

Abstract
Polycystin-1 (PC-1), the PKD1 gene product, is a large receptor whose expression in renal epithelial cells results in resistance to apoptosis and tubulogenesis, a model consistent with the phenotype observed in patients. This study links PC-1 expression to a signaling pathway that is known to be both antiapoptotic and important for normal tubulogenesis. This study found that PC-1 expression results in phosphorylation of Akt and downstream effectors and that phosphatidylinositol 3-kinase (PI3-K) inhibitors prevent this process. In addition, it is shown that dominant negative Akt can revert PC-1-induced protection from apoptosis. Furthermore, it was observed that increased PI3-K β activity in PC-1- expressing MDCK cells seems to be dependent on both tyrosine-kinase activity and heterotrimeric G proteins. It also was found that PC-1-induced tubulogenesis is inhibited by PI3-K inhibitors. Taken together, these data suggest that the PI3-K/Akt cascade may be a central modulator of PC-1 function and that its deregulation might be important in autosomal dominant polycystic kidney disease.
Autosomal dominant polycystic kidney disease (AD- PKD) is one of the most common genetic disorders, affecting 1/1000 (1). The main clinical manifestation is bilateral renal cystic disease, which results in ESRD by age 50 in half of affected individuals. Extrarenal manifestations of the disease include cysts of the biliary tract and pancreas (2) and an assortment of cardiovascular abnormalities (e.g., hypertension, intracranial aneurysms, cardiac valvular defects) (1).
Two genes have been shown definitively to result in ADPKD when mutated: PKD1, which accounts for 85%, and PKD2, which is responsible for the rest (3-6). The 14-kb PKD1 mRNA encodes a 4302-amino acid (aa; 520 kD) protein (polycystin-1 [PC-1]) that is a highly glycosylated plasma membrane receptor (7) with a large (>3000 aa) extracellular N-terminal domain, 11 transmembrane domains (8), and a short intracellular C-terminus of 198 aa (9). The extracellular portion has two leucine-rich repeats, a C-type lectin domain, 16 PKD (IgG-like) repeats, an REJ (receptor for egg jelly) domain, and a proteolytic GPS domain (G protein-coupled receptor [GPCR] proteolytic site) (10) that we recently showed is functionally active (11). The short intracellular C-terminus of PC-1 was reported to interact with the Gαi/Gα0 subunit of heterotrimeric G proteins, suggesting that PC-1 itself might be a GPCR (12-14). PC-1 has also been reported to modulate the activity of protein kinase C and induce activator protein 1 (AP-1), to activate the Wnt signaling pathway, and to modulate mitogen-activates protein kinase activity (15-17). Furthermore, in vitro kinase assays that were performed on the short C-terminus suggest that this portion can be phosphorylated, further suggesting a role for this receptor in cell signaling (18,19). The ligands for PC-1 still remain elusive, although recent work suggests that PC-1 might be able to homodimerize through its PKD (IgG-like) repeats (20). A number of groups have localized the protein to cell-cell junctions and postulated that self-association might be sufficient for its activation (7,20,21).
We previously reported that expression of full-length polycystin-1 in MDCK cells results in reduced growth rates, resistance to apoptosis, and spontaneous tubulogenesis when cells are grown in collagen gels (22). We subsequently found that the Janus activated kinase (JAK)/STAT signaling pathway mediates the growth-suppressive effects in this system (23). In this work, we show that PC-1 expression also induces activation of the phosphatidylinositol 3-kinase (PI3-K) signaling pathway, a system that was implicated previously in regulating these other properties.
Materials and Methods
Antibodies and Inhibitors
Anti-PC-1 antibodies were described previously (7). Anti-P-Ser-Akt, P-Thr-Akt, Akt, P-FKHR, FKHR, P-PTEN, and PTEN were from Cell Signaling Technologies (Danvers, MA). The anti-p110α, anti-p110β, anti-p110γ, and anti-phosphotyrosine (PY20) antibodies were from Santa Cruz Biotechnology (cat. nos. sc-7174, sc-602, sc-7177, and sc-508, respectively; Santa Cruz, CA). Two independent anti-pan-p85 polyclonal antibodies were from Upstate Biotechnology (cat. nos. 06-195 and 06-496; Charlottesville, VA). High-affinity anti hemagglutinin (HA) antibody was from Roche (1867423; Monza, Italy).
LY294002 (LY) was purchased from Cell Signaling Technology. For studies using the LY inhibitor, the cells were initially grown in 10% FCS/DMEM, then switched to DMEM with 0.5% FCS with LY (FCS-/LY+) for 3 to 5 h to reduce background. Medium then was replaced with either FCS-/LY- or FCS-/LY+.
Wortmannin was from Sigma (cat. no. W1628; St. Louis, MO); genistein, herbimycin, and pertussis toxin (PTX) were from Calbiochem (cat. nos. 345834 and 375670, respectively; San Diego, CA); and rapamycin was from Cell Signaling Technologies. All inhibitors were prepared as directed and used at the indicated final concentrations.
Transient and Stable Transfections, Immunoprecipitations, and Immunoblots
Full-length and truncated forms of human PKD1 were described previously (7,11). HepG2 and MDCK cells were transiently transfected using Lipofectamine 2000 (Invitrogen, San Giuliano Milanese, Italy). MDCK cells were cotransfected with green fluorescence protein and sorted using a flow cytometer before analysis. HepG2 stable transfectants were generated as described previously (22) with the pCI-β- PKD1-Flag vector previously described (7) and using Zeocin as a selectable marker. Screening for positive clones was carried out by reverse transcription-PCR using the following primers: Forward gdst5 5’-CTGCTCACCCAGTTTGAC-3’, reverse gdst3 5’-CGTCGTCCTTG- TAGTCAG-3’. For immunoprecipitation (IP) studies, cells were lysed (250 mM sucrose, 20 mM imidazole, and 1 mM EDTA [pH 7.4], and 0.5% Triton-X 100) supplemented with Protease Inhibitors Cocktail (Amersham, Cologno Monzese, Italy) and phosphatase inhibitors (1 mM final of glycerophosphate, sodium orthovanadate, and sodium fluoride). A total of 1 μg/μl antibody was added to the supernatants, and the mix was rocked at 4°C for 2 h. Prewashed G-Sepharose beads were added, and the samples were rocked for an additional 2 h, centrifuged, and washed extensively. Laemmli buffer at 1× final was added for SDS-PAGE and immunoblot studies.
PI3-K Assays
After IP, samples were washed three times in lithium buffer (LiB; 0.5 M LiCl and 100 mM Tris-HCl [pH 7.4]), three times in EDTA buffer (EB; 1 mM EDTA, 100 mM NaCl, and 10 mM Tris-HCl [pH 7.4]), and three times in kinase buffer (KB; 40 mM Tris-HCl [pH 7.6], 150 mM NaCl, and 20 mM MgCl). Beads then were resuspended in KB + 1 mM dithiothreitol in the presence or absence of Wortmannin at a final concentration of 25 nM. l-α-Phosphatidylinositol was used as substrate (Sigma; cat. no. 8443) and resuspended in HEPES (pH 7.6), 1 mM EDTA, and 0.5% deoxycholate at a concentration of 1 μg/μl. A total of 2 μg of lipids was added to each reaction along with 20 mM cold ATP and 20 μCi of γ32ATP with high specific activity (2000 Ci/mmol; Amersham cat. no. AA0018). The reaction was carried out in a final volume of 100μl, at room temperature for 10 min, then stopped using 25 μl of 4 N HCl. The lipids were extracted using 200 μl of chloroform/methanol (1:1) and washed with 100 μl of chloroform:1 N HCl (1:1). The organic phase then was dried, resuspended in chloroform, and spotted on a silicagel-60 plate. The samples were resolved in a thin-layer chromatography chamber using chloroform:methanol:water:hydroxylamine (9:7:2:1). The plate was dried, and then the signal was captured using a PhosphorImager SI (Molecular Dynamics, Eugene, OR). ImageQuant was used for quantification of the reactions.
Apoptosis Assays
Cells were transfected using Lipofectamine 2000 (Invitrogen). The vectors that encode for the different forms of Akt were previously described (24,25).
Cells were transfected, cultured in complete medium for 24 h, and then treated overnight with recombinant hTNF-α/TNFSF1A (210-TA) 25 nM and Cycloheximide 35 μM or using ultraviolet (UV) light at 30 or 60 J/cm2. Cells then were fixed, permeabilized with 0.5% Triton X-100, blocked, and incubated with primary followed by secondary antibodies. Cells then were processed for transferase-mediated dUTP nick-end labeling (TUNEL) using the Apoptosis Detection System (Promega, Madison, WI) following the manufacturer's instructions. Hoechst 33342 (bisbenzimide) was purchased from Sigma (bb2261) and used at a final concentration of 1 μg/ml. All samples were mounted using the Prolong Antifade Kit (Invitrogen) and observed with an Axiophot, Zeiss microscope. Statistical analysis was performed using unpaired t test.
Tubulogenesis Assays
For the tubulogenesis assays, cells were grown to confluence, trypsinized, and then cultured in a collagen type I mixture as described previously (22). Where specified, cultures were treated with 15 μM of LY294002 or 25 nM Wortmannin, replaced daily. Quantitative assays were performed as described previously (22).
Results
PC-1 Induces Activation of Akt
We previously reported that stable expression of PC-1 in MDCK type II cells (MDCKPKD1Zeo) protects them from apoptosis that is induced by serum starvation (22). We also observed resistance to apoptosis induced by UV light or TNF-α but not from anoikis (Supplementary Figures 1, 3, and 4). Because numerous previous studies have shown that Akt can mediate these effects in other cell culture systems (24,26), we queried whether the same pathway might be involved in our system. Akt is activated by phosphorylation on two residues (Ser473 and Thr308) by two distinct kinases: PDK1, which phosphorylates at Thr308 (27), and the recently identified rapamycin-insensitive complex mammalian target of rapamycin (mTOR)/Rictor (28), which phosphorylates Ser473. Both phosphorylation events are necessary to achieve complete activation and can be detected using epitope-specific antibodies (24,26). Therefore, we tested for the presence of phosphorylated Akt in cell lysates that were prepared from three independent MDCK control cell lines and three PKD1+ MDCK cell lines (MDCKPKD1Zeo) after a short serum starvation pulse to reduce basal levels of serum-induced phosphorylation. As shown in Figure 1a, increased levels of phospo-Akt (both Ser 473 and Thr 308) were present in each of the three MDCKPKD1Zeo cell lines compared with the controls (Figure 1a). Stripping and reprobing the same membrane with an anti-total Akt antibody revealed that almost an equal level of protein was present in each sample, excluding the possibility that differences in loading or Akt expression could account for such an effect (Figure 1a). We then tested whether PC-1 was able to activate Akt in MDCK cells without the possible confounding effect of clonal selection. We expressed by transient transfection wild-type (wt) as well as a series of truncation mutants of PC-1 (Figure 1, b and c) in MDCK cells and found that wt but not mutant PC-1 was able to induce phosphorylation of Akt (Figure 1c). In agreement with Akt's being active in MDCKPKD1Zeo cells, we found increased phosphorylation levels of the Forkhead transcription factor FKHR-1, a downstream target of activated Akt, in each of the three MDCKPKD1Zeo cell lines (Figure 1d, top). Consistent with this, FKHR translocation into the nucleus upon serum starvation is greatly reduced in MDCKPKD1Zeo as compared with negative controls (Figure 1d, bottom) (25). We then tested whether PC-1 expression results in activation of Akt in other cell types. We expressed by transient transfection full-length normal and mutant PKD1 cDNA (Figure 1, b and e) in the hepatic cell line, HepG2, and found that, again, only the wt gene resulted in phosphorylation of Akt (Figure 1f).
Figure 1.

Polycystin-1 (PC-1) expression results in activation of Akt. (a) Immunoblot (IB) analysis of PC-1 expression in immunoprecipitation (IP) products of three MDCKPKD1Zeo (C8/68 [68], G7/36 [36], and G3) and control cell lines (MDCKtTA [Mt], F6, and F2). An anti-PC-1 C-term (α-CT) antibody was used for both IP and IB (7). Open arrows indicate PC-1 (top panel). (Bottom) Cell lysates were tested for Akt activation with either anti-phospho-Ser473-Akt or anti-Thr-308 antibodies. Membranes were stripped and reprobed for total Akt to control for loading. (b) Several truncation constructs (top) described previously (11) were expressed by transient transfection in HepG2 cells. Wild-type (WT): pCI-β-PKD1Flag (7); C-terminal cleavage products (CTF): AF66; REJ-CTF: HBF160; PK-REJ-CTF: BF110 (11). (c) MDCK cells were transiently co-transfected with the constructs shown in b along with green fluorescence protein, sorted using a flow cytometer, and assayed using an anti-phospho-Ser473-Akt antibody. The same membrane then was stripped and reprobed using an anti-Akt antibody. (d) Total lysates from two control cell lines (F6 and F2) and three PC-1-expressing cell lines (C8/68, G7/36, and G3) cultured as in a were immunoblotted using an anti-phospho- Ser256-FKHR antibody. The same gel was stripped and reprobed with an anti-FKHR antibody to control for loading. (Bottom) Immunofluorescence staining performed using an anti-FKHR antibody after 24 h of serum starvation revealed relocation of FKHR in the nucleus in F6 as compared to C8/68. (e) Immunoblot (IB) analysis of PC-1 expression in HepG2 cells transfected with the various PKD1 constructs presented in b. M2 beads were used to purify the FLAG-tagged PC-1 variants from an equal quantity of total cell lysate, and the affinity-purified products were analyzed using α-CT in a 5% polyacrylamide gel. The black arrow identifies the CTF that result from cleavage of WT and mutant PC-1 at the GPS site (11). REJ-CTF (*uncleaved form) is cleaved at a markedly reduced rate, explaining the relatively small amount of CTF present in the corresponding lane (11). In contrast, IgG-REJ-CTF (°uncleaved form) and WT are cleaved in a higher proportion. To better visualize the full-length PC-1 (open arrow), two lanes were loaded with samples from IgG-REJ-PKD1 (°right blot) and wt-PKD1 (open arrow, right blot) in a 4% polyacrylamide gel and analyzed using α-CT. (f) HepG2 cells were transiently transfected, serum starved, and total lysates were assayed using an anti-phospho-Ser473-Akt antibody. The same membrane then was stripped and reprobed using an anti-Akt antibody.
Akt Is Necessary for PC-1-Mediated Resistance to Apoptosis
Expression of a constitutively active form of Akt in MDCK cells is sufficient to induce resistance to apoptosis (29). To test whether Akt is mediating the antiapoptotic effects of PC-1 in our MDCK cell culture system, we transiently transfected HA-tagged forms of a kinase-dead Akt (DN-Akt, K179M) and HA-tagged wt Akt (WT-Akt) (25) into MDCK controls (MDCKZeo) and in MDCKPKD1Zeo. Cells then were treated with TNF-α, and the apoptotic rates of mock, WT-Akt, and DN-Akt transfected cells were evaluated using a TUNEL assay as described previously (22). As shown in Figure 2a, higher apoptotic rates were observed in MDCK control cells (F6 mock, 19.66 ± 4.00%) as compared with MDCKPKD1Zeo (C8/68 mock, 4.33 ± 2.88%), confirming that these cells are resistant to apoptosis (P = 0.0059). Expression of WT-Akt in C8/68 did not impair the capability of these cells to survive in the presence of an apoptotic stimulus (Figure 2, a and c), whereas transfection with DN-Akt dramatically increased the apoptotic rates in these cells (Figure 2, a and c) despite that equal expression levels for both forms are achieved as revealed by Western blot analysis (Figure 2b). Quantification of the apoptotic rates (Figure 2c) confirmed that significantly higher rates of apoptosis were observed in C8/68 when transfected with DN-Akt (14.66 ± 1.15%) as compared with mock-transfected (4.33 ± 2.88%; P = 0.0045) or WT-transfected (4.66 ± 4.16%; P = 0.016) cells. The results are representative of three different experiments performed in triplicate. Similar results were observed using two different clones F2 and G7/36 (data not shown). In a similar way, we found that higher apoptotic rates are observed in HepG2Zeo (E19) as compared with HepG2PKD1Zeo (A15) stable transfectants when apoptosis was induced by either TNF or UV light (Supplementary Figure 4b). Quantification of the apoptotic rates (Figure 2d) confirmed that significantly higher rates of apoptosis were observed in E19 as compared with A15 when apoptosis was induced using UV light (20.3 ± 3.9 versus 5.2 ± 1.5%; P = 0.0001; Figure 2d). Furthermore, a significant increase in apoptotic rates was observed in A15 when transfected with DN- Akt (35.6 ± 7.8%) as compared with mock-transfected (5.2 ± 1.5%; P < 0.0001) or WT-transfected (7.1 ± 2.00%; P < 0.0001) cells. The results shown are representative of three different experiments performed in triplicate. We thus conclude from these data that active Akt is a necessary component for PC-1- mediated resistance to apoptosis.
Figure 2.

Akt is necessary for PC-1-mediated resistance to apoptosis. (a) MDCK control (F6) and MDCKPKD1Zeo (C8/68) were transiently transfected using a mock, a WT (WT-Akt), or a dominant negative (DN_Akt) Akt construct. Apoptosis was induced using TNF-α (see Materials and Methods), and cells were stained with an anti-hemagglutin (HA) antibody (in red) to identify transfected cells, with a transferase-mediated dUTP nick-end labeling (TUNEL) assay (in green) to visualize apoptotic cells and counterstained with Hoechst 33342 (blue nuclei) to visualize all of the cells that were present in the field. Very few apoptotic cells are visible in mock-transfected C8/68 cells, whereas a considerable number are visible in F6 cells that are treated under the same conditions. Similar results were observed when the WT-Akt construct was transfected in these two cell lines, whereas higher rates of HA-positive/TUNEL-positive cells are visible in C8/68 when DN-Akt is transfected in these cells. (b) Same experimental design as in a except that cell lysates were prepared from transiently transfected F6 and C8/68 cells and analyzed by Western blot using a mixture of anti-HA and anti-actin antibodies as a loading control. Equal expression levels were achieved with all constructs in all cell lines. (c) Quantification of the results visualized in a. In mock-transfected cells, the percentage of apoptosis is calculated as the number of TUNEL-positive cells over the number of total cells present in the field. In WT-and DN-Akt-transfected cells, the percentage of apoptosis is expressed as the number of TUNEL-positive cells over the number of HA-positive cells. The ANOVA test was used to perform statistical analysis. The results for each pairwise comparison reached statistical significance (see text for details). (d) Same experiment as in a through c performed in HepG2 cells. Both the percentage of apoptosis and the statistical analysis were performed as in c.
PC-1-Mediated Activation of Akt Requires Active PI3-K
We next sought to determine the mechanism by which PC-1 results in activation of Akt. Given that Akt is a well-established target of the PI3-K pathway, we repeated the serum starvation experiment using the three MDCK control and MDCKPKD1Zeo cell lines in the presence or absence of the highly specific PI3-K inhibitor LY294002 (15 μM). We found that this inhibitor was able to prevent Akt phosphorylation in both MDCK (Figure 3a) and HepG2 (Figure 3b) cells that express full-length PC-1. Because LY294002 was reported to inhibit also the kinase activity of the Ser/Thr kinase mTOR, we treated the cells in the presence of another PI3-K inhibitor (Wortmannin, 12 nM) and of a potent mTOR inhibitor (rapamycin, 25 nM). We found that the former but not the latter was able to prevent Akt phosphorylation in our system (data not shown), demonstrating that PI3-K is a necessary component in this pathway.
Figure 3.
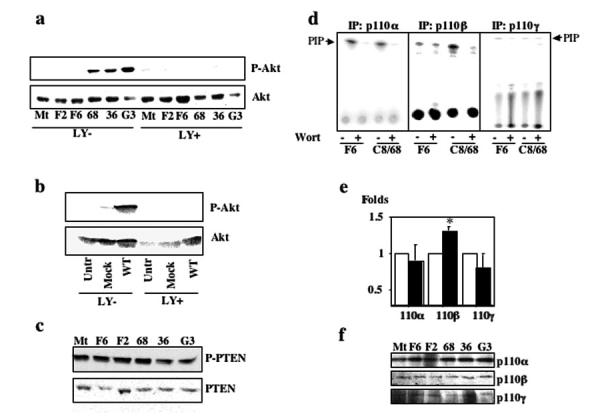
PC-1 associated activation of Akt is phosphatidylinositol 3-kinase (PI3-K) dependent, and PI3-Kβ has increased activity in MDCKPKD1Zeo cell lines. (a) Lysates from MDCK control (1, MDCKtTA; 2, F6; 3, F2) and MDCKPKD1Zeo cell lines (4, C8/68; 5, G7/36; 6, G3), treated with serum starvation in the presence (LY+) or absence (LY-) of LY29400 (15 μM final), were immunoblotted using an antibody specific for phospho-Ser473-Akt (top). The same membrane was stripped and reprobed using an anti-Akt antibody (bottom). (b) Same experimental design as in a except that the lysates were prepared from HepG2 cells that were transfected with PKD1 (WT) and transfected with vector (Mock) and from untransfected controls. (c) The same MDCK control and MDCKPKD1Zeo cell lines described in a were serum starved for 24 h, and then cell lysates were assayed using an anti-phospho-Ser380-PTEN antibody and an anti-PTEN antibody. (d) Analysis of PI3-K activity. MDCK control F6 and MDCKPKD1Zeo C8/68 cell lines were serum-deprived for 36 h and then lysed. Each of the class I p110 subunits was immunoprecipitated with a suitable antibody, and then its PI3-K activity was measured in either the absence (-) or the presence (+) of Wortmannin (25 nM final) as described in Materials and Methods. Only the p110β IP product had increased activity. Similar results were obtained using the other negative control MDCK (Mt and F2) and MDCKPKD1Zeo (G3 and G7/36) cell lines (data not shown). (e) Quantification of in vitro PI3-K assays performed using lysates of all three negative control MDCK (□) and three MDCKPKD1Zeo cell lines (■). The level of activity observed in controls was set at a value of 1, whereas the activity that was observed in MDCKPKD1Zeo cells was expressed as a ratio over that of the controls. The assay for p110β activity was performed in six independent experiments, whereas that for p110α and p110γ was repeated three times. Results are expressed as the average ± SEM. *Statistically significant differences (P = 0.0019; t test unpaired). (f) Expression level of PI3-K p110 subunits in the same MDCK control (Mt, F6, and F2) and MDCKPKD1Zeo (C8/68, G7/36, G3) cell lines used in b. The products were immunoprecipitated and subjected to immunoblot analysis using the same subunit-specific antibody.
The apparent increase in PI3-K/Akt activity observed in association with PKD1 expression could result from either decreased activity of phosphatases that are known to degrade phosphatidylinositol-3,4,5-triphosphate (PIP3), an effector molecule that is involved in Akt activation, or to enhanced activation of PI3-K. PTEN is the primary phosphatase that is known to dephosphorylate the critical D3 position of PIP3, so we examined how PKD1 expression altered PTEN’s properties (30). We found no differences in the level of either total PTEN or its phosphorylated form in MDCKPKD1Zeo cell lines as compared with controls (Figure 3c). These data suggest that Akt activation that was observed with PC-1 expression results from enhanced PI3-K activity.
PC-1 Induces Activation of PI3-Kβ (p85/p110β)
Three classes of PI3-K have been described. Class I PI3-K are major producers of PIP3 and thus primarily implicated in Akt activation. Members of this class of enzymes are heterodimers that are composed of a regulatory subunit and a catalytic subunit that are constitutively associated and inhibited by low doses of inhibitors such as Wortmannin and LY294002 (31,32). The larger subclass (class IA) is formed by various combinations of distinct regulatory (p85α, β and p55α, γ) and catalytic (p110α, β, δ) subunits. Class IB PI3-K has only a single member that is composed of a 110-kD catalytic subunit (p110γ) and a 101-kD regulatory subunit (p101). Most class IA PI3-K are activated by binding to tyrosine phosphorylated receptors (usually tyrosine kinase receptors) (31,32). Exceptions include p110β, which can also be activated by GPCR, and p110α, which can be activated by cytokine receptors via Janus activated kinase 2 (JAK2) (31-34). Class IB PI3-K is activated exclusively by GPCR, via direct binding of the Gβγ subunits (31,32).
We tested for class I kinase activity in lysates of PC-1- expressing versus control cell lines. We immunoprecipitated the p110α, β, γ catalytic subunits from the MDCK control and MDCKPKD1Zeo cell lines and performed in vitro kinase assays. As shown in Figure 3d, only the p110β IP products of cells that expressed PC-1 catalyzed an increased quantity of 32P-labeled PIP versus controls. Quantification of several independent experiments revealed a low but consistent increase in activity associated with p110β immunoprecipitates (1.29 ± 0.07; n = 6) from MDCKPKD1Zeo versus controls but not with either p110α or p110γ (Figure 3e). These data suggest that p110β catalytic activity is increased by PC-1 expression. Consistent with this interpretation, neither the total level of p110β present in lysates as measured by immunoblot nor the amount of IP product used for the kinase reactions differed between the two cell lines (Figure 3f).
Activation of PI3-K by PC-1 Requires Tyrosine-Kinase Activity and Heterotrimeric G Proteins
Class I PI3-K are usually activated by recruitment of the regulatory subunit p85 to tyrosine-phosphorylated receptors or adaptors through its SH2 domain (31,32). We tested for this property by comparing the in vitro kinase activity of anti-phospho-tyrosine immunoprecipitates from MDCKPKD1Zeo versus control lysates. Surprisingly, no differences were observed (Figure 4a). In control studies, we found that both sets of cell lines responded normally to treatment with hepatocyte growth factor (HGF) and EGF, with markedly increased kinase activity in anti-phosphotyrosine immunoprecipitates (Figure 4, a and b). These results exclude the presence of a general inhibitory factor in PKD1-expressing cells or a technical problem as the likely explanations for our findings.
Figure 4.
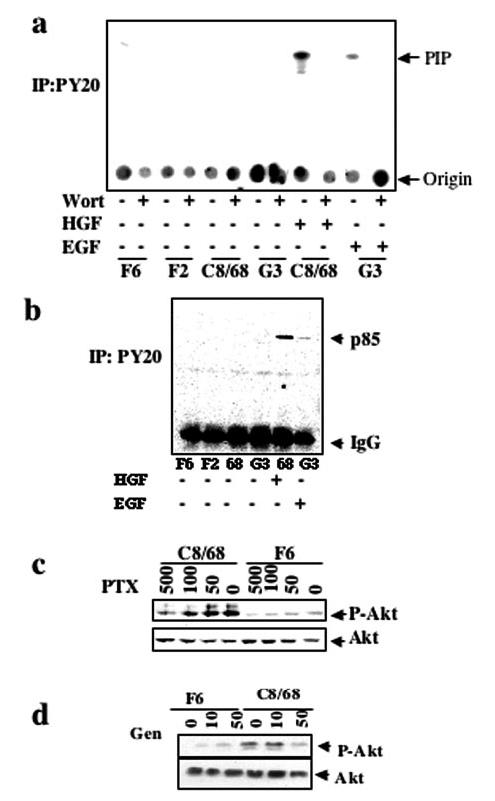
Activated PI3-Kβ is not associated with phospho-tyrosine and is inhibited by pertussis toxin (PTX). (a) Analysis of PI3-K activity of anti-phospho-tyrosine (PY20) immunoprecipitates from MDCK control or MDCKPKD1Zeo cells either in the absence (-) or the presence (+) of Wortmannin (25 nM final) using the same protocol as in Figure 3. Some cultures were treated with hepatocyte growth factor (HGF) or EGF for 5 min before cell harvesting, as indicated, as positive controls. PIP identifies the 32P-labeled phosphatidylinositols. (b). An aliquot of each of the immunoprecipitates used in a was subjected to immunoblot analysis using an anti-p85 antibody (see Materials and Methods). Increased levels of p85 were immunoprecipitated by anti-phosphotyrosine antibodies upon treatment with HGF or EGF but not in untreated cells. (c) The control cell line F6 and the PC-1-expressing clone C8/68 were serum starved for 24 h in the presence or absence of 0, 50, 100, or 500 ng/ml PTX. Cell lysates were immunoblotted using the anti-phospho-Ser473-Akt antibody. Membranes were stripped and reprobed using an anti-Akt antibody. (d) Experimental design similar to that used in c except that genistein was used at the doses (mm) shown.
Previous studies suggested that p110β can be activated by both tyrosine kinase-and heterotrimeric G-protein-dependent pathways (33,34). We therefore tested whether heterotrimeric G proteins could be involved in this process using PTX, a highly specific inhibitor of heterotrimeric G proteins of the Gi subfamily. Indeed, we observed a dose-dependent inhibition of phospho-Akt in response to PTX (Figure 4c) in the C8/68 subclone and minimal basal inhibition in the control F6. These data suggest that the heterotrimeric G proteins play a role in activation of PI3-Kβ that results from PC-1 expression. Given that Kurosu et al. (33) previously reported that p110β activation by Gβγ was markedly enhanced by addition of a phosphotyrosyl peptide, we queried whether the process that we observed was completely independent of tyrosine phosphorylation. We addressed this question by examining the effect of generic inhibitors of tyrosine phosphorylation on Akt activation by PC-1. As shown in Figure 4d, we found that both genistein and herbimycin (data not shown) greatly reduced the level of Akt activation that was induced by PC-1. Similar results were obtained for the other PKD1+ cell lines (data not shown). These results suggest that a dual mechanism of activation is occurring in our system, although at too low a level to be detected by kinase assays of anti-phospho-tyrosine immunoprecipitates.
PI3-K Mediates PC-1-Induced Morphogenesis
PI3-K also has been implicated in the motogenic and tubulogenic properties in the HGF/SF-MDCK model (35); we there-fore tested whether LY294002 or Wortmannin could prevent the morphogenic properties of the MDCKPKD1Zeo cells. As shown in Figure 5, both LY294002 and Wortmannin prevented PC-1-induced tube initiation in a three-dimensional collagen gel assay, strongly suggesting that PI3-K plays a central role in polycystin-1-mediated morphogenesis. Removal of the inhibitor at different time points resulted in further growth of the structures, indicating that treatment with the inhibitors for such short periods did not result in cell death (data not shown).
Figure 5.
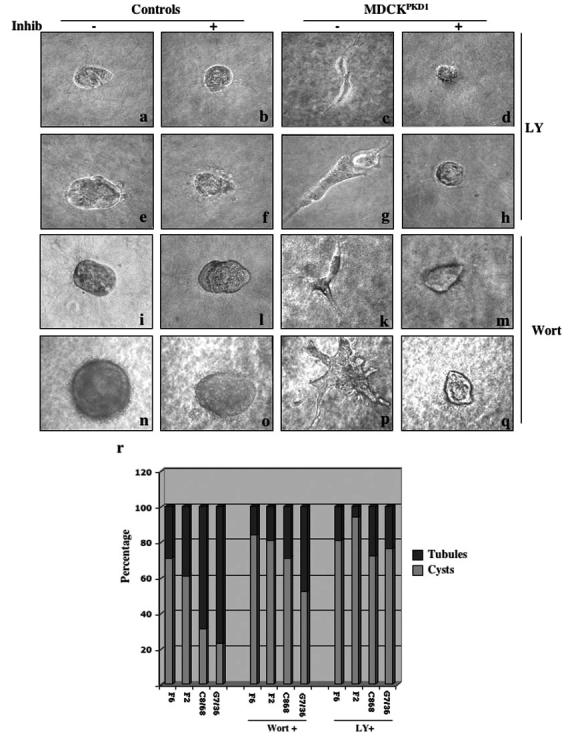
PI3-K inhibition prevents PC-1-induced tubulogenesis. (a through q) PC-1-induced tubulogenesis is inhibited by PI3-K inhibition. MDCK and MDCKPKD1Zeo cell lines were grown suspended in a collagen gel as described previously (22) either in the absence (-) or the presence (+) of 15 μM LY294002 or 25 nM Wortmannin for 2 d (a through d and I through m), 4 d (e through h), or 8 d (n through q). Representative examples of the control cell lines (a, b, e, f, i, l, n, and o) and MDCKPKD1Zeo cell lines (c, d, g, h, k, m, p, and q) are shown. Photographs were taken at ×40 magnification; a through m were magnified further ×4. (r) Quantification of the experiments described in a was performed as described previously (22). The structures observed in three to five wells for each clone were counted. Treatment in the presence of 15 μM LY294002 or 25 nM Wortmannin dramatically reduced, although not completely suppressed, the total number of tubules observed in MDCKPKD1Zeo cultures.
Discussion
Increased proliferation has been reported in cystic kidneys of individuals with ADPKD (36,37). The disease, however, progresses very slowly throughout an affected individual's lifetime, a characteristic that is difficult to explain if increased proliferation is the sole or primary defect (1). Increased rates of apoptosis also have been reported in human cystic kidneys, and it therefore has been proposed that the balance between cell proliferation and death is perturbed in the epithelia lining the cysts (37). We previously showed that expression of PC-1 in renal epithelial cells results in reduced growth rates, resistance to apoptosis, and conversion of a cystic phenotype to one of the branching tubules (22). We then showed that activation of the JAK-STAT signaling system by PC-1/PC-2 is responsible for the growth effects seen in this model (23). In this work, we link PC-1 to a signaling pathway that is known to be both antiapoptotic and protubulogenic (24,31,32,35). We have shown that PC-1 expression results in activation of PI3-Kβ (p85/p110β), Akt, and effector molecules such as FKHR and that these are inhibited by very low doses of highly specific PI3-K inhibitors. Importantly, in this study, we show that activation of Akt is necessary for PC-1-induced resistance to apoptosis in vitro, because MDCKPKD1Zeo as well as HepG2PKD1Zeo that express a dominant negative form of this kinase are no longer resistant to apoptosis.
We note that our results are consistent with recent findings of Yamaguchi et al. (17). These authors and others had previously reported that cAMP had a mitogenic effect on epithelial cells that were derived from ADPKD cysts but inhibited the proliferation of normal renal epithelial cells (38,39). In seeking to determine the mechanism underlying this phenomenon, they found that they could reproduce the phenotype in normal cells by altering intracellular calcium levels and by inhibiting the PI3-K/Akt pathway, which in turn resulted in activation of extracellular signal regulated kinase (ERK). They also found that disruption of endogenous PC-1 function by expression of its short C-terminus in the M1 renal collecting duct cell line resulted in a similar upregulation of the ERK pathway and a mitogenic response to cAMP. The authors suggested a model whereby loss of PC-1 function may result in changes in intracellular calcium, downregulation of PI3-K-Akt activity, and activation of ERK in ADPKD cysts.
It is interesting that a recent report by Ma et al. (40) showed that polycystin-2 (PC-2) channel activity can be enhanced by EGF in a process that requires activity of PI3-K. One might hypothesize that PC-1-induced PI3-K activity might be relevant to regulate the PKD2 channel as well, although direct studies would be needed to test this hypothesis.
Several unusual aspects of our findings require comment. The first surprising observation is that we did not find PI3-K activity associated with the tyrosine phosphorylated pool of proteins. This is in contrast to what is reported for most receptors that induce activation of class IA PI3-K. In most known examples, the SH2 domains of the p85 regulatory subunit are recruited to a phosphorylated tyrosine lying within a signature sequence YXXM, on the receptor itself, or on adaptor molecules. Therefore, PI3-K activity is typically associated with phosphotyrosine pulldowns. It should be noted that there is a precedent for having phosphotyrosine-independent activation of PI3-K when p110β is activated in response to GPCR (41). Because PC-1 has been reported to associate and activate a subset of heterotrimeric G proteins, we speculate that a similar mechanism might be responsible for the results that we observed (12-14). In contrast to our study, however, Murga et al. (41) reported that pretreatment with genistein failed to block Akt activation by GPCR agonists in their cell culture system. Our tyrosine kinase inhibitor studies suggest that activation of Akt may result from synergistic cooperation between two path-ways, consistent with kinetic studies performed in vitro on p110β (33,34).
How do we reconcile these findings with our inability to show PI3-K activity associated with anti-phosphotyrosine immunoprecipitates? We suggest that it is most likely due to the nature of the model system that we have used. The tyrosine-dependent activation of PI3-K of class IA normally is an extremely rapid on-off process, with complete loss of phospho-tyrosine-associated PI3-K activity within 5 to 10 min after exposure to an agonist (42). The lack of a known ligand for PC-1 renders it impossible to study the early phases of activation of PI3-K in our system. Thus, we might miss an initial peak of activation that is dependent on tyrosine phosphorylation. It is also possible that there is a low but continuous level of tyrosine phosphorylation that is below the threshold of detection.
From our data, we could not determine whether PC-1 is directly involved in activation of PI3-K. Because no YXXM sequence is found in the C-terminus or putative intracellular loops of PC-1, it seems that activation is unlikely to occur via direct binding of p85 SH2 domain to PC-1 but rather through an intermediary molecule. One such molecule could be JAK2, because we previously showed it to be activated and bound to PC-1 (23,43,44). Another possibility is that a 14-3-3 adapter protein may mediate the interaction as has been reported for the IL-3 receptor (45). In this example, a protein kinase A- dependent phosphorylation site in the IL-3 receptor binds to a 14-3-3 adapter protein, and the latter mediates binding and activation of p85. A similar mechanism might be occurring in our system as a potential protein kinase A phosphorylation site has been identified in the PC-1 C-terminus (18,19).
How the various in vitro study results relate to PC-1 function in vivo remains to be determined. Nishio et al. (46) recently reported a mouse model mosaic for Pkd1+/+ and Pkd1-/- cells that develops renal cystic disease. In that model system, Akt phosphorylation seems to be increased in both normal and cystic epithelia but does not correlate with the apoptotic rate in either. These results seem to be contrary to the well-established antiapoptotic role of this molecule in most systems studied to date (24). However, the authors do not specify which phosphorylation site they have studied, and, most important, they do not show increased phosphorylation of any downstream effector of Akt. Because phosphorylation of both Ser473 and Thr308 is necessary for Akt to be activated (24,25,27,28), it is impossible on the basis of the currently available data to know whether Akt is also catalytically active in their system. A direct correlation between our findings and theirs therefore is impossible at the moment. However, one could speculate that phosphorylation of Akt in that context might represent an attempt (possibly failed) of the cells to counteract a strong apoptotic stimulus or could be due simply to upregulation of the EGF receptor signaling (46). In addition, Nishio et al. (46) demonstrated that cyst formation in vivo seems to require a number of "transforming" events that occur in a step-wise manner. It is possible that careful examination of the phosphorylation state of Akt at the different stages of cyst formation would indeed reveal reduced activation in the absence of Pkd1 in some but not other of these stages. Systematic evaluation of Akt status at various times after inactivation of Pkd1 will be required to resolve this issue.
One final possible explanation for the seeming discrepancy between the in vitro and in vivo findings is that the down regulation of the PI3-K/Akt pathway that results from loss of PC-1 in vivo is not responsible for increased apoptosis in cystic epithelia but rather might be responsible for some of the other phenotypes observed in Pkd1-/- mice. This hypothesis is consistent with both our findings that specific inhibitors for PI3-K are able to block branching tubulogenesis induced by PC-1 and a growing body of literature that describes a central role for this kinase in the regulation of HGF-induced tubulogenesis in MDCK cells (35,47). We therefore propose a model in which PC-1 activates PI3-K as a central molecule that is capable of inducing both resistance to apoptosis and tubulogenesis. Future study will define the precise mechanism by which PC-1 activates this process as well as the physiologic relevance of this pathway in PC-1 function.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (DK48006, DK57325), the PKD Foundation, and the National Kidney Foundation of Maryland. G.G.G. is the Blum Scholar of the Johns Hopkins University School of Medicine. A.B. is supported by the European Community (MCEXT-CT-2003-002785) and by Telethon-Italy (TCP01018) and is an Assistant Telethon Scientist. We thank J. Hurt and L. Liu for technical assistance and Dr. Greenberg (Boston, MA) for the Akt constructs.
References
- 1.Gabow PA Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332–342. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- 2.Torra R, Nicolau C, Badenas C, Navarro S, Perez L, Estivill X, Darnell A. Ultrasonographic study of pancreatic cysts in autosomal dominant polycystic kidney disease. Clin Nephrol. 1997;47:19–22. [PubMed] [Google Scholar]
- 3.The European Polycystic Kidney Disease Consortium The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;77:881–894. doi: 10.1016/0092-8674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 4.The International Polycystic Kidney Disease Consortium Polycystic kidney disease: The complete structure of the PKD1 gene and its protein. Cell. 1995;81:289–298. doi: 10.1016/0092-8674(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 5.Burn TC, Connors TD, Dackowski WR, Petry LR, Van Raay TJ, Millholland JM, Venet M, Miller G, Hakim RM, Landes GM, Klinger KK, Qian F, Onuchic LF, Watnick T, Germino GG, Doggett NA. Analysis of the genomic sequence for the autosomal dominant polycystic kidney disease (PKD1) gene predicts the presence of a leucine-rich repeat. The American PKD1 Consortium. Hum Mol Genet. 1995;4:575–582. doi: 10.1093/hmg/4.4.575. [DOI] [PubMed] [Google Scholar]
- 6.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 7.Boletta A, Qian F, Onuchic LF, Bragonzi A, Cortese M, Deen PM, Courtoy PJ, Soria MR, Devuyst O, Monaco L, Germino GG. Biochemical characterization of bona fide polycystin-1 in vitro and in vivo. Am J Kidney Dis. 2001;38:1421–1429. doi: 10.1053/ajkd.2001.29282. [DOI] [PubMed] [Google Scholar]
- 8.Nims N, Vassmer D, Maser RL. Transmembrane domain analysis of polycystin-1, the product of the polycystic kidney disease-1 (PKD1) gene: Evidence for 11 membrane-spanning domains. Biochemistry. 2003;42:13035–13048. doi: 10.1021/bi035074c. [DOI] [PubMed] [Google Scholar]
- 9.Sandford R, Sgotto B, Aparicio S, Brenner S, Vaudin M, Wilson RK, Chissoe S, Pepin K, Bateman A, Chothia C, Hughes J, Harris P. Comparative analysis of the polycystic kidney disease 1 (PKD1) gene reveals an integral membrane glycoprotein with multiple evolutionary conserved domains. Hum Mol Genet. 1997;6:1483–1489. doi: 10.1093/hmg/6.9.1483. [DOI] [PubMed] [Google Scholar]
- 10.Sandford R, Mulroy S, Foggensteiner L. The polycystins: A novel class of membrane-associated proteins involved in renal cystic disease. Cell Mol Life Sci. 1999;56:567–579. doi: 10.1007/s000180050454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian F, Boletta A, Bhunia AK, Xu H, Liu L, Ahrabi AM, Watnick TJ, Zhou F, Germino GG. Cleavage of polycys-tin-1 requires the REJ domain and is disrupted by Human ADPKD1-associated Mutations. Proc Natl Acad Sci U S A. 2002;99:16981–16986. doi: 10.1073/pnas.252484899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parnell SC, Magenheimer BS, Maser RL, Rankin CA, Smine A, Okamoto T, Calvet JP. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem Biophys Res Commun. 1998;251:625–631. doi: 10.1006/bbrc.1998.9514. [DOI] [PubMed] [Google Scholar]
- 13.Parnell SC, Magenheimer BS, Maser RL, Zien CA, Frischauf AM, Calvet JP. Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. J Biol Chem. 2002;277:19566–19572. doi: 10.1074/jbc.M201875200. [DOI] [PubMed] [Google Scholar]
- 14.Delmas P, Nomura H, Li X, Lakkis M, Luo Y, Segal Y, Fernandez-Fernandez JM, Harris P, Frischauf AM, Brown DA, Zhou J. Constitutive activation of G-proteins by poly-cystin-1 is antagonized by polycystin-2. J Biol Chem. 2002;277:11276–11283. doi: 10.1074/jbc.M110483200. [DOI] [PubMed] [Google Scholar]
- 15.Arnould T, Kim E, Tsiokas L, Jochimsen F, Gruning W, Chang JD, Walz G. The polycystic kidney disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. J Biol Chem. 1998;273:6013–6018. doi: 10.1074/jbc.273.11.6013. [DOI] [PubMed] [Google Scholar]
- 16.Kim E, Arnould T, Sellin LK, Benzing T, Fan MJ, Gruning W, Sokol SY, Drummond I, Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J Biol Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 18.Li HP, Geng L, Burrow CR, Wilson PD. Identification of phosphorylation sites in the PKD1-encoded protein C-terminal domain. Biochem Biophys Res Commun. 1999;259:356–363. doi: 10.1006/bbrc.1999.0780. [DOI] [PubMed] [Google Scholar]
- 19.Parnell SC, Magenheimer BS, Maser RL, Calvet JP. Identification of the major site of in vitro PKA phosphorylation in the polycystin-1 C-terminal cytosolic domain. Biochem Biophys Res Commun. 1999;259:539–543. doi: 10.1006/bbrc.1999.0810. [DOI] [PubMed] [Google Scholar]
- 20.Ibraghimov-Beskrovnaya O, Bukanov NO, Donohue LC, Dackowski WR, Klinger KW, Landes GM. Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant poly-cystic kidney disease gene, PKD1. Hum Mol Genet. 2000;9:1641–1649. doi: 10.1093/hmg/9.11.1641. [DOI] [PubMed] [Google Scholar]
- 21.Scheffers MS, van der Bent P, Prins F, Spruit L, Breuning MH, Litvinov SV, de Heer E, Peters DJ. Polycystin-1, the product of the polycystic kidney disease 1 gene, co-localizes with desmosomes in MDCK cells. Hum Mol Genet. 2000;9:2743–2750. doi: 10.1093/hmg/9.18.2743. [DOI] [PubMed] [Google Scholar]
- 22.Boletta A, Qian F, Onuchic LF, Bhunia AK, Phakdeekitcharoen B, Hanaoka K, Guggino W, Monaco L, Germino GG. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol Cell. 2000;6:1267–1273. doi: 10.1016/s1097-2765(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 23.Bhunia AK, Piontek K, Boletta A, Liu L, Qian F, Xu P-N, Germino FJ, Germino GG. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell. 2002;109:157–168. doi: 10.1016/s0092-8674(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 24.Datta SR, Brunet A, Greenberg ME. Cellular survival: A play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 25.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 26.Scheid MP, Woodgett JR. PKB/AKT: Functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2:760–768. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- 27.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 28.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 29.Shtivelman E, Sussman J, Stokoe D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr Biol. 2002;12:919–924. doi: 10.1016/s0960-9822(02)00843-6. [DOI] [PubMed] [Google Scholar]
- 30.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 31.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 32.Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-ki-nases: Implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 33.Kurosu H, Maehama T, Okada T, Yamamoto T, Hoshino S, Fukui Y, Ui M, Hazeki O, Katada T. Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide. J Biol Chem. 1997;272:24252–24256. doi: 10.1074/jbc.272.39.24252. [DOI] [PubMed] [Google Scholar]
- 34.Maier U, Babich A, Nurnberg B. Roles of non-catalytic subunits in betagamma-induced activation of class I phosphoinositide 3-kinase isoforms beta and gamma. J Biol Chem. 1999;274:29311–29317. doi: 10.1074/jbc.274.41.29311. [DOI] [PubMed] [Google Scholar]
- 35.Derman MP, Cunha MJ, Barros EJ, Nigam SK, Cantley LG. HGF-mediated chemotaxis and tubulogenesis require activation of the phosphatidylinositol 3-kinase. Am J Physiol. 1995;268:F1211–F1217. doi: 10.1152/ajprenal.1995.268.6.F1211. [DOI] [PubMed] [Google Scholar]
- 36.Grantham JJ, Geiser JL, Evan AP. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 1987;31:1145–1152. doi: 10.1038/ki.1987.121. [DOI] [PubMed] [Google Scholar]
- 37.Woo D. Apoptosis and loss of renal tissue in polycystic kidney diseases. N Engl J Med. 1995;333:18–25. doi: 10.1056/NEJM199507063330104. [DOI] [PubMed] [Google Scholar]
- 38.Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, Rome LA, Sullivan LP, Grantham JJ. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000;57:1460–1471. doi: 10.1046/j.1523-1755.2000.00991.x. [DOI] [PubMed] [Google Scholar]
- 40.Ma R, Li WP, Rundle D, Kong J, Akbarali HI, Tsiokas L. PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol Cell Biol. 2005;25:8285–8298. doi: 10.1128/MCB.25.18.8285-8298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murga C, Fukuhara S, Gutkind JS. A novel role for phosphatidylinositol 3-kinase beta in signaling from G protein-coupled receptors to Akt. J Biol Chem. 2000;275:12069–12073. doi: 10.1074/jbc.275.16.12069. [DOI] [PubMed] [Google Scholar]
- 42.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 43.Yamauchi T, Kaburagi Y, Ueki K, Tsuji Y, Stark GR, Kerr IM, Tsushima T. Growth hormone and prolactin stimulate tyrosine phosphorylation of insulin receptor substrate-1, -2, and -3, their association with p85 phosphatidylinositol 3-kinase (PI3-kinase), and concomitantly PI3-kinase activation via JAK2 kinase. J Biol Chem. 1998;273:15719–15726. doi: 10.1074/jbc.273.25.15719. [DOI] [PubMed] [Google Scholar]
- 44.Al-Shami A, Naccache PH. Granulocyte-macrophage colony-stimulating factor-activated signaling pathways in human neutrophils. Involvement of Jak2 in the stimulation of phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:5333–5338. doi: 10.1074/jbc.274.9.5333. [DOI] [PubMed] [Google Scholar]
- 45.Guthridge MA, Stomski FC, Barry EF, Winnall W, Woodcock JM, McClure BJ, Dottore M, Berndt MC, Lopez AF. Site-specific serine phosphorylation of the IL-3 receptor is required for hemopoietic cell survival. Mol Cell. 2000;6:99–108. [PubMed] [Google Scholar]
- 46.Nishio S, Hatano M, Nagata M, Horie S, Koike T, Tokuhisa T, Mochizuki T. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. J Clin Invest. 2005;115:910–918. doi: 10.1172/JCI22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.