

Abstract
IL-2 is a critical T cell growth factor in vitro, but predominantly mediates tolerance in vivo. IL-2 is mainly produced by CD4+ Th cells, but the role of Th cell-derived IL-2 in vivo is controversial. We demonstrate that during immunity to a tumor/self-Ag, the predominant role of Th cell-derived IL-2 was to maintain IL-2Rα (CD25) on CD4+ T regulatory cells (Treg), which resulted in their maintenance of the Treg cell lineage factor, Forkhead/winged helix transcription factor (Foxp3), and tolerance. However, in the absence of Treg cells, Th cell-derived IL-2 maintained effector T cells and caused autoimmunity. IL-2R signaling was indispensable for Treg cell homeostasis and efficient suppressor function in vivo, but, surprisingly, was not required for their generation, because IL-2−/− and CD25−/− mice both contained Foxp3+ T cells in the periphery. IL-2R signaling was also important for CD8+ T cell immunity, because CD25−/− tumor-reactive CD8+ T cells failed to affect established tumors. Conversely, IL-2R signaling was not required for Th cell function. Lastly, administration of anti-IL-2 plus exogenous IL-15 to tumor-bearing mice enhanced the adoptive immunotherapy of cancer. Therefore, Th cell-derived IL-2 paradoxically controls both tolerance and immunity to a tumor/self-Ag in vivo.
Historically, IL-2 was described as a T cell growth factor because of its ability to grow and expand T cells in culture (1). This fundamental observation lead to its clinical use in patients with cancer (2). However, the subsequent generation of mice deficient in IL-2 (3) or the IL-2Rα (CD25) (4, 5), challenged the idea that this was the principal function of this cytokine in vivo. IL-2−/− and CD25−/− mice and a single human patient with a CD25 mutation develop an autoimmune syndrome characterized by the accumulation of activated CD4+ T cells, production of autoantibodies, and inflammatory bowel disease, which has been termed, the IL-2 deficiency syndrome (4–7). These observations indicated that the nonredundant function of IL-2 in vivo was to maintain self-tolerance, but that the function of IL-2 in vitro was a T cell growth factor. These observations created the IL-2 paradox.
Recently, a number of studies have demonstrated that IL-2 functions to maintain the homeostasis of T regulatory (Treg)3 cells inthe periphery (8–10). In the past decade, Treg cells have emerged as the dominant T cell population governing peripheral tolerance to self-Ags and have been shown to suppress immunity to tumors (11–13). Treg cells develop in the thymus and represent 5–10% of the peripheral CD4+ T cell compartment. They constitutively express CD25, glucocorticoid-induced TNFR (GITR), CTLA-4, and Fork-head/winged helix transcription factor (Foxp3), which directs their lineage specification and is critical for their suppressor function (14). Because Treg cells constitutively express CD25, it became apparent that this molecule was also important for their function.
CD25 is a component of the high-affinity IL-2R, which increases the sensitivity of IL-2 for its receptor 100-fold (15, 16). IL-2 up-regulates the expression of CD25 on recently activated T cells, but why CD25 is constitutively expressed on Treg cells is not well understood. The importance of IL-2 signaling in the homeostasis and/or generation of Treg cells was demonstrated in IL-2−/−, CD25−/−, IL-2 receptor-β−/−, and STAT5−/− mice, which all develop autoimmune disease with age (15, 17, 18). These mice were assumed not to have Treg cells, because they had little or no CD4+CD25+ T cells in the periphery (15, 17, 19).
Nevertheless, we now know that CD25 is not an ideal marker for Treg cells, and its combination with Foxp3 expression and other markers can distinguish Treg cells from activated T cells or T cells lacking IL-2-signaling components (8). With that said, in vitro models have demonstrated the importance of IL-2 signaling to Treg cell function. Treg cells could suppress IL-2 mRNA levels in responder T cells in a culture dish, even in the presence of IL-2, but anti-IL-2 added to the cultures reversed suppression (20). In vivo, CD25 signaling on Treg cells has been suggested to be important for their generation. The adoptive transfer of CD4+ T cells from CD25−/− mice into mice with experimental autoimmune encephalitis led to increased disease, but adoptive transfer of CD4+ T cells from IL-2−/− mice suppressed disease (21). This shows that Treg cells may exist in a precursor form in IL-2−/− mice, but not in CD25−/− mice. However, in this experiment it was not determined whether a Foxp3+ Treg cell precursor population truly existed in IL-2−/− or CD25−/− mice or whether IL-2 was required in generating Treg cells from the thymus or de novo from the periphery.
Recently, it has been shown that Foxp3+ Treg cells lacking CD25 can suppress as well as wild-type Treg cells in vitro (8). Although this experiment shows that IL-2 signaling is not necessary for generation of suppression in vitro, this study is in direct conflict with the observation that CD25−/− mice develop fatal autoimmune disease despite having Foxp3+ T cells. Another study claimed that the source of IL-2 for Treg cells was Th cells expressing CD25low receptor (10). This was shown by in vivo depletion of Treg cells with anti-IL-2 Abs (10). However, the experiment did not test whether Th cell-derived IL-2 directly supported Treg cell function in vivo.
Lastly, the role of IL-2 as a Th cytokine in vivo has come into question (22). Because IL-2−/− mice were shown to mount effective immune responses to different viruses (23), it was assumed that IL-2 was not an important T cell growth factor in vivo. Although CD4+ T cells maintain CD8+ T cell immunity and memory (24–28), the signals that dictate this maintenance are still largely unknown. Therefore, we re-evaluated the role of IL-2 and its receptor in vivo using a novel adoptive transfer system that allows us to monitor immunity or tolerance to a tumor/self-Ag (12). We found that during immunity to established tumors, the predominant role of Th cell-derived IL-2 was to maintain Foxp3 expression by Treg cells, which resulted in tolerance. However, when Treg cells were absent, IL-2 was required to maintain CD8+ T cell-mediated autoimmunity. More importantly, treatment of established tumors in CD25−/− mice was more effective than that in wild-type (WT) mice, suggesting that CD25−/− Treg cell function was impaired in vivo, contrary to in vitro experiments (8) and consistent with the phenotype of CD25−/−mice. These results help explain why Treg cells constitutively express CD25 and require persistent IL-2 signaling in vivo, but also explain the paradoxical observations of IL-2 as a growth factor in vitro and as a mediator of self-tolerance in vivo.
Materials and Methods
Mice, tumor cell lines, recombinant fowlpox virus, and irradiation
Thy1.1 Pmel-1 CD8+ TCR transgenic mice (Thy1a/Cy Tg(TcraTcrb)8Rest/J) have been described previously and are deposited at The Jackson Laboratory (29). C57BL/6J, congenic CD45.1 (Ptprca), RAG 1−/− (Rag1tm1Mom), IL-2−/− (Il2tm1Hor), and CD25−/− (Il2ratm1Dw) mice were obtained from The Jackson Laboratory. All mice were bred and kept at the National Institutes of Health animal facility. All experiments were conducted with the approval of the National Cancer Institute animal use and care committee. B16.F10 (H-2b), hereafter called B16, is a gp100+ spontaneous murine melanoma obtained from the National Cancer Institute tumor repository and was maintained in culture medium as previously described (29). All recombinant viruses encoding human (h) gp100 have been described previously and were provided by Therion Biologics (29). Transient lymphopenia of tumor-bearing mice was induced by sublethal total body irradiation (TBI; 5 Gy).
Generation of hIL-2 transgenic mice
The cDNAs encoding the bovine preprolactin signal peptide, hIL-2 mature coding sequence, human elongation factor 1α promoter, and SV40 polyadenylase signal were amplified by PCR, then ligated to generate an expression construct (pEF-PPLSP-hIL-2-SV40 polyadenylase). The linearized DNA was microinjected into fertilized eggs of C57BL/6 mice to obtain founder mice, and their sera were tested for the presence of hIL-2 by a specific ELISA (Pierce). Hemizygous transgenic mice were generated by mating the serum-positive founder mice with WT C57BL/6 mice. The hIL-2 transgenic mice were bred and kept at National Institutes of Health animal facilities.
Flow cytometry and Abs
CD8α (53-6.7), CD4 (H129.19), Vβ13 (MR12-3), CD45.1, CD45.2, CD25 (PC61), IFN-γ, IL-2, and anti-IL-2 (S4B6) as well as the Fc blocking reagent anti-CD16/CD32 were purchased from BD Biosciences. The FITC anti-mouse/rat Foxp3 staining kit (FJK-16s) was purchased from eBio-science. For most samples, propidium iodide-staining cells were excluded from analysis. Samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences) and FlowJo software (Tree Star).
Purification of CD4+ Th and Treg cell subsets for adoptive transfer
CD4+ Th cells were purified from C57BL/6 mice on an IL-2−/−, CD25−/−, or WT background using a CD4+ enrichment kit (Miltenyi Biotec). CD4+CD25− Th cells were further purified by removal of CD4+CD25− T cells on an LS+ selection column twice using a MACS CD4+CD25− isolation kit (Miltenyi Biotec) to obtain > 99% Th cells. Treg cells were purified from spleens of CD45.1 C57BL/6 mice using MACS CD4+CD25+ isolation kits (Miltenyi Biotec). The resulting populations were >95%. Cells were verified to be Treg cells by Foxp3 staining (eBioscience).
Real-time PCR of adoptively transferred Treg and Th cells
Adoptively transferred CD45.1 Treg cells and CD45.2 Th cells were isolated 3–4 wk after transfer from spleens of tumor-bearing mice. Single-cell suspensions of spleens were passed through a cell strainer twice and lysed with ACK lysis buffer (Invitrogen Life Technologies) to remove RBC. For isolation of CD45.1 Treg cells, spleen cells were labeled with anti-CD45.1 PE (15 μg/108 cells) for 10 min at 4°C, then washed once. Cell staining was followed by anti-PE microbeads (Miltenyi Biotec) for 15 min at 4°C. Labeled cells were sorted twice using LS+ selection columns (Miltenyi Biotec). For Th cells, CD4+ T cells were enriched from spleens of tumor-bearing mice using a CD4+ T cell enrichment kit and sorting twice over LS+ selection columns (Miltenyi Biotec). Total mRNA was extracted from sorted populations using an RNA isolation kit (Qiagen). mRNA was reverse transcribed into cDNA using SuperScript II (Invitrogen Life Technologies). Foxp3 RT-PCR primers, probes, and reagents were purchased from Applied Biosystems, and TaqMan was used according to the manufacturer’s instructions (Applied Biosystems). Serial dilutions of cDNA were made to determine the linear range for amplification. Real-time PCR was performed with appropriate dilutions of cDNA using the Applied Bio-systems PRISM 7700 sequence detector and Assays-on-Demand (Foxp3). PCR amplifications were performed in a total volume of 25 μl according to the manufacturer’s instructions. Data are presented as the fold change relative to CD4+CD25− Th cells.
Lymphocyte activation and intracellular staining
For detection of IFN-γ and IL-2 by intracellular staining, splenocytes from the indicated treatment groups were plated at 2 × 106/well in 24-well plates and stimulated with lymphocyte activating mixture (BD Biosciences) for 6 h at 37°C. Cells were collected, washed twice, and stained with the appropriate Abs to surface molecules, then fixed and permeablized with the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s protocol and subsequently stained for intracellular cytokines using anti-IL-2 or anti-IFN-γ Abs (BD Biosciences) suspended in Permwash buffer (1/20) for 30 min. Cells were washed in Permwash twice, then closed with 0.5% BSA in PBS at 4°C. Samples were analyzed by flow cytometry.
Adoptive cell transfer, administration of cytokines, and tumor measurements
Mice were injected s.c. with 2.5 × 105 B16 melanoma cells and treated 7–10 days later with an i.v. administration of 1.0–2.0 × 106 thy1.1 pmel-1 T cells activated for 1 wk in vitro with 1 μM hgp100 peptide and 30 IU of rIL-2 (Cetus). One-week cultured pmel-1 T cells were purified using CD8+ enrichment kits (Miltenyi Biotec) to a purity >98%. CD45.1 Treg cells (1.0 × 105), CD45.2 Th (WT) cells (1.0 × 106), CD45.2 Th (IL-2−/−) cells (1.0 × 106), CD45.2 Th (CD25−/−) cells (1.0 × 106), or a mixture of CD45.1 Treg/CD45.2 Th (1/10) were coinjected with Thy1.1 pmel-1 T cells as indicated. Mice were vaccinated by i.v. injection of 2.0 × 107 PFU of a recombinant fowlpox virus encoding human gp100 (rFP) on the day of transfer. Where indicated, recombinant human IL-2 or recombinant human IL-15 (36 μg/dose) was administered by i.p. injection twice daily for a total of six doses as previously described (30, 31). Tumors were measured in a blinded fashion, and the products of perpendicular diameters were recorded.
Statistics
Tumor graphs were compared using Wilcoxon’s rank-sum test. Statistical analysis was performed using unpaired t test, and p < 0.05 was considered significant.
Results
CD25+Foxp3+ T cells suppress immunity to a tumor/self-Ag
To study the role of IL-2 and its receptor during immunity to a tumor/self-Ag, we used the pmel-1 model. Pmel-1 TCR transgenic mice carry a rearranged transgene that encodes specificity for the tumor/self-melanocyte differentiation Ag, gp100 (29). Adoptive transfer of self-reactive CD8+ T cells can treat established tumors and cause autoimmunity in RAG-1−/− mice, but requires IL-2 from Th to be most effective. This therapy is inhibited by adoptive transfer of naturally occurring Treg cells (12). However, distinguishing Treg cells from activated Th cells and studying their effects on each other after adoptive transfer were previously impossible (12). Thus, we used congenic markers and Foxp3 expression to distinguish between these two populations.
CD45.1 Treg cells were isolated from WT CD45.1 mice, and CD45.2 Th cells were isolated from WT CD45.2 mice by magnetic bead separation (Fig. 1A) and stained with an Ab to Foxp3. We found that the majority of sorted CD25+ T cells were positive for the Treg cell lineage factor, Foxp3, and that sorted CD25−T cells (which contained a few CD25low T cells) were essentially Foxp3− (Fig. 1B ). Treg cells, Th cells, or a 1/10 mixture were cotransferred i.v. with 1-wk-cultured pmel-1 T cells and rFP encoding human gp100 into 7- to 10-d tumor-bearing RAG-1−/− mice, as shown in the schema (Fig. 1A) (12). As shown in Fig. 1C, adoptive transfer of Treg cells inhibited effective tumor immunotherapy, but pmel-1 T cells, rFP vaccine, and Th cells were able to stabilize aggressive B16 tumors. Transfer of pmel-1 T cells alone with rFP vaccine had a minimal effect on tumor growth in RAG-1−/− mice. These data confirmed that Foxp3+ T cells could suppress immunity to a tumor/self-Ag in vivo.
FIGURE 1.

Foxp3+ T cells suppress immunotherapy of established tumors. A, Treatment schematic and illustration of cell isolation for adoptive cell transfer into tumor-bearing recipients. B, Flow cytometry showing the percentage of Foxp3+ T cells in sorted CD45.1 Treg and CD45.2 Th cell populations at the single-cell level. C, Treg cells suppress and Th cells help immunotherapy of established tumors. Tumor-bearing RAG-1−/− hosts were treated with pmel-1 CD8+ T cells (1 × 106), rFP vaccine (2.0 × 107 PFU), and either 1 × 106 Th cells or 1 × 105 Treg cells and Th cells (1:10 ratio). Tumor graphs represent the average tumor measurement ± SEM with at least three mice per group. Experiments were repeated independently six times.
IL-2 and Treg cells control Th cell numbers in vivo
Next, we studied the CD4+ T cell response in tumor-bearing mice, because it has been shown that IL-2 and IL-2R control the size of the peripheral lymphocyte compartment (5, 32, 33). We used WT, IL-2−/−, and CD25−/− mice as a source of Th cells. We sorted Th cells into CD25− fractions and transferred them along with pmel-1 T cells and rFP vaccine into tumor-bearing RAG-1−/− mice, as shown in Fig. 1A. Some groups also received ~10% Treg cells.
Because we were transferring T cells into a lymphopenic environment, it was possible that we could change the normal biology of these cells with respect to their natural ratios in vivo. Therefore, the relative ratios of transferred Treg to Th cells were determined 3–4 wk after adoptive cell transfer (Fig. 2A). The ratio of adoptively transferred Treg to Th cells (~1:10 before transfer) remained approximately the same (range, 5–15%), showing that Treg and Th (WT) cells expanded at similar rates and engrafted the host (Fig. 2A).
FIGURE 2.
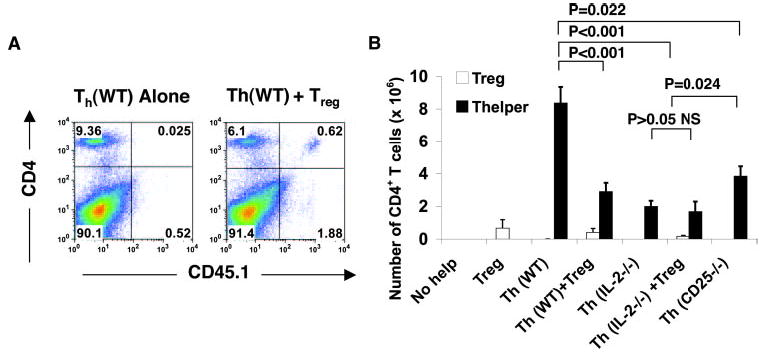
IL-2 and Treg cells control the expansion of CD4+ T cells in vivo. A, Adoptively transferred Treg and Th cells engraft the host at physiologic ratios. Flow cytometry of CD4+ T cells was performed 3–4 wk after adoptive cell transfer, showing the relative ratios of CD45.1 cells and Treg CD45.2 Th cells in spleen. B, Absolute numbers of CD45.1+ Treg (□) and CD45.2 Th cells (▪) from the indicated groups were enumerated 3–4 wk after adoptive cell transfer into tumor-bearing RAG-1−/− hosts. Cells were isolated from spleens of the respective groups.
Once we established that Treg and Th cells engrafted the host at approximately physiological ratios, the absolute number of adoptively transferred Treg cells and Th cells was quantitated. As shown in Fig. 2B, Th (WT) cells expanded 8- to 9-fold, whereas Th (WT) cells with Treg cells were suppressed ( p < 0.001). Th (IL-2−/−) cells expanded significantly less than Th (WT; p < 0.001; Fig. 2B), similar to the transfer of Th (IL-2−/− ) with Treg cells. Unexpectedly, Th (CD25−/−) cells, which could not respond to low doses of IL-2 in vitro (5), expanded ~4-fold in vivo ( p = 0.022; Fig. 2B). This expansion was significantly greater than that of Th (IL-2−/−) cells with or without Treg cells ( p = 0.024; Fig. 2B). Th (CD25−/−) cells expanded ~2-fold less than Th cells (WT), indicating that the high-affinity IL-2R was important for their expansion in vivo. These data supported the idea that IL-2R signaling strength was important for the expansion of Th cells in vivo, but Foxp3+ T cells suppressed this expansion.
IL-2 controls Treg frequency in the periphery and is not required for their generation in the thymus
To obtain an understanding of Treg cell biology and function in vivo, we first looked at the homeostasis of Treg cells in intact nontumor-bearing IL-2−/− and CD25−/− mice. Because CD25 is not an ideal marker for Treg cells in mice that lack IL-2 or IL-2R, we looked at other markers, such as Foxp3, CTLA-4, and the TNFR superfamily molecule, GITR. Recent reports confirm that Foxp3 is the lineage marker for Treg cells (14) and that GITR and CTLA-4 are expressed on resting Treg cells (34, 35). We found that IL-2−/− and CD25−/− mice contained Foxp3+Treg cells (Fig. 3A). However, Foxp3+ T cells were fewer (in frequency) in CD25−/− and IL-2−/− mice and predominantly CD25−/low in IL-2−/− mice (Fig. 3A). GITR and CTLA-4 were increased in Foxp3− T cells from IL-2−/− and CD25−/− mice (Fig. 3), indicating that an activated effector population dominated in these mice, which is consistent with their phenotypes. Surprisingly, absolute numbers of Foxp3+ T cells were similar in WT, IL-2−/−, and CD25−/− mice at 8 wk of age (Fig. 3B).
FIGURE 3.
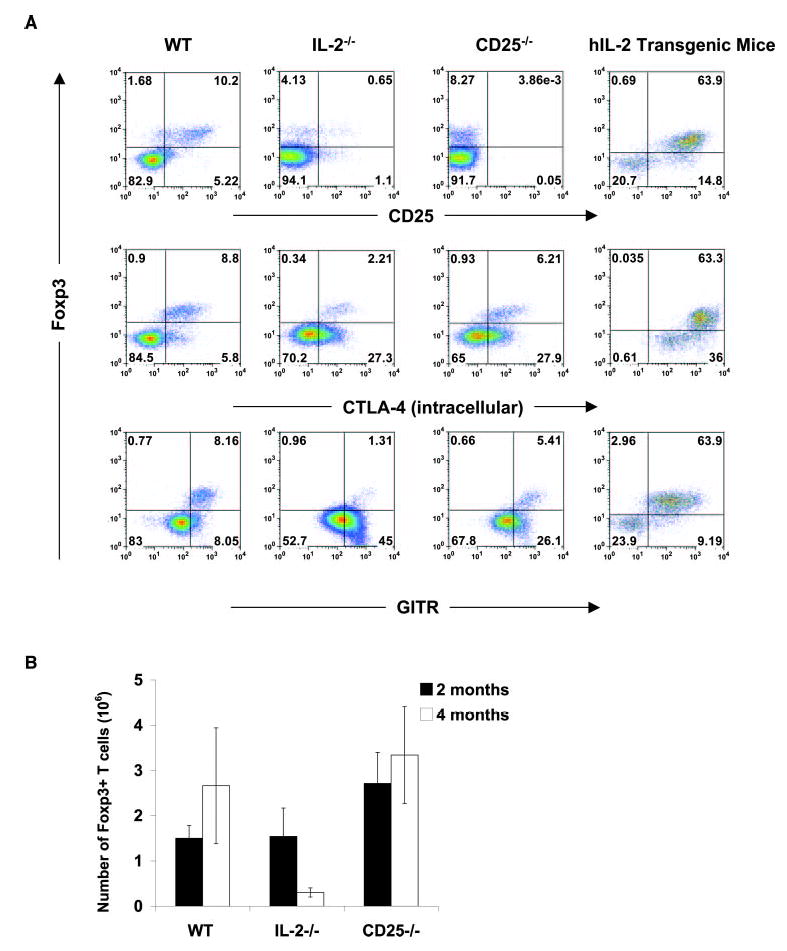
IL-2 controls the frequency of Treg cells in vivo. A, Flow cytometry of phenotypic markers on Foxp3+ Treg cells from WT, IL-2−/−, CD25−/−, and human IL-2 transgenic mice. IL-2−/− and CD25−/− mice have Foxp3− T cells, but have more activated Foxp3− cells than WT mice. CD25, CTLA-4, and GITR are increased proportionally with Foxp3− T cells in human IL-2 transgenic mice. Cells were isolated from spleens and stained with the indicated Abs. Experiments were repeated independently more than three times. B, Absolute number of Foxp3+ T cells from WT, IL-2−/−, and CD25−/− mice 2 and 4 mo after birth.
Because both IL-2−/− and CD25−/− contained Foxp3+ T cells, we directly tested whether IL-2 was needed for the generation and/or maintenance of Treg cells. We first looked at the thymi of IL-2−/− mice and CD25−/− mice and found Foxp3+ T cells (data not shown). Therefore, we analyzed cells in the periphery at different time points, because IL-2−/− and CD25−/− mice develop disease with age, and we found that Foxp3+ T cells were present in CD25−/− and IL-2−/− mice, but their number waned with age in IL-2−/− mice (Fig. 3B). Because of this discrepancy, we directly tested whether IL-2 controls Treg cell number in the periphery by generating IL-2 transgenic mice, which constitutively express human IL-2 at picogram levels in serum (16–22 pg/ml). By flow cytometry, IL-2 transgenic mice had a 6-fold increase in the frequency of Foxp3+CD25+ T cells in the spleen compared with normal WT mice (Fig. 3A) and were healthy like their WT litter-mates. CTLA-4 and GITR expression were also increased 6-fold in Foxp3+ T cells from IL-2 transgenic mice (Fig. 3A), indicating that these cells were bona fide Treg cells. Determination of the absolute number of Treg cells in IL-2 Tg mice indicated that these mice had an ~ 2-fold expansion of Treg cells in the periphery compared with WT controls (data not shown). Therefore, the aforementioned data showed that IL-2 could expand Foxp3+ T cells in vivo, but IL-2 was not necessary for generation of Treg cells in the thymus.
Th cell-derived IL-2 maintains CD25 and Foxp3 expression by Treg cells
Next, we directly tested whether Treg cells were using IL-2 in vivo during tumor immunity by measuring the expression of CD25 in adoptively transferred Treg cells. Because it has been shown that Treg cells lose the expression of CD25 upon adoptive transfer into lymphopenic hosts (36), we wanted to understand whether this loss of expression was related to homeostatic expansion or lack of a source of IL-2. Before transfer, CD25 expression on sorted Treg cells was, on the average, >94% (Fig. 4A). When transferred alone into tumor-bearing RAG-1−/− hosts, Treg cells significantly decreased CD25 expression (Fig. 4A). Foxp3 expression was also down-regulated in Treg cells when transferred alone, but remained high when transferred with Th cells (WT) that could produce IL-2 (Fig. 4B). Treg cells also maintained their CD25 expression by mean fluorescence intensity when transferred with Th cells (WT; Fig. 4C).
FIGURE 4.

Th cell-derived IL-2 controls CD25 and Foxp3 expression by Treg cells. A, CD25 expression on adoptively transferred CD45.1 Treg cells before (▪) and 3–4 wk after transfer (□) into tumor-bearing RAG-1−/− hosts with or without Th cells as indicated. Groups with p values are compared with Treg and Th (WT) cells. B, Foxp3 expression by Treg cells is controlled by Th cells. Foxp3 expression in adoptively transferred CD45.1 Treg cells alone or with WT, IL-2−/−, or CD25−/− Th cells 3–4 wk after treatment of tumor-bearing RAG-1−/− hosts with pmel-1 T cells and rFP vaccine. C, Flow cytometry of CD25 expression on CD45.1 Treg cells 3–4 wk after transfer into RAG-1−/− hosts. The numbers indicates the average mean fluorescence intensity for CD25 expression on CD45.1 Treg cells. Experiments were repeated independently seven times.
To address the source of IL-2 responsible for this regulation, Treg cells were cotransferred with Th (IL-2−/− cells. In the absence of Th cell-derived IL-2, Treg cells decreased CD25 and Foxp3 expression (Fig. 4, A and B). Although Foxp3 expression was not as low as that in Treg cells alone, this indicated that growth factors other than IL-2 might be contributing to Foxp3 expression. Transfer of Treg cells with Th (CD25−/−) cells, which can provide a source of IL-2, only minimally maintained CD25 expression on Treg cells. This may be explained by the lesser amount of IL-2 available from Th (CD25−/−) cells (see Fig. 8). However, Treg cells maintained Foxp3 expression levels (Fig. 4B ) when Treg were compared with Th (WT) cells. In vitro suppression assays also confirmed that a T cell source of IL-2 was required to sustain CD25 expression on Treg cells in vitro (data not shown). Together, these findings indicated that Th cell-derived IL-2 tightly regulated CD25 expression on Treg cells and, as a result, regulated their Foxp3 expression.
FIGURE 8.
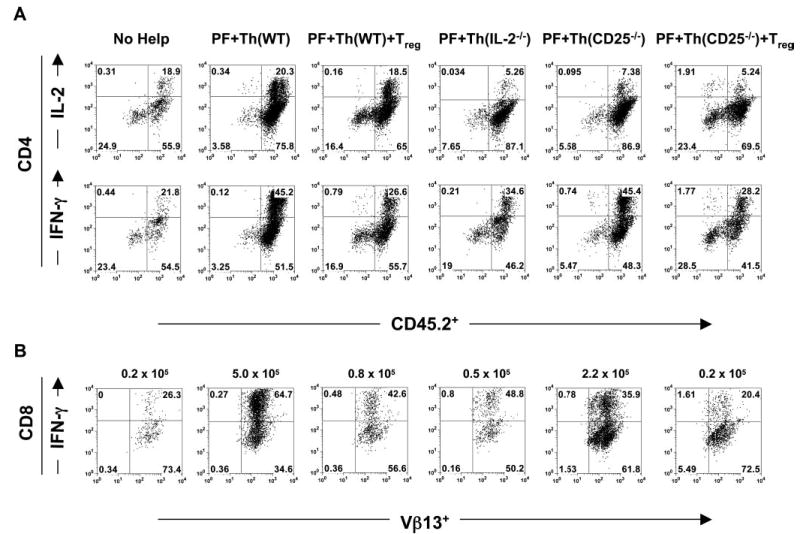
IL-2 sustains and Treg cells suppress effector T cell numbers. A, IL-2 controls effector CD4+ T cells. Intracellular staining of IL-2 and IFN-γ in Th cells from the indicated treatment groups after stimulation with lymphocyte-activating mixture for 6 h. B, IL-2 maintains and Treg cells suppress effector CD8+ T cells. Intracellular staining of IFN-γ in pmel-1 T cells from the indicated treatment groups after stimulation with lymphocyte activating mixture for 6 h is shown. The average absolute number of CD8+Vβ13+IFN-γ+ T cells is indicated above each density plot. The absolute number was calculated by multiplying the cell count (total splenocytes) by the frequency of CD8+ T cells by the frequency of Vβ13+IFN-γ+ T cells. Data are representative of three experiments.
IL-2 signaling is required for the maintenance and function of Foxp3+ T cells in vivo
Because CD25−/− and IL-2−/− mice had approximately normal levels of Foxp3+ Treg cells but still acquired disease, we wanted to understand how IL-2 and IL-2R influenced Treg cell survival in the periphery. Because transferring CD4+CD25− T cells into lym-phopenic hosts leads to the establishment of a Foxp3+CD25+ T cell population (37–39), we looked at the requirements for the induction of Foxp3 in the periphery. IL-2R and Foxp3 levels were determined in CD4+ T cell populations before and after transfer into tumor-bearing RAG-1−/− hosts. Before transfer, sorted populations had negligible CD25 expression (Fig. 5A). However, after 3–4 wk in vivo, Th (WT) cells up-regulated their IL-2Rα by an average of 15% in vivo (Fig. 5A). However, Th (IL-2−/−) cells persistently only expressed 2–6% CD25 (Fig. 5A), even in a host capable of producing IL-2 from dendritic cells (40). Th (CD25−/−) cells were naturally negative for CD25.
FIGURE 5.

IL-2 signaling is required for the maintenance of Foxp3+ Treg cells in the periphery. A, CD25 expression on adoptively transferred CD45.2 Th cells before and 3–4 wk after transfer into tumor-bearing RAG-1−/− hosts. The p values are compared with Th (WT) cells (wk 3–4). B, Foxp3 expression at the single-cell level in CD4+ T cells 3–4 wk after transfer into tumor-bearing RAG-1−/− hosts. C, Foxp3 expression determined by RT-PCR in CD4+ T cells 3–4 wk after transfer into tumor-bearing RAG-1−/− hosts. Data are representative of three experiments.
Next, Foxp3 levels were determined in CD4+ T cells after adoptive transfer at the single-cell level (Fig. 5B) and the mRNA level (Fig. 5C). As shown in Fig. 5B, Foxp3 expression was readily detectable in the pretransfer CD4+ T cells from WT mice (<1%) and was maintained in a population that expanded after transfer (0.96 vs 2.41% posttransfer; Fig. 5B). This Foxp3+ population was contained solely in the CD25+ fraction (Th (WT) CD25+ cells; Fig. 5C). However, CD4+ T cells from IL-2−/− mice, which also were <1% Foxp3+ before transfer, had undetectable levels of Foxp3 after transfer (0.91 vs 0.18%; Fig. 5B). Th (IL-2−/−) cells that had converted to CD25+ did not express Foxp3 (Fig. 5C). However, CD4+ T cells from CD25−/− mice, which had high levels of Foxp3 before transfer (4.57%; Fig. 5B), became completely negative after transfer (0.15%; Fig. 5B). This was also con-firmed at the mRNA level (Fig. 5C).
To gain additional insight into whether IL-2 signaling on Treg cells was responsible for either their homeostasis and/or their function in vivo, we tested whether Foxp3+-expressing cells from CD25−/− mice could suppress tumor immunity in vivo. We found that before transfer, ~5–10% of CD4+ T cells from CD25−/− mice expressed Foxp3 (Fig. 6A). However, 4 wk after adoptive transfer into tumor-bearing RAG-1−/− hosts, CD25−/− Foxp3+ T cells were undetectable in the periphery (Fig. 6A). Therefore, treatment of established tumors in three independent experiments with CD4+CD25−/− T cells was not compromised by this ~5–10% Foxp3+ contaminating population (Fig. 6B), and CD8+ T cells numbers were not significantly lower than those of WT cells (Fig. 6C). However, when CD25+/+ Foxp3+ T cells were cotransferred with CD25−/− T cells, antitumor immunity was compromised (Fig. 6B).
FIGURE 6.
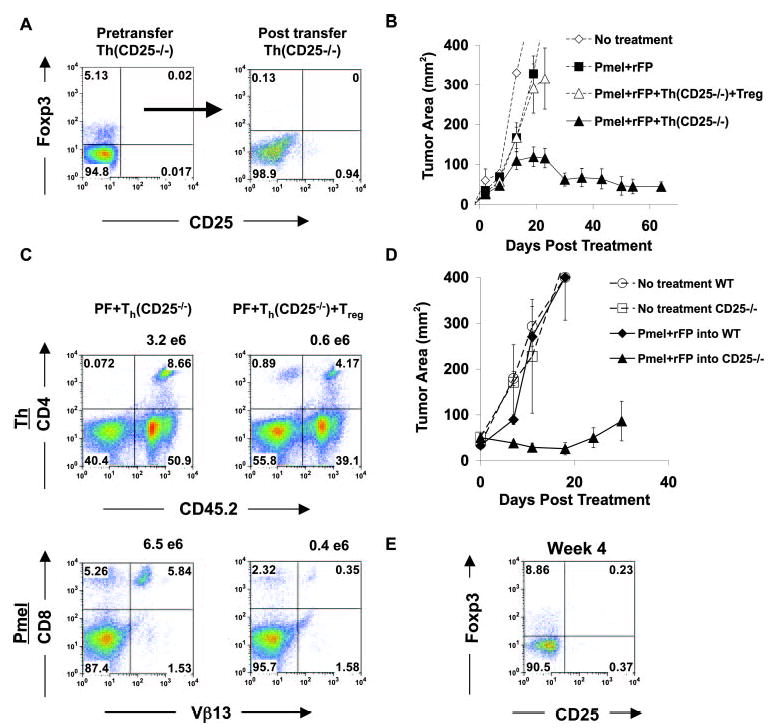
IL-2 signaling is required for the efficient function of Foxp3+ Treg cells in the periphery. A, Foxp3 expression in CD25−/− T cells at the single-cell level before and 3–4 wk after transfer into tumor-bearing RAG-1-deficient hosts. Gates are based on WT controls. B, CD8+ T cells are amenable to suppression only by CD25+/+ Foxp3+ T cells. RAG-1−/− mice bearing 7- to 10-day B16 tumors were treated with pmel-1 CD8+ T cells, rFP vaccination, and Th (CD25−/−) cells (~5–10% Foxp3+) with and without CD25+/+ Foxp3+ T cells. Error bars represents ±SEM. C, CD25+/+ Foxp3+ Treg cells, but not CD25−/− Foxp3+ T cells, can suppress Th (CD25−/−) cells in vivo and prevent effective expansion of CD8+ T cells (pmel-1). The frequency of Th (CD25−/−) cells (upper panel) and pmel-1 T cells (lower panel) with CD45.1 Treg cells (right) or without (left) after adoptive transfer and rFP vaccination into tumor-bearing RAG-1−/− hosts is shown. The number above each density plot represents the absolute number of T cells. D, Treatment of established tumors is enhanced in CD25−/− hosts. Ten-day tumor-bearing CD25−/− and WT mice received 5 Gy TBI and pmel-1 T cells and rFP vaccine on the day of treatment. E, Flow cytometry of Foxp3+ T cells in tumor-bearing CD25−/− mice 4 wk after treatment.
To ascertain the in vivo effects of the transferred CD25+/+ Treg cells, we analyzed the frequency and absolute number of pmel-1 and Th cells (Fig. 6C). With the addition of CD25+/+ Treg cells in three independent experiments, the average numbers of CD25−/− Th cells and pmel-1 T cells were decreased (Fig. 6C). Because CD25−/− Foxp3+ T cells disappeared after adoptive transfer, we tested our immunotherapy regimen in intact CD25−/− hosts, because they have a consistent supply of Treg cells from thymic output. This would allow us to test their in vivo suppressive ability. Therefore, 10-d tumor-bearing WT mice and CD25−/− mice underwent 5-Gy TBI and were treated with pmel-1 T cells and rFP vaccine. As shown in Fig. 6D, treatment was enhanced in CD25−/− mice compared with WT mice. Compared with RAG-1−/− mice (Fig. 6B), pmel-1 T cells transferred with rFP vaccine into CD25−/− mice were better at controlling tumor immunity. To confirm that Treg cells were present, mice were analyzed for the presence of Foxp3+ T cells. As shown in Fig. 6E, CD25−/− Foxp3+ T cells were present at normal levels in CD25−/− mice 4 wk after treatment (~5–10%). This showed that Treg function in CD25−/− mice was not as efficient as that in WT mice, which is contradictory to the in vitro results reported by others (8). This also demonstrates the contribution of Th (CD25−/−) cells in CD25−/− mice, which we show can help pmel-1 T cells in RAG-1−/− mice upon adoptive transfer (Fig. 6B). In conclusion, in the absence of the high-affinity IL-2R, CD25−/− Treg cells were not able to compete in the periphery to initiate their survival program; however, in intact hosts, which have a constant supply of CD25−/− Treg cells, IL-2R signaling was shown to be important for efficient suppressor function. These results showed that IL-2R signaling was required for both the homeostatic maintenance and the efficient suppressive function of Treg cells in the periphery.
IL-2 signaling is critical for CD8+ T cell immunity in vivo
Next, we looked at CD8+ T cell immunity. The role of IL-2 in vivo with regard to in CD8+ T cells has not been clearly elucidated, but recent work has shown that IL-2 is important for their expansion (12, 22, 41, 42). Therefore, we transferred T (WT), Th (IL-2−/−), or Th (CD25−/−) cells with pmel-1 T cells and vaccination into tumor-bearing RAG-1−/− mice with or without Treg cells as indicated and examined the expansion and treatment ability of CD8+ T cells. As previously shown (12), Th (IL-2−/−) cells did not help pmel-1 T cells expand or treat established tumors; however, in five independent experiments, Th (CD25−/−) cells helped CD8+ T cells maintain tumor immunity similarly to Th (WT) cells (Fig. 7A) and were suppressed by Treg cells (Fig. 6B). IL-2 appeared to play a role in the maintenance of pmel-1 T cells, because the frequency and absolute numbers of CD8+ T cells were higher in groups that had access to Th cell-derived IL-2, but were suppressed in all groups receiving Treg cells (Fig. 7, B and C).
FIGURE 7.

IL-2 signaling is dispensable for Th cell function, but is indispensable for CD8+ T cell immunity. A, Treatment of established tumors in RAG-1−/− mice requires Th cell-derived IL-2, but not CD25 expression on Th cells. B, Th cells maintain the persistence of CD8+ T cells. The frequency of adoptively transferred CD8+Vβ13+ (pmel-1) T cells 3–4 wk after treatment with the indicated CD4+ T cells is shown. C, IL-2 sustains and Treg cells suppress CD8+ T cell numbers in vivo. The absolute number of pmel-1 CD8+ T cells 3–4 wk after adoptive transfer and vaccination with rFPVhgp100 into tumor-bearing RAG-1−/− hosts is shown. D, IL-2R signaling is important for CD8+ T cell function in vivo. Naive pmel-1 (WT) T cells or naive pmel-1 (CD25−/−) T cells were enriched from splenocytes by CD8 negative selection and transferred i.v. with rFP vaccine into tumor-bearing RAG-1−/− mice on day 7 after tumor inoculation. Error bars represent ±SEM.
Because IL-2 appeared to be supporting CD8+ T cell maintenance in vivo, we created pmel-1 T cells, which were CD25−/−, to block the IL-2 signal to determine whether other cytokines could compensate for IL-2 in vivo, because it has been reported that IL-2−/− mice can mount normal antiviral immune responses (23). To this end, naive splenocytes from pmel-1(CD25−/−) transgenic mice were enriched for CD8+ T cells and transferred into tumor-bearing RAG-1−/− hosts along with rFP vaccination. As a control, we transferred naive pmel-1 (WT) T cells into tumor-bearing mice along with rFP vaccination. Because naive pmel-1 (WT) T cells have the propensity to make autocrine IL-2 at much higher levels than cultured T cells (43, 44), it was possible that tumor immunity could become Th cell independent. However, even though naive T cells could produce IL-2 and treat tumors in RAG−/− mice in the absence of T cell help, it became apparent that IL-2 signaling was important for CD8+ T cell function, because pmel-1(CD25−/−) T cells could not treat established tumors (Fig. 7D). The pmel-1(CD25−/−) T cells transferred with Th cells were also unable to treat tumors (data not shown). This experiment showed that our system was not artificially IL-2 dependent, and in vivo IL-2 was indispensable for CD8+ T cell immunity, but was not required for Th cell function.
Treg cells prevent the maintenance of IFN-γ+ effector T cells
To investigate how Treg cells prevented antitumor immunity in vivo, we studied the effector function of pmel-1 T cells and Th cells ex vivo. Splenocytes from treated groups were activated ex vivo with a lymphocyte-activating mixture for 6 h and were stained with Abs to the appropriate congenic markers to distinguish Th (CD45.2) from Treg (CD45.1) and pmel-1 CD8+T (Vβ13) cells (Fig. 8). Subsequently, cells were stained for intra-cellular cytokines, IFN-γ and IL-2. As shown in Fig. 8, IFN-γ expression in CD8+ T cells was highest (3-fold higher) in the group that received Th (WT) or Th (CD25−/−) cells and was markedly suppressed in all other groups, especially groups with Treg cells. CD4+ T cells that produced IL-2 made more IFN-γ and helped sustain CD8+IFN-γ+ T cell numbers (Fig. 8,). Although Th (IL-2−/−) cells could make IFN-γ, it was not at the same level as Th (WT) cells (Fig. 8A), and this did not support CD8+ T cell expansion (Fig. 8B). Th (CD25−/−) cells made high amounts of IFN-γ, but repeatedly made lower amounts of IL-2 (Fig. 8A), indicating that full IL-2 signaling may be required for the efficient production of IL-2 in vivo. However, this did not affect CD8+ T cell function with respect to tumor immunity (Fig. 7A), but did slightly affect their numbers (Figs. 7C and 8B). When Th (WT) cells were cotransferred with Treg cells, the frequency and number of IL-2-producing cells were also decreased (Fig. 8A and data not shown). These results suggested that CD4+ T cells were required to maintain both effector CD4+ and CD8+ T cells through IL-2 production. These findings support our initial finding that exogenous IL-2 can enhance IFN-γ-producing CD8+ T cells in mice undergoing regression of established tumors (29). Therefore, Th cell-derived IL-2 was essential in maintaining effector T cell function and number, and this effect was compromised when Treg cells expressing high-affinity IL-2R were present.
IL-2 neutralization in vivo enhances immunotherapy of cancer
Because we showed that IL-2R signaling was essential for Treg cell homeostasis and function, and that this signaling was important for the maintenance of Foxp3 expression, we hypothesized that neutralizing IL-2 may be an important means of eliminating Treg cell function in vivo. However, because IL-2 was also important for effector T cells, we hypothesized that anti-IL-2 would not work alone. Therefore, we would also have to provide a supportive cy-tokine, such as IL-15, which has been shown to help CD8+ T cells cause tumor regression, but not support Treg cell function (31, 45). We used immunocompetent tumor-bearing mice and administered 5 Gy TBI, as previously reported (12, 31, 46). Using TBI enhances immunotherapy when using pmel-1 T cells, rFP vaccine, and IL-2 or IL-15 (30, 31). This information combined with our new understanding of Treg cell function in vivo helped us develop an unconventional approach to immunotherapy (16). Using anti-IL-2 to neutralize Treg cell function (8, 10) and replacing IL-2 with exogenous IL-15 to enhance CD8 immunity (30, 31), we were able to enhance immunotherapy beyond that seen with IL-2 or IL-15 alone (Fig. 9). Anti-IL-2 therapy alone had no effect, indicating that FcR-bound Abs containing cytokine or antibody-cytokine immune complexes were not stimulating T cell in trans.
FIGURE 9.
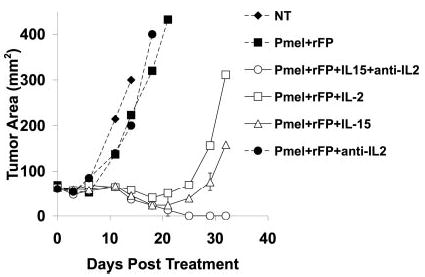
Anti-IL-2 therapy plus exogenous IL-15 enhances immu-notherapy of cancer. Tumor-bearing B6 mice were irradiated with 5 Gy and treated with pmel-1 T cells, rFP vaccine, and IL-2, IL-15, or anti-IL-2 as indicated. Anti-IL-2 (0.1 mg) was given every other day i.p. 4 days after therapy for 3 wk. IL-2 and IL-15 (36 μg/dose) were injected twice a day for 3 days after adoptive transfer. The tumor graph represents the average tumor measurement ±SEM for at least three mice per group. The experiment was repeated twice.
Discussion
Described almost 30 years ago, IL-2 has long been thought to be a T cell growth factor. It was also labeled a Th cytokine based on in vitro work. These proposals are not far from the truth. In fact, as shown in this study and elsewhere, IL-2 is a T cell growth factor. However, the outcome of that growth depends on which cell is responding. We propose that the main role of Th cell-derived IL-2 in vivo is to support Treg cell homeostasis and function, which leads to the mediation of self-tolerance. However, in the absence of Treg cells, Th cell-derived IL-2 supports immunity.
In this study we show that the mechanism by which IL-2 supports Treg cells is related to regulation of the expression of CD25 on Treg cells and the maintenance of Foxp3 expression in the periphery. Because IL-2 up-regulates its own receptor (47), these results show that IL-2 was constantly being used by Treg cells. This paracrine action of Th cell-derived IL-2 is related to Treg survival and the suppressive mechanism, because it appears that other Th cell-derived cytokines are not sufficient to fully compensate for these important functions. However, there is evidence that other common γ-chain (γc) cytokines, such as IL-4, are critical for the induction of Foxp3 expression (20, 45, 48), and that in the absence of γc signaling, Foxp3+ T cells do not exist (8). Therefore, the nonredundant function of IL-2 may be attributed to the maintenance of Foxp3 expression in the periphery. This paracrine effect of IL-2 signaling may help explain why adoptively transferred WT Treg cells can prevent autoimmunity in CD25−/− and IL-2Rβ−/− mice, which have a source of IL-2, but not in IL-2−/− mice (18, 33). These data also explain why CD25−/− Foxp3+ T cells cannot survive after transfer into a lymphopenic setting. Our data show a critical link between IL-2 signaling and Foxp3 expression in Treg cells in the periphery. However, whether IL-2 supports CD25+Foxp3+ T cells that come from CD25− precursors that were Foxp3−, Foxp3+, or both is still not clear.
Our findings also suggest that constitutive expression of CD25 on Treg cells allows competition for IL-2 in vivo to be an important mechanism that Treg cells use to acquire this growth factor. High expression of CD25 increases the sensitivity of IL-2 binding to the IL-2R ~100-fold compared with the low-affinity receptor (16). This may confer a survival advantage to Treg cells when they need to traffic to extralymphoid tissue sites where IL-2 is limiting (14). This is supported by the fact that ectopic expression of Foxp3 in CD4+ or CD8+ T cells and naturally expressing Foxp3+ T cells always induces the high-affinity IL-2R (14, 49, 50).
It has been shown in vitro that the mechanism of suppression by Treg cells requires IL-2 from responding T cells, and that anti-IL-2 or exogenous IL-2 or IL-4 added to cultures can abrogate suppression (20). This seems paradoxical, but in the case of high doses of IL-2 or IL-4, Treg cells could suppress IL-2 mRNA in responder T cells, even though T responders could still divide. This emphasizes the importance of signaling through CD25 and IL-4R on Treg cell function during states of inflammation.
In cases of higher amounts of IL-2, as in exogenous administration to cancer patients (51) or elimination of cytokine sinks (46), proliferation of effector T cells can still occur. However, in the long term, this may be the Achilles’ heel of immunotherapy, because Treg cells may compensate their levels due to the long exposure to IL-2, as we demonstrate in IL-2 transgenic mice and recently demonstrated in patients (52, 53). This may help explain the fixed 15–20% response rate to high dose IL-2 therapy in patients with cancer (51).
Therefore, to prevent activation of Treg cells, we used anti-IL-2, which decreases Treg cell Foxp3 expression (8, 10), and replaced exogenous IL-2 with IL-15, a cytokine that does not support Treg cells (45), but instead supports CD8+ T cell immunity (31). Addition of IL-15 was necessary, because CD8+ T cells also need IL-2 signaling to cause effective immunity. As a result, this treatment regimen enhanced adoptive immunotherapy more than IL-15 or IL-2 alone. These conditions may have provoked CD8+ T cells to take on a central memory, rather than an effector memory, phenotype, which we have shown preserves their antitumor efficacy in vivo (30, 43).
This is an important finding for immunotherapy. We believe that IL-2 consumption by Treg cells is coupled to their suppressive mechanism in vivo. This feedback loop maintains immunological balance in the immune system, keeping Treg cells alive and auto-reactive T cells in check (16). IL-2 signaling, in addition to TCR stimuli to self Ags (54), probably activates the program of suppression, which may include mechanisms such as reverse signaling through engagement of B7 molecules on effector T cells (55), competition for cytokines (56–58), secretion of immunosuppressive cytokines (56, 57, 59), or direct killing (60). Treg cells may also negatively condition APCs (61, 62). Regardless, the results presented in this study help explain why Treg cells constitutively express the high-affinity IL-2Rα and why blocking their access to IL-2 can inhibit their suppressive ability.
With regard to CD25−/− mice, the lack of full IL-2 signaling on Treg cells probably explains the disease in these mice. Although CD25−/− mice had normal levels of Foxp3+ T cells, these probably represent cells that are either recent thymic emigrants or are being supported by other Th cell-derived γc cytokines during a state of active disease. However, it is unclear why CD25−/− mice still have disease. Although recent evidence suggests that Treg cells are unable to keep up with cells mediating disease (8), these mice have normal levels of Foxp3+ T cells, which, surprisingly, are just as suppressive as WT cells in vitro. So why do CD25−/− mice develop fatal autoimmune disease? Are CD25−/− Treg cells dysfunctional in vivo? We believe that our results show that IL-2 signaling has an important role in suppression in vivo. We show that pmel-1 T cells were able to treat established tumors much better in CD25−/− hosts than in WT hosts. In fact, 1-wk-cultured pmel-1 cells transferred alone with rFP vaccine into RAG-1−/− mice or WT mice have never treated established tumors. Because, we were able to achieve treatment in CD25−/− mice, this shows that CD25−/− Treg cells were not as efficient suppressors as WT cells and possibly that Th (CD25−/−) cells contributed to the treatment effect. Considering that Th (CD25−/−) cells were able to expand significantly better than Th (IL-2−/−) cells and could help CD8+ T cells induce tumor regression probably indicates that Th cells may be able to signal through the IL-2Rβγ in vivo. Therefore, these low-affinity signals may be sufficient to reach a threshold of activation to help CD8+ T cells, but not enough to activate CD25−/− Foxp3+Treg cells. This is also supported by the fact that Treg cells could not suppress IL-2−/− Th cells in vivo as efficiently as Th (WT) cells. Because Th cells do not need high-affinity IL-2R to generate help, but Treg cells need high-affinity IL-2R to survive and initiate their suppressor program, CD25−/−Foxp3+ Treg cells may be less efficient at suppressing in vivo; this explains why CD25−/− mice develop autoimmune disease with age. These data are most consistent with a study that showed that Ag-specific CD25−/− Treg cells were not as efficient suppressors as WT cells in an in vitro assay (9). Therefore, the suppression seen with co-transfer of WT CD25+/+ Treg cells with CD25−/− Th cells may indicate that IL-2 from Th (CD25−/−) cells was supporting Treg cell homeostasis and suppressor function. This was confirmed by higher amounts of CD25 and Foxp3 expression by Treg cells mixed with Th (CD25−/−) or Th (WT) cells.
Therefore, our data suggest that there may be two types of au-toimmunity that can occur in vivo: IL-2-independent autoimmu-nity, as seen in IL-2−/− mice, which may be primarily CD4+ T cell mediated due to the absence of Treg and CD8+ T cell function, and IL-2-dependent autoimmunity, which is primarily CD8+ T cell mediated and requires the absence of Treg cells. Although recent work shows a kinetic relationship between the requirement for IL-2 to activate T effector and Treg cells in vivo (63), this system only studied CD4+ T cell-mediated disease and did not link IL-2 to CD8+ T cell immunity. Our results link IL-2 with CD8+ T cells and identify the source of IL-2 in vivo. Previous data from our group also showed that Th cells from IL-2−/− mice administered along with exogenous IL-2 did not support CD8 immunity in tumor-bearing RAG-1−/− (12). Therefore, for CD8 immunity, it appears that IL-2 may be important throughout the immune response.
Together, the results show a dual role for Th cell-derived IL-2 in vivo. Treg cells maintained self tolerance by using IL-2 from Th cells through their high-affinity IL-2R. This appeared to be directly linked to Foxp3 expression, which is known to be required for Treg cell function. Therefore, Th cell-derived IL-2 may be enhancing the transcription of Foxp3 in Treg cells, which is directly related to their suppressive characteristics in vivo. This action subsequently leads to the lack of effector T cell expansion, which paradoxically also requires IL-2. Although the precise mechanism of suppression is unclear, our work helps explain the IL-2 paradox: that is, IL-2 is important for both tolerance and immunity in vivo. The use of anti-IL-2 Abs (and/or anti-IL-4 Abs) along with other γc-signaling cytokines, which support CD8+ T cell immunity (IL-15 and IL-21), may offer an advantage over current therapies for advanced cancer and chronic infections. In contrast, IL-2 therapy may be useful for treating many autoimmune diseases.
Acknowledgments
We thank Ethan Shevach for critical comments and suggestions, and Katherine Bergstrong, Paul Spiess, and Debbie Surman for technical work. We thank Luca Gattinoni and the National Cancer Institute Fellows Editorial Board for critically reviewing this manuscript.
Footnotes
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Abbreviations used in this paper: Treg, T regulatory; Foxp3, Forkhead/winged helix transcription factor; γc, common γ-chain; GITR, glucocorticoid-induced TNFR; h, human; rFP, recombinant fowlpox virus; TBI, total body irradiation; WT, wild type.
Note added in proof. The lack of Treg cell function in CD25−/− mice was also supported recently by a paper showing that Abs to CD25 do not deplete but actually inhibit the function of Treg cells in vivo (64).
Disclosures
The authors have no financial conflict of interest.
References
- 1.Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. 1976;193:1007–1008. doi: 10.1126/science.181845. [DOI] [PubMed] [Google Scholar]
- 2.Mule JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science. 1984;225:1487–1489. doi: 10.1126/science.6332379. [DOI] [PubMed] [Google Scholar]
- 3.Schorle H, Holtschke T, Hunig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352:621–624. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, Schmits R, Simard JJ, Ohashi PS, Griesser H, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- 5.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 6.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 7.Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the α chain of the interleukin-2 receptor. Proc Natl Acad Sci USA. 1997;94:3168–3171. doi: 10.1073/pnas.94.7.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 9.D’Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol. 2005;6:1152–1159. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
- 10.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+CD25+CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 12.Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, Palmer DC, Chan CC, Klebanoff CA, Overwijk WW, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–782. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol. 2004;172:3983–3988. doi: 10.4049/jimmunol.172.7.3983. [DOI] [PubMed] [Google Scholar]
- 16.Antony PA, Restifo NP. CD4+CD25+ T regulatory cells, immunotherapy of cancer, and interleukin-2. J Immunother. 2005;28:120–128. doi: 10.1097/01.cji.0000155049.26787.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antov A, Yang L, Vig M, Baltimore D, Van Parijs L. Essential role for STAT5 signaling in CD25+CD4+ regulatory T cell homeostasis and the maintenance of self-tolerance. J Immunol. 2003;171:3435–3441. doi: 10.4049/jimmunol.171.7.3435. [DOI] [PubMed] [Google Scholar]
- 18.Malek T, Yu A, Vincek V, Scibelli P, Kong L. CD4 Regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice: implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 19.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol. 2004;4:665–674. doi: 10.1038/nri1435. [DOI] [PubMed] [Google Scholar]
- 20.Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172:6519–6523. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- 21.Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J Exp Med. 2002;196:851–857. doi: 10.1084/jem.20020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bevan MJ. Helping the CD8+ T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 23.Kundig TM, Schorle H, Bachmann MF, Hengartner H, Zinkernagel RM, Horak I. Immune responses in interleukin-2-deficient mice. Science. 1993;262:1059–1061. doi: 10.1126/science.8235625. [DOI] [PubMed] [Google Scholar]
- 24.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 26.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 27.Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, Fein S, Schoenberger S, Levitsky HI. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med. 1999;5:780–787. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- 28.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, De Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leung DT, Morefield S, Willerford DM. Regulation of lymphoid homeostasis by IL-2 receptor signals in vivo. J Immunol. 2000;164:3527–3534. doi: 10.4049/jimmunol.164.7.3527. [DOI] [PubMed] [Google Scholar]
- 33.Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2Rα and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169:4850–4860. doi: 10.4049/jimmunol.169.9.4850. [DOI] [PubMed] [Google Scholar]
- 34.McHugh RS, Whitters MJ, Piccirillo C, Young DA, Shevach EM, Collins M, Byrne MC. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–309. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4+CD25+ suppressor T cells in vivo. Nat Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]
- 37.Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25− T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol. 2004;173:7259–7268. doi: 10.4049/jimmunol.173.12.7259. [DOI] [PubMed] [Google Scholar]
- 38.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang S, Alard P, Zhao Y, Parnell S, Clark SL, Kosiewicz MM. Conversion of CD4+ CD25− cells into CD4+CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J Exp Med. 2005;201:127–137. doi: 10.1084/jem.20041201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Granucci F, Vizzardelli C, Pavelka N, Feau S, Persico M, Virzi E, Rescigno M, Moro G, Ricciardi-Castagnoli P. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat Immunol. 2001;2:882–888. doi: 10.1038/ni0901-882. [DOI] [PubMed] [Google Scholar]
- 41.Teague RM, Tempero RM, Thomas S, Murali-Krishna K, Nelson BH. Proliferation and differentiation of CD8+ T cells in the absence of IL-2/15 receptor β-chain expression or STAT5 activation. J Immunol. 2004;173:3131–3139. doi: 10.4049/jimmunol.173.5.3131. [DOI] [PubMed] [Google Scholar]
- 42.D’Souza WN, Lefrancois L. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J Immunol. 2003;171:5727–5735. doi: 10.4049/jimmunol.171.11.5727. [DOI] [PubMed] [Google Scholar]
- 43.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thornton AM, Piccirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol. 2004;34:366–376. doi: 10.1002/eji.200324455. [DOI] [PubMed] [Google Scholar]
- 46.Gattinoni L, Klebanoff CA, Finkelstein SE, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Depper JM, Leonard WJ, Drogula C, Kronke M, Waldmann TA, Greene WC. Interleukin 2 (IL-2) augments transcription of the IL-2 receptor gene. Proc Natl Acad Sci USA. 1985;82:4230–4234. doi: 10.1073/pnas.82.12.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skapenko A, Kalden JR, Lipsky PE, Schulze-Koops H. The IL-4 receptor α-chain-binding cytokines, IL-4 and IL-13, induce forkhead box P3-expressing CD25+CD4+ regulatory T cells from CD25−CD4+ precursors. J Immunol. 2005;175:6107–6116. doi: 10.4049/jimmunol.175.9.6107. [DOI] [PubMed] [Google Scholar]
- 49.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 50.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 51.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 52.Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, Long LM, Bernstein D, Hill BJ, Douek DC, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 53.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+CD25hiFoxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–2414. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 55.Paust S, Lu L, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc Natl Acad Sci USA. 2004;101:10398–10403. doi: 10.1073/pnas.0403342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De La Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2480–2488. doi: 10.1002/eji.200425274. [DOI] [PubMed] [Google Scholar]
- 57.Barthlott T, Moncrieffe H, Veldhoen M, Atkins CJ, Christensen J, O’Garra A, Stockinger B. CD25+CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int Immunol. 2005;17:279–288. doi: 10.1093/intimm/dxh207. [DOI] [PubMed] [Google Scholar]
- 58.Barthlott T, Kassiotis G, Stockinger B. T cell regulation as a side effect of homeostasis and competition. J Exp Med. 2003;197:451–460. doi: 10.1084/jem.20021387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, Khazaie K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc Natl Acad Sci USA. 2005;102:419–424. doi: 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–1786. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- 61.Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur J Immunol. 2000;30:1538–1543. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 62.Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 63.Knoechel B, Lohr J, Kahn E, Bluestone JA, Abbas AK. Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J Exp Med. 2005;202:1375–1386. doi: 10.1084/jem.20050855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kohm AP, McMahon JS, Podojil JR, Smith Begolka W, Degutes M, Kasprowicz DJ, Ziegler SF, Miller SD. Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J Immunol. 2006;176:3301–3305. doi: 10.4049/jimmunol.176.6.3301. [DOI] [PubMed] [Google Scholar]