

Abstract
Protein expression after delivery of plasmid DNA to the cell nucleus depends on the processes of transcription and translation. Cytotoxic gene-delivery systems may compromise these processes and limit protein expression. This situation is perhaps most prevalent in current nonviral polycationic gene-delivery systems in which the polycationic nature of the delivery system can lead to cytotoxicity. To approach the problem of creating nontoxic but effective gene-delivery systems, we hypothesized that by optimizing the balance between polymer cationic density with endosomal escape moieties, effective gene transfer with low cytotoxicity could be created. As a model system, we synthesized a series of polymers whose side-chain termini varied with respect to the balance of cationic centers and endosomal escape moieties. Specifically, by polymer-analogous amidation we conjugated imidazole groups to the ɛ-amines of polylysine in varying mole ratios (73.5 mol % imidazole, 82.5 mol % imidazole, and 86.5 mol % imidazole). The primary ɛ-amine terminus of polylysine served as a model for the cationic centers, whereas the imidazole groups served as a model for the endosomal escape moieties. These polymers condensed plasmid DNA into nanostructures <150 nm and possessed little cytotoxicity in vitro. Transfection efficiency, as measured by luciferase protein expression, increased with increasing imidazole content of the polymers in a nonlinear relationship. The polymer with the highest imidazole content (86.5 mol %) mediated the highest protein expression, with levels equal to those mediated by polyethylenimine, but with little to no cytotoxicity.
Protein expression after delivery of plasmid DNA to the cell nucleus depends on the processes of transcription and translation. Cytotoxic gene-delivery systems may compromise these processes and potentially limit protein expression. This situation is perhaps the most prevalent in the nonviral polycationic gene-delivery class in which the polycationic nature of the delivery system can lead to cytotoxicity (1–8). To drive gene therapy ultimately into the clinic, improved delivery systems, or vectors, must deliver DNA to the cell in a transcriptionally active form and must fulfill all regulatory agency mandates to be considered safe for use in humans. Gene-delivery vectors are classified routinely as viral or nonviral, and the advantages and disadvantages of each category are well documented (9). To deliver DNA successfully to the nucleus, vectors in both categories must overcome several physicochemical and cellular barriers including DNA condensation into nanometer-scale structures <150 nm to allow cellular internalization, escape of DNA degradation by nucleases, intranuclear localization, and separation of the DNA from the carrier (10). Gene-delivery systems that are able to overcome these barriers without eliciting adverse effects on both the organism and cellular levels have potential to be safe and effective vectors.
Polycations are a leading class of nonviral gene-delivery vehicles in part because of their molecular diversity that can be modified to fine tune their physicochemical properties. It was determined that through coulombic interactions, polycations, such as polylysine, can condense DNA into toroidal nanostructures (11). When properly formulated, the radius of gyration of these polycation–DNA complexes is <150 nm, effectively overcoming the first barrier to successful gene delivery.
The second barrier to gene delivery, nuclease degradation in the lysosomal compartment, has been the focus of many investigators. Important research has been moving toward the development of polycation-based gene-delivery systems (such as polylysine conjugates) designed to minimize nuclease degradation through the design of vectors with the capacity to escape the endosomal–lysosomal pathway (12–14). Behr and others introduced the concept of the “proton sponge” and hypothesized that polymers with buffering capacities between 7.2 and 5.0, such as polyethylenimine (PEI) and imidazole-containing polymers, could buffer the endosome and potentially induce its rupture (15–18). Protein-expression levels mediated by the polycationic proton-sponge polymer, PEI, were at least 10-fold greater than polylysine alone. However, one drawback of PEI is its inherent cytotoxicity.
Although polymers with high cationic density condense DNA into structures amenable to cellular internalization via endocytosis, the high charge density is also one factor that contributes to their cytotoxicity (1, 12, 19). On the other hand, low cationic-charge density can reduce or eliminate DNA condensation capability (20, 21). The balance between cationic density and DNA condensation is complicated further when conjugates with endosomal escape moieties are considered. It occurred to us that optimizing the balance between charged and endosomal-escape moieties could serve as the basis for the rational design and subsequent synthesis of a polycationic gene-delivery system with low cytotoxicity, DNA condensation capability, and endosomal-escape potential. Here we describe a model polycationic gene-delivery system to evaluate this hypothesis.
Experimental Procedures
Polylysine (Mr = 34,300, lot #4745546), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), and MES were purchased from Sigma. N-hydroxysuccinimide, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, 4-imidazoleacetic acid sodium salt dihydrate, and PEI (Mr = 25,000 by light scattering) were purchased from Aldrich and used without further purification. The HepG2 hepatoblastoma and CRL1476 smooth-muscle cell lines were purchased from the American Type Culture Collection and grown according to their respective ATCC protocols. P388D1 mouse macrophage cell line was a kind gift from Zycos (Cambridge, MA) and grown in RPMI medium 1640 and 10% (vol/vol) FBS with penicillin (100 units/ml) and streptomycin (100 μ g/ml) added. Cytomegalovirus (CMV)-luc was grown in STBL Escherichia coli (a kind gift from Zycos). Plasmids were isolated by using Qiagen Giga-preps according to the manufacturer's directions (Qiagen, Chatsworth, CA). Plasmids were purified by ethanol precipitation until their 260-nm:280-nm absorption ratios were 1.8 or greater. All plasmids used in transfection experiments contained greater than 90% supercoiled DNA.
Polymer Conjugate Synthesis.
Polylysine-graft imidazoleacetic acid was synthesized by amidation of the ɛ-amine of polylysine with 4-imidazoleacetic acid by using an 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide(EDAC)/N-hydroxysuccinimide (NHS) condensation system (22). The general synthetic procedure is described below for the 75 mol % substituted polymer, and the method was adapted easily for other percents of imidazole substitution. At room temperature and in a 15-ml polypropylene tube, 200 mg of polylysine (1.5625 × 10−3 mol ɛ-NH2) was dissolved in 4 ml of 25-mM MES (pH 6.5) buffer to which was added 75 mol % (173.5 mg, 1.17 × 10−3 mol) 4-imidazoleacetic acid (sodium salt, dihydrate). This solution was used to dissolve EDAC (449 mg, 2.34 × 10−3 mol). NHS (18.1 mg, 1.56 × 10−3 mol) was dissolved in 2 ml of MES buffer and was added immediately to the polylysine solution. The reaction tube was sealed and inverted for 24 h at room temperature, after which the reaction solution was added to a Centriprep filtration unit (molecular weight cut-off 10,000). The Centriprep units were centrifuged at 4,500 RPM for 2 h to separate the polymer from low-molecular-weight components. Distilled water was added to the polymer solution after each centrifugation. This process of centrifugation and polymer-fraction dilution was repeated a total of 10 times, after which the polymer solution was frozen and the water removed by lyophilization.
Characterization.
The imidazole content of each polymer synthesized was determined through quantitation of the free amines remaining on the polymer by using the well characterized ninhydrin method (23, 24), followed by subtraction of the amine signal.
Gel Retardation Assay.
DNA–polymer complexes were formed by mixing 50 μl of plasmid DNA (200 μg/ml CMV-luc in 10 mM Hepes buffer, pH 7.2) with 50 μl of polymer solution containing varying amounts of polymer. Complexes were allowed to form for 20 min at room temperature, after which 20 μl was run on a 1% agarose gel (90 V, 1 h). Bands were visualized with ethidium-bromide staining.
Complex Size and Zeta-Potential Measurement.
DNA–polymer complexes were formed in 1.5-ml Eppendorf tubes by adding 50 μl of plasmid DNA (200 μg/ml CMV-luc in 10 mM Hepes buffer, pH 7.2) to 50 μl of vortexing polymer solution containing varying amounts of polymer. Complexes were allowed to form for 15 min at room temperature. The complexes then were diluted to 1 ml with filtered Hepes buffer. The size and zeta potentials of the complexes were measured at 25°C by using a ZetaPALS dynamic light-scattering detector (Brookhaven Instruments, Holtsville, NY) with a 15-mW laser and incident laser beam at 676 nm. Correlation functions were collected at a scattering angle of 90°, and particle sizes were calculated by using the multiangle sizing option of the instrument's particle-sizing software (version 2.30). The particle sizes are expressed as effective diameters assuming a lognormal distribution. Zeta potentials were calculated by using the Smoluchowsky model for aqueous suspensions.
Cytotoxicity Assay.
Cytotoxicity was evaluated by using both the MTT assay (25) as well as total-cell count. Cells were grown in 96-well plates at an initial density of 10,000 cells per well in 0.2 ml of growth medium for 24 h, after which the growth medium was removed and replaced with 0.18 ml OPTI-MEM I (GIBCO/BRL). Then polymer in 20 μl of Hepes buffer was added to the cells (Hepes without polymer was added to control wells). After a 4-h incubation at 37°C, the polymer containing medium was removed and replaced with working medium. The metabolic activity of the cells was measured 24 h later by using the MTT assay. Briefly, 25 μl of a 5-mg/ml MTT stock in sterile PBS was added to each well. After incubating the cells in the presence of MTT for 2 h, 100 μl of extraction buffer [20% wt/vol SDS in DMF/water (1:1), pH = 4.7] was added to each well, and the plate was incubated at 37°C overnight. The optical densities of each well were measured at 560 nm by using a microplate reader (MR5000, Dynatech) and expressed as a percentage relative to control cells. In all experiments, cell counting paralleled the trends generated by the MTT assay.
Transfection Experiments.
Cells were grown in 6-well plates at an initial density of 1 × 105 to 3 × 105 cells per well in 2 ml of working medium to be 60–70% confluent at the time of transfection, which was 24 h after plating. DNA–polymer complexes were formed by adding 50 μl of DNA (200 μg/ml CMV-luc in 10 mM Hepes buffer, pH 7.2) to 50 μl of polymer (containing varying amounts of polymer) while vortexing. Each sample of complex contained 10 μg of DNA and was incubated at room temperature for 20 min to complete complex formation. The cell-growth medium was removed and replaced with OPTI-MEM just before the complexes were added. After incubating the cells with the complexes for 4 h at 37°C in humidified 5% CO2 atmosphere, the complex-containing medium was removed and replaced with 2 ml of growth medium. After 24 h, cell lysates were formed and analyzed for luciferase activity (by using a luciferase assay kit, Promega) and total-protein content (BCA assay, Pierce).
Results
Polymer Synthesis.
Polylysine-graft-imidazoleacetic acid conjugates substituted with imidazole groups in three different mole percentages (75, 85, and 95 input mol %) were synthesized to give insight into the possible effect of the amine/imidazole ratio on DNA condensation, cytotoxicity, and transfection efficiency. The conjugates were made in a one-step reaction shown schematically in Fig. 1. The conjugation of 4-imidazoleacetic acid to the polylysine was nearly quantitative with the input molar ratios with the exception of the 95 mol % feed ratio, which had a final imidazoleacetic acid content of 86.5 mol %. The polymers are designated “1,” “2,” and “3” containing 73.5, 82.5, and 86.5 mol % imidazole substitution, respectively.
Figure 1.

Synthetic route to polylysine-graft-imidazole acetic acid. Numbering corresponds to the mole percentage of imidazole on each polymer.
Each amidation effectively replaces an ɛ-amine of lysine with an imidazole functional group, resulting in the randomization of cationic charges along the polymer as well as an overall decrease in cationic density of each polymer chain at pH 7.2. It is this replacement of the lysine ɛ-amine with the imidazole functionality that will help determine the structure/function relationships that exist in the system.
Characterization of Polymer–DNA Complexes.
Of initial importance was to determine whether the imidazole-containing conjugates could complex electrostatically with and condense plasmid DNA. Reduction of the polycationic density may decrease the DNA-condensation capacity of the polymer (26). Also, reports of DNA delivery with polylysine show that transfection efficiency decreases with a decrease in the degree of polymerization (27, 28). On the other hand, some investigators have demonstrated that low-molecular-weight polylysine can condense DNA into small (≈30-nm) complexes (29), whereas others have demonstrated increasing DNA transcriptional activity with decreasing polylysine molecular weight (30). With these somewhat disparate reports in the literature, it was difficult to predict the biophysical interactions between DNA and the imidazole-containing polymers, particularly with respect to DNA condensation. Previously, we determined that carbohydrate-modified polyhistidine alone was unable to condense DNA into structures less than 150 nm, and that ternary complexation with polylysine was necessary to condense the DNA adequately (17). In contrast, the polymers reported herein contain cationic centers built into the polymer structure to introduce condensation properties into the polymer.
As an initial characterization of the complexes, the electrostatic interaction between plasmid DNA and the polymers as a function of both polymer imidazole content and DNA:polymer ratio was determined by gel electrophoretic retardation (31). The results in Fig. 2 show that polymers 1–3 electrostatically neutralize plasmid DNA and eliminate its electrophoretic mobility.
Figure 2.

Electrophoretic retardation of DNA with polylysine and polymers 1, 2, and 3. Lane assignments correspond to DNA/polymer weight ratios and are as follows with DNA/polymer weight ratios in parenthesis: lane A (1:0) without vortex; lane B (1:0) with vortex; lane C (1:0.1); lane D (1:0.2); lane E (1:0.5); lane F (1:1); lane G (1:2); lane H (1:3); lane I (1:4); lane J (1:5); lane K (1:7.5); and lane L (1:10).
To determine the size distribution of the complexes, dynamic-laser light scattering was used. As Fig. 3 shows, all of the imidazole-containing polymers condensed DNA into structures below 150 nm, a size necessary for efficient cellular uptake (32), and the complex sizes are a function of the DNA/polymer ratio. At polymer-cation concentrations exceeding DNA-anion concentrations, the light-scattering signal was strong and nonfluctuating for each of the DNA/polymer ratios tested, suggesting that the complexes were discrete particles.
Figure 3.

Effective solution diameter of DNA–polymer complexes with polylysine and polymers 1, 2, and 3 (n = 3 ± SD).
The zeta potentials of the complexes formed between polymers 1–3 are shown in Fig. 4. The trend of initially negative zeta-potential complexes becoming positively charged with increasing polycation content is typical for DNA–polycation complexes (20). These zeta-potential results correlate closely with trends observed in both the electrophoretic-mobility and particle-size analysis. The DNA/polymer wt:wt ratio at which the zeta potential of the imidazole-containing complexes becomes positive is larger than the ratio at which the polylysine complexes become positive, which is consistent with conjugation of the imidazole groups to the ɛ-amine of the polylysine.
Figure 4.

Zeta potential of DNA–polymer complexes with polylysine and polymers 1, 2, and 3 (n = 3 ± SD).
Cytotoxicity.
The in vitro cytotoxicity of the polylysine-graft-imidazoleacetic acid polymers was measured as a function of polymer concentration by using the MTT assay (25). Results from the CRL 1476 cell line in Fig. 5 show that for both polylysine and PEI, cytotoxicity increases with increasing polymer concentration, with greater than half the cell population metabolically inactive at the highest polymer concentration (55 μg/ml). In contrast, cells incubated with polymers 1–3 retained greater than 80% of their metabolic activity. The results are consistent among all of the cell lines tested (CRL 1476 smooth-muscle cells, P388D1 macrophages, and HepG2 hepatoblastoma, data not shown for the latter two). Because the MTT assay is a measure only of cell metabolic activity, similar cytotoxicity experiments were conducted, and cell viability was determined by direct cell-count analysis. The data obtained by using the MTT assay directly correlate to the data obtained by using cell-count analysis (data not shown).
Figure 5.

In vitro metabolic activity of CRL 1476 smooth-muscle cells as a function of polymer concentration (n = 3 ± SD).
In Vitro Transfection of Model Cell Lines.
Fig. 6 shows the luciferase-expression levels mediated by polymer 3 as a function of DNA/polymer ratio. The goal of the experiment was to compare the maximal protein-expression capacity of the imidazole-containing polymers in different cell lineages. Three model cell lines were used to evaluate the transfection efficiency of the imidazole-containing polymers. Each cell line was chosen to approximate target cell types of potential clinical significance. The HepG2 hepatoblastoma cell line was used to approximate hepatocytes to mimic liver transfection for diseases such as hemophilia, the P388D1 macrophage cell line was used to approximate transfection of antigen-presenting cells for DNA-based vaccines, and the CRL 1476 muscle cell line was used to mimic muscle transfection after intramuscular injection. In addition to their relationship to potential in vivo-cell targets, the differences in the cell lineages allow the determination of generalized polymer effects on transfection rather than isolated events specific for one type of cell. Luciferase expression mediated by DNA delivery with PEI was determined also in the same experiment to directly compare the luciferase expression levels mediated by the two polymer types.
Figure 6.

Comparison of luciferase expression mediated by both polymer 3 and PEI in different cell lines. (A) CRL 1476 smooth muscle; (B) P388D1 macrophage; (C) HepG2 hepatoblastoma (n = 3 ± SD).
Whereas Fig. 6 shows the influence of DNA/polymer ratio on protein expression, Fig. 7A shows luciferase expression as a function of the primary amine/imidazole group balance. At each DNA/polymer ratio tested, luciferase expression increased with increasing imidazole content of the polymers. The cell line used to demonstrate this trend in Fig. 7A is HepG2, but it is a trend apparent with the other cell types tested (P388D1, CRL 1476; data not shown). From Fig. 7 it is clear that the polymer containing 86.5 mol % imidazole-group substitution (polymer 3) produces maximal protein expression; therefore, this polymer was used to generate the data in Fig. 6 to compare the maximum protein-expression levels.
Figure 7.

Luciferase expression measured in light units as a function of imidazole content and DNA/polymer weight ratio (A) and by polymer 3 and PEI at equal zeta potentials (B) (n = 3 ± standard deviation).
Nonspecific interactions exist between the positively charged DNA–polymer complexes and negatively charged cell surface potentially leading to differences in transfection efficiency between polymer types. To normalize for nonspecific interactions and allow direct comparison between the imidazole-containing polymers and PEI, transfections were conducted by using complexes with equal zeta potentials. The results are shown in Fig. 7B. In general, protein expression mediated by polymers 3 and PEI are equal, with slight differences depending on cell type.
Discussion
Approximately 400 gene-therapy clinical trials are underway worldwide (see http://www.wiley.co.uk/genetherapy/clinical/countries.html). A key component to the outcome of these trials is the effectiveness of the delivery system. Clinical utility of a gene-delivery system requires, at a minimum, a threshold level of protein expression at an acceptable level of safety. Approximately one-fourth of the clinical trials currently underway use nonviral means of DNA delivery, and an excellent review article profiles these systems in greater detail (33). In general, synthetic DNA-delivery systems have lower delivery efficiency than viruses but surpass viral vectors in terms of safety.
In this report, we evaluate the hypothesis that the optimization of the balance between polymer cationic density with endosomal-escape moieties can lead to effective gene transfer with low cytotoxicity. We describe the synthesis of an imidazole-containing polycation as a model to evaluate the hypothesis. What makes the polymer unique is that it delivers the DNA without adversely influencing the metabolic activity of the cell as determined by the MTT assay and corroborated by cell-count analysis. The polymer, polylysine-graft-imidazoleacetic acid (Fig. 1), is designed to balance the relative contributions of cationic centers (primary-amine groups) with pH-sensitive centers (imidazole groups) to control their collective effects on DNA complexation, condensation, endosomal escape, and transfer to the nucleus. High cationic density is one approach to condense DNA into structures less than 150 nm to allow efficient cell uptake through endocytosis. Once internalized, the complex is trafficked through the endosomal pathway, and ultimately the complex resides within the lysosome (34). Because the lysosomal compartment contains nucleases that can degrade and inactivate DNA, enhancing DNA escape from the endosomal pathway is one mechanism through which the transfection efficiency of a delivery system can be increased.
Polymers that have a buffering capacity between pH 7.2 and 5.0, such as PEI, are hypothesized to mediate escape from the endosomal pathway through the proton-sponge effect where, after acidification of the endosome, the vesicle is ruptured by an increased osmotic pressure, by direct membrane perturbation, or by mechanical swelling of the membrane-limited organelle (35–37). Although cells are transfected successfully with PEI-based complexes and express high levels of protein, one disadvantage of PEI is its cytotoxicity (31). Our approach to the design and synthesis of a high-efficiency DNA-delivery system with low cytotoxicity was to mimic the hypothesized mechanism of action of PEI, but do so with biocompatible functional groups to minimize cytotoxicity. The imidazole functionality, which is the principal side-chain component of histidine, has an apparent pKa of 6.5, and as such, polymers containing this group may serve as a basis for the design of biocompatible molecularly engineered materials with high gene-transfer efficiency. It should be noted that our endpoint for analysis of the model polymer is protein expression. The careful elucidation of the mechanism of action of the imidazole group is not the focus of this paper and is yet to be investigated.
Varying models exist to help explain the parameters that govern the condensation of DNA via polycationic species (for review see ref. 38). In general, discrete polycation–polyanion complexes are formed spontaneously in aqueous solutions by ionic bonds between the polymers. Cooperativity of the ionic bonds depends on the density of the charge repeat units on each polymer with a minimum of 6 bonds, also called the lower critical chain length, required for a cooperative system (39–41). Given the randomized amidation of the imidazole on the polylysine in polymers 1–3, it is unlikely that the repeat cationic units are spaced equally throughout the polymer, potentially reducing the number of long cationic repeat units among the polymer side chains, thus reducing cooperativity between the polymer and DNA. However, polymers 1–3 form nanoscale structures with DNA, suggesting that there is sufficient ionic-bond cooperativity between the polymers and DNA. One potential reason for these results is that the imidazole group also may contribute to the condensation of DNA. By using the equation reported by Wang and Huang (42) for the protonation state of a polycation at a specific pH:
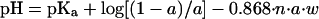 |
where pKa = intrinsic pK of the protonatable moiety (pKa = 6.5 for imidazole), n = the average number of protonatable moieties per polymer chain (n = 231 for polymer 3), w = an electrostatic interaction factor (w = 0.0575 as per ref. 42), and a = fraction of protonated moieties. Solving for a in an iterative fashion, approximately 5% of the imidazole groups on the polymer are protonated at pH 7.2 and are available potentially to assist in the condensation of DNA by polymers 1–3.
The mechanisms by which polycations induce cytotoxicity are understood poorly. There are examples reported in the literature of both biochemically and biophysically induced cytotoxicity of polycations, such as polylysine (1–8). The mechanism(s) by which conjugation of imidazole groups to polylysine decreases cytotoxicity could be investigated further.
The balance between ɛ-amine and imidazole side chains on the polymer directly influences the protein expressed by the model cell lines with a general trend that protein expression increases with increasing imidazole content. Interestingly, with a 9% increase in polymer imidazole content (from 73.5 to 82.5 mol %), the level of luciferase expression approximately doubles at each DNA/polymer ratio. However, with a further 4% increase in imidazole content (from 82.5 to 86.5 mol %), luciferase expression levels approximately double again. These results suggest a nonlinear relationship between polymer imidazole content and protein expression.
The results from this model system may help to design future materials for gene delivery with the ultimate goal to create gene-delivery systems with robust transfection efficiencies suitable for a range of clinical applications.
Acknowledgments
We thank Umaima Marvi, Dave Ting, and Dave Nguyen for their technical help. This research was supported in part by a National Research Service Award from the National Institutes of Health (NIH) to D.P., an NIH training grant to C.G. from the Joint Harvard–Massachusetts Institute of Technology (MIT) Program in Biomaterials administered by the Harvard School of Dental Medicine, NIH Grant GM-26698, National Science Foundation funding for the MIT Biotechnology Process Engineering Center, and through the Center for Innovative Minimally Invasive Therapy (DOD Grant no. DAMD17-99-2-9-001).
Abbreviations
- PEI
polyethylenimine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- CMV
cytomegalovirus
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.031577698.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.031577698
References
- 1.Elferink J G R. Inflammation. 1985;9:321–331. doi: 10.1007/BF00916280. [DOI] [PubMed] [Google Scholar]
- 2.Elfrink J G R. Inflammation. 1991;15:103–115. doi: 10.1007/BF00917505. [DOI] [PubMed] [Google Scholar]
- 3.Antonelli M, Olate J, Allende C C, Allende J E. Comp Biochem Physiol B Biochem Mol Biol. 1991;99:827–832. doi: 10.1016/0305-0491(91)90149-8. [DOI] [PubMed] [Google Scholar]
- 4.Gatica M, Allende C C, Antonelli M, Allende J E. Proc Natl Acad Sci USA. 1987;84:324–328. doi: 10.1073/pnas.84.2.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Cruijff B, Rietveld A, Telders N, Vaandrager B. Biochim Biophys Acta. 1985;820:295–304. doi: 10.1016/0005-2736(85)90124-5. [DOI] [PubMed] [Google Scholar]
- 6.Kleinschmidt J H, Marsh D. Biophys J. 1997;73:2546–2555. doi: 10.1016/S0006-3495(97)78283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gad A E, Silver B L, Eytan G D. Biochim Biophys Acta. 1982;690:124–132. doi: 10.1016/0005-2736(82)90246-2. [DOI] [PubMed] [Google Scholar]
- 8.Kato T, Lee S, Ono S, Agawa Y, Aoyagi H, Ohno M, Nishino N. Biochim Biophys Acta. 1991;1063:191–196. doi: 10.1016/0005-2736(91)90370-n. [DOI] [PubMed] [Google Scholar]
- 9.Lehn P, Fabrega S, Oudrhiri N, Navarro J. Adv Drug Delivery Rev. 1998;30:5–11. doi: 10.1016/s0169-409x(97)00102-6. [DOI] [PubMed] [Google Scholar]
- 10.Pouton C W, Seymour L W. Adv Drug Delivery Rev. 1998;34:3–19. doi: 10.1016/s0169-409x(98)00048-9. [DOI] [PubMed] [Google Scholar]
- 11.Haynes M, Garrett R A, Gratzer W B. Biochemistry. 1970;9:4410–4416. doi: 10.1021/bi00824a600. [DOI] [PubMed] [Google Scholar]
- 12.Plank C, Zatloukal K, Cotton M, Mechtler K, Wagner E. Bioconjugate Chem. 1992;3:533–539. doi: 10.1021/bc00018a012. [DOI] [PubMed] [Google Scholar]
- 13.Plank C, Oberhauser B, Mechtler K, Koch C, Wagner E. J Biol Chem. 1994;269:12918–12924. [PubMed] [Google Scholar]
- 14.Wagner E, Plank C, Zatloukal K, Cotton M, Birnstiel M L. Proc Natl Acad Sci USA. 1992;89:7934–7938. doi: 10.1073/pnas.89.17.7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behr J-P. Chimica. 1997;51:34–36. [Google Scholar]
- 16.Boussif O, Lezoualc'h F, Zanta M A, Mergny M D, Scherman D, Demeneix B, Behr J-P. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pack D, Putnam D, Langer R. Biotechnol Bioeng. 2000;67:217–223. [PubMed] [Google Scholar]
- 18.Midoux P, Monsigny M. Bioconjugate Chem. 1999;10:406–411. doi: 10.1021/bc9801070. [DOI] [PubMed] [Google Scholar]
- 19.Clark R A, Olsson I, Klebanoff S J. J Cell Biol. 1976;70:719–723. doi: 10.1083/jcb.70.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolfert M A, Seymour L W. Gene Ther. 1996;3:269–273. [PubMed] [Google Scholar]
- 21.Wadhwa M S, Collard W T, Adami R C, McKenzie D L, Rice K G. Bioconjugate Chem. 1997;8:81–88. doi: 10.1021/bc960079q. [DOI] [PubMed] [Google Scholar]
- 22.Sehgal D, Vijay I K. Anal Biochem. 1994;218:87–91. doi: 10.1006/abio.1994.1144. [DOI] [PubMed] [Google Scholar]
- 23.Moore S. J Biol Chem. 1968;243:6281–6283. [PubMed] [Google Scholar]
- 24.Yeh P-Y, Kopeckova P, Kopecek J. J Polym Sci Pt A Polym Chem. 1994;32:1627–1637. [Google Scholar]
- 25.Hansen M B, Neilsen S E, Berg K. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 26.Plank C, Tang M X, Wolfe A R, Szoka F C., Jr Hum Gene Ther. 1999;10:319–332. doi: 10.1089/10430349950019101. [DOI] [PubMed] [Google Scholar]
- 27.Taxman D J, Lee E S, Mojchowski D M. Anal Biochem. 1993;213:97–103. doi: 10.1006/abio.1993.1391. [DOI] [PubMed] [Google Scholar]
- 28.Wagner E, Zenke M, Cotton M, Beug H, Birnstiel M L. Proc Natl Acad Sci USA. 1990;87:3410–3414. doi: 10.1073/pnas.87.9.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfert M A, Seymour L W. Gene Ther. 1996;3:269–273. [PubMed] [Google Scholar]
- 30.Schaffer D V, Fidelman N A, Dan N, Lauffenburger D A. Biotechnol Bioeng. 2000;67:598–606. doi: 10.1002/(sici)1097-0290(20000305)67:5<598::aid-bit10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 31.Putnam D, Langer R. Macromolecules. 1999;32:3658–3662. [Google Scholar]
- 32.Guy J, Drabek D, Antoniou M. Mol Biotechnol. 1995;3:237–248. doi: 10.1007/BF02789334. [DOI] [PubMed] [Google Scholar]
- 33.Luo D, Saltzman W M. Nat Biotechnol. 2000;18:33–37. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- 34.Felgner J H, Barenholtz Y, Behr J-P, Cheng S H, Cullis P, Huang L, Jessee J A, Seymour L, Szoka F, Thierry A R, et al. Hum Gene Ther. 1997;8:511–512. doi: 10.1089/hum.1997.8.5-511. [DOI] [PubMed] [Google Scholar]
- 35.Behr J-P. Chimia. 1997;51:34–36. [Google Scholar]
- 36.Godbey W T, Wu K K, Mikos A G. J Controlled Release. 1999;60:149–160. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- 37.Haensler J J, Szoka F C., Jr Bioconjugate Chem. 1993;4:372–379. doi: 10.1021/bc00023a012. [DOI] [PubMed] [Google Scholar]
- 38.Tang M X, Szoka F C., Jr . In: Self-Assembling Complexes for Gene Delivery. Kabanov A V, Felgner P L, Seymour L W, editors. New York: Wiley; 1998. pp. 27–50. [Google Scholar]
- 39.Kabanov A V, Astafieva I V, Chikindas M L, Rosenblat G F, Kiselev V I, Severin E S, Kabanov VA. Biopolymers. 1991;31:1437–1443. doi: 10.1002/bip.360311210. [DOI] [PubMed] [Google Scholar]
- 40.Papisov I M, Litmanovich A A. Adv Polym Sci. 1988;90:139–179. [Google Scholar]
- 41.Harada K, Kataoka K. J Macromol Sci, Chem. 1997;34:2119–2133. [Google Scholar]
- 42.Wang C Y, Huang L. Biochemistry. 1984;23:4409–4416. doi: 10.1021/bi00314a026. [DOI] [PubMed] [Google Scholar]