

Abstract
Clan CA, family C1 cysteine peptidases (CPs) are important virulence factors and drug targets in parasites that cause neglected diseases. Natural CP inhibitors of the I42 family, known as ICP, occur in some protozoa and bacterial pathogens, but are absent from metazoa. They are active against both parasite and mammalian CPs, despite having no sequence similarity with other classes of CP inhibitor. Recent data suggest that L. mexicana ICP plays an important role in host-parasite interactions. We have now solved the structure of ICP from L. mexicanaby NMR and shown that it adopts a type of immunoglobulin-like fold not previously reported in lower eukaryotes or bacteria. The structure places three loops containing highly conserved residues at one end of the molecule, one loop being highly mobile. Interaction studies with CPs confirm the importance of these loops for the interaction between ICP and CPs and suggest the mechanism of inhibition. Structure-guided mutagenesis of ICP has revealed that residues in the mobile loop are critical for CP inhibition. Data-driven docking models support the importance of the loops in the ICP-CP interaction. This study provides structural evidence for the convergent evolution from an immunoglobulin fold of CP inhibitors with a cystatin-like mechanism.
Characterisation of the structure and mechanisms of action of natural inhibitors of cysteine peptidases (CPs)1 has provided important insights into the functional roles of the inhibitors themselves and also those of the target CPs. CP inhibitors occur widely in nature, and several distinct groups with unrelated primary structures are recognised (1). Members of the cystatin super-family (clan IH, family I25 (2)) have been extensively characterised at the molecular level (3–5). A second group of natural CP inhibitors, assigned to clan IX, family I31, are similar to cystatin in inhibiting CPs of clan CA, family C1 such as papain but are distinguished by the presence of a thyroglobulin type I domain (6). The recently discovered chagasin family of CP inhibitors, designated ICP for Inhibitor of Cysteine Peptidases (clan I-, family I42), also inhibit papain-like CPs and yet have no significant sequence identity with the other groups of CP inhibitors (7). This raises the intriguing question: Have the different groups of CP inhibitors evolved related tertiary structures in order to interact with the same target CPs in a similar fashion, as predicted from in silico analysis by Rigden and co-workers (8), or does ICP act in a different way?
ICP family members appear to occur in species from a very limited phylogenetic range (some parasitic protozoa, bacteria [including Pseudomonas aeruginosa] and archaea, but no metazoa), suggesting that the genes have been acquired by horizontal gene transfer or retained for some special function. Whilst there is good evidence that the in vivo target for ICPs are clan CA, family C1 CPs, it remains uncertain whether the inhibitors’ main function is to modulate the activity of enzymes of the parasite itself (as is suggested for protozoan parasite Trypanosoma cruzi (9)) or the host (as suggested for the related parasite Leishmania (10)). Interestingly, no clan CA, family C1 CPs appear to be present in the Pseudomonas aeruginosa genome, which provides further support for the suggestion that one role of ICPs in pathogens might be to regulate host CP activity and so facilitate infection.
This has been investigated with Leishmania by gene targeting to create parasite lines that either lack, or over-express, the ICP gene (10). ICP null mutants grow normally axenically in vitro and are as infective to macrophages in vitro as wild type parasites. However, they have reduced infectivity to mice. Lines that over-express ICP also show markedly reduced virulence in vivo. Thus, ICP may be important after it is released in the parasite’s interaction with its mammalian host or as a mechanism of preventing damage from host CPs taken up by the parasite through endocytosis (e.g. from the parasitophorous vacuole, which contains host lysosomal enzymes).
The ICP proteins are small (~13 kDa) and very divergent, with L. mexicana ICP having only 31% identity with ICP of T. cruzi and 24% identity with ICP of P. aeruginosa (11). Nevertheless, there are highly conserved motifs that suggest important functional regions. This has facilitated the identification of predicted ICP from genome data and recombinant ICPs have produced from the L. mexicana, T. brucei, Entamoeba histolytica and P. aeruginosa genes and confirmed to have potent inhibitory activity towards CPs, notably cathepsin L homologues (7,11,12)
To date, the structural basis of the inhibitory activity of ICP is unknown. Previous threading studies have suggested that the binding site of T. cruzi ICP may be located on the loops between β-strands in a fold that resembles immunoglobulin light-chain variable domains (8,13). Another study drew parallels between the sequence conservation in predicted loops of the ICP family and the peptidase-binding regions of the cystatin family (12). We have now determined the structure of L. mexicana ICP in solution by NMR spectroscopy, confirmed residues key for its inhibitory activity using site-directed mutagenesis, and investigated how the key residues may bind- to the model clan CA, family C1 peptidase papain and an important L. mexicana CP, known as CPB (14).
EXPERIMENTAL PROCEEDURES
Protein production
Recombinant L. mexicana ICP was expressed from a pET28 (Novagen) derived plasmid in Escherichia coli BL21 (DE3) cells as described previously (11). 15N,13C-labelled protein was produced by growing the cells in M9 medium using 15NH4Cl and 13C-glucose (Spectra Stable Isotopes) as the sole nitrogen and carbon sources. The fusion protein was purified by nickel chelate chromatography and digested with thrombin (Novagen). The cleaved histidine tag and thrombin were removed by nickel chelate and benzamidine sepharose (Sigma) affinity chromatography. The protein comprising the complete native sequence (Q868H12;CAD689753) with the addition of three residues (GSH) at the N-terminus (designated ICP-2–113) was buffer exchanged into 25 mM sodium phosphate pH 4.5, 50 mM NaCl, 0.001% NaN3 by extensive diafiltration using a 5,000 MWCO centrifugal concentrator (Vivascience) and concentrated to approximately 1 mM. D2O was added to a final concentration of 10% (v/v).
NMR samples of L. mexicana ICP-2–113 underwent proteolysis over two to three days under NMR sample conditions to produce an N-terminally truncated protein starting at residue serine 6 (ICP6–113) as confirmed by mass spectrometry, which then remained stable. No difference in Ki for L. mexicana CPB could be detected between ICP-2–113 and ICP6–113.
Interaction studies were carried out using papain from Papaya latex (Sigma) and L. mexicana CPB2.8ΔCTE, produced as described previously (14). In each case, peptidase was mixed with an excess of 15N-labelled ICP in NMR sample buffer and the complex isolated by gel filtration on a Superdex 75 HR10/30 column (APBiotech) and then concentrated using a 10,000 MWCO centrifugal concentrator.
NMR spectroscopy and data analysis
Resonance assignments were determined using standard triple resonance NMR techniques and have been deposited as described (15). Distance restraints for structure calculation were derived from 3D 15N- and 13C-HSQC-NOESY spectra recorded with 100 ms mixing times recorded on an 800 MHz Bruker Avance spectrometer. Slowly exchanging amide protons were identified by re-dissolving a lyophilised sample in D2O and recording a series of 15N HSQC spectra. Spectra were processed with AZARA (http://www.bio.cam.ac.uk/azara) and analysed using CCPN analysis (16).
Structure calculation
Assigned, partially assigned and ambiguous NOESY crosspeaks were used to generate distance constraints within CCPN analysis that were exported directly to CNS/XPLOR format and used as input for structure calculations using CNS v1.1 (17) using a modified version of the PARALLHDG 5.3 forcefield (18) with IUPAC recommended nomenclature (19). Structures were generated from random atomic coordinates following the scheme of the rand, dgsa and refine scripts from the XPLOR 3.1 manual (20) re-implemented in CNS and modified to incorporate floating chirality at prochiral centres (21) using a metropolis acceptance criterion. The tools provided by the ARIA (22) module within CNS were used to identify consistently violated restraints for checking, to reduce the ambiguity of ambiguous distance restraints based on the ensemble of structures calculated, to remove duplicate restraints, and to recalibrate the distance-intensity mapping. Distance restraints representing hydrogen bonds were incorporated for slowly exchanging amides where corroborating NOEs existed and an acceptor atom could be unambiguously identified. Atomic coordinates have been deposited at the PDB (PDB code 2c344) and structural statistics are summarised in Table I.
Table I.
NMR structural statistics for L. mexicana ICP.
| NMR distance constraints | |
| Distance constraints | |
| Total NOE | 2370 |
| Ambigous | 812 |
| Unambiguous | 1558 |
| Intra-residue | 869 |
| Inter-residue | |
| Sequential (|i−j| = 1) | 302 |
| Medium-range (|i−j| < 4) | 83 |
| Long-range (|i−j > 5) | 304 |
| Hydrogen bonds | 22 |
| Structure Statistics | |
| Violations (mean and s.d.) | |
| Distance constraints (Å) | 0.052±0.002 |
| Distance constraint violations > 0.5 Å | 1.3±0.98 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 2.54e−3±1.00e−4 |
| Bond angles (º) | 0.382±1.69e−2 |
| Impropers (º) | 0.354±1.71e−2 |
| R.m.s.d. to the unbiased mean structure** (Å) | |
| Heavy | 0.854±0.189 |
| Backbone | 0.586±0.181 |
| Ramachandran plot (% residues in) | |
| Most favoured regions | 52.5 |
| Additionally allowed regions | 44.4 |
| Generously allowed | 2.7 |
| Disallowed regions | 0.5 |
R.m.s.d. to the unbiased mean structure was calculated over residues 14–60 and 73–112 among 38 refined structures.
15N Relaxation measurements
Protein dynamics were probed through 15N relaxation measurements of the backbone amide groups. 15N T1, T2 and 1H,15N-heteronuclear NOE experiments were recorded at 600.13MHz (1H) and the data analysed according to the Lipari-Szabo model-free formalism using the programs curvefit and modelfree (23).
Docking
Data-driven molecular docking was carried out using HADDOCK v1.3 (24) using the default parameters and using chemical shift perturbation data as input. In the HADDOCK terminology, the “active” ICP residues are those whose backbone or side-chain amide chemical shifts are significantly perturbed in the complex with papain and which are surface exposed. The “passive” ICP residues are their immediate, surface exposed, neighbours. In the absence of chemical shift perturbation data for the peptidase, and given the superficial similarity between the ICP and stefin B structures, the “active” papain residues were defined as those residues in close contact with stefin B in the crystal structure of the stefin B:papain complex. The “passive” papain residues are their immediate, surface exposed neighbours. “Semi-flexible” regions are defined to be two residues either side of the active and passive residues in the primary sequences of both proteins. The ICP D-E loop was made “fully flexible” to reflect its high mobility in unbound ICP.
Mutations
Mutations of L. mexicana ICP were incorporated into the expression vector using the QuickChange site-directed mutagenesis kit (Stratagene) and the following pairs of complementary primers (mutated sites in lower case): NT300, CCCGACCACTGGAgcCATGTGGACGCGC and NT301, GCGCGTCCACATGgcTCCAGTGGTCGGG to generate pBP191 (encoding Y34A); NT282, CATCCTCGACTCCTgacGacGGAGaTGGTG GCATCTAC and NT283, GTAGATGCCACCAtCTCCgtCgtcAGGAGTC GAGGATG to generate pBP193 (encoding M64D, V68D, V70D); NT298, CCTCGACTCCTATGGTGccAGTTccTcccAT CTACGTTGTGCTCG and NT299, CGAGCACAACGTAGATgggAggAACTggCA CCATAGGAGTCGAGG to generate pBP194, (encoding G69P, G71P, G72P); NT284, CTGGTCTACACGgcCCCCgcCGAGGGCATC AAGC and NT285, GCTTGATGCCCTCGgcGGGGgcCGTGTAG ACCAG to generate pBP197, (encoding R94A, F96A); NT280, GAAGGGCAACCCGggCggTGGATACATGT GGACG and NT281, CGTCCACATGTATCCAccGccCGGGTTGCC CTTC to generate pBP199 (encoding T31,T32G). The mutated plasmids were verified by nucleotide sequencing.
Ki determinations
Kis for L. mexicana ICP and its mutants against L. mexicana CPB2.8ΔCTE were determined essentially as described previously (11). In summary, concentrations of ICP stock solutions were determined by titration against a known concentration of CPB2.8ΔCTE in a colorimetric assay using N-benzoyl-PFR-p-nitroanilide hydrochloride (Sigma) as a substrate under pseudo-irreversible conditions. For mutants with a detectable inhibitor activity, IC50s were determined in a fluorimetric assay using 10 μM benzyloxycarbonyl-FR-7-amino-4-methylcoumarin hydrochloride (ZFR-AMC, Sigma) as a substrate and Kis calculated using the relationship Ki=IC50(1/(1+[S]/Km) where Km = 0.7 μM (25).
Circular Dichrosim
Near (250–320 nm) and far (190–240 nm) UV CD analyses were performed with 0.6mg/ml protein in 0.5 and 0.02cm pathlength quartz cells respectively, using a JASCO J-810 spectropolarimeter. Eight scans were collected for each protein using a bandwidth of 1.0 nm, a scanning speed of 50 nm min−1 and a response time of 0.25 sec. Data were corrected for cell path length and protein concentration and smoothed using the Savitsky-Golay algorithm (convolution width 9.0). The secondary structure analyses of wild type and mutant proteins were obtained using the CDSSTR method (26) available from the DICHROWEB website, at the University of Birkbeck. All three proteins gave similar secondary structure estimates with low NRMSD values (~0.025).
RESULTS
ICP adopts an immunoglobulin fold
In solution, ICP adopts an immunoglobulin-like (Ig) fold with seven β-strands (fig 1). One β-sheet is formed by anti-parallel strands B, E and D, while the other is formed by anti-parallel strands G, F and C with strand A parallel to strand G. A search for structural homologues using the DALI server (27) identified the N-terminal Ig domain from α-dystroglycan (PDB:1u2c(28)) as the closest match with a Z-score of 5.1 and an RMSD of 3.2Å over 82 residues. The SCOP database (29) classifies this as a cadherin-like superfamily fold, which also closely resembles the I-set immunoglobulin-like fold (30,31).
Fig. 1.
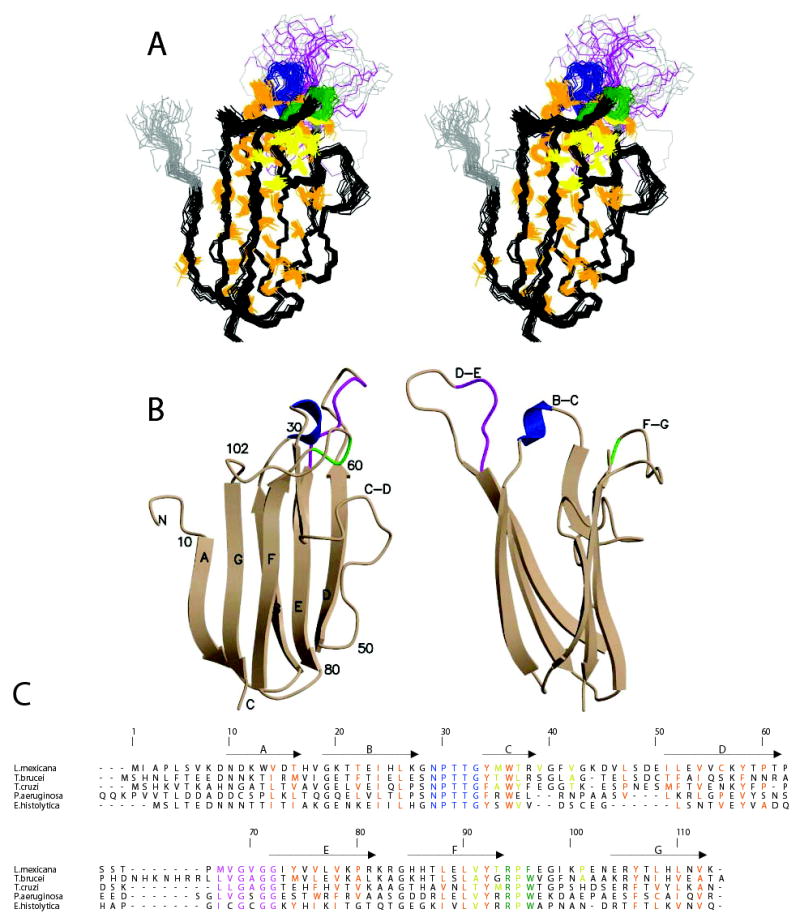
The solution structure of L. mexicana ICP6–113. Conserved regions of the B-C, F-G and D-E loops are coloured blue, green and magenta, respectively. Residues of the central hydrophobic core are coloured orange and of the secondary hydrophobic cluster yellow. A, A stereo-view of the ensemble of 38 structures superposed on Cα atoms of residues 14–60 and 73–112. The heavy atoms of the residues that make up the two hydrophobic clusters are shown. B, Orthogonal views of a cartoon of the structure. C, Structure based sequence alignment of ICPs confirmed as inhibitors numbered according to L. mexicana ICP sequence. The locations of the β-strands are indicated by arrows.
The β-sheets enclose a well-packed core of principally hydrophobic residues that correspond to the alternating pattern of conserved residues along the strands (see fig 2). The C-D loop of ICP folds back on strand C with a number of hydrophobic side-chains making contacts that are not part of the main core of the molecule. These interactions involving residues M35, T37, V39, V42, V91, T93, P95 and I99 serve to pin the F-G loop back to the outer face of one β-sheet. The prediction of the ICP family structure based on threading (13) correctly identified the Ig-like fold, but the details of the predicted strands and loops were out of register after the C-D loop – possibly due to the misidentification of the hydrophobic residues in the C-D loop as representative of an extended C strand.
Fig. 2.
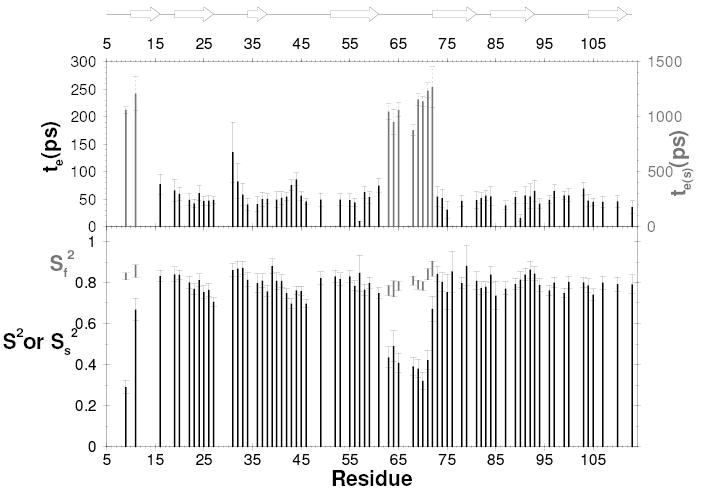
15N backbone dynamics of L. mexicana ICP6–113. Lipari-Szabo order parameters and internal correlation times plotted as a function of residue number. Two order parameters are shown for the residues that were best fit with two timescales of internal motion. The locations of the β-strands are indicated by arrows.
The three highly conserved groups of residues previously noted (8,11,12) are all located in loops at one end of the molecule (fig 1b). The GNPTTGY motif lies in the B-C loop, the GXGG motif lies in the highly mobile D-E loop and the RPW/F motif in the F-G loop. The co-location of all three motifs, and the finding that none of the residues appear to have roles that are key to the integrity of the protein’s fold, suggests that they together form the CP-binding site.
Residues 30–32 in the B-C loop form a turn of 310-helix projecting the sidechains of N29, T31 & T32 towards the solvent. G28 and G33 allow the loop to be accommodated between the B and C strands by adopting conformations with positive φ dihedral angles. The conserved Y34, which caps the main hydrophobic core of the protein, interacts with residues in the F-G loop, and contributes to the solvent exposed surface at this end of the protein. Pinned back to the GFC β-sheet, the 11-amino acid F-G loop (residues 93–103) forms a broad saddle with residues 96–101 running roughly perpendicular to the direction of the β-strands. The side-chain of conserved R94 interacts with Y34, and along with Y96 projects out of this end of the protein accompanied by E97, K100 and E102. The D-E loop (residues 61–72) projects from this end of the protein on the other side of the B-C loop and is poorly defined by the experimental data.
The D-E loop is flexible
15N relaxation measurements were used to probe the backbone dynamics of ICP6–113. 15N NOE, 15N T1 and 15N T2 were measured at 60.8 MHz (15N), the derived order parameters and correlation times are shown in fig 2. The majority of the residues display uniform relaxation rates close to the average values typical of a compactly folded monomeric protein of this size and their behaviour could be modelled using either a single order parameter (S2) or S2 and an internal correlation time. The exceptions are the N-terminus up to residue 13 and residues 61–72 in the D-E loop, which display depressed NOE values and lengthened T2 values. These residues were best modelled with two order parameters representing internal motion on distinct timescales and an internal correlation time of the order of a nanosecond. In addition, residue N29, which could not be well fitted by any model, has a significantly shortened T2 value indicative of millisecond timescale chemical exchange which may reflect backbone flexibility, or given the proximity of Y92, may simply be due to the combination of local motion and the ring current shift from this aromatic side-chain.
The conserved motifs in the loops are involved in CP-binding
Chemical shift perturbation mapping was used to identify the CP-binding site on ICP. Complexes of 15N-labelled ICP with L. mexicana CPB and with Papaya latex papain were purified and their 15N HSQC spectra compared with free ICP. A similar subset of backbone HN and sidechain crosspeaks are perturbed from their free ICP chemical shifts in both complexes, and the most distinctive are shown in fig 3 for the papain complex. It is clear that all the most perturbed residues fall in the ranges 28–37 (covering the B-C loop), 59–74 (covering the D-E loop) and 94–104 (covering the F-G loop). Chemical shifts are sensitive to changes in chemical environment caused either by proximity to a ligand, or by propagated structural changes. The co-location of all the significant changes in the vicinity of the three conserved loops strongly suggests that this is due to their involvement in binding to target CPs.
Fig. 3.
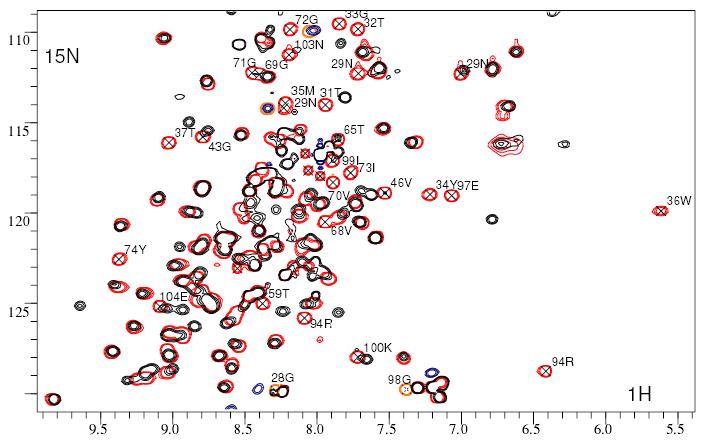
Chemical shift perturbations on binding papain. 15N,1H HSQC spectra of L. mexicana ICP free (red) and bound to papain (black). The most unambiguously perturbed resonances are indicated with their sequence-specific assignments.
Residues in the D-E loop are critical for CP binding
Based on our L. mexicana ICP structure and on sequence similarity between ICP family members, we made a limited number of mutant ICPs to test their effect on ICP’s inhibitory activity. In the highly conserved B-C loop, we mutated the pair of threonines to glycine (T31G,T32G) and mutated the conserved tyrosine at the base of the loop to alanine (Y34A). In the flexible D-E loop, we made two triple mutants: one to replace the semi-conserved hydrophobic residues with charged residues (M67D,V68D,V70D) and the other to replace the glycines by more conformationally restricted residues (G69P,G71P,G72P). In the F-G loop, we made a double mutant of the conserved arginine and aromatic residues (R94A,F96A).
The D-E loop mutants lacked inhibitory activity against L. mexicana CPB. To ensure that these mutants had retained their three-dimensional fold we analysed them using CD spectroscopy. Wild-type ICP and mutant M67D,V68D,V70D possess superimposable CD spectra in both the near and far UV regions indicating very similar, if not identical secondary and tertiary structures. Mutant G69P,G71P,G72P gave rise to a slightly different far UV spectrum, whilst retaining similar spectral features in the near UV region, and CDSSTR analysis placed the regular secondary structure estimates within a few percent of the wild-type ICP. Of the other mutants, the B-C loop mutations resulted in somewhat lower Kis, while the Ki of the F-G loop mutant was approximately three-fold higher (see Table II).
Table II.
Ki values for L. mexicana ICP mutants against L. mexicana CPB. ND, not determined.
| Loop | Ki (pM) | |
|---|---|---|
| L. mexicana ICP | - | 132.8 ± 21.1 |
| T31G,T32G | B–C | 53.0 ± 8.3 |
| Y34A | B–C | 46.4 ± 6.5 |
| M67D,V68D,V70D | D–E | ND – no inhibition observed |
| G69P,G71P,G72P | D–E | ND – no inhibition observed |
| R94A,F96A | F–G | 428.5 ± 86.5 |
A model for the interaction between ICP and CPs
The identification of the three conserved loops as the CP-binding site prompted a comparison of the ICP structure with other CP inhibitors. A slight, but suggestive, structural similarity was detected with the cystatin, stefin B (vide infra), where the CP-binding site is formed by a short central loop flanked by a flexible N-terminal peptide and a longer flattened loop that incorporates a proline (see fig 4). We speculated that ICP might bind target CPs in a similar fashion with the flexible D-E loop binding in place of stefin B’s N-terminal peptide. In order to produce a model for the interaction between ICP and a CP, we used the chemical shift perturbation data for ICP in complex with papain, together with information from the crystal structure of the complex between papain and stefin B, to drive a docking simulation using HADDOCK (24). The input data dictated that ICP should bind into the active site of papain, but did not, a priori, dictate that ICP should bind the peptidase in a stefin-like orientation. Of the 200 lowest energy structures from the HADDOCK run, all adopted a stefin-like mode of binding (fig 4) with residues 67–70 from the D-E loop filling the “unprimed” sites of the active site cleft, while the B-C loop approaches the active site cysteine-histidine pair, and sidechains from residues in the F-G loop fill the “primed” end of the active site cleft. The preferred orientation for the interaction additionally brings the C-D loop into contact with the short helix (residues 139–143) on the top of the R domain of papain. The feasibility of this contact is born out by the observation that the chemical shifts of residues 43 and 46 in the C-D loop can clearly be seen to change on complex formation (fig 3).
Fig. 4.
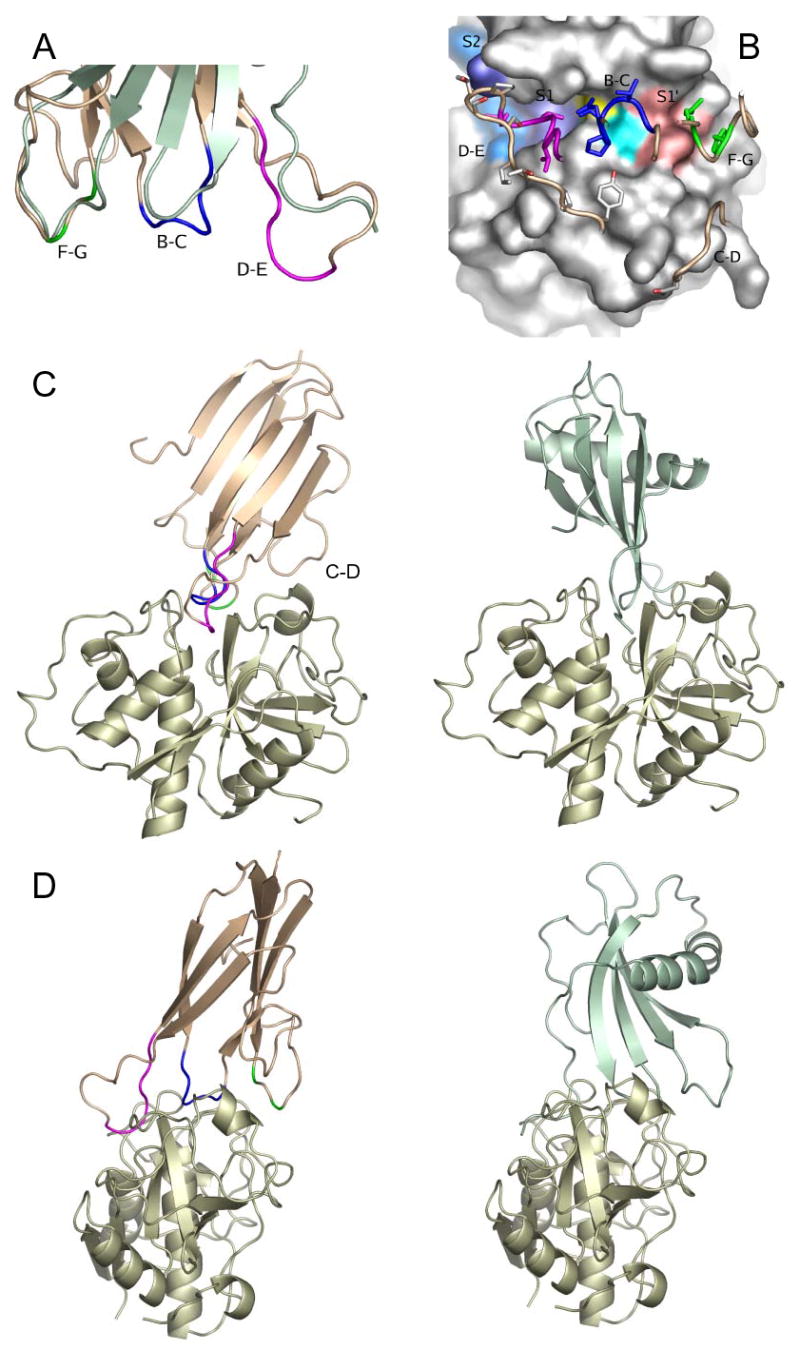
Model for the interaction of ICP and cysteine peptidases (colouring as in fig 1). A, Structural similarity of the ICP CP-binding loops to stefin B (green). B, The interface between ICP and papain modelled using HADDOCK. Peptidase substrate recognition sites and active site residues are coloured on the surface of the peptidase: S2, light blue; S1, dark blue; cysteine, yellow; histidine, cyan; S1′, pink. ICP loops are labelled and coloured. C and D, Two views of the stefin-papain complex (4) in comparison with the predicted ICP-papain interaction. The complexes are shown in the standard view (5) C and rotated 90° D such that the peptidase substrate would run from left to right.
DISCUSSION
This study has shown that ICP has an Ig fold that acts as a scaffold for three inter-strand loops carrying the most highly conserved residues in the chagasin family of ICP proteins. These three loops are located adjacent to one another at one end of the protein with the most highly conserved B-C loop lying between the structured F-G loop and the flexible D-E loop. Together, the three loops form a ridge that is of the correct dimensions to be complementary to the active sites of clan CA, family C1 CPs. Chemical shift perturbation studies identify these loops as forming the binding interface with the two representative CPs papain and L. mexicana CPB.
Comparison of the ICP structure with CP inhibitors of other families reveals no similarity at the level of the overall fold. However, there are suggestive similarities between the CP-interacting regions of ICP and the cystatin family of inhibitors. The ICP F-G loop (residues 93–103) bears a structural resemblance to the second binding loop of the tripartite wedge of stefins A and B (4,32) and superposing the structures on these features brings the B-C loop and C-terminal end of ICP strand B into the vicinity of the first interacting loop and strand B of the stefins (fig 4a). Superposition of both these structural features places ICP’s flexible D-E loop in the vicinity of the stefin N-terminal trunk (fig 4a). This suggests that the three conserved ICP loops may form a similar tripartite wedge to that of the cystatin family, both adapted to fit the active site cleft of clan CA, family C1 CPs. The implications of this similarity would be that ICP recognises target CPs with the flexible D-E loop adapting to the recognition sites for the substrate residues N-terminal to the cleavage site (thought to be a main determinant of specificity for this group of CPs) and the F-G loop binding to the distal end of the substrate recognition groove. The importance of these interactions for the binding of ICP is demonstrated by our data for the ICP mutants (Table II). In particular, the changes to the D-E loop abrogated inhibitory activity. It should be highly informative to express this “dead” mutant in L. mexicana so that the role of ICP in the parasite and its interaction with its host can be studied. Additional support for the importance of the F-G loop is provided by the observation that ICP inhibits cathepsin B considerably less well than cathepsin L (7). This is also a feature of stefins ((33) and can be explained by the steric hindrance afforded by the occluding loop, that is characteristic of cathepsin B-like CPs and is partially responsible for their substrate specificity.
To investigate the feasibility of this model, we carried out data-driven docking between ICP and papain with the assumption that ICP would contact the same region of the active site as the cystatins since the flexibility of the D-E loop make the results of any less biased approach such as rigid-body docking difficult to evaluate. The HADDOCK modelling surprisingly revealed just one clearly favoured orientation for the interaction interaction in which the fold of ICP places the highly conserved NPTT sequence motif in the B-C loop close to catalytic residues of the target CP. The ICP B-C loop is longer and bulkier than the cystatin family’s first interacting loop and, given its disposition relative to the loops on either side, may interact intimately with the CP active site residues. Thus it is surprising that the two B-C loop mutations had no deleterious effect on inhibitory activity despite the high level of sequence conservation between bacterial and protozoan ICPs in this region. However, the changes introduced into the mutants both decreased the bulk of the B-C loop. It is known that CP active sites are expanded when in complex with stefins (4,5), and so, taking into account the bulkiness of the ICP B-C loop, wild-type ICP may be larger than is optimal for the tightest interaction with this peptidase active site. The most likely explanation for this is that the loop is optimised for inhibition of an as yet untested a CP other than those investigated, such as a host CP that that has a wider active site. This suggestion is be consistent with previous findings derived by generating mutants of Leishmania that lack or over-express ICP (10).
Proteins with Ig folds are rare in species other than higher eukaryotes and the viruses that infect them. Hitherto they seemed to be entirely absent from protozoa. ICP is the first protein with a cadherin-like Ig domain to be discovered in a non-metazoan. This raises the possibility that ICPs were acquired by the pathogens from their hosts, although for this to be the case the event would have had to have happened on multiple occasions as gene transfer between the parasites seems unlikely. A more likely explanation is that a crucial role in the host-pathogen interaction has ensured retention of an otherwise unimportant gene by the pathogens.
Most cadherin-like Ig domains in cell-adhesion molecules mediate protein-protein interactions. However, for the cadherin domains themselves, these principally involve the faces of the β-sheets (34). This differs from the binding mode of ICP, which is more akin to antigen recognition by Ig variable domains. The finding that ICP has an inhibitory mechanism potentially rather similar to the cystatins, despite the unrelated primary structures and folds of the proteins, clearly demonstrates the likelihood that there has been convergent evolution. This apparent similarity also suggests that this type of protein-protein interaction is efficient for the regulatory functions of these inhibitors and, moreover, perhaps, that such a tripartite binding mechanism is required.
The discovery of the binding mode of ICP may also be informative for the design of chemical inhibitors of CPs that could have therapeutic value, both against pathogens and also in the treatment of CP-related mammalian disorders. Perhaps a compound that mimics the binding of the D-E loop and also has an active site-directed nucleophile would show good activity against the CPs. Indeed the peptidyl vinylsulphone compound currently in development for use against T. cruzi infections (35,36) presumably is acting in just this way. Discovery of the physiological targets of each ICP should allow design of inhibitors that have optimal specificity for these target CPs, and so potentially have value in therapies directed against them.
Footnotes
This work was supported by a grant from the Wellcome Trust. NMR data were acquired at the University of Edinburgh Biomolecular NMR Unit. We thank Sharon Kelly and Tommy Jess for assistance with the CD spectroscopy and analysis.
Abbreviations used: CP, cysteine peptidase; ICP, inhibitor of cysteine peptidase.
UniProt # Q868H1
NCBI Accession # CAD68975
PDB # 2c34
References
- 1.Rawlings ND, Tolle DP, Barrett AJ. Biochem J. 2004;378:705–716. doi: 10.1042/BJ20031825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawlings ND, Tolle DP, Barrett AJ. Nucleic Acids Res. 2004;32:D160–D164. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bode W, Engh R, Musil D, Thiele U, Huber R, Karshikov A, Brzin J, Kos J, Turk V. EMBO J. 1988;7:2593–2599. doi: 10.1002/j.1460-2075.1988.tb03109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stubbs MT, Laber B, Bode W, Huber R, Jerala R, Lenarcic B, Turk V. EMBO J. 1990;9:1939–1947. doi: 10.1002/j.1460-2075.1990.tb08321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenko S, Dolenc I, Guncar G, Dobersek A, Podobnik M, Turk D. J Mol Biol. 2003;326:875–885. doi: 10.1016/s0022-2836(02)01432-8. [DOI] [PubMed] [Google Scholar]
- 6.Guncar G, Pungercic G, Klemencic I, Turk V, Turk D. EMBO J. 1999;18:793–803. doi: 10.1093/emboj/18.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monteiro ACS, Abrahamson M, Lima APCA, Vannier-Santos MA, Scharfstein J. J Cell Sci. 2001;114:3933–3942. doi: 10.1242/jcs.114.21.3933. [DOI] [PubMed] [Google Scholar]
- 8.Rigden DJ, Mosolov VV, Galperin MY. Protein Sci. 2002;11:1971–1977. doi: 10.1110/ps.0207202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santos CC, Sant’Anna C, Terres A, Cunha-e-Silva N, Scharfstein J, de Lima A. J Cell Sci. 2005;118:901–915. doi: 10.1242/jcs.01677. [DOI] [PubMed] [Google Scholar]
- 10.Besteiro S, Coombs GH, Mottram JC. Mol Microbiol. 2004;54:1224–1236. doi: 10.1111/j.1365-2958.2004.04355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanderson SJ, Westrop GD, Scharfstein J, Mottram JC, Coombs GH. FEBS Lett. 2003;542:12–16. doi: 10.1016/s0014-5793(03)00327-2. [DOI] [PubMed] [Google Scholar]
- 12.Riekenberg S, Witjes B, Saric M, Bruchhaus I, Scholze H. FEBS Lett. 2005;579:1573–1578. doi: 10.1016/j.febslet.2005.01.067. [DOI] [PubMed] [Google Scholar]
- 13.Rigden DJ, Monteiro ACS, De Sá MFG. FEBS Lett. 2001;504:41–44. doi: 10.1016/s0014-5793(01)02753-3. [DOI] [PubMed] [Google Scholar]
- 14.Sanderson SJ, Pollock KG, Hilley JD, Meldal M, St Hilaire PM, Juliano MA, Juliano L, Mottram JC, Coombs GH. Biochem J. 2000;347:383–388. doi: 10.1042/0264-6021:3470383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith, B. O., Westrop, G. D., Mottram, J. C., and Coombs, G. H. (2005) Journal of Biomolecular NMR in press. [DOI] [PMC free article] [PubMed]
- 16.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED. Proteins: Struct Funct Bioinf. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 17.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 18.Linge JP, Williams MA, Spronk CAEM, Bonvin AMJJ, Nilges M. Proteins: Struct Funct, Genet. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 19.Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wuthrich K. Pure Appl Chem. 1998;70:117–142. [Google Scholar]
- 20.Brunger, A. T. (1992) X-PLOR 3.1, Yale University Press, New Haven, CT,
- 21.Folmer RHA, Hilbers CW, Konings RNH, Nilges M. J Biomol NMR. 1997;9:245–258. doi: 10.1023/a:1018670623695. [DOI] [PubMed] [Google Scholar]
- 22.Nilges M, Macias MJ, ODonoghue SI, Oschkinat H. J Mol Biol. 1997;269:408–422. doi: 10.1006/jmbi.1997.1044. [DOI] [PubMed] [Google Scholar]
- 23.Mandel AM, Akke M, Palmer AG. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 24.Dominguez C, Boelens R, Bonvin AMJJ. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 25.Alves LC, Judice WAS, St Hilaire PM, Meldal M, Sanderson SJ, Mottram JC, Coombs GH, Juliano L, Juliano MA. Mol Biochem Parasitol. 2001;116:1–9. doi: 10.1016/s0166-6851(01)00290-0. [DOI] [PubMed] [Google Scholar]
- 26.Manavalan P, Johnson WC. Anal Biochem. 1987;167:76–85. doi: 10.1016/0003-2697(87)90135-7. [DOI] [PubMed] [Google Scholar]
- 27.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 28.Bozic D, Sciandra F, Lamba D, Brancaccio A. J Biol Chem. 2004;279:44812–44816. doi: 10.1074/jbc.C400353200. [DOI] [PubMed] [Google Scholar]
- 29.Murzin AG, Brenner SE, Hubbard T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 30.Harpaz Y, Chothia C. J Mol Biol. 1994;238:528–539. doi: 10.1006/jmbi.1994.1312. [DOI] [PubMed] [Google Scholar]
- 31.Halaby DM, Poupon A, Mornon JP. Protein Eng. 1999;12:563–571. doi: 10.1093/protein/12.7.563. [DOI] [PubMed] [Google Scholar]
- 32.Martin JR, Jerala R, Kroonzitko L, Zerovnik E, Turk V, Waltho JP. Eur J Biochem. 1994;225:1181–1194. doi: 10.1111/j.1432-1033.1994.1181b.x. [DOI] [PubMed] [Google Scholar]
- 33.Pol E, Bjork I. Biochim Biophys Acta. 2003;1645:105–112. doi: 10.1016/s1570-9639(02)00526-5. [DOI] [PubMed] [Google Scholar]
- 34.Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 35.Sajid M, McKerrow JH. Mol Biochem Parasitol. 2002;120:1–21. doi: 10.1016/s0166-6851(01)00438-8. [DOI] [PubMed] [Google Scholar]
- 36.Engel JC, Doyle PS, Hsieh I, McKerrow JH. J Exp Med. 1998;188:725–734. doi: 10.1084/jem.188.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]