

Abstract
Adoptive cell transfer after host preconditioning by lymphodepletion represents an important advance in cancer immunotherapy. Here, we describe how a lymphopaenic environment enables tumour-reactive T cells to destroy large burdens of metastatic tumour and how the state of differentiation of the adoptively transferred T cells can affect the outcome of treatment. We also discuss how the translation of these new findings might further improve the efficacy of adoptive cell transfer through the use of vaccines, haematopoietic-stem-cell transplantation, modified preconditioning regimens, and alternative methods for the generation and selection of the T cells to be transferred.
Substantial progress has been made in our understanding of the molecular and cellular bases of T-cell-mediated antitumour responses. CD8+ T cells have been identified as potent effectors of the adaptive antitumour immune response1,2. The target antigens that are recognized by tumour-reactive CD8+ T cells have been shown to be mostly non-mutated self-antigens that are also expressed by tumour cells (and these antigens are denoted here self/tumour antigens)1,2. Tumour-specific CD4+ T cells have been also identified, but their functionality can be manifold because CD4+ T cells can help or hinder anti-tumour immune responses3–5. The molecular signals that modulate T-cell activation, function and memory are currently being elucidated. Both positive and negative signals from co-stimulatory molecules have been shown to shape the antitumour response6,7. Cytokines, including those signalling through receptors that contain the common cytokine-receptor γ-chain (γc), have been shown to alter the programming of effector CD8+ T cells8,9.
Despite a wealth of knowledge relevant to basic aspects of tumour immunology, the clinical realization of effective therapeutic vaccines for solid tumours has not yet been convincingly achieved. Enthusiasm about the effectiveness of cancer vaccines has often been grounded in surrogate and subjective endpoints, rather than reliable objective cancer regressions using standard oncological criteria. In a recent review of 1,306 vaccine treatments, including those conducted in the Surgery Branch at the National Cancer Institute (NCI), Maryland, USA, a 3.3% overall objective response rate was observed10. Results such as these highlight the need to improve current cancer vaccines11 and to develop alternative immunotherapeutic strategies for the treatment of metastatic cancer10.
Cancer vaccines aim to stimulate the adaptive arm of the immune system directly in vivo. ‘Active immunotherapy’ with therapeutic vaccines has been attempted despite the presence of many redundant negative influences of the host immune system5,12 and tumour microenvironment13,14. By contrast, adoptive cell transfer (ACT) therapies achieve T-cell stimulation ex vivo by activating and expanding autologous tumour-reactive T-cell populations to large numbers of cells that are then transferred back to the patient15–17. Early attempts of ACT therapies using tumour-infiltrating lymphocytes (TILs) and immunoreplete patients met with some success18. However, previous preclinical studies indicated that immune ablation is an effective preconditioning regimen that can increase T-cell responses after adoptive transfer19,20. We have now reported that adoptive transfer of TILs after non-myeloablative, but lymphodepleting, systemic chemotherapy (FIG. 1) can induce clear and reproducible responses in a substantial percentage (~50%) of patients21,22. Notably, dramatic tumour regressions can be elicited in patients with multivisceral, bulky melanoma that is refractory to standard treatments including chemotherapy, radiation and cytokine therapies (FIG. 2).
Figure 1. Current clinical protocols for adoptive cell therapy.
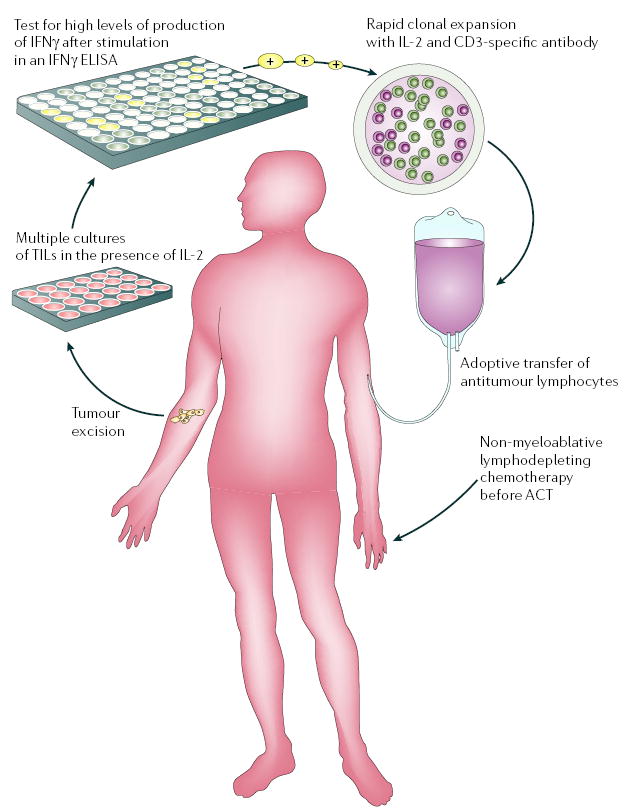
Adoptive cell therapy (ACT) requires the generation of highly avid tumour-antigen-reactive T cells. Tumour-specific T cells, derived from tumour-infiltrating lymphocytes (TILs), can be efficiently isolated ex vivo from melanoma lesions using high levels of interleukin-2 (IL-2). TILs are successively selected for their ability to secrete high levels of interferon-γ (IFNγ) when cultured with autologous or allogeneic MHC-matched tumour-cell lines. Alternatively, cell-mediated lysis has been used to identify tumour-reactive T cells for transfer. Highly avid, tumour-antigen-reactive T-cell populations selected for ACT are rapidly expanded (to up to 1011 cells) using CD3-specific antibody, exogenously supplied IL-2 and irradiated allogeneic peripheral-blood mononuclear ‘feeder’ cells, and are validated for activity before transfer. Patients now receive systemic immunosuppression before the adoptive transfer of antitumour lymphocytes. Published lymphodepleting regimens consist of a non-myeloablative, but lymphodepleting, conditioning chemotherapy comprised of cyclophosphamide and fludarabine before administration of T cells. Newer, as yet unpublished, regimens also include total body irradiation. ELISA, enzyme-linked immunosorbent assay. This figure is reproduced with permission from REF. 12 © (2005) Elsevier Science.
Figure 2. Antitumour response induced by lymphodepletion and adoptive cell therapy.
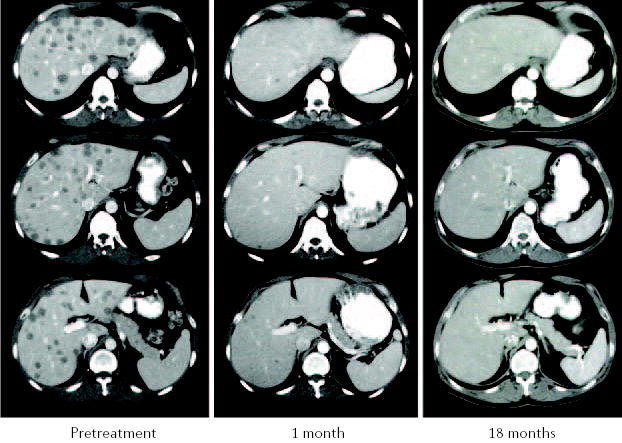
Computed tomography (CT) scans of the liver in a patient with metastatic melanoma show dramatic tumour regression of liver metastases after the administration of tumour-reactive tumour-infiltrating lymphocytes (TILs) following lymphodepletion. The patient is still disease-free after 27 months.
Here, we describe the mechanisms by which the transfer of tumour-reactive T cells into a lymphopaenic recipient mediates tumour regression, and the phenotypic and functional characteristics of tumour-specific T cells that induce antitumour responses in vivo. These factors provide the bases for rational design of new ACT-based immunotherapies that incorporate vaccines, increased intensity preconditioning regimens with haematopoietic stem cell (HSC) transplantation, and alternative methods for the generation and selection of T cells for transfer.
Lymphodepletion increases the efficacy of ACT
It has long been observed in mice that depletion of immune cells before ACT can markedly improve the antitumour efficacy of transferred CD8+ T cells19,20, but the specific mechanisms that contribute to this increased immunity have only recently begun to be elucidated. Although it seems counter-intuitive that the efficacy of ACT-based tumour immunotherapy can be improved by the removal of the host immune system, several mechanisms might underlie the augmented efficacy of tumour-reactive T cells in the lymphopaenic environment. These factors include the elimination of immunosuppressive cells such as CD4+CD25+ regulatory T (TReg) cells, the depletion of endogenous cells that compete for activating cytokines, and the increased function and availability of antigen-presenting cells (APCs) (FIG. 3).
Figure 3. Mechanisms underlying the impact of lymphodepletion on adoptively transferred T cells.
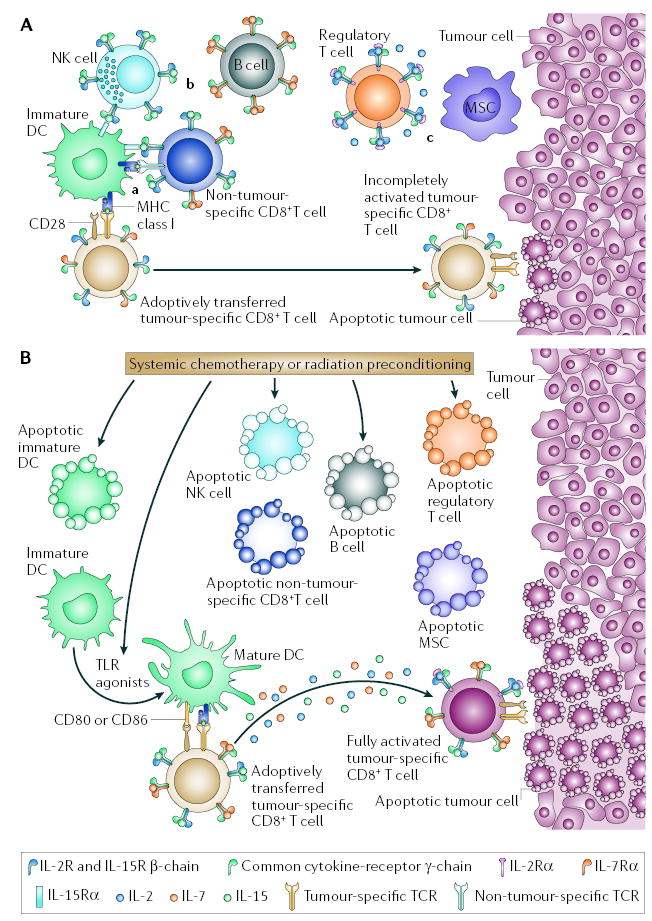
A | Adoptive cell therapy (ACT) in a lymphoreplete host. In a lymphoreplete environment, antitumour responses mediated by adoptively transferred tumour-reactive CD8+ T cells might be reduced because of: a | competition for antigen at the surface of antigen-presenting cells (APCs) and inefficient lymphocyte activation in the absence of co-stimulatory molecules by immature dendritic cells (DCs); b | reduced availability of activating cytokines (including interleukin-2 (IL-2), IL-7 and IL-15) by cellular ‘sinks’ for these cytokines, which include B cells, T cells and natural killer (NK) cells; and c | the suppressive activities of regulatory T (TReg) cells, myeloid suppressor cells (MSCs) and possibly NK cells. TReg-cell suppression is mediated by direct T-cell contact and possibly by the release of inhibitory cytokines such as IL-10 and transforming growth factor-β. MSCs mediate T-cell inhibition through direct T-cell contact and the use of enzymes involved in L-arginine metabolism such as the inducible forms of arginase and nitric-oxide synthase, ARG1 and NOS2. B | Systemic chemotherapy or radiation before ACT might modify the tumour-bearing host. APCs are reduced in number by direct killing but there might be a net increase in lymphocyte activation because of reduced competition for antigen at the APC surfaces. At the same time, as a result of the liberation of Toll-like receptor (TLR) agonists after mucosal damage, DCs might be mature, increasing lymphocyte activation. Activating cytokines, such as IL-2, IL-7 and IL-15 might be increased because of the removal of cellular ‘sinks’; and TReg cells, MSCs, NK cells and their suppressive activities are decreased. These modifications might promote the full activation of adoptively transferred tumour-reactive CD8+ T cells and ultimately tumour destruction.
Elimination of immunosuppressive cells
TReg cells are crucial for the maintenance of peripheral self-tolerance and for the suppression of antitumour responses5. TReg cells represent a unique T-cell lineage that is characterized by expression of the transcription factor forkhead box P3 (FOXP3) and high levels of expression of cell-surface molecules associated with T-cell activation, including CD25 (also known as IL-2Rα ), glucocorticoid-induced tumour-necrosis factor (TNF)-receptor-related-protein (GITR) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA4)5. However, exclusive molecular signatures for human TReg cells do not currently exist because activation of CD4+ T cells can also result in upregulation of FOXP3 expression23. Experiments using mice lacking TReg cells, owing to specific gene defects, as well as the ‘add-back’ of these cells, have convincingly shown that they suppress the antitumour activities of adoptively transferred self/tumour-reactive T cells24. Augmented antitumour responses were observed after ACT of self/tumour-reactive effector CD8+ T cells to tumour-bearing Cd4−/ −, but not Cd8−/ −, mice, indicating that the immunoregulatory cells are contained in the CD4+ T-cell population. The suppressive activity was restricted to the CD25+ T-cell subset because transfer of CD4+CD25+ TReg cells alone, or in combination with CD4+CD25− T helper (TH) cells, inhibited effective immunotherapy in lymphopaenic mice. By contrast, transfer of TH cells alone resulted in profound autoimmunity and eradication of established tumour. Interstingly, the maintenance and function of effector CD8+ T cells required the presence of TH cells that were able to produce interleukin-2 (IL-2)24.
The immunosuppressive role of TReg cells in patients with cancer has only recently begun to be explored. TReg cells, which are over-represented in tumour lesions from patients with melanoma and lung cancer, can inhibit the function of infiltrating T cells25,26, and TReg cells specific for melanoma antigens have been described4. Reduced survival was reportedly associated with increased tumour infiltration by TReg cells in patients with ovarian cancer27, although these findings have been recently contradicted28. Therefore, at present, no conclusive data link the in vivo function of TReg cells and the progression of cancer. Nevertheless, the suppressive effects of TReg cells might contribute to the poor clinical response rates reported in patients with cancer who receive immunotherapy in non-lymphodepleting settings. Selective elimination of TReg cells29 from TILs of patients might further improve the efficacy of ACT approaches in the lymphodepleting setting, because TReg-cell proliferation can be increased by the lymphopaenic environment and the presence of exogenous IL-2 (REFS 30,31). Furthermore, removal of TReg cells from peripheral-blood lymphocytes (PBLs) might generate a population of cells that is enriched for TH cells that are able to bolster the response of self/tumour-specific CD8+ T cells in vivo24.
Other immune cells, including natural killer (NK) cells, natural killer T (NKT) cells and CD11b+Gr1+ myeloid suppressor cells (MSCs), have been shown to dampen T-cell function32–34. Little is known about the immunosuppressive activities of NK and NKT cells, although a perforin-dependent immunosuppressive mechanism has recently been reported for NK cells33. More is known about MSCs, which are a heterogeneous population of cells that comprises myeloid cells at various stages of differentiation, including monocytes, granulocytes and a subset of immature myelo-monocytic cells34. Increased frequencies of MSCs are found in conditions characterized by impaired T-cell function, including tumours and chronic infections34. In mice and humans, MSCs can infiltrate the tumour bed and inhibit T-cell responses through mechanisms involving direct contact with tumour-reactive T cells, L-arginine consumption and the release of L-arginine metabolism products35,36. Depletion of MSCs using a Gr1-specific antibody can result in protection from tumour challenge37. Therefore, removal of MSCs, and thereby their suppressive activity, might contribute to the increased antitumour T-cell responses observed after ACT in patients that have been lymphodepleted.
Minimizing cellular cytokine sinks
Transfer of small numbers of antigen-specific T cells into a lymphopaenic host results in the expansion and activation of the transferred T-cell population, a process that is known as homeostatic proliferation38–40. In the lymphopaenic environment, antigen-experienced T cells proliferate independently of self-peptide–MHC complexes40. However, either co-transferring an ‘irrelevant’ population of T cells or increasing the number of transferred cells can reduce the level of homeostatic proliferation in a dose-dependent manner, showing that other elements exist that limit homeostatic proliferation39–41. Although host-mediated inhibition of the proliferation of adoptively transferred T cells might involve direct cellular contact, competition might also exist between transferred and host T cells for a limited amount of the cytokines that are required to support CD8+ T-cell homeostasis; such competition is known as the ‘cytokine sink’ effect12,42.
The importance of the availability of these cytokines has been shown in experiments in which mice deficient for IL-7 or IL-15 showed impaired homeostatic maintenance and proliferation of memory CD8+ T cells43–45. Conversely, transgenic mice overexpressing IL-7 or IL-15 have increased numbers of T cells, owing to the preferential expansion of the memory CD8+CD44hi T-cell population46,47. In the pmel-1 mouse model of ACT therapy48, lymphodepletion before cell transfer increased the persistence of self/tumour-specific T cells, as well as their effector function and tumour regression, compared with immunoreplete hosts42,49. In mice deficient for both IL-7 and IL-15, the beneficial effect of ablation was completely abrogated42. Conversely, increased antitumour responses were seen when these cytokines were exogenously administered and when the host lymphocytes competing for these cytokines were removed by using mice lacking both recombination-activating gene 2 (Rag2) and γc42. These findings show that a key mechanism underlying the improved efficacy of ACT therapies after lymphodepletion is the transient eradication of endogenous lymphocytes, which serve as cellular cytokine sinks42.
Elucidating the specific endogenous cellular components that function as cytokine sinks is important for understanding the mechanism by which lymphodepletion augments the effectiveness of ACT-based therapies. In Rag1−/ − mice (which, unlike Rag2−/ −γc−/ − mice, lack B cells and T cells but do have NK cells), more extensive tumour regression was observed in irradiated recipients than in non-irradiated recipients, whereas in Rag2−/ −γc −/ − hosts, ACT treatment became so efficacious that it was difficult to detect the effects of irradiation42. This finding, coupled with the observation that depletion of cells expressing NK1.1 (using an NK1.1-specific antibody) improves the efficacy of ACT therapy in tumour-bearing Rag1−/ − mice42, implicates NK cells as key effectors of the cytokine sink effect, a process that might be mediated by consumption of IL-15, a crucial cytokine for the survival and proliferation of NK cells in vivo50,51.
IL-2, another cytokine that signals through a receptor containing γc, is a T-cell growth factor that is commonly used to promote the expansion and function of tumour-specific T-cell populations in vitro and in vivo15. Perhaps more importantly, IL-2 is essential for the maintenance of peripheral self-tolerance52. Mice deficient in either IL-2 or components of the IL-2 receptor spontaneously develop lymphoproliferative and autoimmune disorders52. These observations have been linked to impaired TReg-cell homeostasis and ‘metabolic fitness’ in vivo, rather than suppressive function, because TReg cells from either IL-2- or IL-2Rα (CD25)-deficient mice are competent when tested in in vitro assays of suppressive function53. However, more recent findings have shown that in vivo IL-2 signalling is important not only for maintaining TReg-cell ‘fitness’ but also for their suppressive function54,55. Antitumour activity of adoptively transferred self/tumour-specific CD8+ T cells was inhibited in wild-type, but not IL-2Rα -deficient, mice despite both having comparable numbers of FOXP3+ TReg cells54. Furthermore, blockade of IL-2Rα with specific antibody can induce profound autoimmunity resulting from impaired TReg-cell function, rather than depletion of these cells55. These results indicate that in vivo immunoregulation by TReg cells might, in part, be a product of their constitutive expression of the component of the IL-2 receptor that confers the highest affinity for IL-2, IL-2Rα , and their increased capacity to consume IL-2 (REFS 54,56). Therefore, removal of TReg cells by lymphodepletion might result in increased antitumour reactivity of adoptively transferred CD8+ T cells, not only by the elimination of direct cellular inhibition but also through increased availability of IL-2.
Improved APC function and availability
Systemic chemotherapy and total body irradiation have both been used before ACT to deplete the lymphoid compartment of the host and create a niche for the transferred cells. Investigators have long hypothesized that these treatments might also cause necrosis or apoptosis of tumour cells, resulting in APC uptake of tumour antigens and the subsequent cross-presentation of these antigens to the adoptively transferred tumour-reactive CD8+ T cells57. Although lymphodepletion can reduce the absolute number of APCs in vivo, it can also promote their transition to an activated state58,59. In mice, the expression of the activation markers CD86 and I-Ab (an MHC class II molecule) has been reported to be upregulated on the surface of splenic dendritic cells (DCs) after irradiation59. Furthermore, DCs that were isolated after irradiation released substantially more IL-12 than DCs that were isolated from non-irradiated mice59. Activation of DCs after chemotherapy or irradiation might be triggered by translocation of bacterial products, such as lipopolysaccharide (LPS) and other Toll-like receptor (TLR) agonists, into the blood following damage to the integrity of mucosal barriers60. The production of pro-inflammatory cytokines such as TNF, IL-1 and IL-4 by host cells might also be involved in mediating DC maturation58,61–63. In addition, the lymphopaenic environment might facilitate the activation of transferred self/tumour-reactive T cells through decreased competition at the surface of antigen-bearing APCs64. Although the net effect of lymphodepletion on APC function is less clear than its impact on TReg cells and cellular cytokine sinks, ablation might ultimately increase the antitumour reactivity of transferred T cells by increasing the activation and availability of APCs.
T-cell differentiation state and ACT
Lymphodepletion can have a marked impact on treatment with ACT-based immunotherapies, but it is not the only factor responsible for affecting clinical responses. Emerging findings from both mouse studies and clinical trials indicate that intrinsic properties related to the differentiation state of the adoptively transferred T-cell populations are crucial to the success of ACT-based approaches65–68.
CD8+ T-cell subsets in both mice and humans can be categorized into distinct differentiation states on the basis of phenotypic and functional attributes69,70 (FIG. 4). A progressive pathway of CD8+ T-cell differentiation69,70 has gained acceptance based on the findings of ex vivo phenotypic analyses of virus-specific T cells69, measurement of telomere length71, gene-expression profiling72 and in vitro differentiation studies65,71. Within this model, activation of naive CD8+ T cells results in proliferation and progressive differentiation through early, intermediate and late effector stages depending on signal strength70 (FIG. 4). Memory CD8+ T cells might reflect T cells arrested at intermediate stages of the differentiation pathway73,74, but there remains some debate regarding the pathways by which effector and memory T cells develop75.
Figure 4. Inverse relationship of in vitro and in vivo antitumour functions of adoptively transferred naive and effector T-cell subsets.
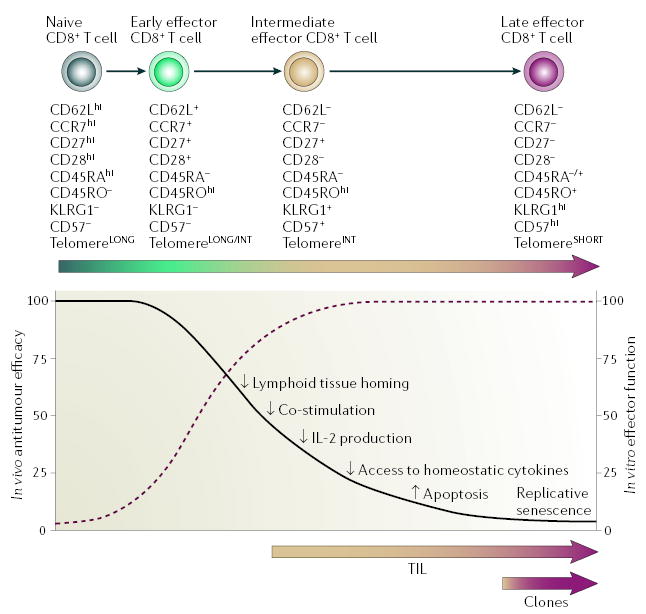
At increasing strength of stimulation, naive CD8+ T cells proliferate and progressively differentiate through early, intermediate and late effector stages. The phenotypic and functional changes that characterize this process are illustrated as no expression (–), intermediate expression (+) and high expression (hi) of the various markers. T cells progressively lose telomere length and proliferative potential, and subsequently become senescent and undergo apoptosis. The progressive acquisition of full effector functions (dashed burgundy line) is associated with a decreased ability of T cells to cause tumour regression after adoptive transfer (black line). The molecular mechanisms underlying this inverse correlation might be comprised of: decreased expression by T cells of lymph-node homing and co-stimulatory molecules, which reduce activation of T cells in vivo; the inability of terminally differentiated T cells to produce interleukin-2 (IL-2); a reduction in the amount of receptors required to receive activating signals from homeostatic cytokines; and finally, an inversion of the expression of pro- and anti-apoptotic molecules with the corresponding acquisition of replicative senescence. Adoptively transferred tumour-infiltrating lymphocytes (TILs) contain several clonotypes with a differentiation state ranging between intermediate and late effector stages, whereas tumour-reactive CD8+ T-cell clones are uniformly late effector T cells. KLRG1, killer-cell lectin-like receptor G1. This figure is reproduced with permission from REF. 65 © (2005) Highwire Press.
The phenotypic and functional characteristics of self/tumour-specific CD8+ T cells that are associated with optimal in vivo tumour responses in the pmel-1 mouse model of ACT therapy have recently been elucidated65. Self/tumour-specific CD8+ T cells at progressive stages of differentiation were generated using multiple in vitro stimulations with antigen. Surprisingly, CD8+ T cells that acquired terminal effector properties and had increased antitumour activity in vitro were found to be less effective at triggering tumour regression in vivo. Terminally differentiated CD8+ T cells were nearly 100-fold less effective in vivo on a per-cell basis than T cells at an early stage of differentiation. Similar findings have been reported by other groups using different mouse tumour76,77 and allogeneic HSC transplantation78,79 models. In vitro expansion of T cells for ACT — as it is currently performed for clinical use — induces progressive CD8+ T-cell differentiation towards a late effector state, resulting in phenotypic and functional changes that make T cells less ‘fit’ to mediate antitumour responses in vivo and less able to benefit from the activating cues present in the lymphopaenic host (FIG. 4). For example, less-differentiated, central-memory-like T cells have a high proliferative potential, are less prone to apoptosis than more differentiated cells and have a higher ability to respond to homeostatic cytokines, ,ecause they express receptors such as the IL-7 receptor α -chain (IL-7Rα)65,75,80,81. Therefore, less-differentiated, central-memory-like T cells might proliferate and become fully activated in the lympho paenic environment, which is rife with homeostatic cytokines such as IL-7.
Similar to mouse studies, ACT of human tumour-reactive CD8+ T-cell clones that were generated and expanded ex vivo through multiple stimulations did not meditate objective responses in either immunoreplete16 or immunodepleted patients82. T-cell clones used for therapy were highly avid and showed potent tumour-specific cytolytic activity in vitro, but they did not persist after infusion, indicating that they were in a state of terminal differentiation16,82 (FIG. 4).
The importance of trafficking to lymph nodes
Tumour immunologists have long sought to cause T cells to specifically traffic to their tumour targets83,84. The loss of expression of the lymphoid homing molecule CD62L and the acquisition of CD44 expression were reported to be associated with increased antitumour effects of adoptively transferred T cells76,85. However, it is now clear, in both tumour and viral models, that T cells that can home to secondary lymph nodes, where they can be effectively stimulated by DCs, are more effective in adoptive immunotherapy65,75,81. Indeed, tumours alone are inefficient at triggering effective immune responses65,81,86. Antitumour responses were abrogated in hosts devoid of peripheral lymphoid tissues and with a disrupted splenic structure81. Furthermore, after transfer, CD62L-deficient self/ tumour-reactive CD8+ T cells were markedly impaired in their ability to inhibit tumour growth compared with CD62L-sufficient T cells65,81. Similarly, CD62L-deficient T cells were less effective at mediating alloresponses in an allogeneic HSC transplantation model78. Therefore, downregulation of expression of lymph-node homing molecules at the intermediate and late stages of effector CD8+ T-cell differentiation can result in impairment of their antitumour capacity. However, the principle that T cells must home to lymph nodes to be effective has not been established in humans. Despite the lack of expression of lymph-node homing molecules, adoptively transferred CD62L−CCR7− TILs87 were able to engraft, proliferate and ultimately induce objective responses in ~50% of patients21,22.
Co-stimulatory molecules and T-cell persistence
Transition from an early to an intermediate effector stage is marked by downregulation of CD28 expression. Interaction of CD28 with CD80 and/or CD86 on APCs amplifies T-cell receptor (TCR)-mediated T-cell activation and proliferation88. Secretion of IL-2, induction of anti-apoptotic molecules and accelerated cell-cycle progression have been reported for CD28-expressing T cells88,89. The role of CD28 expression in ACT-based clinical trials has been recently investigated in detail. Tumour-specific TILs express low, but detectable, levels of CD28 (REF. 87). After cell infusion, immediate and high expression of CD28 was detected on circulating tumour-reactive T cells, indicating that either rapid upregulation of CD28 expression or early selective expansion and survival of the CD28+ T-cell population had occurred. Analysis of persisting and non-persisting TIL clones indicates preferential survival of the clonotypes expressing the highest levels of CD28, implicating a survival advantage for transferred T cells with an early effector phenotype67,68.
Engagement of the co-stimulatory molecule CD27 can also augment TCR-induced T-cell proliferation and is required for the generation and maintenance of memory T cells in vivo90,91. Consistent with a late effector state, T cells lacking CD27 have been shown to have potent cytolytic function and secrete little IL-2 (REF. 75). In the pmel-1 mouse model, self/tumour-specific late effector cells were less effective at mediating tumour regression after adoptive transfer relative to early effector T cells that express high levels of CD27 (REF. 65). Moreover, the administration of soluble CD27 ligand, CD70, augmented in vivo CD8+ T-cell responses to viral infection and tumour challenge by increasing the expansion and maintenance of the antigen-specific T-cell population, indicating that CD27 expression is not only a marker of less-differentiated T cells but also functionally crucial for optimal immune responses92. In the clinical arena, a statistically significant difference in the frequency and number of CD27-expressing cells could be found in bulk TIL populations from responding versus non-responding patients when IL-2 was withdrawn93. After ACT, the frequency of TILs expressing CD27 gradually increased and was associated with the long-term maintenance of stable numbers of tumour-specific T cells in responding patients87. This result, and findings from viral studies, predicts that T cells that express CD27 selectively persist in vivo, giving rise to a stable population of memory CD8+ T cells87,94.
Homeostatic cytokine signals and T-cell persistence
Increased access to homeostatic cytokines has been shown to be crucial for the enhanced antitumour responses that occur following ACT to lymphodepleted hosts8,12,42. Homeostatic signals can be regulated by both the availability of cytokines in the host and the level of expression of the cytokine receptors on the surface of transferred CD8+ T cells. Expression of IL-7Rα by a subset of effector CD8+ T cells might identify precursors that are destined to become long-lived memory cells80. IL-7Rα low self/tumour-specific late effector CD8+ T cells transferred to tumour-bearing mice persisted at decreased numbers and were less effective at inducing antitumour responses than were IL-7Rα hi early effector CD8+ T cells65. In patients, IL-7Rα was expressed at low levels on all TIL populations at the time of ACT, but it was upregulated immediately after infusion on the surface of robustly proliferating tumour-specific T cells that persisted87. Therefore, IL-7 signalling seems to be important for the immediate and long-term survival of tumour-specific T cells after ACT.
IL-15Rα was weakly expressed by most TILs used for ACT and, unlike IL-7Rα, was not upregulated on persisting tumour-specific T cells after ACT87. However, IL-15 signalling might remain intact because trans-presentation of IL-15 by APCs and stromal cells can occur95.
T-cell persistence and antitumour response
Because IL-2 is provided both in vitro during expansion of T-cell populations and in vivo in the immediate aftermath of cell infusion, tumour-reactive CD8+ T cells might undergo apoptosis after IL-2 withdrawal96. Because early effector T cells have the capacity to release IL-2, selective survival of these cells might occur in an autocrine fashion65. In addition, early effector T cells have survival advantages over intermediate and late effector T cells, as reflected by the expression of lower levels of the pro-apoptotic molecules BID (B-cell lymphoma 2 (BCL-2)-homology domain 3 (BH3)-interacting-domain death agonist), BAD (BCL-2-antagonist of cell death) and CD95L (CD95 ligand; also known as FASL), and higher levels of anti-apoptotic molecules65,81. The intrinsic proliferative capacity of adoptively transferred T cells might also affect their ability to engraft and persist. Increased proliferation of the early effector T-cell subset has been seen in vitro and in vivo following stimulation with cognate antigen65. In parallel with T-cell proliferation and progressive differentiation, gradual telomere erosion occurs until a critical degree of shortening (termed the Hayflick limit) results in chromosomal abnormalities, and cell death or senescence97. This process might be partially compensated for by telomerase activity97. Therefore, telomere length and telomerase activity can influence T-cell replicative capacity. Interestingly, recent analyses of human TILs have shown a correlation between the length of the telomeres of the transferred cells and persistence of T cells in vivo following ACT, indicating that in addition to tumour-antigen recognition, the intrinsic proliferative capacity of adoptively transferred T cells might also be a factor affecting persistence and successful tumour treatment68.
Optimizing tumour-reactive T cells for ACT
The finding that less-differentiated, central-memory-like T cells might be the optimal population for ACT-based immunotherapies raises a clinical problem. Data from animal studies indicate a direct correlation between the number of adoptively transferred T cells and antitumour responses in vivo, leading to the idea that large numbers of tumour-reactive T cells must be administered to patients to obtain therapeutically effective antitumour responses65,76. Therefore, in clinical trials, tumour-reactive CD8+ T-cell populations are expanded to large numbers in vitro with CD3-specific antibody plus IL-2 or with specific-antigen plus IL-2, which drives differentiation of T cells to intermediate and late effector stages of differentiation16,22,82. New findings in mice emphasize that the quantity of transferred T cells is an important factor when T cells with the same quality and fitness are being used for ACT. Increased antitumour responses were observed after adoptive transfer of low numbers of ‘fit’ (early effector) T cells compared with high numbers of ‘unfit’ (late effector) T cells65,76. Therefore, one of the greatest challenges in the field is currently the generation of large numbers of ‘fit’ T cells for ACT.
Modifications of current in vitro protocols for expanding T-cell populations
Using a standard rapid expansion protocol, TILs for transfer are selected and populations are expanded for about 2 weeks with CD3-specific antibody, high doses of IL-2 and irradiated allogeneic feeder cells22 (FIG. 1). This procedure results in the differentiation of tumour-specific CD8+ T cells to an intermediate and late effector state. Limiting the in vitro expansion phase to a short duration might markedly improve the ‘fitness’ of the transferred T cells because a greater percentage of tumour-reactive T cells express CCR7, co-stimulatory molecules and IL-7Rα , and are actively dividing in the first week of growth98. The question remains whether this improved fitness can compensate for the reduced number of cells generated soon after activation.
Cytokines, acting in concert with signals through the TCR and co-stimulatory molecules, can function as accelerators or brakes for T-cell proliferation and differentiation70. IL-2 has been shown to be an effective T-cell growth factor but has undesirable effects, including the ability to decrease the expression of lymph-node homing molecules and to promote the terminal differentiation of T cells, predisposing them to activation-induced cell death65,99. Other cytokines that signal through a receptor that contains γc, such as IL-15, can analogously induce the in vitro expansion of tumour-reactive CD8+ T-cell populations for ACT8,65,100. IL-15 supports the growth of similar numbers of T cells as IL-2, but it does not induce the detrimental T-cell differentiation and apoptosis that IL-2 does65,101,102. T-cell populations that had been expanded in the presence of IL-15 were shown to have a superior ability to elicit tumour regression in vivo after ACT to tumour-bearing mice, compared with T-cell populations that had been expanded in the presence of IL-2 (REFS 8,65,81). Other cytokines that signal through a receptor that contains γc (including IL-7 and IL-21) that were evaluated in a similar manner did not promote robust proliferation or differentiation of self/ tumour-reactive CD8+ T cells in vitro, but they had a greater antitumour efficacy than IL-2 treated cells in vivo (Hinrichs C. S., unpublished observations). By contrast, no differences in the differentiation state of tumour-reactive T cells from vaccinated patients were detected when the cells were stimulated ex vivo with cognate antigen in the presence of IL-2, IL-7 or IL-15 (REF. 103). Results obtained using human cells probably reflect the use of antigen-experienced T cells that have already differentiated into intermediate and late effector stages, instead of the naive populations that are used in mouse studies104. Indeed, stimulation of naive human tumour-reactive T cells in the presence of IL-21 induced the preferential expansion of a less-differentiated CD28hiCD45RO+ T-cell population that could release IL-2 after stimulation with cognate antigen9. Therefore, the ability to obtain naive tumour-specific CD8+ T cells might be of paramount importance to improving current ACT-based therapies.
Genetic modification of T cells for ACT
Naturally occurring self/tumour-specific T cells have been described in patients with cancer, as well as in healthy individuals105,106. Antigen-experienced CCR7−CD45RA−CD45RO+ self/tumour-specific T cells are preponderant in the metastatic lymph nodes and are uniformly present at tumour sites, whereas naive self/tumour-specific T cells are predominantly found in the blood106. Unfortunately, these naive cells are mainly characterized by a low TCR avidity, thereby making them unsuitable for ACT107. To circumvent this issue, high-affinity TCRs, derived from TILs that mediate strong in vivo tumour regression, have been identified, cloned and transduced into the PBLs of patients with cancer108–110. These TCR-transduced PBLs have cytolytic activity, secrete cytokines in vitro after stimulation with melanoma-cell lines and are currently being clinically evaluated109,110.
The affinity of the TCR selected for transduction, the level of transduced TCR expressed on the cell surface and the differentiation state of the transduced T cells that are used for ACT might contribute to the success of trials following TCR transduction. Naturally occurring T cells that express high-affinity TCRs specific for self/ tumour antigens might be difficult to obtain owing to intrathymic deletion. However, high-affinity TCRs can be generated in vivo in immunized HLA-A2-transgenic mice111,112 or in vitro by phage display of TCRs containing degenerate complementarity-determining regions113.
Integration of retrovirally delivered sequences requires active division of target cells, a process that also promotes T-cell differentiation (FIG. 5a). As PBLs contain T cells at multiple stages of differentiation, inducing activation and proliferation of PBLs guarantees that TCR-transduced T cells are largely devoid of naive T cells. Alternatively, as lentiviral vectors are less dependent on active cell division, they might be used to transduce high-affinity TCRs into T cells without driving differentiation114. Lentiviral transduction of T cells that are pre-selected for specific markers might therefore be a way of generating large numbers of naive tumour-specific T cells for ACT (FIG. 5b).
Figure 5. Generation of less-differentiated, central-memory-like tumour-antigen-specific CD8+ T cells by TCR transduction.
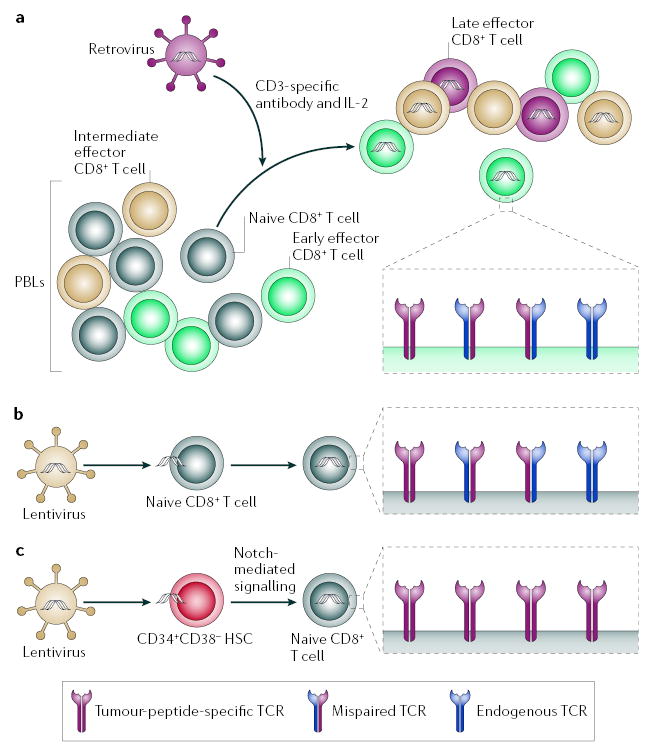
a | Retroviral transduction of peripheral-blood lymphocytes (PBLs). PBLs at different stages of differentiation, naive (grey), early (green), intermediate (beige) and late effector (burgundy) are activated in vitro with CD3-specific antibody in the presence of interleukin-2 (IL-2) to promote integration of tumour-specific T-cell receptor (TCR) retroviral constructs. This procedure results in the generation of more-differentiated TCR transductants. Pairing with endogenous receptor can reduce the number of tumour-specific TCRs. b | Lentiviral transduction of naive CD8+ T cells. Naive CD8+ T cells isolated through selective sorting can be transduced with tumour-specific TCR by using lentiviral constructs that do not require activation and consequent differentiation. Pairing with endogenous receptor can reduce the number of tumour-specific TCRs. c | Lentiviral transduction of haematopoietic stem cells (HSCs). CD34+CD38− HSCs isolated though selective sorting can be transduced with tumour-specific TCR using lentiviral constructs. HSCs can be induced to differentiate into naive CD8+ T cells in vitro through Notch-mediated signalling. Repression of recombination-activating genes by the transduced tumour-specific TCR allows for the uniform expression of tumour-specific TCRs.
Delivery of both the TCR α -chain (TCRα) and β-chain (TCRβ) directs the expression of the intact TCR; however, pairing with endogenous TCRα and TCRβ can occur, thereby reducing the surface density of tumour-specific TCR (FIG. 5a,b). Transduction of HSCs followed by T-cell-lineage differentiation — through in vitro Notch signalling115,116 or through natural development in vivo in immunodeficient mice117 — is an attractive approach to overcome this problem. Forced expression of transduced TCRs by differentiating HSCs facilitates the repression of expression of the Rag genes, such that endogenous TCRβ are not expressed118 (FIG. 5c). Alternative approaches to overcome the problem of mispairing with endogenous TCRα and TCRβ might include the manipulation of the transmembrane-association domains of TCRα and TCRβ119, the use of chimeric receptors with antibody specificity (known as T-bodies)120 and TCR transduction into T cells that lack an α βTCR, such as γδ T cells121.
Several other genes have been proposed for transduction of tumour-reactive T cells to improve their quality and functionality122. These include co-stimulatory molecules89, anti-apoptotic molecules123, pro-inflammatory or homeostatic cytokines96,102 and chemokine receptors84. Although these manipulations are able to alter specific cell functions in differentiated tumour-specific T cells, the TCR approach confers self/tumour-specificity to cells that might have all the desired characteristics. Transduction with genes encoding TCRs specific for known epitopes allows the concurrent use of vaccines to potentiate the antitumour response of adoptively transferred T cells124–126. Another interesting approach includes the modulation of transcription factors such as BCL-6 (REFS 127,128), BCL-6B129, lymphoid-enhancer-binding factor 1 (LEF1) and T-cell factor 7 (TCF7)130 in intermediate and late effector tumour-reactive T cells that might revert T cells to a less-differentiated state131.
Concluding remarks
ACT to a lymphodepleted host has emerged as a promising advance in cancer immunotherapy. Preclinical and clinical studies have identified multiple mechanisms contributing to successful adoptive immunotherapies, including host-related factors, as well as the phenotypic and functional characteristics of the tumour-reactive T cells used for transfer. These findings provide the rationale for the design of new clinical protocols for the treatment of patients with cancer.
The improved effectiveness of immunotherapy following a non-myeloablative lymphodepleting regimen provides the rational basis for the evaluation of more intensive conditioning regimens such as a myeloablative regimen in conjunction with autologous HSC transplantation132. In the pmel-1 mouse model of ACT therapy, the use of a myeloablative regimen profoundly depleted host immunosuppressive cells and cellular sinks for activating cytokines, resulting in an increased ratio of effector cells to endogenous cells and increased anti-tumour responses compared with non-myeloablative conditioning (C. Wrzesinski, unpublished observations). The improved therapeutic effect was independent of antigen-specific vaccination but required the transfer of HSCs, which increased the proliferation and survival of co-administered self/tumour-reactive T cells, possibly through the release of cytokines, growth factors and anti-apoptotic factors (C. Wrzesinski, unpublished observations). The finding that myeloablative conditioning with a HSC transplant removed the need for specific vaccination has important implications for ACT-based immunotherapies in humans, which use polyclonal TILs for which the specificity is often unknown and for which effective vaccines are not currently available10. The use of a myeloablative preconditioning regimen involving chemotherapy and total body irradiation together with HSC transplantation in humans is currently under evaluation.
Increased immunity might be achieved with the use of more selective approaches to lymphodepletion to eliminate the toxicities associated with the use of non-specific preconditioning regimens based on chemotherapy and radiation. For example, TReg cells and other immunosuppressive cells might be selectively depleted with directed immunotoxins or suppressed by administering a cytokine such as TNF133–136. To overcome the sink effect of competing endogenous cells, saturating levels of activating cytokines might be provided exogenously137. Because IL-2 can promote TReg-cell proliferation and suppressive function, other cytokines that signal through a receptor that contains γc, such as IL-7, IL-15 and IL-21, might be preferable12,42. Alternatively, administration of IL-2–IL-2-specific antibody complexes could be used to selectively stimulate effector T cells rather than TReg cells138. Moreover, TH cells that can produce many cytokines might be co-transferred with self/tumour-reactive T cells24. Finally, APCs might be activated through selective ligation of activation-associated molecules such as TLRs139. The use of combinatorial approaches might be of greater clinical benefit than single modality strategies.
Mouse models have now shown that early effector T cells mediate better in vivo antitumour responses than intermediate and late effector T cells on the basis of their increased proliferative and survival potential, receptiveness to homeostatic and co-stimulatory signals, homing to secondary lymphoid tissues and ability to secrete IL-2 (REF. 65). In humans, mounting evidence seems to support the preclinical finding that less-differentiated T cells are the ideal cells for ACT66–68. Taken together, these findings indicate that the current criteria for selection of T cells for ACT, including release of interferon-γ or cytolysis alone, are sub-optimal. Consideration of other important factors for selection such as phenotype, telomere length, alternative cytokine production (such as IL-2) and TCR affinity are currently being investigated. The next generation of ACT-based immunotherapies might rely on the ability to endow ‘fit’ cells with elevated cell-surface expression of high-affinity, self/tumour-specific TCRs by gene-transfer technology that can be used in conjunction with specific vaccines48,124–126. Ultimately, the TCR gene-therapy approach might hold the key to the widespread application of ACT-based therapy to the treatment of cancers of multiple histologies110,112.
Note added in proof
It has recently been shown that T cells also express the transcriptional repressor B-lymphocyte-induced maturation protein 1 (BLIMP1)140,141. This provides a further potential transcription-factor target to modulate in an attempt to generate less differentiated T cells.
Common cytokine-receptor γ-chain
(γc). A signalling subunit of the receptors for interleukin-2 (IL-2), IL-4, IL-7, IL-9, IL-15 and IL-21.
Standard oncological criteria
Clinical criteria that determine whether or not a treatment for cancer is effective. The World Health Organization originally defined an objective clinical response as a 50% decrease in the sum of the products of perpendicular diameters of all lesions without an increase greater than 25% in any lesions or appearance of new lesions. Subsequent updated criteria are known as response evaluation criteria in solid tumours (RECIST). RECIST defines an objective clinical response as a 30% decrease in the sum of the longest diameters of target lesions, without an increase greater than 20% in any target lesions or appearance of new lesions.
Tumour-infiltrating lymphocytes
(TILs). The heterogeneous population of T cells found in a tumour bed. These cells are characterized by a diversity of phenotypes, antigen specificities, avidities and functional characteristics. They can be activated and expanded ex vivo and re-infused into the tumour-bearing host.
Non-myeloablative regimen
Treatment that induces a severe, but transient, leukopaenia without permanent damage to haematopoietic stem cells, thereby allowing spontaneous recovery of the haematological function of the host.
Homeostatic proliferation
A process of activation and proliferation of leukocytes in the lymphopaenic environment. T-cell homeostatic proliferation is driven by T-cell receptor interactions with self-peptide–MHC complexes and T-cell responsiveness to cytokines such as interleukin-7 (IL-7), IL-15 and possibly IL-21.
Pmel-1 mouse model of ACT
A mouse model of adoptive cell transfer (ACT) therapy for established B16 melanomas and autoimmunity against the melanocyte-associated differentiation antigen gp100. Treatment consists of adoptive transfer of gp100-specific CD8+ T cells derived from the T-cell receptor (TCR) transgenic mouse pmel-1 in combination with altered ligand vaccine and cytokines that signal through a receptor that contains the common cytokine-receptor γ-chain (γc).
Cross-presentation
The process whereby antigen-presenting cells take up, process and present extracellular antigens, in association with MHC class I molecules, to CD8+ T cells.
Toll-like receptor
A member of the family of evolutionarily conserved receptors that was first described in Drosophila melanogaster. These receptors mediate innate immunity and inflammatory responses that can subsequently modulate adaptive immunity in mammals.
Trans-presentation
A process by which the interleukin-15 receptor α -chain (IL-15Rα ) presents active IL-15 in trans to opposing cells expressing a complex, with a low affinity for IL-15, that contains IL-15Rα and the common cytokine-receptor γ-chain (γc), thereby transducing a signal.
Telomere
The segment at the end of chromosome arms, which consists of a series of repeated DNA sequences (TTAGGG in all vertebrates) that regulate chromosomal replication at each cell division.
Telomerase
A ribonucleoprotein enzyme that uses its internal RNA component as a template to synthesize telomeric DNA directly onto the ends of chromosome arms.
Phage display
A technique in which bacteriophages are engineered to express on their cell surface a fusion protein comprised of a foreign peptide or protein and their capsid proteins.
Complementarity-determining region
The hypervariable amino-acid sequences in T-cell-receptor variable regions that interact with complementary amino acids on the peptide–MHC complex.
Myeloablative regimen
Treatment that causes severe bone-marrow suppression requiring administration of haematopoietic stem cells to reconstitute the haematological function of the host and to assure host survival.
Acknowledgments
This work was supported by the Intramural Research Program of the Center for Cancer Research, NCI, NIH. The authors would like to thank all the members of the translational immunology team at the NCI especially M. E. Dudley, J. C. Yang, P. F. Robbins, R. A. Morgan, R. M. Sherry, M. R. Parkhurst, J. R Wunderlich and S. L. Topalian.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
DATABASES
The following terms in this article are linked online to:
Entrez Gene:
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene γc | CD4 | CD8 | CD25 | CD28 | CD62L | CCR7 | FOXP3 | IL-2 | IL-7 | IL-15 | IL-7α | IL-15α
FURTHER INFORMATION
Nicholas Restifo’s laboratory
http://ccr.cancer.gov/staff/staff.asp?profileid=5762
Steven Rosenberg’s laboratory
http://ccr.cancer.gov/staff/staff.asp?profileid=5757
Access to this links box is available online.
References
- 1.Boon T, Coulie PG, Van Den Eynde BJ, Van Der BP. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 3.Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumour immunity. Curr Opin Immunol. 1998;10:588–594. doi: 10.1016/s0952-7915(98)80228-8. [DOI] [PubMed] [Google Scholar]
- 4.Wang RF, Peng G, Wang HY. Regulatory T cells and Toll-like receptors in tumour immunity. Semin Immunol. 2006;18:136–42. doi: 10.1016/j.smim.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nature Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 6.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell co-stimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 7.Chambers CA, Allison JP. Co-stimulation in T cell responses. Curr Opin Immunol. 1997;9:396–404. doi: 10.1016/s0952-7915(97)80087-8. [DOI] [PubMed] [Google Scholar]
- 8.Klebanoff CA, et al. IL-15 enhances the in vivo antitumour activity of tumour-reactive CD8+ T cells. Proc Natl Acad Sci USA. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. 2005;175:2261–2269. doi: 10.4049/jimmunol.175.4.2261. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature Med. 2004;10:909–915. doi: 10.1038/nm1100. This paper highlights the ineffectiveness of current cancer vaccine strategies and the need to develop alternative immunotherapeutic strategies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waldmann TA. Effective cancer therapy through immunomodulation. Annu Rev Med. 2006;57:65–81. doi: 10.1146/annurev.med.56.082103.104549. [DOI] [PubMed] [Google Scholar]
- 12.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumour immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schreiber H, Wu TH, Nachman J, Kast WM. Immunodominance and tumour escape. Semin Cancer Biol. 2002;12:25–31. doi: 10.1006/scbi.2001.0401. [DOI] [PubMed] [Google Scholar]
- 14.Khong HT, Restifo NP. Natural selection of tumour variants in the generation of ‘tumour escape’ phenotypes. Nature Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nature Rev Cancer. 2003;3:666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yee C, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumour effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bollard CM, et al. Cytotoxic T lymphocyte therapy for Epstein–Barr virus+ Hodgkin’s disease. J Exp Med. 2004;200:1623–1633. doi: 10.1084/jem.20040890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg SA, et al. Treatment of patients with metastatic melanoma with autologous tumour-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–1166. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 19.Cheever MA, Greenberg PD, Fefer A. Specificity of adoptive chemoimmunotherapy of established syngeneic tumours. J Immunol. 1980;125:711–714. This pioneering paper reports the increased antitumour efficacy of tumour-reactive T cells in a lymphodepleted host. [PubMed] [Google Scholar]
- 20.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumour depends on elimination of tumour-induced suppressor T cells. J Exp Med. 1982;155:1063–1074. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudley ME, et al. Cancer regression and autoimmunity in patients after clonal repopulation with ntitumour lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. This paper describes the first successful clinical trail of ACT with TILs following non-myeloablative chemotherapy for the treatment of patients with melanoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dudley ME, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker MR, et al. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+ J Clin Invest. 2003;112:1437–1443. doi: 10.1172/JCI19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antony PA, et al. CD8+ T cell immunity against a tumour/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. This paper elucidates the role of CD4+CD25+T cells in preventing an otherwise productive antitumour immune response against an established syngeneic tumour. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo EY, et al. Regulatory CD4+CD25+T cells in tumours from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–4772. [PubMed] [Google Scholar]
- 26.Viguier M, et al. Foxp3 expressing CD4+CD25high regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;173:1444–1453. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- 27.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Med. 2004;10:942–949. doi: 10.1038/nm1093. This paper was the first to correlate the presence of TRegcells and clinical outcome in patients with cancer. [DOI] [PubMed] [Google Scholar]
- 28.Sato E, et al. Intraepithelial CD8+ tumour-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favourable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powell DJ, Jr, Parker LL, Rosenberg SA. Large-scale depletion of CD25+ regulatory T cells from patient leukapheresis samples. J Immunother. 2005;28:403–411. doi: 10.1097/01.cji.0000170363.22585.5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nature Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 31.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+CD25hi Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–2414. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol. 2005;23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742. [DOI] [PubMed] [Google Scholar]
- 33.Beilke JN, Kuhl NR, Van Kaer L, Gill RG. NK cells promote islet allograft tolerance via a perforin-dependent mechanism. Nature Med. 2005;11:1059–1065. doi: 10.1038/nm1296. [DOI] [PubMed] [Google Scholar]
- 34.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nature Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez PC, et al. Arginase I production in the tumour microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 36.Bronte V, et al. Boosting antitumour responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. 2005;201:1257–1268. doi: 10.1084/jem.20042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seung LP, Rowley DA, Dubey P, Schreiber H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumour rejection. Proc Natl Acad Sci USA. 1995;92:6254–6258. doi: 10.1073/pnas.92.14.6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho BK, Rao VP, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med. 2000;192:549–556. doi: 10.1084/jem.192.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 41.Dummer W, Ernst B, LeRoy E, Lee D, Surh C. Autologous regulation of naive T cell homeostasis within the T cell compartment. J Immunol. 2001;166:2460–2468. doi: 10.4049/jimmunol.166.4.2460. [DOI] [PubMed] [Google Scholar]
- 42.Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumour-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. This paper establishes the direct role of the endogenous homeostatic cytokines IL-7 and IL-15 in increasing CD8+T-cell effector functions in a lymphodepleted environment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nature Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 44.Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 2000;288:675–678. doi: 10.1126/science.288.5466.675. [DOI] [PubMed] [Google Scholar]
- 45.Tan JT, et al. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. 2002;195:1523–1532. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kieper WC, et al. Overexpression of interleukin (IL)-7 leads to IL-15-independent generation of memory phenotype CD8+ T cells. J Exp Med. 2002;195:1533–1539. doi: 10.1084/jem.20020067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marks-Konczalik J, et al. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci USA. 2000;97:11445–11450. doi: 10.1073/pnas.200363097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Overwijk WW, et al. Tumour regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang LX, et al. Interleukin-7-dependent expansion and persistence of melanoma-specific T cells in lymphodepleted mice lead to tumour regression and editing. Cancer Res. 2005;65:10569–10577. doi: 10.1158/0008-5472.CAN-05-2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prlic M, Blazar BR, Farrar MA, Jameson SC. In vivo survival and homeostatic proliferation of natural killer cells. J Exp Med. 2003;197:967–976. doi: 10.1084/jem.20021847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koka R, et al. Interleukin (IL)-15Rα -deficient natural killer cells survive in normal but not IL-15R α -deficient mice. J Exp Med. 2003;197:977–984. doi: 10.1084/jem.20021836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signalling is required for CD4+regulatory T cell function. J Exp Med. 2002;196:851–857. doi: 10.1084/jem.20020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nature Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 54.Antony, P. A. et al. Interleukin-2 dependent mechanisms of tolerance and immunity in vivo. J. Immunol. (in the press)References 53 and 54 highlight the role of IL-2 in maintaining the homeostasis and competitive fitness of TRegcellsin vivo. [DOI] [PMC free article] [PubMed]
- 55.Kohm AP, et al. Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J Immunol. 2006;176:3301–3305. doi: 10.4049/jimmunol.176.6.3301. [DOI] [PubMed] [Google Scholar]
- 56.de la RM, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2480–2488. doi: 10.1002/eji.200425274. [DOI] [PubMed] [Google Scholar]
- 57.Russo V, et al. Dendritic cells acquire the MAGE-3 human tumour antigen from apoptotic cells and induce a class I-restricted T cell response. Proc Natl Acad Sci USA. 2000;97:2185–2190. doi: 10.1073/pnas.040540197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown S, Konopa J, Zhou D, Thompson J. Expression of TNFα by CD3+ and F4/80+ cells following irradiation preconditioning and allogeneic spleen cell transplantation. Bone Marrow Transplant. 2004;33:359–365. doi: 10.1038/sj.bmt.1704362. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Louboutin JP, Zhu J, Rivera AJ, Emerson SG. Preterminal host dendritic cells in irradiated mice prime CD8+ T cell-mediated acute graft-versus-host disease. J Clin Invest. 2002;109:1335–1344. doi: 10.1172/JCI14989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hill GR, et al. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90:3204–3213. [PubMed] [Google Scholar]
- 61.Sherman ML, Datta R, Hallahan DE, Weichselbaum RR, Kufe DW. Regulation of tumour necrosis factor gene expression by ionizing radiation in human myeloid leukemia cells and peripheral blood monocytes. J Clin Invest. 1991;87:1794–1797. doi: 10.1172/JCI115199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xun CQ, Thompson JS, Jennings CD, Brown SA, Widmer MB. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood. 1994;83:2360–2367. [PubMed] [Google Scholar]
- 63.Rigby SM, Rouse T, Field EH. Total lymphoid irradiation nonmyeloablative preconditioning enriches for IL-4-producing CD4+-TNK cells and skews differentiation of immunocompetent donor CD4+ cells. Blood. 2003;101:2024–2032. doi: 10.1182/blood-2002-05-1513. [DOI] [PubMed] [Google Scholar]
- 64.Kedl RM, et al. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. 2000;192:1105–1113. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gattinoni L, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumour efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. This paper elucidates the gene-expression, phenotypic and functional profiles of CD8+T cells that mediate a highly effective antitumour responsein vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robbins PF, et al. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang J, et al. Survival, persistence, and progressive differentiation of adoptively transferred tumour-reactive T cells associated with tumour regression. J Immunother. 2005;28:258–267. doi: 10.1097/01.cji.0000158855.92792.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou J, et al. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumour regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175:7046–7052. doi: 10.4049/jimmunol.175.10.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Appay V, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nature Med. 2002;8:379–385. doi: 10.1038/nm0402-379. This paper shows the progressive differentiation of CD8+T cells from patients with acute and chronic viral infections. [DOI] [PubMed] [Google Scholar]
- 70.Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nature Rev Immunol. 2002;2:982–987. doi: 10.1038/nri959. [DOI] [PubMed] [Google Scholar]
- 71.Papagno L, et al. Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 Infection. PLoS Biol. 2004;2:e20. doi: 10.1371/journal.pbio.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. 2005;175:5895–5903. doi: 10.4049/jimmunol.175.9.5895. [DOI] [PubMed] [Google Scholar]
- 73.Lanzavecchia A, Sallusto F. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. 2000;290:92–97. doi: 10.1126/science.290.5489.92. [DOI] [PubMed] [Google Scholar]
- 74.Fearon DT, Manders P, Wagner SD. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science. 2001;293:248–250. doi: 10.1126/science.1062589. [DOI] [PubMed] [Google Scholar]
- 75.Wherry EJ, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nature Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 76.Wang LX, et al. Adoptive immunotherapy of cancer with polyclonal, 108-fold hyperexpanded, CD4+ and CD8+ T cells. J Transl Med. 2004;2:41. doi: 10.1186/1479-5876-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sussman JJ, Parihar R, Winstead K, Finkelman FD. Prolonged culture of vaccine-primed lymphocytes results in decreased antitumour killing and change in cytokine secretion. Cancer Res. 2004;64:9124–9130. doi: 10.1158/0008-5472.CAN-03-0376. [DOI] [PubMed] [Google Scholar]
- 78.Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of allogeneic CD62L− memory T cells without graft-versus-host disease. Blood. 2004;103:1534–1541. doi: 10.1182/blood-2003-08-2987. [DOI] [PubMed] [Google Scholar]
- 79.Bondanza A, et al. Suicide gene therapy of graft-versus-host disease induced by central memory human T lymphocytes. Blood. 2006;107:1828–1836. doi: 10.1182/blood-2005-09-3716. [DOI] [PubMed] [Google Scholar]
- 80.Kaech SM, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nature Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. This paper prospectively identifies the sub-population of antigen-specific effector CD8+T cells expressing IL-7Rα that will persist as a pool of memory T cells. [DOI] [PubMed] [Google Scholar]
- 81.Klebanoff CA, et al. Central memory self/tumour-reactive CD8+ T cells confer superior antitumour immunity compared with effector memory T cells. Proc Natl Acad Sci USA. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dudley ME, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumour antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang H, Li F, Gordon JR, Xiang J. Synergistic enhancement of antitumour immunity with adoptively transferred tumour-specific CD4+ and CD8+ T cells and intratumoural lymphotactin transgene expression. Cancer Res. 2002;62:2043–2051. [PubMed] [Google Scholar]
- 84.Kershaw MH, et al. Redirecting migration of T cells to chemokine secreted from tumours by genetic modification with CXCR2. Hum Gene Ther. 2002;13:1971–1980. doi: 10.1089/10430340260355374. [DOI] [PubMed] [Google Scholar]
- 85.Kagamu H, Touhalisky JE, Plautz GE, Krauss JC, Shu S. Isolation based on L-selectin expression of immune effector T cells derived from tumour-draining lymph nodes. Cancer Res. 1996;56:4338–4342. [PubMed] [Google Scholar]
- 86.Speiser DE, et al. Self antigens expressed by solid tumours do not efficiently stimulate naive or activated T cells: implications for immunotherapy. J Exp Med. 1997;186:645–653. doi: 10.1084/jem.186.5.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Powell DJ, Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late stage effector T cells to CD27+ CD28+ tumour-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105:241–250. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nature Rev Immunol. 2003;3:939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 89.Topp MS, et al. Restoration of CD28 expression in CD28− CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. J Exp Med. 2003;198:947–955. doi: 10.1084/jem.20021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hendriks J, et al. CD27 is required for generation and long-term maintenance of T cell immunity. Nature Immunol. 2000;1:433–440. doi: 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- 91.Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med. 2003;198:1369–1380. doi: 10.1084/jem.20030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arens R, et al. Tumour rejection induced by CD70-mediated quantitative and qualitative effects on effector CD8+ T cell formation. J Exp Med. 2004;199:1595–1605. doi: 10.1084/jem.20031111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang J. et al. Modulation by IL-2 of CD70 and CD27 expression on CD8+ T cells: importance for the therapeutic effectiveness of cell transfer immunotherapy. J. Immunol. (in the press). [DOI] [PMC free article] [PubMed]
- 94.Ochsenbein AF, et al. CD27 expression promotes long-term survival of functional effector-memory CD8+ cytotoxic T lymphocytes in HIV-infected patients. J Exp Med. 2004;200:1407–1417. doi: 10.1084/jem.20040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Rα recycles and presents IL-15 in trans to neighboring cells. Immunity. 2002;17:537–547. doi: 10.1016/s1074-7613(02)00429-6. [DOI] [PubMed] [Google Scholar]
- 96.Liu K, Rosenberg SA. Interleukin-2-independent proliferation of human melanoma-reactive T lymphocytes transduced with an exogenous IL-2 gene is stimulation dependent. J Immunother. 2003;26:190–201. doi: 10.1097/00002371-200305000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hodes RJ, Hathcock KS, Weng NP. Telomeres in T and B cells. Nature Rev Immunol. 2002;2:699–706. doi: 10.1038/nri890. [DOI] [PubMed] [Google Scholar]
- 98.Speiser DE, Romero P. Toward improved immunocompetence of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1467–1469. doi: 10.1172/JCI25427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–623. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- 100.Teague RM, et al. Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumours. Nature Med. 2006;12:335–341. doi: 10.1038/nm1359. [DOI] [PubMed] [Google Scholar]
- 101.Opferman JT, et al. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 102.Hsu C, et al. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J Immunol. 2005;175:7226–7234. doi: 10.4049/jimmunol.175.11.7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu, S., Riley, J. L., Rosenberg, S. A. & Parkhurst, M. R. Comparison of common γ-chain cytokines, interleukin-2, interleukin-7, and interleukin-15 for the in vitro generation of human tumour-reactive T lymphocytes for adoptive cell transfer therapy. J. Immunother. (in the press). [DOI] [PubMed]
- 104.Powell DJ, Jr, Rosenberg SA. Phenotypic and functional maturation of tumour antigen-reactive CD8+ T lymphocytes in patients undergoing multiple course peptide vaccination. J Immunother. 2004;27:36–47. doi: 10.1097/00002371-200401000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pittet MJ, et al. High frequencies of naive Melan-A/ MART-1-specific CD8+ T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med. 1999;190:705–715. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zippelius A, et al. Effector function of human tumour-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 2004;64:2865–2873. doi: 10.1158/0008-5472.can-03-3066. [DOI] [PubMed] [Google Scholar]
- 107.Dutoit V, et al. Degeneracy of antigen recognition as the molecular basis for the high frequency of naive A2/Melan-A peptide multimer+ CD8+ T cells in humans. J Exp Med. 2002;196:207–216. doi: 10.1084/jem.20020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roszkowski JJ, et al. Simultaneous generation of CD8+ and CD4+ melanoma-reactive T cells by retroviral-mediated transfer of a single T-cell receptor. Cancer Res. 2005;65:1570–1576. doi: 10.1158/0008-5472.CAN-04-2076. [DOI] [PubMed] [Google Scholar]
- 109.Hughes MS, et al. Transfer of a TCR gene derived from a patient with a marked antitumour response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhao Y, et al. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumour cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kuball J, et al. Cooperation of human tumour-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2. 1-specific TCR. Immunity. 2005;22:117–129. doi: 10.1016/j.immuni.2004.12.005. This paper describes an effective way to generate highly avid TCRs specific for self/tumour antigens using HLA-A2-transgenic mice. [DOI] [PubMed] [Google Scholar]
- 112.Cohen CJ, et al. Recognition of fresh human tumour by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol. 2005;175:5799–5808. doi: 10.4049/jimmunol.175.9.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li Y, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nature Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 114.Cavalieri S, et al. Human T lymphocytes transduced by lentiviral vectors in the absence of TCR activation maintain an intact immune competence. Blood. 2003;102:497–505. doi: 10.1182/blood-2003-01-0297. [DOI] [PubMed] [Google Scholar]
- 115.Schmitt TM, et al. Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nature Immunol. 2004;5:410–417. doi: 10.1038/ni1055. [DOI] [PubMed] [Google Scholar]
- 116.Clark RA, Yamanaka KI, Bai M, Dowgiert R, Kupper TS. Human skin cells support thymus-independent T cell development. J Clin Invest. 2005;115:3239–3249. doi: 10.1172/JCI24731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ishikawa F, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chain (null) mice. Blood. 2005;106:1565–1573. doi: 10.1182/blood-2005-02-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schlissel MS. Regulating antigen-receptor gene assembly. Nature Rev Immunol. 2003;3:890–899. doi: 10.1038/nri1225. [DOI] [PubMed] [Google Scholar]
- 119.Willemsen RA, et al. Grafting primary human T lymphocytes with cancer-specific chimeric single chain and two chain TCR. Gene Ther. 2000;7:1369–1377. doi: 10.1038/sj.gt.3301253. [DOI] [PubMed] [Google Scholar]
- 120.Pinthus JH, et al. Adoptive immunotherapy of prostate cancer bone lesions using redirected effector lymphocytes. J Clin Invest. 2004;114:1774–1781. doi: 10.1172/JCI22284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carding SR, Egan PJ. γδ T cells: functional plasticity and heterogeneity. Nature Rev Immunol. 2002;2:336–345. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- 122.Kershaw MH, Teng MW, Smyth MJ, Darcy PK. Supernatural T cells: genetic modification of T cells for cancer therapy. Nature Rev Immunol. 2005;5:928–940. doi: 10.1038/nri1729. [DOI] [PubMed] [Google Scholar]
- 123.Charo J, et al. Bcl-2 overexpression enhances tumour-specific T-cell survival. Cancer Res. 2005;65:2001–2008. doi: 10.1158/0008-5472.CAN-04-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Palmer DC, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumour destruction. J Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hwang LN, Yu Z, Palmer DC, Restifo NP. The in vivo expansion rate of properly stimulated transferred CD8+ T cells exceeds that of an aggressively growing mouse tumour. Cancer Res. 2006;66:1132–1138. doi: 10.1158/0008-5472.CAN-05-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rapoport AP, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nature Med. 2005;11:1230–1237. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 127.Ichii H, et al. Role for Bcl-6 in the generation and maintenance of memory CD8+ T cells. Nature Immunol. 2002;3:558–563. doi: 10.1038/ni802. [DOI] [PubMed] [Google Scholar]
- 128.Ichii H, Sakamoto A, Kuroda Y, Tokuhisa T. Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J Immunol. 2004;173:883–891. doi: 10.4049/jimmunol.173.2.883. [DOI] [PubMed] [Google Scholar]
- 129.Manders PM, et al. Inaugural article: BCL6b mediates the enhanced magnitude of the secondary response of memory CD8+ T lymphocytes. Proc Natl Acad Sci USA. 2005;102:7418–7425. doi: 10.1073/pnas.0501585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Willinger T, et al. Human naive CD8 T cells downregulate expression of the WNT pathway transcription factors lymphoid enhancer binding factor 1 and transcription factor 7 (T cell factor-1) following antigen encounter in vitro and in vivo. J Immunol. 2006;176:1439–1446. doi: 10.4049/jimmunol.176.3.1439. [DOI] [PubMed] [Google Scholar]
- 131.Fujita N, et al. MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell. 2004;119:75–86. doi: 10.1016/j.cell.2004.09.014. This paper represents the proof of principle that lymphocyte differentiation states can be reverted by manipulation of key transcriptional factors. [DOI] [PubMed] [Google Scholar]
- 132.Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumour immunotherapy. Curr Opin Immunol. 2005;17:195–201. doi: 10.1016/j.coi.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dannull J, et al. Enhancement of vaccine-mediated antitumour immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother. 2005;28:582–592. doi: 10.1097/01.cji.0000175468.19742.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Attia P, et al. Selective elimination of human regulatory T lymphocytes in vitro with the recombinant immunotoxin LMB-2. J Immunother. 2006;29:208–214. doi: 10.1097/01.cji.0000187959.45803.0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Valencia, X. et al TNF down-modulates the function of human CD4+CD25hi T regulatory cells. Blood 14 Mar 2006 (doi:1182/blood-2005-11-4567). [DOI] [PMC free article] [PubMed]
- 137.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6:S11–S14. [PubMed] [Google Scholar]
- 138.Boyman, O., Kovar, M., Rubinstein, M., Surh, C. D. & Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 15 Feb 2006 (doi:10.1126/science.1122927). [DOI] [PubMed]
- 139.Speiser DE, et al. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest. 2005;115:739–746. doi: 10.1172/JCI23373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kallies, A. et al Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nature Immunol. 26 Mar 2006 (doi:1038/ni1321). [DOI] [PubMed]
- 141.Martins, G. A. et al. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nature Immunol. 26 Mar 2006 (doi:1038/ni1320). [DOI] [PubMed]