix. The rate of dephosphorylation of AZT-MP by the mitochondrial 5′-nucleotidase is only about 2% of the rate of TMP dephosphorylation (19).
Time-Course of Thymidine and AZT Phosphorylation in Isolated Heart Mitochondria
To better understand the conversion of thymidine and AZT to their phosphorylated forms, we determined the time-course of mitochondrial phosphorylation. Having demonstrated in the above section that phosphorylation occurs in the matrix, a more simplified approach was used in these experiments in which an aliquot of the total sample was analyzed without separating the mitochondria from the medium. The time-course of phosphorylation was determined by incubating mitochondria with [3H]-thymidine or [3H]-AZT as described above under “Direct Precipitation.” Aliquots of the total incubation were removed at various times and prepared as described in Materials and Methods. The results are shown in Fig. 3 for [3H]-thymidine (left) and for [3H]-AZT (right). After a short lag period, the levels of [3H]-TMP and [3H]-AZT-MP, respectively, increased linearly with time, with [3H]-TMP formation being about double that of [3H]-AZT-MP. The [3H]-TMP was further phosphorylated slowly to [3H]-TDP, which was rapidly phosphorylated to [3H]-TTP. The total amount of [3H]-thymidine phosphorylated was equal to the sum of [3H]-TMP + [3H]-TDP + [3H]-TTP and the rate of thymidine phosphorylation was about four times higher than the phosphorylation rate of AZT to AZT-MP. As noted above (Fig. 2), no AZT-DP or AZT-TP was detected. These data demonstrate that the rate-limiting step of thymidine phosphorylation in isolated mitochondria is the conversion of [3H]-TMP to [3H]-TDP by TMPK.
Fig. 3.
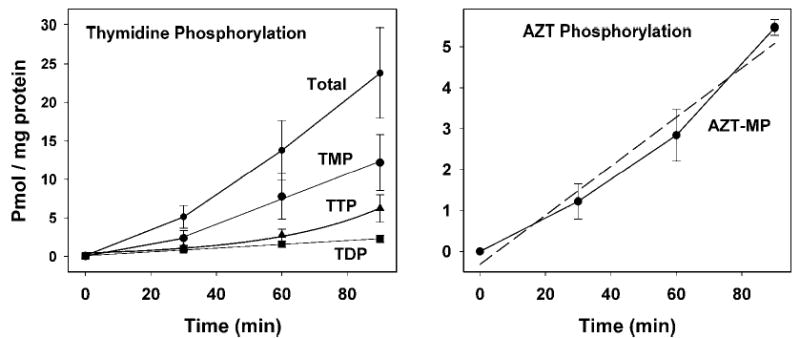
Time-course of phosphorylation of thymidine and AZT in isolated heart mitochondria. Mitochondria were incubated with [3H]-thymidine (2.5 μCi/mL, 1 μM), left, or [3H]-AZT (5 μCi/mL, 1 μM), right, as described in Materials and Methods. Aliquots were removed and a trichloroacetic acid extract of the total sample prepared as described in Materials and Methods. Labeled nucleosides and nucleotides in the extracts were separated and quantitated by HPLC and in-line scintillation counting. Results are presented as picomoles of labeled product per milligram mitochondrial protein by dividing by the specific radioactivity of the thymidine or AZT pool and are presented as the mean ± SEM of five to six experiments.
Compartmentalization of TMP
The results described previously demonstrate that thymidine is phosphorylated in the matrix and suggest that TMP is not transported across the inner mitochondrial membrane. To confirm this point, isolated mitochondria were incubated with 1 μM [3H]-thymidine in the presence and absence of a 10-fold excess of unlabeled TMP in the medium. As shown in Fig. 4, the addition of unlabeled TMP had no effect on the conversion of labeled TMP to TDP and TTP, clearly demonstrating that the labeled TMP arising from labeled thymidine is sequestered in the matrix, while the unlabeled TMP remains in the medium. Incubation of mitochondria with 1 μM [3H]-TMP in the medium led to a relatively low percent conversion to TDP and TTP. About 6–8% of the labeled TMP was dephosphorylated to labeled thymidine (data not shown); hence, it was possible that TMP was converted to thymidine via 5′-nucleotidase, with the labeled thymidine entering the matrix where it was converted to TMP and on to TDP and TTP. To address this possible scenario, we incubated labeled TMP with a 10-fold excess of unlabeled thymidine. The unlabeled thymidine would significantly reduce the specific radioactivity of thymidine arising from labeled TMP in the medium and would reduce this pathway of labeling. Although a small reduction in the relative amounts of labeled TDP and TTP was observed in Fig. 4, these data nevertheless suggest the presence of a small amount of TMPK activity outside the matrix. This may represent a slight leakage of matrix enzymes during the incubation period. The addition of 1 μM [3H]-AZT-MP to the medium with isolated mitochondria was quite stable. Neither phosphorylation nor dephosphorylation was detected (data not shown).
Fig. 4.

Compartmentalization of TMP in isolated heart mitochondria. Mitochondria were incubated for 1 h as described in Materials and Methods with either [3H]-thymidine (5 μCi/mL, 1 μM) in the presence or absence of 10 μM TMP, or with [3H]-TMP (5 μCi/mL, 1 μM) in the presence or absence of 10 μM thymidine. Results were calculated as described in Fig. 3 and are expressed here as the percentage of radioactivity in TDP and TTP relative to the radioactivity in TMP + TDP + TTP.
Kinetics of Thymidine and AZT Phosphorylation in Isolated Heart Mitochondria
The kinetics of thymidine and AZT phosphorylation were determined by measuring the rate of total phosphorylation at increasing concentrations of the labeling precursor. For [3H]-thymidine (Fig. 5), total phosphorylation was calculated as the sum of labeled TMP + TDT + TTP. For [3H]-AZT (Fig. 6), the only phosphorylated form detected was AZT-MP. The top plots in Figs. 5 and 6 were fitted to the standard Michaelis-Menton equation and values for the apparent Km and Vmax for both thymidine and AZT were obtained. For thymidine phosphorylation, Km = 2.4 ± 0.4 μM with a Vmax of 42.1 ± 1.8 pmol/mg/h; and for AZT, Km = 7.5 ± 1.6 μM with a Vmax of 20.0 ± 1.9 pmol/mg/h. Because other investigators have demonstrated negative cooperativity for the interaction of both thymidine and AZT with the purified and cloned TK2 (23,28), we investigated this possibility for the enzyme in isolated mitochondria by replotting the data as Eadie-Hofstee and Hill plots (Figs. 5, thymidine, and 6, AZT, bottom). The biphasic nature of the Eadie-Hofstee plots suggests negative cooperativity for both thymidine and AZT in isolated heart mitochondria, similar to the results with the purified TK2. This was confirmed by Hill plots in which the slope of the line for both compounds was about 0.7 (<1). From these data, two apparent Kms for both thymidine and AZT can be discerned, a Km1 of 0.25 μM for thymidine and 0.41 μM for AZT, and Km2 values of 2.6 μM for thymidine and 8.8 μM for AZT. Qualitatively, the complex kinetics determined here for isolated heart mitochondria are quite similar to those determined for purified and cloned TK2 (23,28), demonstrating that the kinetics in isolated mitochondria reflect TK2 activity. The apparent negative cooperativity is subject to some debate because it has not been clearly shown that TK2 is an oligomer. However, it has been demonstrated that closely related deoxynucleoside kinases are homodimers and this seems likely for TK2 (28). If TK2 is a monomer, then these data would require further investigation.
Fig. 5.
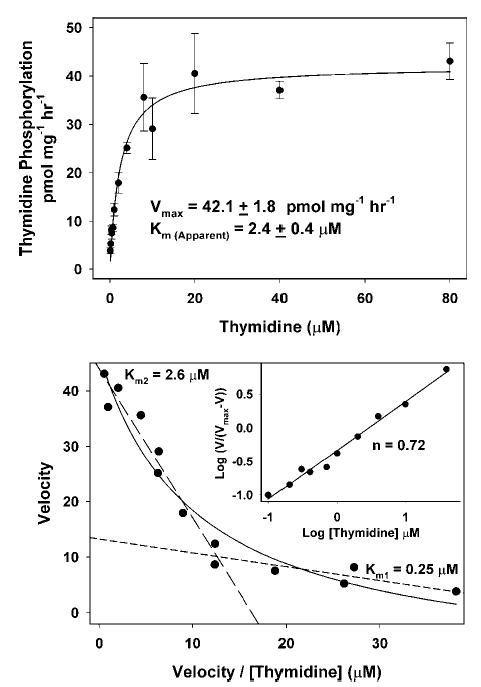
Kinetics of thymidine phosphorylation in isolated heart mitochondria. Mitochondria were incubated as described in the Fig. 3 legend except that the concentration of thymidine was varied between 0.2 and 80 μM (specific radioactivity 3500–11,000 DPM/pmol). (Top) The rate of total thymidine phosphorylation was determined as the sum of TMP, TDP, and TTP formed over time as calculated in Fig. 3. The results from five to six experiments are expressed in a standard Michaelis-Menton plot ± SEM and the data fitted to the Michaelis-Menton equation V = Vmax [S]/(Km + [S]), yielding an apparent Vmax of 42.1 ± 1.8 pmol/mg/h and an apparent Km of 2.4 ± 0.4 μM. (Bottom) The Michaelis-Menton equation was rearranged to the Eadie-Hofstee equation in which V = −Km V/[S] + Vmax. These results indicate that the kinetics of thymidine phosphorylation are more complex than can be accounted for by standard Michaelis-Menton treatment. Rather, the results suggest negative cooperativity of thymidine interaction, which has been observed by others studying purified and cloned TK2 (23,28). These data can be approximated with two straight lines with slopes corresponding to Km1 = 0.25 μM and Km2 = 2.6 μM. (Inset) The results are plotted as a Hill plot in which log [V/(Vmax −V)] = n log S − log K and is used to detect cooperativity in multisubunit enzyme systems. The negative cooperativity is further supported by a Hill plot slope of <1.
Fig. 6.
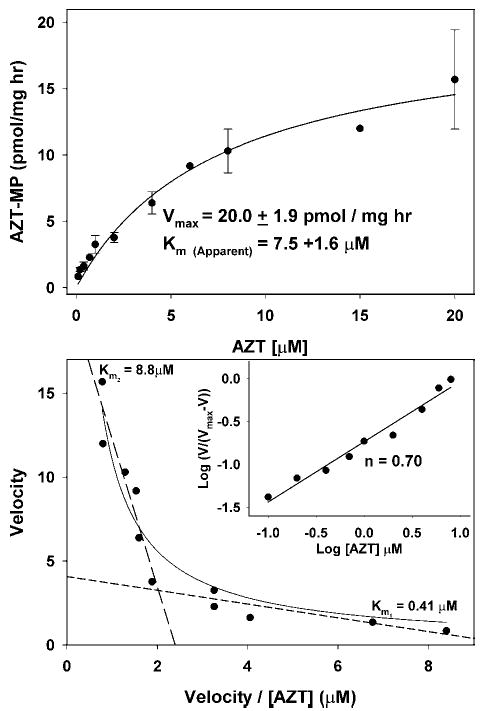
Kinetics of AZT phosphorylation in isolated heart mitochondria. Mitochondria were incubated as described in the Fig. 5 legend, except that [3H]-AZT was used instead of [3H]-thymidine and the concentration of AZT was varied between 0.2 and 20 μM (specific radioactivity 3500–11,000 DPM/pmol). (Top) The rate of AZT phosphorylation was determined by the amount of AZT-MP formed over time as calculated in Fig. 3. The results from three to four experiments are expressed in a standard Michaelis-Menton plot ± SEM and the data fitted to the Michaelis-Menton equation V = Vmax [S]/(Km + [S]), yielding an apparent Vmax of 20.0 ± 1.8 pmol/mg/h and an apparent Km of 7.5 ± 0.4 μM. (Bottom) The results for AZT phosphorylation were treated as described in the Fig. 5 legend for thymidine phosphorylation and indicate that the kinetics of AZT phosphorylation are quite similar to thymidine phosphorylation in Fig. 5, suggesting a negative cooperativity of AZT interaction, which has also been observed by others studying purified and cloned TK2 (23,28). These data can be approximated with two straight lines with slopes corresponding to Km1 = 0.41 μM, and Km2 = 8.8 μM. (Inset) The results are plotted as in Fig. 5 and the negative cooperativity is further supported by a Hill plot slope of <1.
Inhibition of [3H]-Thymidine Phosphorylation by AZT
Recognizing that the mitochondrial phosphorylation pathway is likely to represent the only mechanism of thymidine phosphorylation in myocytes, and recognizing that AZT and AZT-MP are substrates for these enzymes, it seemed likely that these analogs would inhibit thymidine phosphorylation. The effect of AZT on the conversion of [3H]-thymidine to [3H]-TTP in isolated heart mitochondria is shown in Fig. 7. The results clearly demonstrate that the presence of AZT potently inhibited the phosphorylation of thymidine at the TMP step with an IC50 of 7.0 ± 0.9 μM. Thymidine had no effect on the conversion of AZT to AZT-MP (data not shown). Earlier data demonstrated that AZT is slowly converted to AZT-MP, with AZT-MP levels rising with time of incubation (Fig. 3, right). Thus, inhibition of thymidine phosphorylation could be caused by AZT, AZT-MP, or both. However, if AZT-MP were solely responsible for inhibiting thymidine phosphorylation, then inhibition would be greater in the second hour than in the first hour. The observed results show that the percent inhibition in the first hour was about the same as that observed in the second hour of incubation (Fig. 7, top). Further, preincubation of mitochondria with AZT for 30 min to allow for the synthesis of AZT-MP prior to addition of [3H]-thymidine did not alter the observed amount of inhibition (data not shown). Thus, inhibition of thymidine phosphorylation correlated more closely with AZT than with AZT-MP, suggesting that AZT itself is the toxic compound.
Fig. 7.
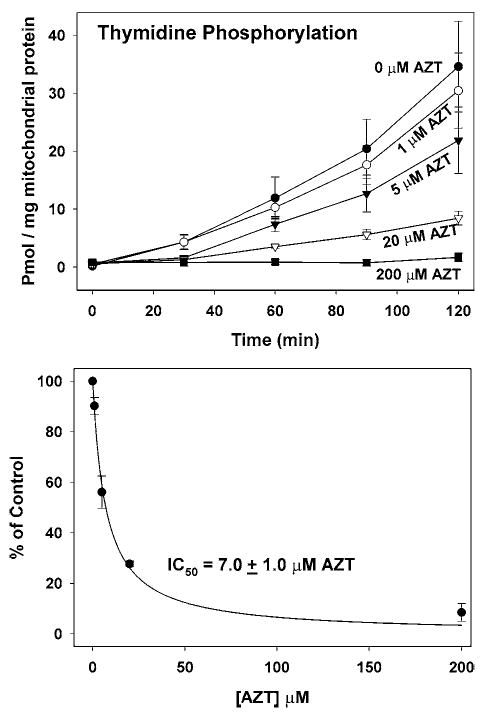
AZT inhibition of thymidine phosphorylation in isolated heart mitochondria. (Top) Mitochondria were incubated with [3H]-thymidine as described in the Fig. 3 legend under conditions in which AZT levels were varied from 0 to 200 μM. Results were calculated for the formation of TMP, TDP, and TTP as described in the Fig 3 legend. Results represent the mean and SEM of five to six experiments. (Bottom) The values obtained at the 90-min time-point were expressed as a percent of control in each experiment and are plotted here as the mean ± SEM as a function of AZT concentration, yielding an IC50 = 7.0 ± 1.0 μM.
Nonmitotic tissues such as the heart do not express a de novo synthesis pathway or cytosolic TK1. As a result, mitochondrial DNA replication would appear to depend solely on the mitochondrial pathway that is inhibited by AZT. Thus it is possible that mitochondrial DNA replication might be affected by limiting levels of TTP. This hypothesis is supported by recent work showing that individuals with inherited deficiencies in TK2 suffer from a severe myopathy associated with marked mitochondrial DNA depletion (24,29).
Discussion
We have demonstrated that isolated heart mitochondria are capable of transporting thymidine into the matrix and phosphorylating it to its TTP form. This implies that normal heart mitochondria express TK2, a TMPK (not characterized), and an NDPK. Although the synthesized TTP and probably TDP can be transported out of the matrix, the TMP intermediate does not appear to cross the inner membrane. This decreases the potential role of cytosolic TMPK in TTP phosphorylation and suggests that all TTP in the myocyte is manufactured by the mitochondrial pathway. Under these same conditions, isolated heart mitochondria synthesized only AZT-MP from AZT and did not allow the transport of AZT-MP out of the matrix. Because cytosolic TK1 is not expressed in myocytes, these data suggest that myocytes would be unable to synthesize AZT beyond the monophosphate. This has recently been confirmed in studies with [3H]-AZT in the perfused rat heart in which the presence of labeled AZT and AZT-MP only could be detected in the heart after 2 h of perfusion with [3H]-AZT (McKee, E.E., Bentley, A.T., and Susan-Resiga, D., personal observations). These results suggest that the toxicity of AZT in the normal adult rat heart is not mediated by AZT-TP inhibition of DNA polymerase γ as originally proposed (7–9). Other investigators have suggested that AZT-MP is more likely to be the toxic compound because the cellular levels of AZT-MP are typically much higher than the levels of AZT-TP (12). In a series of five cell lines, Sales et al. (11) reported that cytotoxicity of AZT correlated with AZT-MP levels but not AZT-TP levels. AZT-MP has also been reported to inhibit the exonuclease activity of DNA polymerase γ, with an IC50 of 2–3 mM (8), which is about 350 times higher than the IC50 reported for AZT inhibition of thymidine phosphorylation. Using the Vmax of 20 pmol/mg/h for AZT-MP synthesis from Fig. 6, the maximal level of AZT-MP expected in our 2-h experiment is 40 pmol/mg mitochondrial protein. If AZT-MP is entirely contained in the approx 1 μL/mg protein water space of the matrix, its concentration would be approx 0.04 mM, well below levels that would inhibit DNA polymerase γ. However, our data indicate that AZT-MP is not transported from the matrix and not further phosphorylated. Its only route of removal is via mitochondrial 5′-nucleotidase, in which the relative rate of AZT-MP dephosphorylation is reported to be only 2% of TMP (19). Thus, AZT-MP levels in the matrix are likely to rise substantially on long-term treatment with AZT and could reach levels consistent with inhibition of DNA polymerase γ, which could contribute to the toxic nature of AZT. Levels of AZT-MP in the millimolar range have been reported for mitotic cells (11–13). The major question that emerges from studies correlating AZT-MP levels with AZT cytotoxicity is the cellular compartmentalization of the AZT-MP. In the five cell lines studied by Sales et. al. (11), it was demonstrated that the different levels of AZT-MP in each cell line was a function of cytosolic TK1 activity rather than mitochondrial TK2. Thus, most AZT-MP in these cell lines is formed in the cytosol. The present study shows that in adult heart, AZT-MP is not transported across the inner membrane. Transport of AZT-MP was not addressed in the study of Sales et. al. (11) and it is unknown if mitotic cells that express TK1 also express a mitochondrial transporter that is not present in adult heart. If a carrier is not present, then the cytotoxicity of AZT-MP must be mediated from the outside of the inner membrane. Alternatively, as noted by Sales et. al. (11), it is possible that the correlation of AZT toxicity with AZT-MP levels in these mitotic cells lines is coincidental. Perhaps the cell line with the highest TK1 activity, and thus the highest level of AZT-MP, is more sensitive to AZT for other reasons. Additional work is required to differentiate among these possibilities.
As an alternative to the AZT-MP-mediated toxicity (discussed above) and the DNA polymerase γ hypothesis, we demonstrate in this paper that AZT itself is a potent inhibitor of thymidine phosphorylation in isolated heart mitochondria. Because the heart does not express TK1, and TMP is not transported across the inner membrane, mitochondrial synthesis of TTP is the only route available to the cell. We have recently confirmed this point by demonstrating that AZT also inhibits thymidine phosphorylation in the intact perfused rat heart (McKee, E.E., Bentley, A., and Susan-Resiga, D., personal observations) and is thus observed in the presence of all cellular enzymes. The importance of a balanced deoxynucleotide pool in mitochondrial replication has recently been strongly supported by three inherited enzyme deficiencies in the deoxynucleoside anabolic and catabolic pathways. Individuals with mutations in TK2 have a severe myopathy associated with marked mitochondrial DNA depletion (24,29). Mutations in the mitochondrial dGK are associated with marked hepatocerebral mitochondrial DNA depletion (30). Lastly, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is caused by loss-of-function mutations in the gene encoding thymidine phosphorylase (TP), leading to an overabundance of thymidine and mitochondrial DNA deletions as well as depletion (31).
The results obtained here are valid only for the normal adult rat heart. Other tissues may express different levels of phosphorylation enzymes and/or potentially different mitochondrial inner membrane transporters. These results would be most relevant to other nonmitotic tissues, because they are unlikely to express cytosolic TK1. It is also possible that in long-term AZT treatment, tissues such as the heart may respond to the toxicity by altering the expression of salvage enzymes and inducing expression of enzymes not found in the normal adult rat heart. This could substantially alter the results presented here and indicates that these experiments should be repeated on heart mitochondria from rats undergoing long-term treatment and displaying cardiotoxicity.
Many of the deoxynucleoside analogs used in HAART, including AZT, stavudine (d4T), lamivudine (ddC), and didanosine (ddI), are associated with mitochondrial DNA depletion and their triphosphates have been shown to varying degrees to inhibit DNA polymerase γ (8,9,10). Each of these compounds displays a significant tissue preference with regard to toxicity (1). In the pre-HAART period, when these compounds were used at higher concentrations by themselves, AZT was most often associated with cardiac and skeletal myopathies, didanosine with peripheral neuropathies, and stavudine with lipodystrophy (1,2). Because the mitochondrial DNA polymerase is the same in all cells, the tissue preference implies a tissue-specific pattern of phosphorylation and/or inner membrane transport such that the matrix levels of the triphosphates of each compound vary according to tissue. For example, didanosine might be expected to have the highest concentration of ddI triphosphate in peripheral neural tissue, whereas d4T might be expected to have the highest concentration of d4-TP in peripheral adipocytes. Alternatively, some of these compounds may act like AZT and inhibit the phosphorylation of their related endogenous deoxynucleosides in a tissue-specific pattern. Although some of the toxicities noted above have become relatively rare as a result of HAART, clarification of these points is crucial, because identifying the target of toxicity is critical in directing development of new drugs. Toward this goal, the present work brings a novel contribution to the understanding of the potential primary target of AZT toxicity.
Acknowledgments
This work was supported by grants from the American Heart Association, Midwest Affiliate, and from the National Institute of Health, HL 72710, to E.E.M.
References
- 1.Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nat Med. 1995;1:417–421. doi: 10.1038/nm0595-417. [DOI] [PubMed] [Google Scholar]
- 2.Benbrik E, Chariot P, Bonavaud S, Ammi-Satd M, Frisdal E, Rey C, et al. Cellular and mitochondrial toxicity of zidovudine (AZT), didanosine (ddI) and zalcitabine (ddC) on cultured human muscle cells. Neurol Sci. 1997;149:19–25. doi: 10.1016/s0022-510x(97)05376-8. [DOI] [PubMed] [Google Scholar]
- 3.Barile M, Valenti D, Quagliariello E, Passarella S. Mitochondrial as cell targets of AZT (Zidovudine) Gen Pharmacol. 1998;31:531–538. doi: 10.1016/s0306-3623(98)00041-x. [DOI] [PubMed] [Google Scholar]
- 4.Arnaudo E, Dalakas M, Shanske S, Moraes CT, Dimauro S, Schon EA. Depletion of muscle mitochondrial DNA in AIDS patients with zidovudine-induced myopathy. Lancet. 1991;337:508–510. doi: 10.1016/0140-6736(91)91294-5. [DOI] [PubMed] [Google Scholar]
- 5.Simpson MV, Chin CD, Keilbaugh SA, Lin TS, Prusof WH. Studies on the inhibition of mitochondrial DNA replication by 3′-azido-3′-deoxythymidine and other dideoxynucleoside analogs which inhibit HIV-1 replication. Biochem Pharmacol. 1989;38:1033–1036. doi: 10.1016/0006-2952(89)90245-1. [DOI] [PubMed] [Google Scholar]
- 6.Lewis W, Gonzalez B, Chomyn A, Papoian TJ. Zidovudine induces molecular, biochemical, and ultrastructural changes in rat skeletal muscle mitochondria. Clin Invest. 1992;89:1354–1360. doi: 10.1172/JCI115722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis W, Simpson JF, Meyer RR. Cardiac mitochondrial DNA polymerase-γ is inhibited competitively and noncompetitively by phosphorylated zidovudine. Circ Res. 1994;74:344–348. doi: 10.1161/01.res.74.2.344. [DOI] [PubMed] [Google Scholar]
- 8.Lim SE, Copeland WC. Differential incorporation and removal of antiviral deoxynucleosides by human DNA polymerase γ. J Biol Chem. 2001;276:23616–23623. doi: 10.1074/jbc.M101114200. [DOI] [PubMed] [Google Scholar]
- 9.Martin JL, Brown CE, Matthews-Davis N, Reardon JE. Effects of antiviral nucleoside analogs on human DNA polymerases and mitochondrial DNA synthesis. Antimicrob Agents Chemother. 1994;38:2743–2749. doi: 10.1128/aac.38.12.2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, et al. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem. 2001;276:40847–40857. doi: 10.1074/jbc.M106743200. [DOI] [PubMed] [Google Scholar]
- 11.Sales SD, Hoggard PG, Sunderland D, Khoo S, Hart CA, Back DJ. Zidovudine phosphorylation and mitochondrial toxicity in vitro. Toxicol Appl Pharmacol. 2001;177:54–58. doi: 10.1006/taap.2001.9288. [DOI] [PubMed] [Google Scholar]
- 12.Lavie A, Schlichting I, Vetter IR, Konrad M, Reinstein J, Goody RS. The bottleneck in AZT activation. Nat Med. 1997;3:922–924. doi: 10.1038/nm0897-922. [DOI] [PubMed] [Google Scholar]
- 13.Lavie A, Vetter IR, Konrad M, Goody RS, Reinstein J, Schlichting L. Structure of thymidylate kinase reveals the cause behind the limiting step in AZT activation. Nat Struct Biol. 1997;4:601–604. doi: 10.1038/nsb0897-601. [DOI] [PubMed] [Google Scholar]
- 14.Frick LW, Nelson DJ, St Clair MH, Furman PA, Krenitsky TA. Effects of 3′azido-3′deoxythymidine on the deoxynucleotide triphosphate pools of cultured human cells. Biochem Biophys Res Commun. 1988;154:124–129. doi: 10.1016/0006-291x(88)90659-6. [DOI] [PubMed] [Google Scholar]
- 15.Hobbs GA, Keilbaugh SA, Rief PM, Simpson MV. Cellular targets of 3′azido-3′-deoxythymidine: an early (non-delayed) effect on oxidative phosphorylation (1995) Biochem Pharmacol. 1995;50:381–390. doi: 10.1016/0006-2952(95)00141-l. [DOI] [PubMed] [Google Scholar]
- 16.Johansson M, Van Rompay AR, Degreve B, Balzarini J, Karlsson A. Cloning and characterization of the multisubstrate deoxyribonucleoside kinase of Drosophila melanogaster. J Biol Chem. 1999;274:23814–23819. doi: 10.1074/jbc.274.34.23814. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Karlsson A, Arner ESJ, Eriksson S. Substrate specificity of mitochondrial 2′deoxyguanosine kinase. J Biol Chem. 1993;268:22847–22852. [PubMed] [Google Scholar]
- 18.Wang L, Munch-Petersen B, Herrstrom Sjoberg A, Hellman U, Bergman T, Jornvall H, et al. Human thymidine kinase 2: molecular cloning and characterization of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS Lett. 1999;443:170–174. doi: 10.1016/s0014-5793(98)01711-6. [DOI] [PubMed] [Google Scholar]
- 19.Mazzon C, Rampazzo C, Chaira Scaini M, Gallinaro L, Karlsson A, Meir C, et al. Cytosolic and mitochondrial deoxyribonucleotidases: activity with substrate analogs, inhibitors and implications for therapy. Biochem Pharmacol. 2003;66:471–479. doi: 10.1016/s0006-2952(03)00290-9. [DOI] [PubMed] [Google Scholar]
- 20.Van Rompay AR, Johansson M, Karlsson A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol Ther. 2000;87:189–198. doi: 10.1016/s0163-7258(00)00048-6. [DOI] [PubMed] [Google Scholar]
- 21.Kowluru A, Tannous M, Chen HQ. Localization and characterization of the mitochondrial isoform of the nucleoside diphosphate kinase in the pancreatic β cell: evidence for its complexation with mitochondrial succinyl-CoA synthetase. Arch Biochem Biophys. 2002;398:160–169. doi: 10.1006/abbi.2001.2710. [DOI] [PubMed] [Google Scholar]
- 22.Dolce V, Fiermonte G, Runswick MJ, Palmieri F, Walker JE. The human mitochondrial deoxynucleotide carrier and tis role in the toxicity of nucleoside antivirals. Proc Natl Acad Sci USA. 2000;98:2284–2288. doi: 10.1073/pnas.031430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munch-Petersen B, Cloos L, Tyrsted G, Eriksson S. Diverging substrate specificity of pure human thymidine kinases 1 and 2 against antiviral dideoxynucleosides. J Biol Chem. 1991;266:9032–9038. [PubMed] [Google Scholar]
- 24.Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342–344. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- 25.McKee EE, Grier BL, Thompson GS, McCourt JD. Isolation and incubation conditions to study heart mitochondrial protein synthesis. Am J Physiol. 1990;258:E492–E502. doi: 10.1152/ajpendo.1990.258.3.E492. [DOI] [PubMed] [Google Scholar]
- 26.McKee EE, Bentley AT, Smith RM, Ciaccio CE. Origin of guanine nucleotides in isolated heart mitochondria. Biochem Biophys Res Commun. 1999;257:466–472. doi: 10.1006/bbrc.1999.0489. [DOI] [PubMed] [Google Scholar]
- 27.McKee EE, Bentley AT, Smith RM, Kraas JR, Ciaccio CE. Guanine nucleotide transport by atractyloside-sensitive and -insensitive carriers in isolated heart mitochondria. Am J Physiol. 2000;279:C1870–C1879. doi: 10.1152/ajpcell.2000.279.6.C1870. [DOI] [PubMed] [Google Scholar]
- 28.Wang I, Eriksson S. Cloning and characterization of full-length mouse thymidine kinase 2: the N-terminal sequence directs import of the precursor protein into mitochondria. Biochem J. 2000;351:469–476. [PMC free article] [PubMed] [Google Scholar]
- 29.Saada A, Ben-Shalom E, Zyslin R, Mandel H, Elpeleg O. Mitochondrial deoxyribonucleoside triphosphate pools in thymidine kinase 2 deficiency. Biochem Biophys Res Commun. 2003;310:963–966. doi: 10.1016/j.bbrc.2003.09.104. [DOI] [PubMed] [Google Scholar]
- 30.Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Anbinder Y, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 31.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]